
Interpenetrating Polymer Networks of Poly(2-hydroxyethyl methacrylate) and Poly(N, N-dimethylacrylamide) as Potential Systems for Dermal Delivery of Dexamethasone Phosphate

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. IPN Synthesis
2.3. Swelling Properties
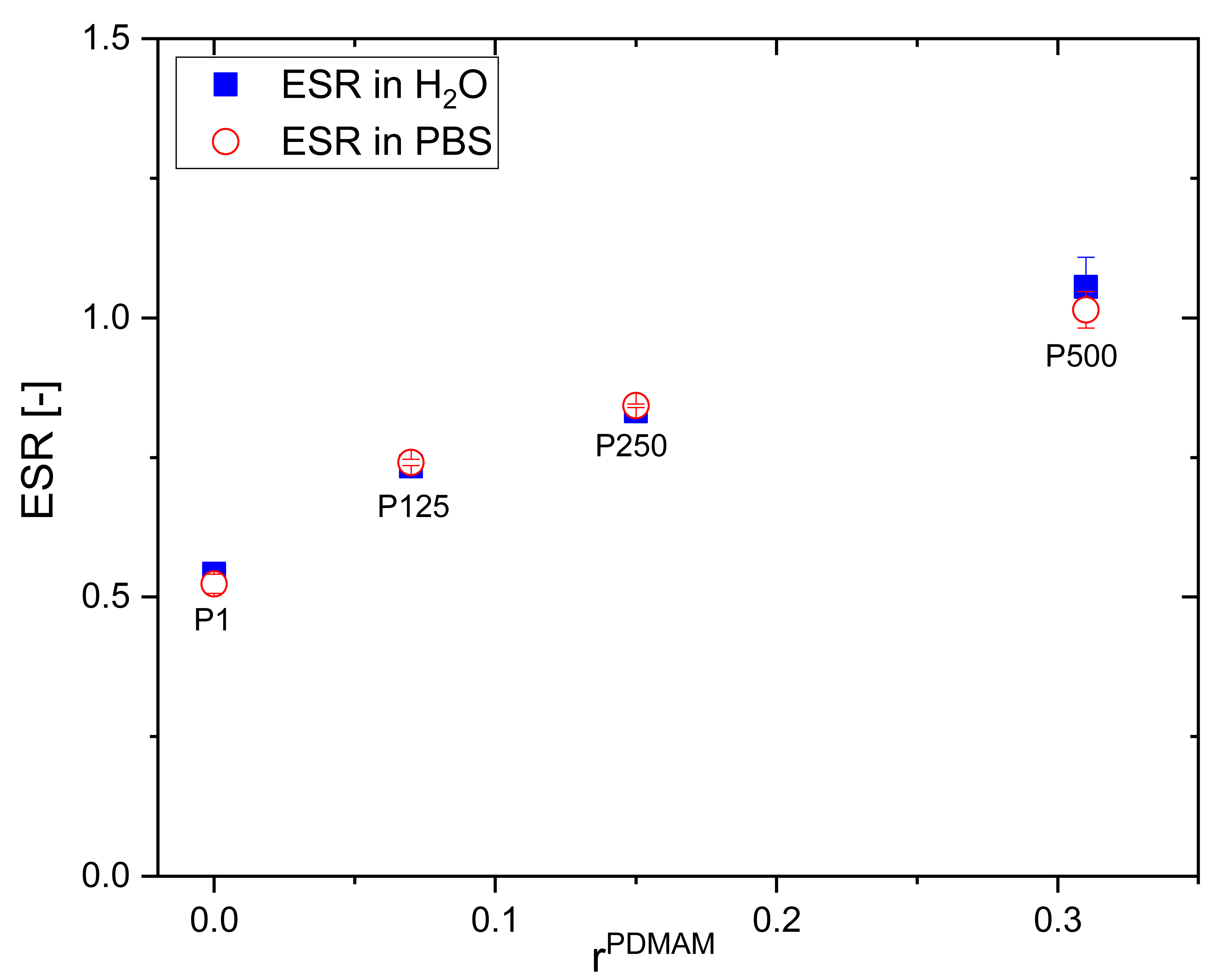
2.3.1. Equilibrium Swelling Ratio (ESR)
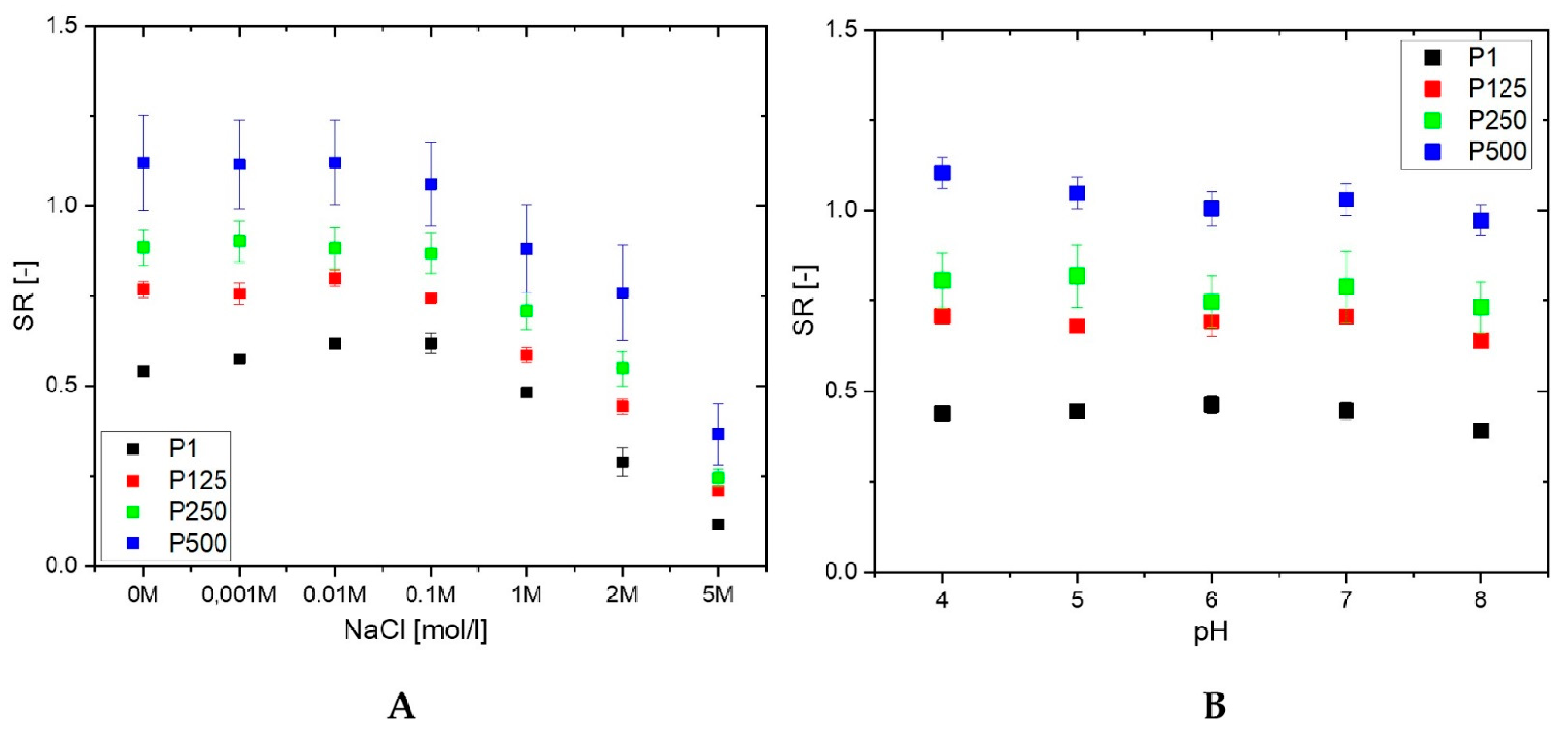
2.3.2. Ionic Strength Responsiveness
2.3.3. pH Responsiveness
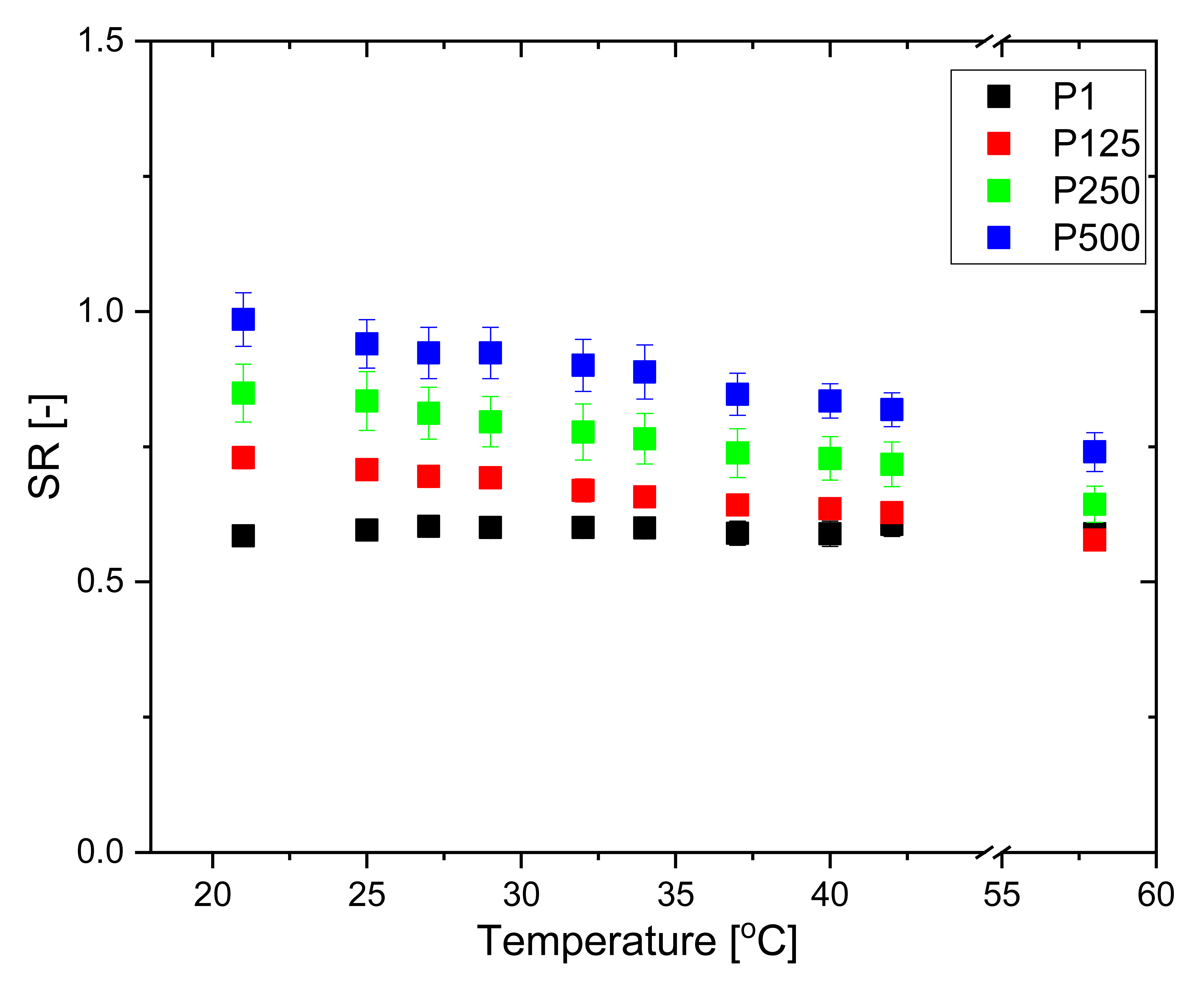
2.3.4. Temperature Responsiveness
2.3.5. Scanning Electron Microscopy (SEM)
2.3.6. Energy-Dispersive X-ray (EDX) Spectroscopy
2.3.7. X-ray Diffraction (XRD)
2.3.8. Temperature-Modulated Differential Scanning Calorimetry (TMDSC)
2.3.9. Attenuated Total Reflectance–Fourier Transform Infrared Spectroscopy (ATR-FTIR)
2.3.10. DXP Loading
2.3.11. DXP Release
2.3.12. DXP Release Kinetics
3. Results and Discussion
3.1. Swelling Properties
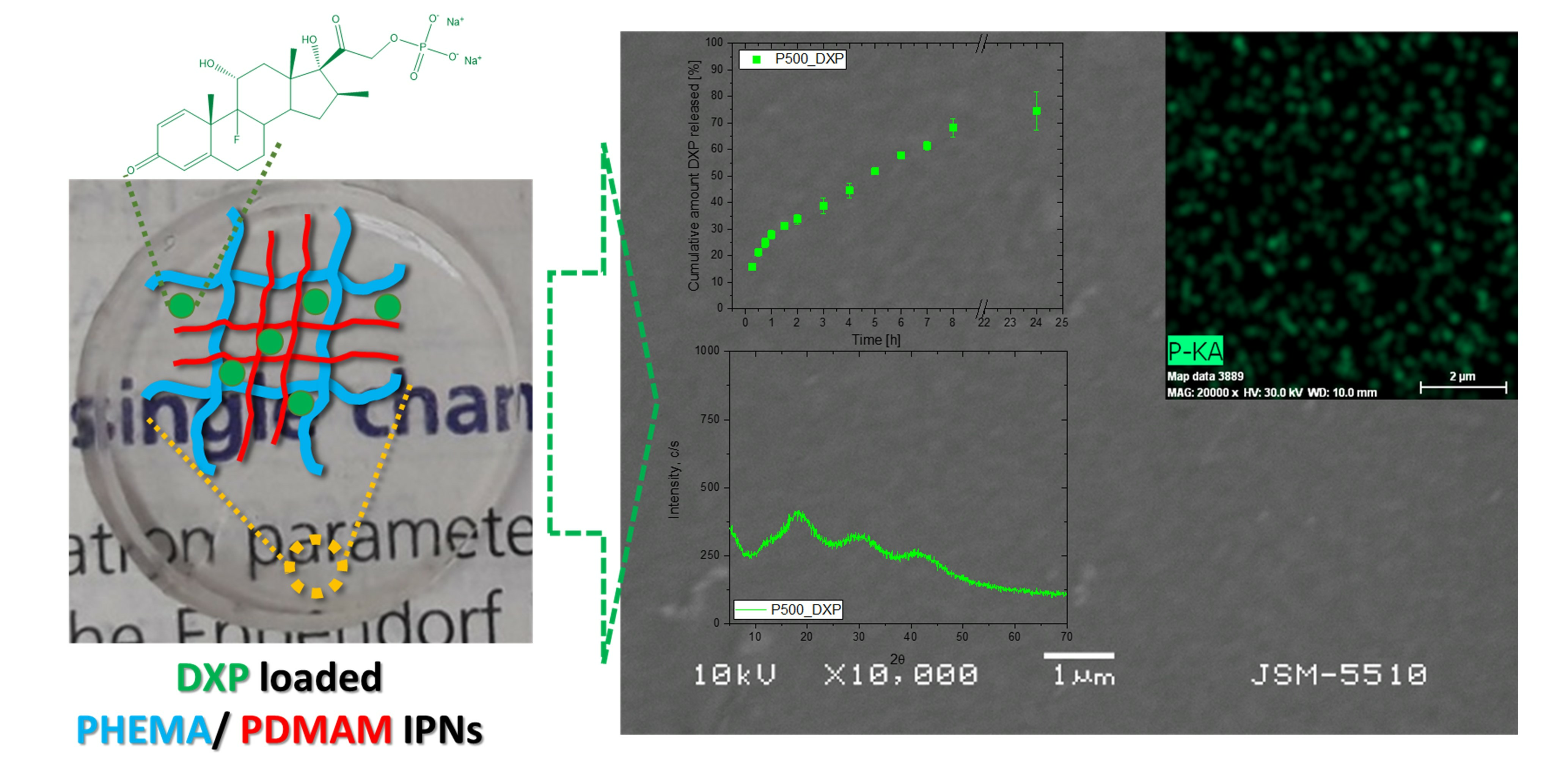
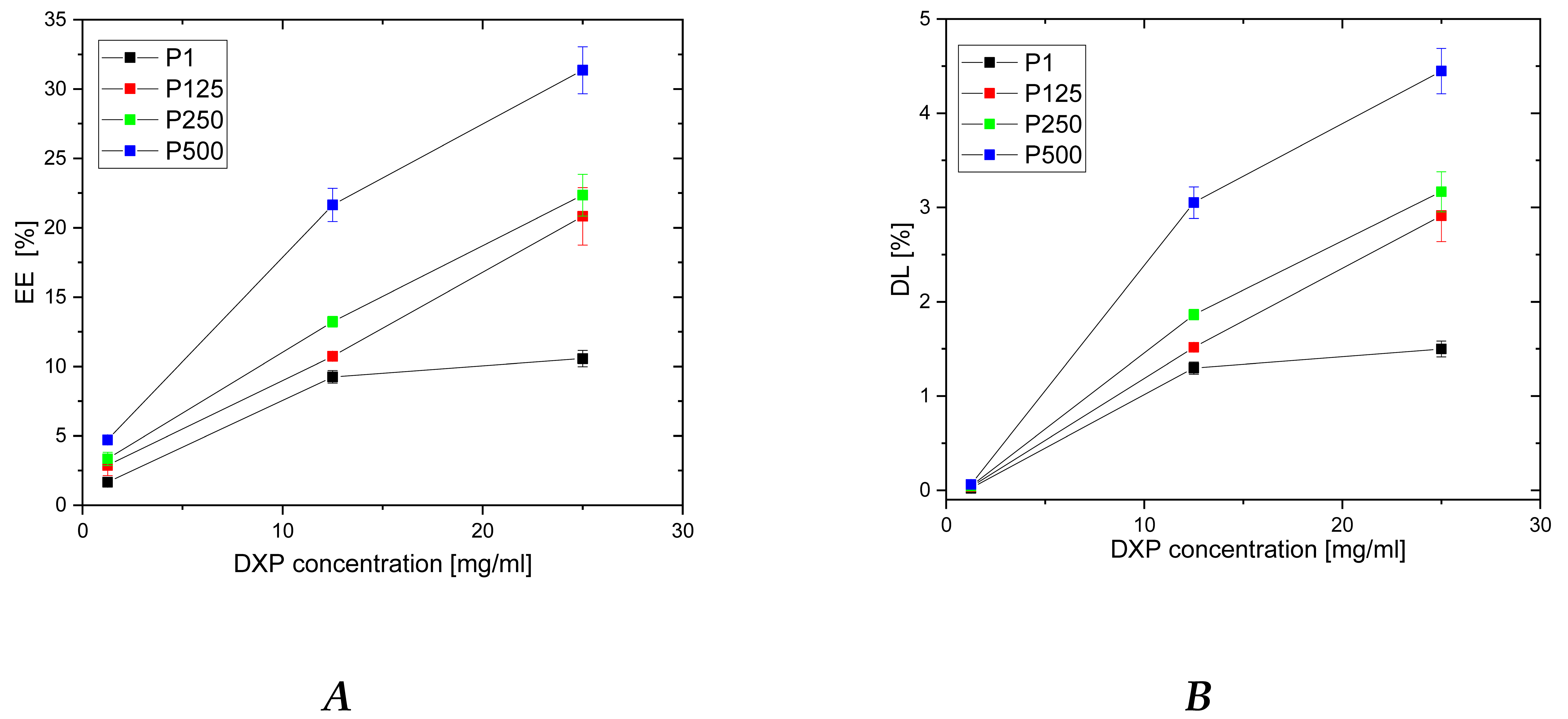
3.2. DXP Loading
3.3. Scanning Electron Microscopy (SEM)
3.4. EDX Analysis
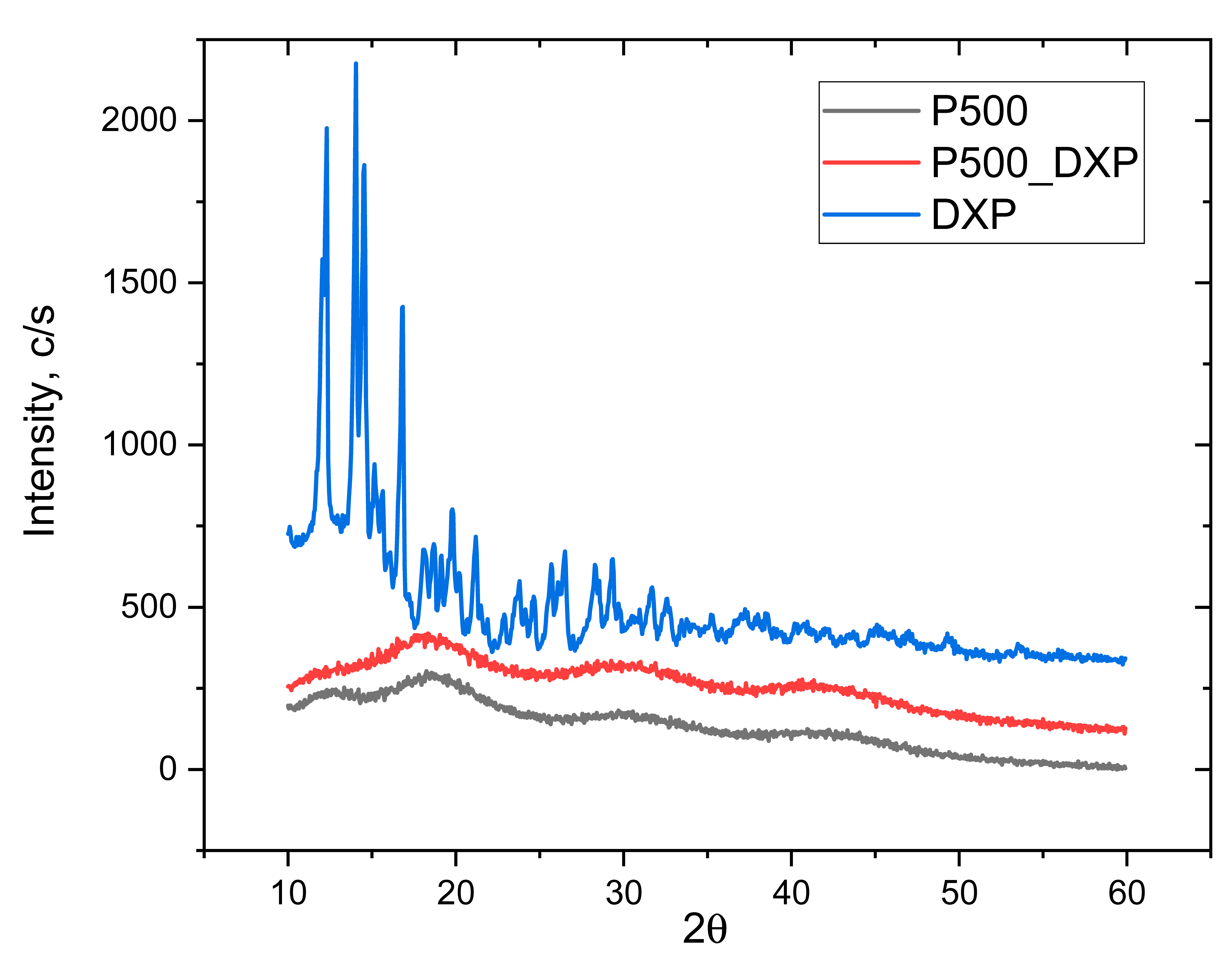
3.5. X-ray Diffraction (XRD)
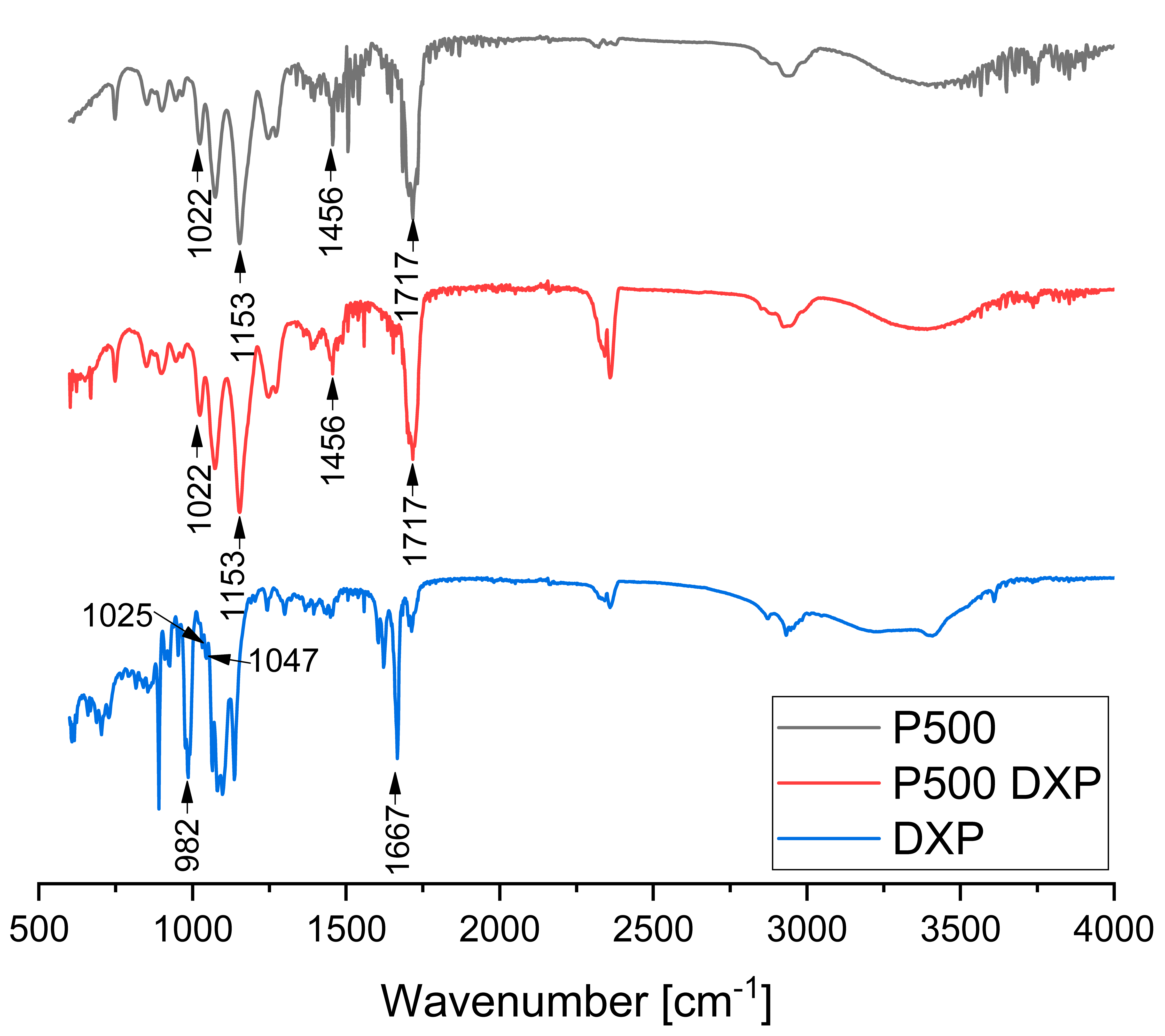
3.6. ATR-FTIR
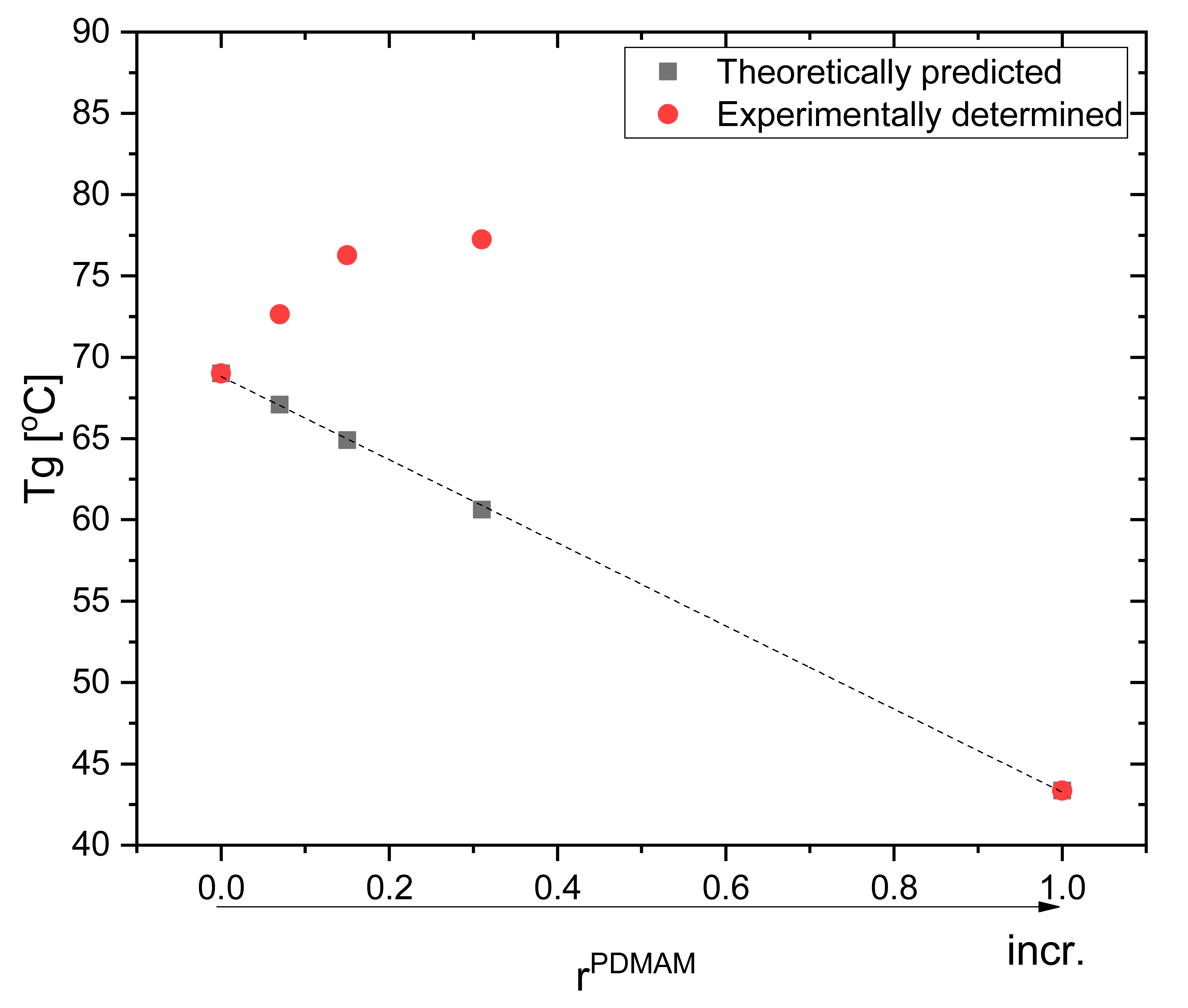
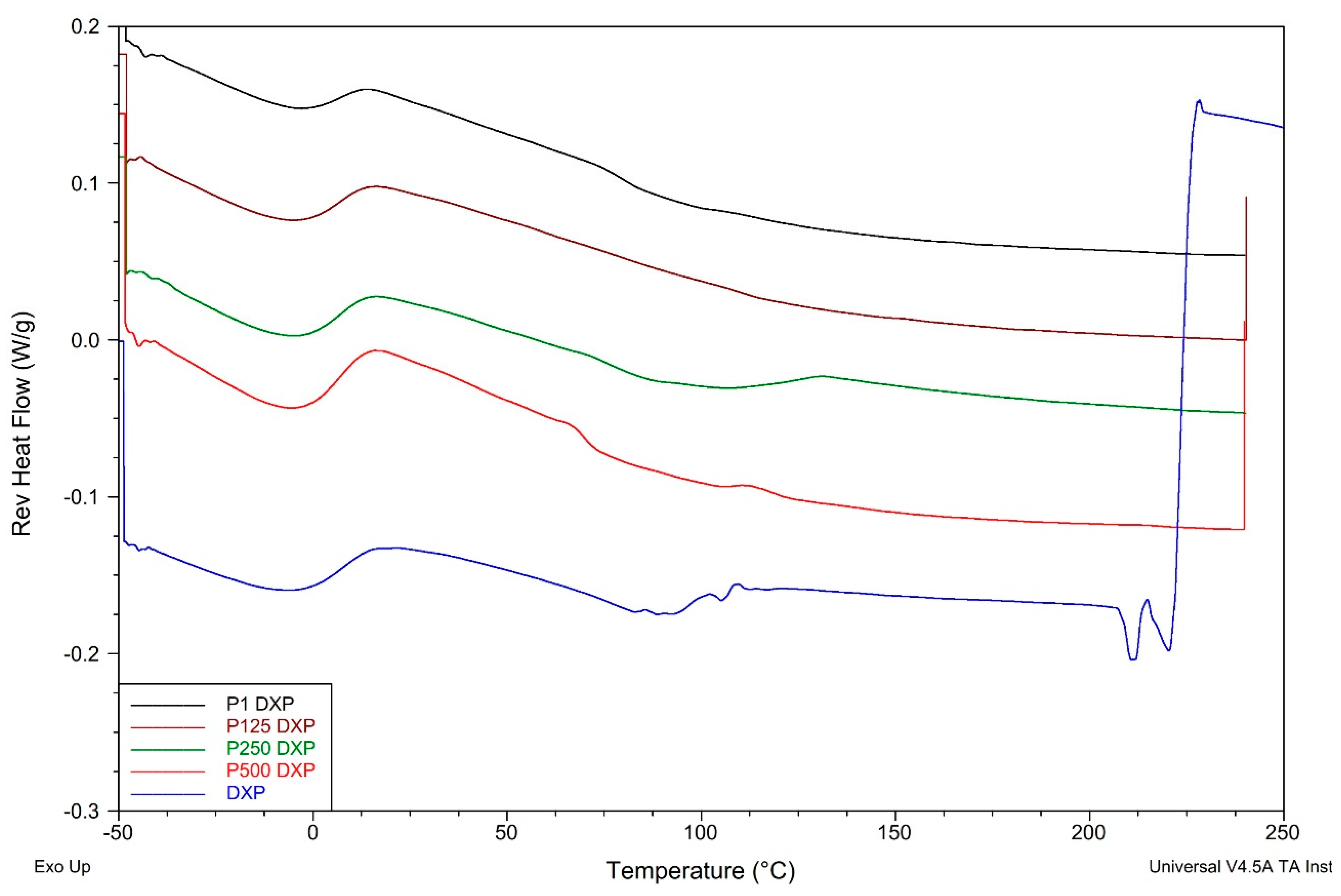
3.7. Thermal Properties of PHEMA/PDMAM IPNs, Non-Loaded and DXP-Loaded, as Revealed by TMDSC
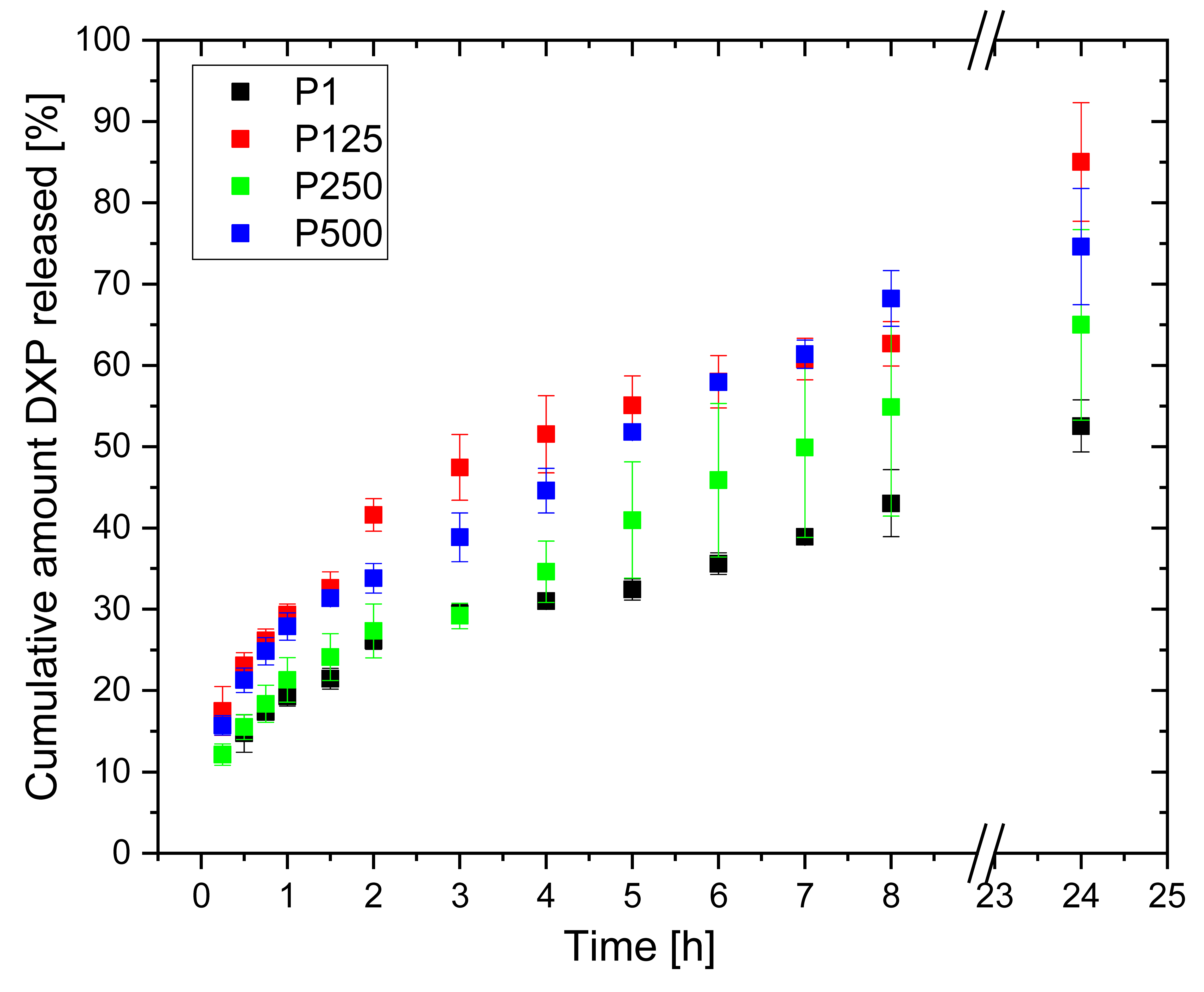
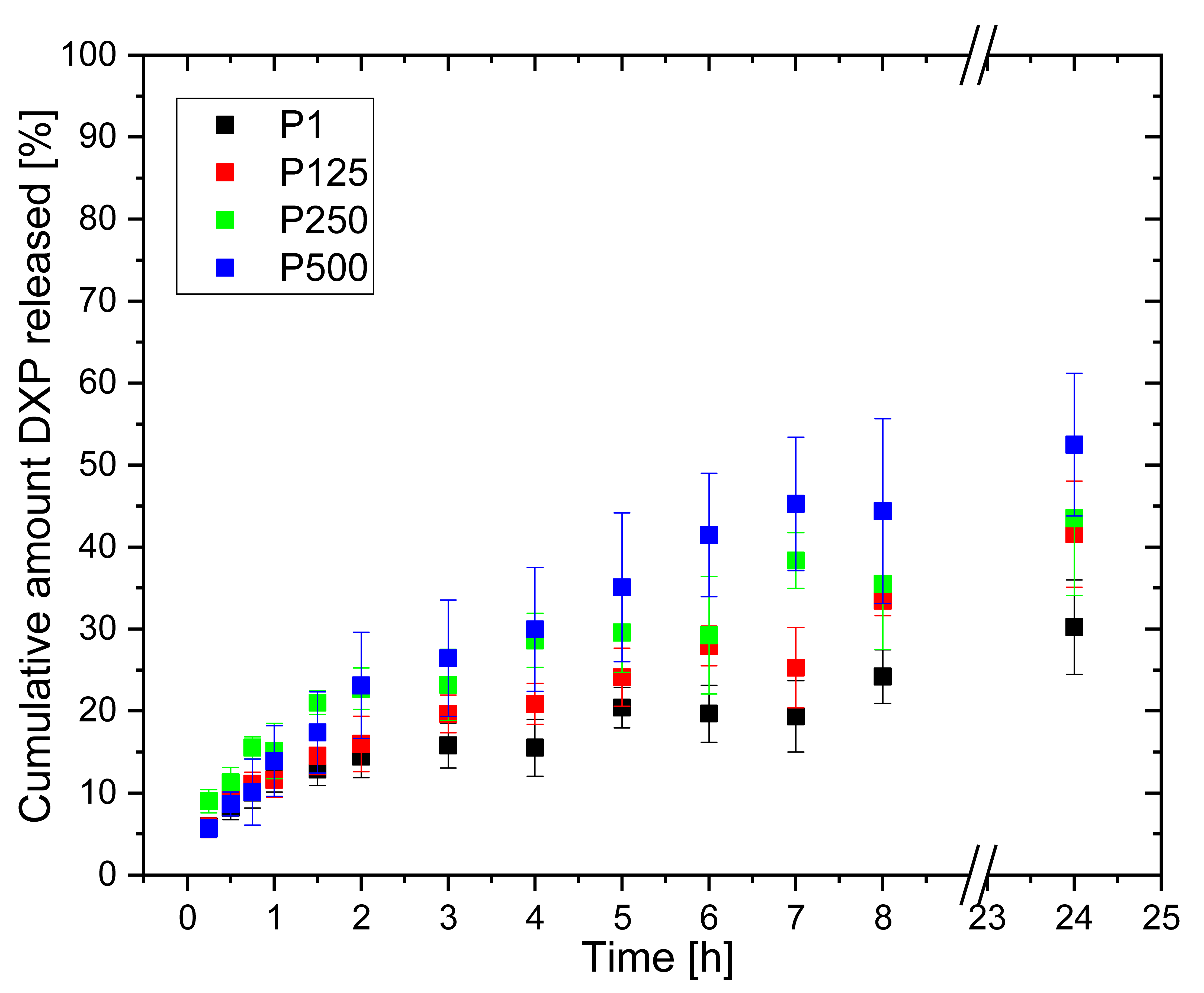
3.8. DXP Release
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DL | drug loading; |
| DMAM | N,N-Dimethylacrylamide; |
