
Triphenylphosphonium Analogs of Short Peptide Related to Bactenecin 7 and Oncocin 112 as Antimicrobial Agents
 , , , , , , , , , ,
, , , , , , , , , ,
Abstract
:1. Introduction

2. Materials and Methods
2.1. Chemicals and Materials
2.2. Chromatography
2.3. Liquid Chromatography–Mass Spectrometry
2.4. Mass Spectrometry
2.5. 1H and 13C NMR
2.6. Molecular Modeling
2.7. Synthetic Procedures
2.8. In Vitro Binding Assay
2.9. In Vitro Translation Inhibition Assays
2.10. Bacterial Inhibition Assays
2.10.1. Detection of Translation Inhibitors Using the pDualrep2 Reporter Strain
2.10.2. Antibacterial Activity of Substances on Agar Plates
2.10.3. Minimum Inhibitory Concentration (MIC) Determination
2.11. Determination of Membrane Potential in B. subtilis
2.12. In Vitro Survival Assay (MTT Assay)
3. Results and Discussion
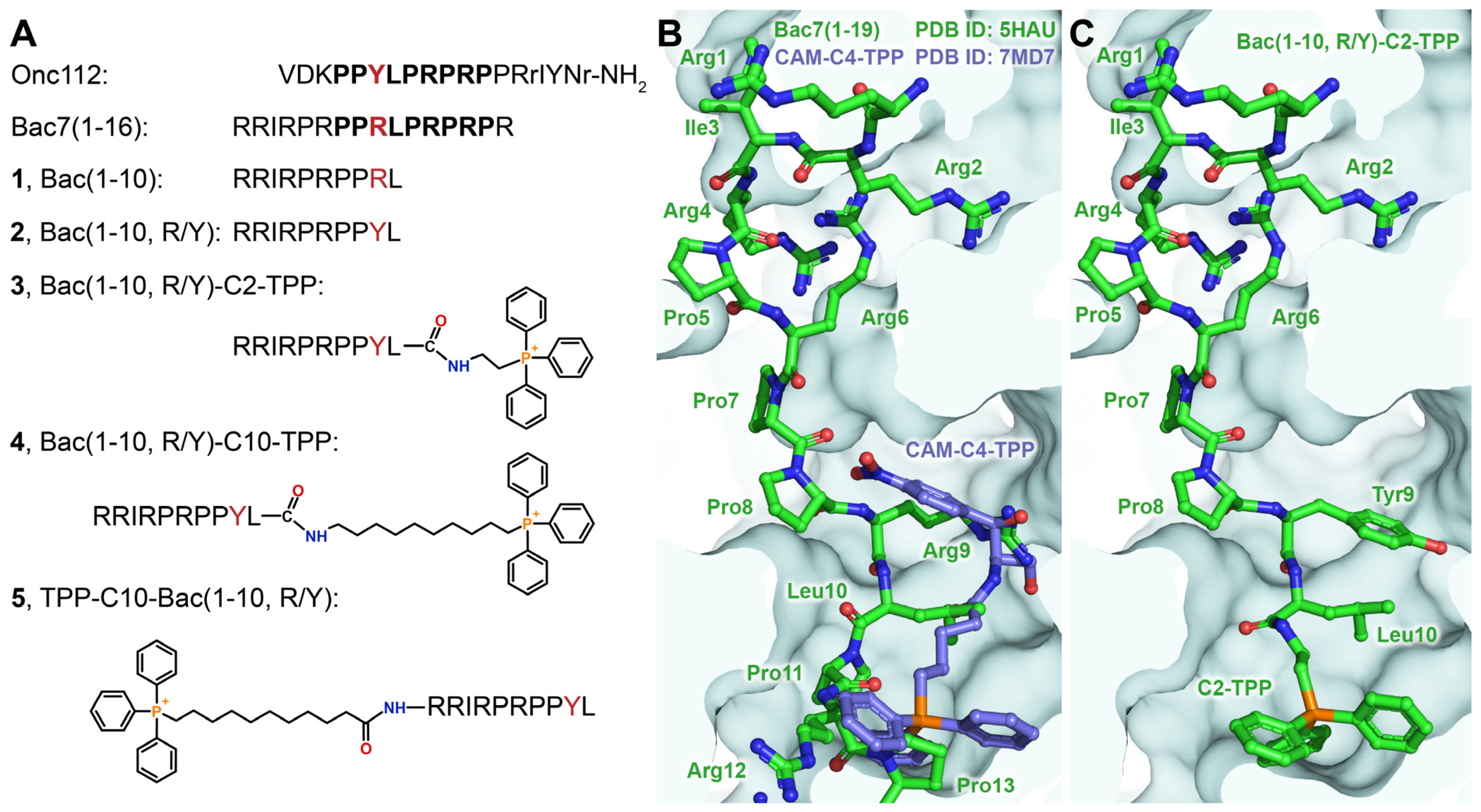
3.1. Modeling of TPP Analogs of Bac7
3.2. Synthesis of TPP Analogs of Bac7
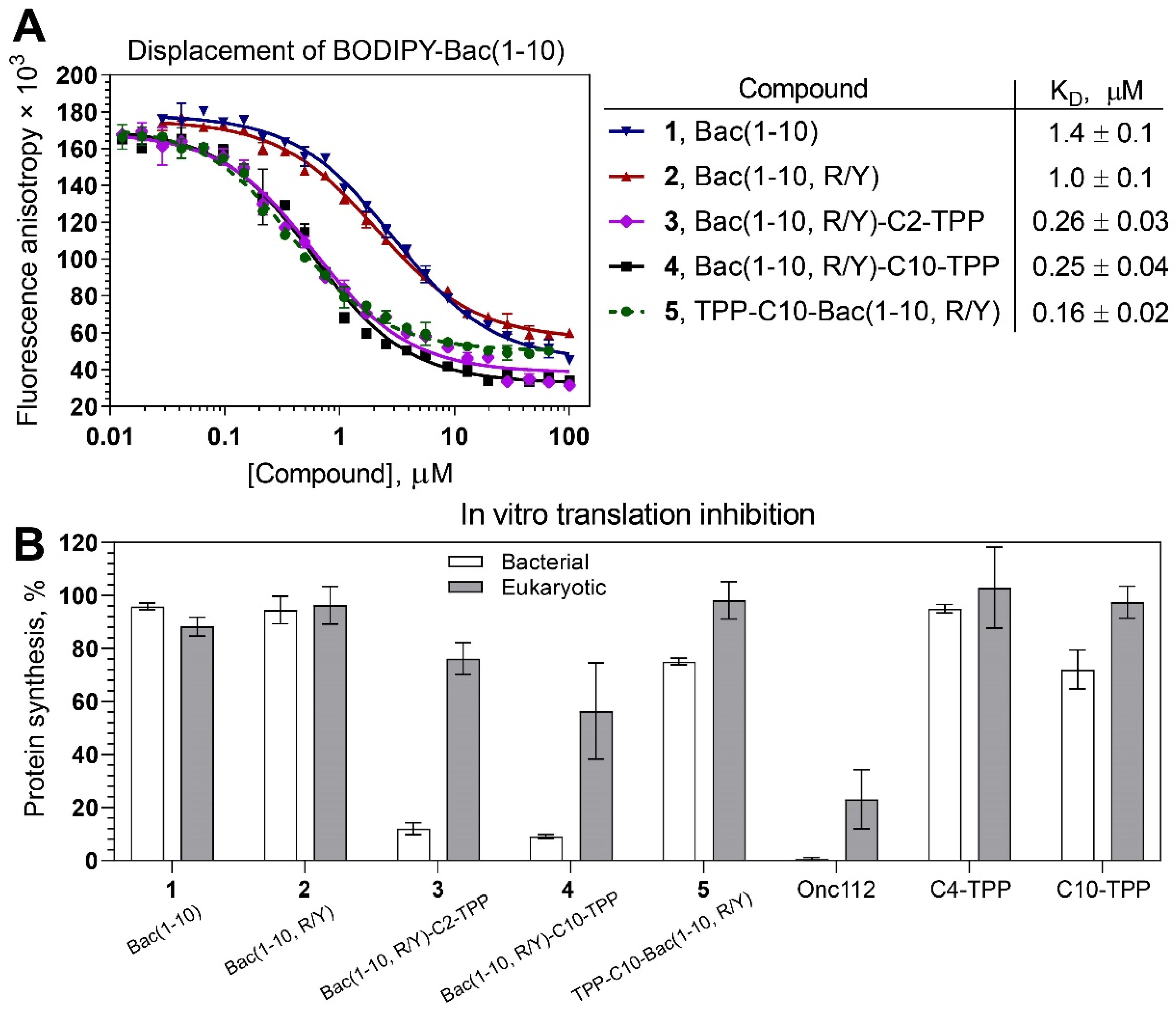
3.3. TPP Derivatives of Decapeptide from N-Terminal Sequence of Bac7 Bind to the Bacterial Ribosome
3.4. Bac(1–10, R/Y)-C2-TPP and Bac(1–10, R/Y)-C10-TPP Selectively Inhibit Prokaryotic Translation
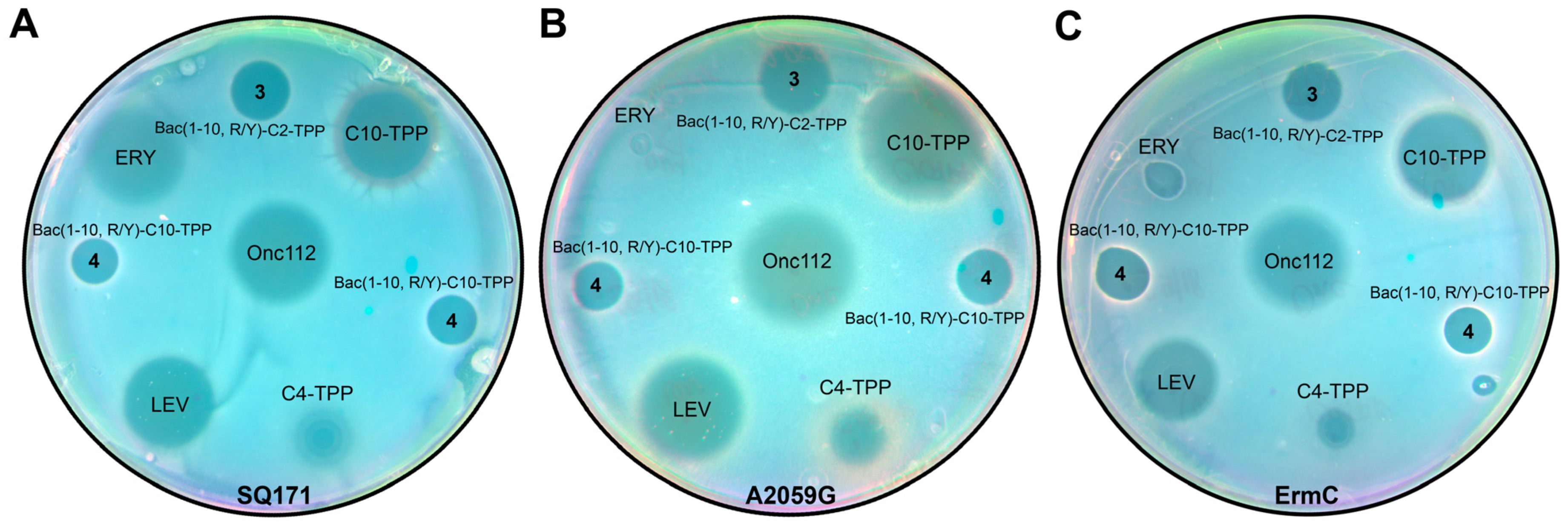
3.5. TPP Derivatives of Decapeptide Related to Bac7 and Onc112 Exhibit Antibacterial Activity against Various Strains
3.5.1. Using the Double-Reporter System pDualrep2 for Preliminary Assessment of the Mechanism of Action
3.5.2. TPP Derivatives of Decapeptide from N-terminal Sequence of Bac7 Exhibit Antibacterial Activity against Various Strains including Some Resistant Laboratory Strains
3.5.3. Bac(1–10, R/Y)-C2-TPP Penetrates into the E. coli Cells via the SbmA Transporter Protein
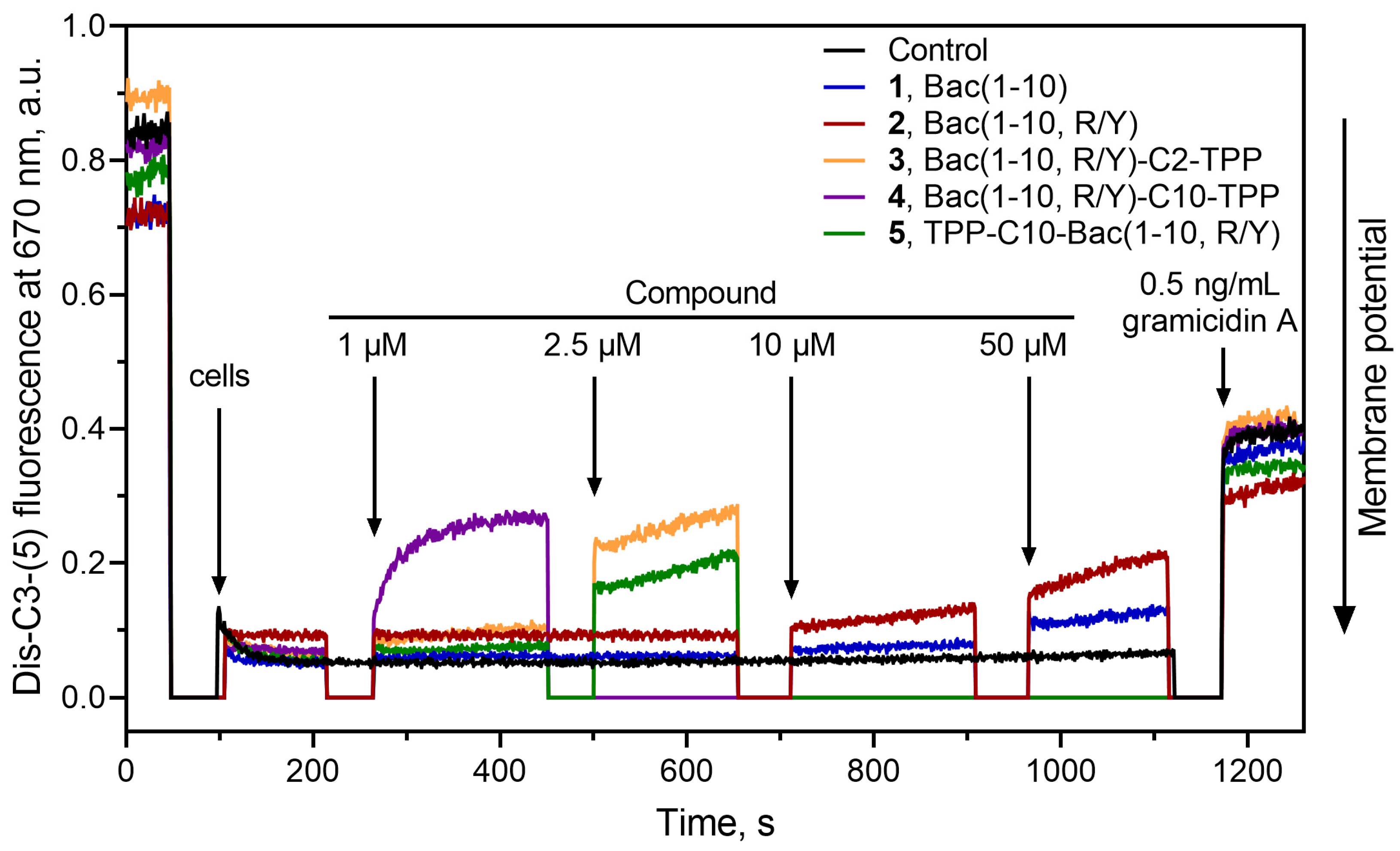
3.6. Bac(1–10, R/Y)-Cn-TPP Cause a Decrease in the Membrane Potential of B. subtilis
3.7. Bac(1–10, R/Y)-C2-TPP Is Non-Toxic for Mammalian Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mahlapuu, M.; Björn, C.; Ekblom, J. Antimicrobial Peptides as Therapeutic Agents: Opportunities and Challenges. Crit. Rev. Biotechnol. 2020, 40, 978–992. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kizhakkedathu, J.; Straus, S. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility in Vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Otvos, L. The short proline-rich antibacterial peptide family. Cell. Mol. Life Sci. 2002, 59, 1138–1150. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Tailhades, J.; O’Brien-Simpson, N.; Separovic, F.; Otvos, L., Jr.; Hossain, M.; Wade, J. Proline-rich antimicrobial peptides: Potential therapeutics against antibiotic-resistant bacteria. Amino Acids 2014, 46, 2287–2294. [Google Scholar] [CrossRef] [PubMed]
- Graf, M.; Mardirossian, M.; Nguyen, F.; Seefeldt, A.C.; Guichard, G.; Scocchi, M.; Innis, C.A.; Wilson, D.N. Proline-Rich Antimicrobial Peptides Targeting Protein Synthesis. Nat. Prod. Rep. 2017, 34, 702–711. [Google Scholar] [CrossRef]