| DSC | Differential scanning calorimetry; |
| DXP | Dexamethasone sodium phosphate; |
| EDX | Energy-dispersive X-ray (EDX) spectroscopy; |
| EE | entrapment efficiency; |
| ESR | Equilibrium swelling ratio; |
| HCHPK | 1-Hydroxycyclohexyl phenyl ketone; |
| HEMA | 2-Hydroxyethyl methacrylate; |
| IPNs | Interpenetrating polymer networks; |
| PDMAM | poly(N,N′-dimethylacrylamide); |
| PEGDA | poly(ethylene glycol) diacrylate; |
| PHEMA | poly(2-hydroxyethyl methacrylate); |
| poly(HEMA-co-VP) | poly[(2-hydroxyethyl methacrylate)-co- vinylpyrrolidone]; |
| PVA | poly(vinyl alcohol); |
| PVP | polyvinyl pyrrolidone; |
| SEM | Scanning electron microscopy; |
| SNs | Single networks; |
| SR | Swelling ratios; |
| Tg | glass transition; |
| TMDSC | Temperature-modulated differential scanning calorimetry; |
| VP | vinylpyrrolidone; |
| XRD | X-ray diffraction; |
References
- Macnaught, A.D.; Wilkinson, A.; Union, I. Compendium of Chemical Terminology: IUPAC Recommendations; Oxford Blackwell Science: Hoboken, NJ, USA, 1997. [Google Scholar]
- Lohani, A.; Singh, G.; Bhattacharya, S.S.; Verma, A. Interpenetrating Polymer Networks as Innovative Drug Delivery Systems. J. Drug Deliv. 2014, 2014, 1–11. [Google Scholar] [CrossRef]
- Yasir, M.; Goyal, A.; Sonthalia, S. Corticosteroid Adverse Effects. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Trautmann, A.; Akdis, M.; Schmid-Grendelmeier, P.; Disch, R.; Bröcker, E.-B.; Blaser, K.; Akdis, C.A. Targeting Keratinocyte Apoptosis in the Treatment of Atopic Dermatitis and Allergic Contact Dermatitis. J. Allergy Clin. Immunol. 2001, 108, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Mizukoshi, K.; Oyobikawa, M.; Ohshima, H.; Sakai, Y.; Tagami, H. Objective Evaluation of the Efficacy of Daily Topical Applications of Cosmetics Bases Using the Hairless Mouse Model of Atopic Dermatitis. Skin Res. Technol. 2005, 11, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Dexamethasone Sodium Phosphate Injection, USP. Dailymed.Nlm.Nih.Gov. Available online: https://dailymed.nlm.nih.gov/dailymed/fda/fdaDrugXsl.cfm?setid=2b626a48-c7aa-4443-9164-a08ff069ccd1&type=display (accessed on 9 August 2023).
- Mokry, J.; Karbanová, J.; Lukáš, J.; Palečková, V.; Dvořánková, B. Biocompatibility of HEMA Copolymers Designed for Treatment of CNS Diseases with Polymer-Encapsulated Cells. Biotechnol. Prog. 2000, 16, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Prasitsilp, M.; Siriwittayakorn, T.; Molloy, R.; Suebsanit, N.; Siriwittayakorn, P.; Veeranondha, S. Cytotoxicity study of homopolymers and copolymers of 2-hydroxyethyl methacrylate and some alkyl acrylates for potential use as temporary skin substitutes. J. Mater. Sci. Mater. Med. 2003, 14, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Balavigneswaran, C.K.; Singh, M. Polymeric Gels for the Controlled Drug Delivery Applications; Woodhead: Sawston, UK, 2018. [Google Scholar] [CrossRef]
- Li, C.C.; Chauhan, A. Ocular Transport Model for Ophthalmic Delivery of Timolol through P-HEMA Contact Lenses. J. Drug Deliv. Sci. Technol. 2007, 17, 69–79. [Google Scholar] [CrossRef]