- Scocchi, M.; Tossi, A.; Gennaro, R. Proline-Rich Antimicrobial Peptides: Converging to a Non-Lytic Mechanism of Action. Cell. Mol. Life Sci. 2011, 68, 2317–2330. [Google Scholar] [CrossRef]
- Polikanov, Y.S.; Aleksashin, N.A.; Beckert, B.; Wilson, D.N. The Mechanisms of Action of Ribosome-Targeting Peptide Antibiotics. Front. Mol. Biosci. 2018, 5, 48. [Google Scholar] [CrossRef]
- Mardirossian, M.; Grzela, R.; Giglione, C.; Meinnel, T.; Gennaro, R.; Mergaert, P.; Scocchi, M. The Host Antimicrobial Peptide Bac71-35 Binds to Bacterial Ribosomal Proteins and Inhibits Protein Synthesis. Chem. Biol. 2014, 21, 1639–1647. [Google Scholar] [CrossRef]
- Krizsan, A.; Volke, D.; Weinert, S.; Sträter, N.; Knappe, D.; Hoffmann, R. Insect-Derived Proline-Rich Antimicrobial Peptides Kill Bacteria by Inhibiting Bacterial Protein Translation at the 70 S Ribosome. Angew. Chem. Int. Ed. 2014, 53, 12236–12239. [Google Scholar] [CrossRef]
- Seefeldt, A.C.; Nguyen, F.; Antunes, S.; Pérébaskine, N.; Graf, M.; Arenz, S.; Inampudi, K.K.; Douat, C.; Guichard, G.; Wilson, D.N.; et al. The Proline-Rich Antimicrobial Peptide Onc112 Inhibits Translation by Blocking and Destabilizing the Initiation Complex. Nat. Struct. Mol. Biol. 2015, 22, 470–475. [Google Scholar] [CrossRef]
- Roy, R.N.; Lomakin, I.B.; Gagnon, M.G.; Steitz, T.A. The Mechanism of Inhibition of Protein Synthesis by the Proline-Rich Peptide Oncocin. Nat. Struct. Mol. Biol. 2015, 22, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Seefeldt, A.C.; Graf, M.; Pérébaskine, N.; Nguyen, F.; Arenz, S.; Mardirossian, M.; Scocchi, M.; Wilson, D.N.; Innis, C.A. Structure of the Mammalian Antimicrobial Peptide Bac7(1–16) Bound within the Exit Tunnel of a Bacterial Ribosome. Nucleic Acids Res. 2016, 44, 2429–2438. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Yang, M.; Sun, R.N.; Liu, Y.; Wang, W.; Xi, Q.; Gong, H.; Chen, C. Mechanism of Actions of Oncocin, a Proline-Rich Antimicrobial Peptide, in Early Elongation Revealed by Single-Molecule FRET. Protein Cell 2018, 9, 890–895. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, M.G.; Roy, R.N.; Lomakin, I.B.; Florin, T.; Mankin, A.S.; Steitz, T.A. Structures of Proline-Rich Peptides Bound to the Ribosome Reveal a Common Mechanism of Protein Synthesis Inhibition. Nucleic Acids Res. 2016, 44, 2439–2450. [Google Scholar] [CrossRef]
- Florin, T.; Maracci, C.; Graf, M.; Karki, P.; Klepacki, D.; Berninghausen, O.; Beckmann, R.; Vázquez-Laslop, N.; Wilson, D.N.; Rodnina, M.V.; et al. An Antimicrobial Peptide That Inhibits Translation by Trapping Release Factors on the Ribosome. Nat. Struct. Mol. Biol. 2017, 24, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Graf, M.; Huter, P.; Maracci, C.; Peterek, M.; Rodnina, M.V.; Wilson, D.N. Visualization of Translation Termination Intermediates Trapped by the Apidaecin 137 Peptide during RF3-Mediated Recycling of RF1. Nat. Commun. 2018, 9, 3053. [Google Scholar] [CrossRef] [PubMed]