- Bengani, L.C.; Chauhan, A. Extended Delivery of an Anionic Drug by Contact Lens Loaded with a Cationic Surfactant. Biomaterials 2013, 34, 2814–2821. [Google Scholar] [CrossRef] [PubMed]
- Faccia, P.A.; Pardini, F.M.; Amalvy, J.I. Uptake and Release of Dexamethasone Using PH-Responsive Poly(2-Hydroxyethyl Methacrylate-Co-2-(Diisopropylamino)Ethyl Methacrylate) Hydrogels for Potential Use in Ocular Drug Delivery. J. Drug Deliv. Sci. Technol. 2019, 51, 45–54. [Google Scholar] [CrossRef]
- Effendy, I.; Maibach, H.I. Surfactants and Experimental Irritant Contact Dermatitis. Contact Derm. 1995, 33, 217–225. [Google Scholar] [CrossRef]
- Mann, J.L.; Grosskopf, A.K.; Smith, A.A.A.; Appel, E.A. Highly Branched Polydimethylacrylamide Copolymers as Functional Biomaterials. Biomacromolecules 2020, 22, 86–94. [Google Scholar] [CrossRef]
- Georgieva, D.; Alexandrova, M.; Ivanova, S.; Christova, D.; Kostova, B. Conceptualization and Investigation of Multicomponent Polymer Networks as Prospective Corticosteroid Carriers. Gels 2023, 9, 470. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, X.; Sun, F. Preparation and Evaluation of a Contact Lens Vehicle for Puerarin Delivery. J. Biomater. Sci. Polym. Ed. 2010, 21, 271–288. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Yu, J.-R.; Chen, L.; Zhu, J.; Hu, Z.-M. Preparation and Properties of Morphology Controlled Poly(2-Hydroxyethyl Methacrylate)/Poly(N-Vinyl Pyrrolidone) Double Networks for Biomedical Use. Curr. Appl. Phys. 2010, 10, 766–770. [Google Scholar] [CrossRef]
- Yang, H.; Zheng, Q.; Cheng, R. New Insight into “Polyelectrolyte Effect”. Colloids Surf. A Physicochem. Eng. 2012, 407, 1–8. [Google Scholar] [CrossRef]
- D’Agostino, A.; Colella, M.; De Rosa, M.; Lanza, A.; Schiraldi, C. Chemico-Physical Characterization of Hybrid Composites Based on Hydroxyethyl Methacrylate and Nanosilica. J. Polym. Res. 2008, 16, 561–567. [Google Scholar] [CrossRef]
- Shamsuddin, A.K.M.; Yanagimoto, S.; Kuwahara, T.; Zhang, Y.; Nomura, C.; Kondo, N. Changes in the Index of Sweat Ion Concentration with Increasing Sweat during Passive Heat Stress in Humans. Eur. J. Appl. Physiol. 2005, 94, 292–297. [Google Scholar] [CrossRef]
- Lara, B.; Gallo-Salazar, C.; Puente, C.; Areces, F.; Salinero, J.J.; Del Coso, J. Interindividual Variability in Sweat Electrolyte Concentration in Marathoners. J. Int. Soc. Sports Nutr. 2016, 13. [Google Scholar] [CrossRef]
- Mueller, K.F. Thermotropic Aqueous Gels and Solutions of N,N-Dimethylacrylamide-Acrylate Copolymers. Polymer 1992, 33, 3470–3476. [Google Scholar] [CrossRef]
- Simeonov, M.; Kostova, B.; Vassileva, E. Interpenetrating Polymer Networks of Poly(Methacrylic Acid) and Polyacrylamide: Synthesis, Characterization and Potential Application for Sustained Drug Delivery. RSC Adv. 2016, 6, 64239–64246. [Google Scholar] [CrossRef]
- Ajaz, N.; Khan, I.U.; Irfan, M.; Khalid, S.H.; Asghar, S.; Mehmood, Y.; Asif, M.; Usra; Hussain, G.; Shahzad, Y.; et al. In Vitro and Biological Characterization of Dexamethasone Sodium Phosphate Laden PH-Sensitive and Mucoadhesive Hydroxy Propyl β-Cyclodextrin-g-Poly(Acrylic Acid)/Gelatin Semi-Interpenetrating Networks. Gels 2022, 8, 290. [Google Scholar] [CrossRef]