- Skowron, K.J.; Baliga, C.; Johnson, T.; Kremiller, K.M.; Castroverde, A.; Dean, T.T.; Allen, A.C.; Lopez-Hernandez, A.M.; Aleksandrova, E.V.; Klepacki, D.; et al. Structure–Activity Relationships of the Antimicrobial Peptide Natural Product Apidaecin. J. Med. Chem. 2023, 66, 11831–11842. [Google Scholar] [CrossRef]
- Mangano, K.; Klepacki, D.; Ohanmu, I.; Baliga, C.; Huang, W.; Brakel, A.; Krizsan, A.; Polikanov, Y.S.; Hoffmann, R.; Vázquez-Laslop, N.; et al. Inhibition of Translation Termination by the Antimicrobial Peptide Drosocin. Nat. Chem. Biol. 2023, 19, 1082–1090. [Google Scholar] [CrossRef]
- Schneider, M.; Dorn, A. Differential Infectivity of Two Pseudomonas Species and the Immune Response in the Milkweed Bug, Oncopeltus Fasciatus (Insecta: Hemiptera). J. Invertebr. Pathol. 2001, 78, 135–140. [Google Scholar] [CrossRef]
- Knappe, D.; Piantavigna, S.; Hansen, A.; Mechler, A.; Binas, A.; Nolte, O.; Martin, L.L.; Hoffmann, R. Oncocin (VDKPPYLPRPRPPRRIYNR-NH2): A Novel Antibacterial Peptide Optimized against Gram-Negative Human Pathogens. J. Med. Chem. 2010, 53, 5240–5247. [Google Scholar] [CrossRef]
- Knappe, D.; Kabankov, N.; Hoffmann, R. Bactericidal Oncocin Derivatives with Superior Serum Stabilities. Int. J. Antimicrob. Agents 2011, 37, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Knappe, D.; Ruden, S.; Langanke, S.; Tikkoo, T.; Ritzer, J.; Mikut, R.; Martin, L.L.; Hoffmann, R.; Hilpert, K. Optimization of Oncocin for Antibacterial Activity Using a SPOT Synthesis Approach: Extending the Pathogen Spectrum to Staphylococcus Aureus. Amino Acids 2016, 48, 269–280. [Google Scholar] [CrossRef]
- Benincasa, M.; Scocchi, M.; Podda, E.; Skerlavaj, B.; Dolzani, L.; Gennaro, R. Antimicrobial Activity of Bac7 Fragments against Drug-Resistant Clinical Isolates. Peptides 2004, 25, 2055–2061. [Google Scholar] [CrossRef] [PubMed]
- Mattiuzzo, M.; Bandiera, A.; Gennaro, R.; Benincasa, M.; Pacor, S.; Antcheva, N.; Scocchi, M. Role of the Escherichia Coli SbmA in the Antimicrobial Activity of Proline-rich Peptides. Mol. Microbiol. 2007, 66, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Krizsan, A.; Knappe, D.; Hoffmann, R. Influence of the yjiL-mdtM Gene Cluster on the Antibacterial Activity of Proline-Rich Antimicrobial Peptides Overcoming Escherichia Coli Resistance Induced by the Missing SbmA Transporter System. Antimicrob. Agents Chemother. 2015, 59, 5992–5998. [Google Scholar] [CrossRef]
- Ghilarov, D.; Inaba-Inoue, S.; Stepien, P.; Qu, F.; Michalczyk, E.; Pakosz, Z.; Nomura, N.; Ogasawara, S.; Walker, G.C.; Rebuffat, S.; et al. Molecular Mechanism of SbmA, a Promiscuous Transporter Exploited by Antimicrobial Peptides. Sci. Adv. 2021, 7, eabj5363. [Google Scholar] [CrossRef]