- Wang, W.-R.; Li, A.; Mei, W.; Zhu, R.; Li, K.; Sun, X.; Qian, Y.; Wang, S. Dexamethasone Sodium Phosphate Intercalated Layered Double Hydroxides and Their Therapeutic Efficacy in a Murine Asthma Model. RSC Adv. 2015, 5, 23826–23834. [Google Scholar] [CrossRef]
- Roorda, W.E.; Bouwstra, J.A.; de Vries, M.A.; Junginger, H.E. Thermal Behavior of Poly Hydroxy Ethyl Methacrylate (pHEMA) Hydrogels. Pharm. Res. 1988, 5, 722–725. [Google Scholar] [CrossRef] [PubMed]
- González-Henríquez, C.M.; Pizarro, G.d.C.; Sarabia-Vallejos, M.A.; Terraza, C.A.; López-Cabaña, Z.E. In Situ-Preparation and Characterization of Silver-HEMA/PEGDA Hydrogel Matrix Nanocomposites: Silver Inclusion Studies into Hydrogel Matrix. Arab. J. Chem. 2019, 12, 1413–1423. [Google Scholar] [CrossRef]
- Krause, S.; Gormley, J.J.; Roman, N.; Shetter, J.A.; Watanabe, W.H. Glass Temperatures of Some Acrylic Polymers. J. Polym. Sci. 1965, 3, 3573–3586. [Google Scholar] [CrossRef]
- Keim, T.; Gall, K. Synthesis, Characterization, and Cyclic Stress-Influenced Degradation of a Poly(Ethylene Glycol)-Based Poly(Beta-Amino Ester). J. Biomed. Mater. Res. A 2010, 92 Pt A, 702–711. [Google Scholar] [CrossRef]
- Prata, A.I.; Coimbra, P.; Pina, M.E. Preparation of Dexamethasone Ophthalmic Implants: A Comparative Study of in Vitro Release Profiles. Pharm. Dev. Technol. 2017, 23, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.; Aikawa, S.; Kashima, Y.; Kikuchi, J.; Ida, Y.; Tanino, T.; Kadota, K.; Tozuka, Y. Anti-Plasticizing Effect of Amorphous Indomethacin Induced by Specific Intermolecular Interactions with PVA Copolymer. J. Pharm. Sci. 2014, 103, 2829–2838. [Google Scholar] [CrossRef]
- Fosca, M.; Rau, J.V.; Uskoković, V. Factors Influencing the Drug Release from Calcium Phosphate Cements. Bioact. Mater. 2022, 7, 341–363. [Google Scholar] [CrossRef]
- Buxadera-Palomero, J.; Fricke, K.; Reuter, S.; Javier, F.; Rodriguez, D.; Canal, C. One-Step Liquid Phase Polymerization of HEMA by Atmospheric-Pressure Plasma Discharges for Ti Dental Implants. Appl. Sci. 2021, 11, 662. [Google Scholar] [CrossRef]
- Lee, J.H.; Han, W.J.; Jang, H.S.; Choi, H.J. Highly Tough, Biocompatible, and Magneto-Responsive Fe3O4/Laponite/PDMAAm Nanocomposite Hydrogels. Sci. Rep. 2019, 9, 15024. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Designation | CDMAM * (mol/L) | ** |
|---|---|---|
| P1 | PHEMA SNs | 0 |
| P125 | 1.25 | 0.07 |
| P250 | 2.50 | 0.15 |
| P500 | 5.00 | 0.31 |
| PDMAM | PDMAM SNs | 1 |
| P1 | P125 | P250 | P500 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Zero order | k0 | R2 | k0 | R2 | k0 | R2 | k0 | R2 | ||||
| 0.106 | 0.930 | 0.241 | 0.833 | 0.228 | 0.973 | 0.388 | 0.989 | |||||
| First order | k1 | R2 | k1 | R2 | k1 | R2 | k1 | R2 | ||||
| −0.046 | 0.939 | −0.09 | 0.959 | −0.074 | 0.978 | −0.097 | 0.985 | |||||
| Higuchi | kH | R2 | kH | R2 | kH | R2 | kH | R2 | ||||
| 0.32 | 0.981 | 0.741 | 0.973 | 0.684 | 0.994 | 1.28 | 0.992 | |||||
| Korsmeyer-Peppas | kKP | n | R2 | kKP | n | R2 | kKP | n | R2 | kKP | n | R2 |
| 0.193 | 0.338 | 0.987 | 0.266 | 0.358 | 0.991 | 0.196 | 0.386 | 0.979 | 0.263 | 0.421 | 0.986 | |