- Graf, M.; Wilson, D.N. Intracellular Antimicrobial Peptides Targeting the Protein Synthesis Machinery. In Antimicrobial Peptides: Basics for Clinical Application; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1117, pp. 73–89. ISBN 9789811335877. [Google Scholar]
- Stalmans, S.; Wynendaele, E.; Bracke, N.; Knappe, D.; Hoffmann, R.; Peremans, K.; Polis, I.; Burvenich, C.; Spiegeleer, B. Blood-Brain Barrier Transport of Short Proline-Rich Antimicrobial Peptides. Protein Pept. Lett. 2014, 21, 399–406. [Google Scholar] [CrossRef]
- Panteleev, P.V.; Safronova, V.N.; Kruglikov, R.N.; Bolosov, I.A.; Ovchinnikova, T.V. Genomic Insights into Bacterial Resistance to Proline-Rich Antimicrobial Peptide Bac7. Membranes 2023, 13, 438. [Google Scholar] [CrossRef]
- Bagheri, M.; Arasteh, S.; Haney, E.F.; Hancock, R.E.W. Tryptic Stability of Synthetic Bactenecin Derivatives Is Determined by the Side Chain Length of Cationic Residues and the Peptide Conformation. J. Med. Chem. 2016, 59, 3079–3086. [Google Scholar] [CrossRef]
- Shaikh, A.Y.; Björkling, F.; Zabicka, D.; Tomczak, M.; Urbas, M.; Domraceva, I.; Kreicberga, A.; Franzyk, H. Structure-Activity Study of Oncocin: On-Resin Guanidinylation and Incorporation of Homoarginine, 4-Hydroxyproline or 4,4-Difluoroproline Residues. Bioorg. Chem. 2023, 141, 106876. [Google Scholar] [CrossRef]
- Kolano, L.; Knappe, D.; Berg, A.; Berg, T.; Hoffmann, R. Effect of Amino Acid Substitutions on 70S Ribosomal Binding, Cellular Uptake, and Antimicrobial Activity of Oncocin Onc112. ChemBioChem 2022, 23, e202100609. [Google Scholar] [CrossRef] [PubMed]
- Khairullina, Z.Z.; Makarov, G.I.; Tereshchenkov, A.G.; Buev, V.S.; Lukianov, D.A.; Polshakov, V.I.; Tashlitsky, V.N.; Osterman, I.A.; Sumbatyan, N.V. Conjugates of Desmycosin with Fragments of Antimicrobial Peptide Oncocin: Synthesis, Antibacterial Activity, Interaction with Ribosome. Biochemistry 2022, 87, 871–889. [Google Scholar] [CrossRef] [PubMed]
- Samizadeh, M.; Zhang, X.; Gunaseelan, S.; Nelson, A.G.; Palombo, M.S.; Myers, D.R.; Singh, Y.; Ganapathi, U.; Szekely, Z.; Sinko, P.J. Colorectal Delivery and Retention of PEG-Amprenavir-Bac7 Nanoconjugates—Proof of Concept for HIV Mucosal Pre-Exposure Prophylaxis. Drug Deliv. Transl. Res. 2016, 6, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.M.; Bonke, G.; Larsen, C.J.; Yavari, N.; Nielsen, P.E.; Franzyk, H. Antibacterial Peptide Nucleic Acid–Antimicrobial Peptide (PNA–AMP) Conjugates: Antisense Targeting of Fatty Acid Biosynthesis. Bioconjugate Chem. 2016, 27, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Goldbach, T.; Knappe, D.; Reinsdorf, C.; Berg, T.; Hoffmann, R. Ribosomal Binding and Antibacterial Activity of Ethylene Glycol-bridged Apidaecin Api137 and Oncocin Onc112 Conjugates. J. Pept. Sci. 2016, 22, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Koch, P.; Schmitt, S.; Heynisch, A.; Gumpinger, A.; Wüthrich, I.; Gysin, M.; Shcherbakov, D.; Hobbie, S.N.; Panke, S.; Held, M. Optimization of the Antimicrobial Peptide Bac7 by Deep Mutational Scanning. BMC Biol. 2022, 20, 114. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-W.; Pavlova, J.A.; Lukianov, D.A.; Tereshchenkov, A.G.; Makarov, G.I.; Khairullina, Z.Z.; Tashlitsky, V.N.; Paleskava, A.; Konevega, A.L.; Bogdanov, A.A.; et al. Binding and Action of Triphenylphosphonium Analog of Chloramphenicol upon the Bacterial Ribosome. Antibiotics 2021, 10, 390. [Google Scholar] [CrossRef]
- Pavlova, J.A.; Khairullina, Z.Z.; Tereshchenkov, A.G.; Nazarov, P.A.; Lukianov, D.A.; Volynkina, I.A.; Skvortsov, D.A.; Makarov, G.I.; Abad, E.; Murayama, S.Y.; et al. Triphenilphosphonium Analogs of Chloramphenicol as Dual-Acting Antimicrobial and Antiproliferating Agents. Antibiotics 2021, 10, 489. [Google Scholar] [CrossRef]
- Cochrane, E.J.; Hulit, J.; Lagasse, F.P.; Lechertier, T.; Stevenson, B.; Tudor, C.; Trebicka, D.; Sparey, T.; Ratcliffe, A.J. Impact of Mitochondrial Targeting Antibiotics on Mitochondrial Function and Proliferation of Cancer Cells. ACS Med. Chem. Lett. 2021, 12, 579–584. [Google Scholar] [CrossRef]
- Fuentes-Retamal, S.; Sandoval-Acuña, C.; Peredo-Silva, L.; Guzmán-Rivera, D.; Pavani, M.; Torrealba, N.; Truksa, J.; Castro-Castillo, V.; Catalán, M.; Kemmerling, U.; et al. Complex Mitochondrial Dysfunction Induced by TPP+-Gentisic Acid and Mitochondrial Translation Inhibition by Doxycycline Evokes Synergistic Lethality in Breast Cancer Cells. Cells 2020, 9, 407. [Google Scholar] [CrossRef]
- Khailova, L.S.; Nazarov, P.A.; Sumbatyan, N.V.; Korshunova, G.A.; Rokitskaya, T.I.; Dedukhova, V.I.; Antonenko, Y.N.; Skulachev, V.P. Uncoupling and Toxic Action of Alkyltriphenylphosphonium Cations on Mitochondria and the Bacterium Bacillus Subtilis as a Function of Alkyl Chain Length. Biochemistry 2015, 80, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Nazarov, P.A.; Osterman, I.A.; Tokarchuk, A.V.; Karakozova, M.V.; Korshunova, G.A.; Lyamzaev, K.G.; Skulachev, M.V.; Kotova, E.A.; Skulachev, V.P.; Antonenko, Y.N. Mitochondria-Targeted Antioxidants as Highly Effective Antibiotics. Sci. Rep. 2017, 7, 1394. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, J.A.; Tereshchenkov, A.G.; Nazarov, P.A.; Lukianov, D.A.; Skvortsov, D.A.; Polshakov, V.I.; Vasilieva, B.F.; Efremenkova, O.V.; Kaiumov, M.Y.; Paleskava, A.; et al. Conjugates of Chloramphenicol Amine and Berberine as Antimicrobial Agents. Antibiotics 2022, 12, 15. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kim, I.H.; Roche, E.D.; Beeman, D.; Lynch, A.S.; Ding, C.Z.; Ma, Z. Design, Synthesis, and Biological Evaluation of BODIPY®–Erythromycin Probes for Bacterial Ribosomes. Bioorganic Med. Chem. Lett. 2006, 16, 794–797. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Dicker, D.W.; Whiting, M.C. 407. Synthetical Studies on Terpenoids. Part I. The Synthesis of Squalene. J. Chem. Soc. 1958, 1994–2000. [Google Scholar] [CrossRef]
- Khan, S.M.; Gadwood, R.; Romero, A.G. Modified Creatine Compounds. U.S. Patent 20150005258, 18 September 2012. [Google Scholar]
- Antonenko, Y.N.; Avetisyan, A.V.; Bakeeva, L.E.; Chernyak, B.V.; Chertkov, V.A.; Domnina, L.V.; Ivanova, O.Y.; Izyumov, D.S.; Khailova, L.S.; Klishin, S.S.; et al. Mitochondria-Targeted Plastoquinone Derivatives as Tools to Interrupt Execution of the Aging Program. 1. Cationic Plastoquinone Derivatives: Synthesis and in Vitro Studies. Biochemistry 2008, 73, 1273–1287. [Google Scholar] [CrossRef]
- Hansen, P.R.; Oddo, A. Fmoc solid–phase peptide synthesis. Methods Mol. Biol. 2015, 1348, 33–50. [Google Scholar] [CrossRef]
- Eissler, S.; Kley, M.; Bächle, D.; Loidl, G.; Meier, T.; Samson, D. Substitution determination of Fmoc-substituted resins at different wavelengths. J. Pept. Sci. 2017, 23, 757–762. [Google Scholar] [CrossRef]
- Kaiser, E.; Colescott, R.L.; Bossinger, C.D.; Cook, P.I. Color test for detection of free terminal amino groups in the solidphase synthesis of peptides. Anal. Biochem. 1970, 34, 595–598. [Google Scholar] [CrossRef]
- Rodnina, M.V.; Wintermeyer, W. GTP Consumption of Elongation Factor Tu during Translation of Heteropolymeric mRNAs. Proc. Natl. Acad. Sci. USA 1995, 92, 1945–1949. [Google Scholar] [CrossRef]
- Yan, K.; Hunt, E.; Berge, J.; May, E.; Copeland, R.A.; Gontarek, R.R. Fluorescence Polarization Method to Characterize Macrolide-Ribosome Interactions. Antimicrob. Agents Chemother. 2005, 49, 3367–3372. [Google Scholar] [CrossRef]
- Wang, Z.-X. An Exact Mathematical Expression for Describing Competitive Binding of Two Different Ligands to a Protein Molecule. FEBS Lett. 1995, 360, 111–114. [Google Scholar] [CrossRef]
- Terenin, I.M.; Andreev, D.E.; Dmitriev, S.E.; Shatsky, I.N. A Novel Mechanism of Eukaryotic Translation Initiation That Is Neither m7G-Cap-, nor IRES-Dependent. Nucleic Acids Res. 2013, 41, 1807–1816. [Google Scholar] [CrossRef]
- Baba, T.; Ara, T.; Hasegawa, M.; Takai, Y.; Okumura, Y.; Baba, M.; Datsenko, K.A.; Tomita, M.; Wanner, B.L.; Mori, H. Construction of Escherichia coli K-12 In-frame, Single-gene Knockout Mutants: The Keio Collection. Mol. Syst. Biol. 2006, 2, 2006.0008. [Google Scholar] [CrossRef]
- Osterman, I.A.; Komarova, E.S.; Shiryaev, D.I.; Korniltsev, I.A.; Khven, I.M.; Lukyanov, D.A.; Tashlitsky, V.N.; Serebryakova, M.V.; Efremenkova, O.V.; Ivanenkov, Y.A.; et al. Sorting Out Antibiotics’ Mechanisms of Action: A Double Fluorescent Protein Reporter for High-Throughput Screening of Ribosome and DNA Biosynthesis Inhibitors. Antimicrob. Agents Chemother. 2016, 60, 7481–7489. [Google Scholar] [CrossRef]
- Hardy, K.; Haefeli, C. Expression in Escherichia Coli of a Staphylococcal Gene for Resistance to Macrolide, Lincosamide, and Streptogramin Type B Antibiotics. J. Bacteriol. 1982, 152, 524–526. [Google Scholar] [CrossRef]