| P1 | P125 | P250 | P500 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Zero order | k0 | R2 | k0 | R2 | k0 | R2 | k0 | R2 | ||||
| 0.046 | 0.871 | 0.073 | 0.937 | 0.108 | 0.873 | 0.229 | 0.975 | |||||
| First order | k1 | R2 | k1 | R2 | k1 | R2 | k1 | R2 | ||||
| 0.08 | 0.887 | −0.142 | 0.943 | −0.211 | 0.928 | −0.494 | 0.969 | |||||
| Higuchi | kH | R2 | kH | R2 | kH | R2 | kH | R2 | ||||
| 0.141 | 0.956 | 0.224 | 0.981 | 0.319 | 0.969 | 0.663 | 0.982 | |||||
| Korsmeyer-Peppas | kKP | n | R2 | kKP | n | R2 | kKP | n | R2 | kKP | n | R2 |
| 0.142 | 0.357 | 0.988 | 0.151 | 0.426 | 0.991 | 0.202 | 0.408 | 0.998 | 0.179 | 0.550 | 0.989 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simeonov, M.; Kostova, B.; Vassileva, E. Interpenetrating Polymer Networks of Poly(2-hydroxyethyl methacrylate) and Poly(N, N-dimethylacrylamide) as Potential Systems for Dermal Delivery of Dexamethasone Phosphate. Pharmaceutics 2023, 15, 2328. https://doi.org/10.3390/pharmaceutics15092328
Simeonov M, Kostova B, Vassileva E. Interpenetrating Polymer Networks of Poly(2-hydroxyethyl methacrylate) and Poly(N, N-dimethylacrylamide) as Potential Systems for Dermal Delivery of Dexamethasone Phosphate. Pharmaceutics. 2023; 15(9):2328. https://doi.org/10.3390/pharmaceutics15092328
Chicago/Turabian StyleSimeonov, Marin, Bistra Kostova, and Elena Vassileva. 2023. "Interpenetrating Polymer Networks of Poly(2-hydroxyethyl methacrylate) and Poly(N, N-dimethylacrylamide) as Potential Systems for Dermal Delivery of Dexamethasone Phosphate" Pharmaceutics 15, no. 9: 2328. https://doi.org/10.3390/pharmaceutics15092328