- Orelle, C.; Carlson, S.; Kaushal, B.; Almutairi, M.M.; Liu, H.; Ochabowicz, A.; Quan, S.; Pham, V.C.; Squires, C.L.; Murphy, B.T.; et al. Tools for Characterizing Bacterial Protein Synthesis Inhibitors. Antimicrob. Agents Chemother. 2013, 57, 5994–6004. [Google Scholar] [CrossRef]
- Antonenko, Y.N.; Denisov, S.S.; Khailova, L.S.; Nazarov, P.A.; Rokitskaya, T.; Tashlitsky, V.N.; Firsov, A.M.; Korshunova, G.A.; Kotova, E.A. Alkyl-Substituted Phenylamino Derivatives of 7-Nitrobenz-2-Oxa-1,3-Diazole as Uncouplers of Oxidative Phosphorylation and Antibacterial Agents: Involvement of Membrane Proteins in the Uncoupling Action. Biochim. Biophys. Acta—Biomembr. 2017, 1859, 377–387. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Ludwig, T.; Krizsan, A.; Mohammed, G.K.; Hoffmann, R. Antimicrobial Activity and 70S Ribosome Binding of Apidaecin-Derived Api805 with Increased Bacterial Uptake Rate. Antibiotics 2022, 11, 430. [Google Scholar] [CrossRef]
- Krizsan, A.; Prahl, C.; Goldbach, T.; Knappe, D.; Hoffmann, R. Short Proline-Rich Antimicrobial Peptides Inhibit Either the Bacterial 70S Ribosome or the Assembly of Its Large 50S Subunit. ChemBioChem 2015, 16, 2304–2308. [Google Scholar] [CrossRef]
- Tereshchenkov, A.G.; Dobosz-Bartoszek, M.; Osterman, I.A.; Marks, J.; Sergeeva, V.A.; Kasatsky, P.; Komarova, E.S.; Stavrianidi, A.N.; Rodin, I.A.; Konevega, A.L.; et al. Binding and Action of Amino Acid Analogs of Chloramphenicol upon the Bacterial Ribosome. J. Mol. Biol. 2018, 430, 842–852. [Google Scholar] [CrossRef]
- Koller, T.; Morici, M.; Berger, M.; Safdari, H.; Lele, D.; Beckert, B.; Kaur, K.; Wilson, D. Structural basis for translation inhibition by the glycosylated drosocin peptide. Nat. Chem. Biol. 2023, 19, 1072–1081. [Google Scholar] [CrossRef]
- Runti, G.; Lopez Ruiz, M.D.C.; Stoilova, T.; Hussain, R.; Jennions, M.; Choudhury, H.G.; Benincasa, M.; Gennaro, R.; Beis, K.; Scocchi, M. Functional Characterization of SbmA, a Bacterial Inner Membrane Transporter Required for Importing the Antimicrobial Peptide Bac7(1-35). J. Bacteriol. 2013, 195, 5343–5351. [Google Scholar] [CrossRef]
- Nikaido, H. Molecular Basis of Bacterial Outer Membrane Permeability Revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef]
- Severin, F.F.; Severina, I.I.; Antonenko, Y.N.; Rokitskaya, T.I.; Cherepanov, D.A.; Mokhova, E.N.; Vyssokikh, M.Y.; Pustovidko, A.V.; Markova, O.V.; Yaguzhinsky, L.S.; et al. Penetrating Cation/Fatty Acid Anion Pair as a Mitochondria-Targeted Protonophore. Proc. Natl. Acad. Sci. USA 2010, 107, 663–668. [Google Scholar] [CrossRef]
- Mardirossian, M.; Sola, R.; Beckert, B.; Valencic, E.; Collis, D.; Borišek, J.; Armas, F.; Stasi, A.; Buchmann, J.; Syroegin, E.; et al. Peptide Inhibitors of Bacterial Protein Synthesis with Broad Spectrum and SbmA-Independent Bactericidal Activity against Clinical Pathogens. J. Med. Chem. 2020, 63, 9590–9602. [Google Scholar] [CrossRef]
- Scheinpflug, K.; Krylova, O.; Nikolenko, H.; Thurm, C.; Dathe, M. Evidence for a Novel Mechanism of Antimicrobial Action of a Cyclic R-,W-Rich Hexapeptide. PLoS ONE 2015, 10, e0125056. [Google Scholar] [CrossRef]
- Epand, R.M.; Walker, C.; Epand, R.F.; Magarvey, N.A. Molecular Mechanisms of Membrane Targeting Antibiotics. Biochim. Biophys. Acta Biomembr. 2016, 1858, 980–987. [Google Scholar] [CrossRef]
- Sokolov, S.; Zyrina, A.; Akimov, S.; Knorre, D.; Severin, F. Toxic Effects of Penetrating Cations. Membranes 2023, 13, 841. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| E. coli BW25113 | E. coli ∆sbmA | B. subtilis 168 | B. subtilis-CFR | |
|---|---|---|---|---|
| Bac(1–10) (1) | - | - | >50 | >50 |
| Bac(1–10, R/Y) (2) | >100 | >100 | - | - |
| Bac(1–10, R/Y)-C2-TPP (3) | 26.3 | >100 | 12.5 | 50 |
| Bac(1–10, R/Y)-C10-TPP (4) | 100 | 100 | 1.6 | 0.8 |
| TPP-C10-Bac(1–10, R/Y) (5) | 5.3 | 5.3 | 12.5 | 12.5 |
| Onc112 | 11.6 | >90 | >50 | >50 |
| C4-TPP | >100 | >100 | >100 | >100 |
| C10-TPP | 50 | 50 | 1.6 | 0.8 |
| ERY | 170 | 170 | <0.1 | <0.1 |
| Chloramphenicol 2 | - | - | 12 | 90 |
| HEK293T | MCF7 | VA13 | A549 | |
|---|---|---|---|---|
| Bac(1–10, R/Y) (2) | >50 | >20 | >50 | >50 |
| Bac(1–10, R/Y)-C2-TPP (3) | >50 | >20 | >50 | >50 |
| Bac(1–10, R/Y)-C10-TPP (4) | 3.1 ± 0.5 | 10 ± 1 | 12 ± 1 | 9.2 ± 0.9 |
| TPP-C10-Bac(1–10, R/Y) (5) | 36 ± 4 | >20 | >50 | >50 |
| Onc112 | >50 | >20 | >50 | >50 |
| C4-TPP | 2.9 ± 0.5 | 8 ± 2 | 11 ± 2 | 6.2 ± 0.9 |
| C10-TPP | <0.16 | 0.25 ± 0.07 | <0.16 | <0.16 |
| Doxorubicin | <1.6 | 1.7 ± 0.2 | 1.2 ± 0.3 | 1.2 ± 0.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tereshchenkov, A.G.; Khairullina, Z.Z.; Volynkina, I.A.; Lukianov, D.A.; Nazarov, P.A.; Pavlova, J.A.; Tashlitsky, V.N.; Razumova, E.A.; Ipatova, D.A.; Timchenko, Y.V.; et al. Triphenylphosphonium Analogs of Short Peptide Related to Bactenecin 7 and Oncocin 112 as Antimicrobial Agents. Pharmaceutics 2024, 16, 148. https://doi.org/10.3390/pharmaceutics16010148
Tereshchenkov AG, Khairullina ZZ, Volynkina IA, Lukianov DA, Nazarov PA, Pavlova JA, Tashlitsky VN, Razumova EA, Ipatova DA, Timchenko YV, et al. Triphenylphosphonium Analogs of Short Peptide Related to Bactenecin 7 and Oncocin 112 as Antimicrobial Agents. Pharmaceutics. 2024; 16(1):148. https://doi.org/10.3390/pharmaceutics16010148
Chicago/Turabian StyleTereshchenkov, Andrey G., Zimfira Z. Khairullina, Inna A. Volynkina, Dmitrii A. Lukianov, Pavel A. Nazarov, Julia A. Pavlova, Vadim N. Tashlitsky, Elizaveta A. Razumova, Daria A. Ipatova, Yury V. Timchenko, and et al. 2024. "Triphenylphosphonium Analogs of Short Peptide Related to Bactenecin 7 and Oncocin 112 as Antimicrobial Agents" Pharmaceutics 16, no. 1: 148. https://doi.org/10.3390/pharmaceutics16010148