
Discriminative Dissolution Method Using the Open-Loop Configuration of the USP IV Apparatus to Compare Dissolution Profiles of Metoprolol Tartrate Immediate-Release Tablets: Use of Kinetic Parameters
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Dissolution Profile Studies
2.2.1. Test 1: Selection of Dissolution Media
2.2.2. Test 2: Dissolution Profiles in Apparatus II USP
2.2.3. Test 3: Dissolution Profiles in Apparatus IV USP (Open-Loop Configuration)
2.3. Similarity Evaluation
2.3.1. f1, f2, and Bootstrap f2 Approaches
2.3.2. ANOVA-Based Method
2.3.3. Dissolution Efficiency
2.3.4. Multivariate Statistical Distance (MSD)
2.3.5. Time Series Approach
2.3.6. Dependent Model: Fit to Mathematical Models
2.3.7. Kinetic Parameters: Non-Cumulative Dissolution Profiles
3. Results and Discussion
3.1. Selection of Dissolution Media (Test 1)
3.2. Dissolution Profiles Obtained in the USP II Apparatus (Test 2)
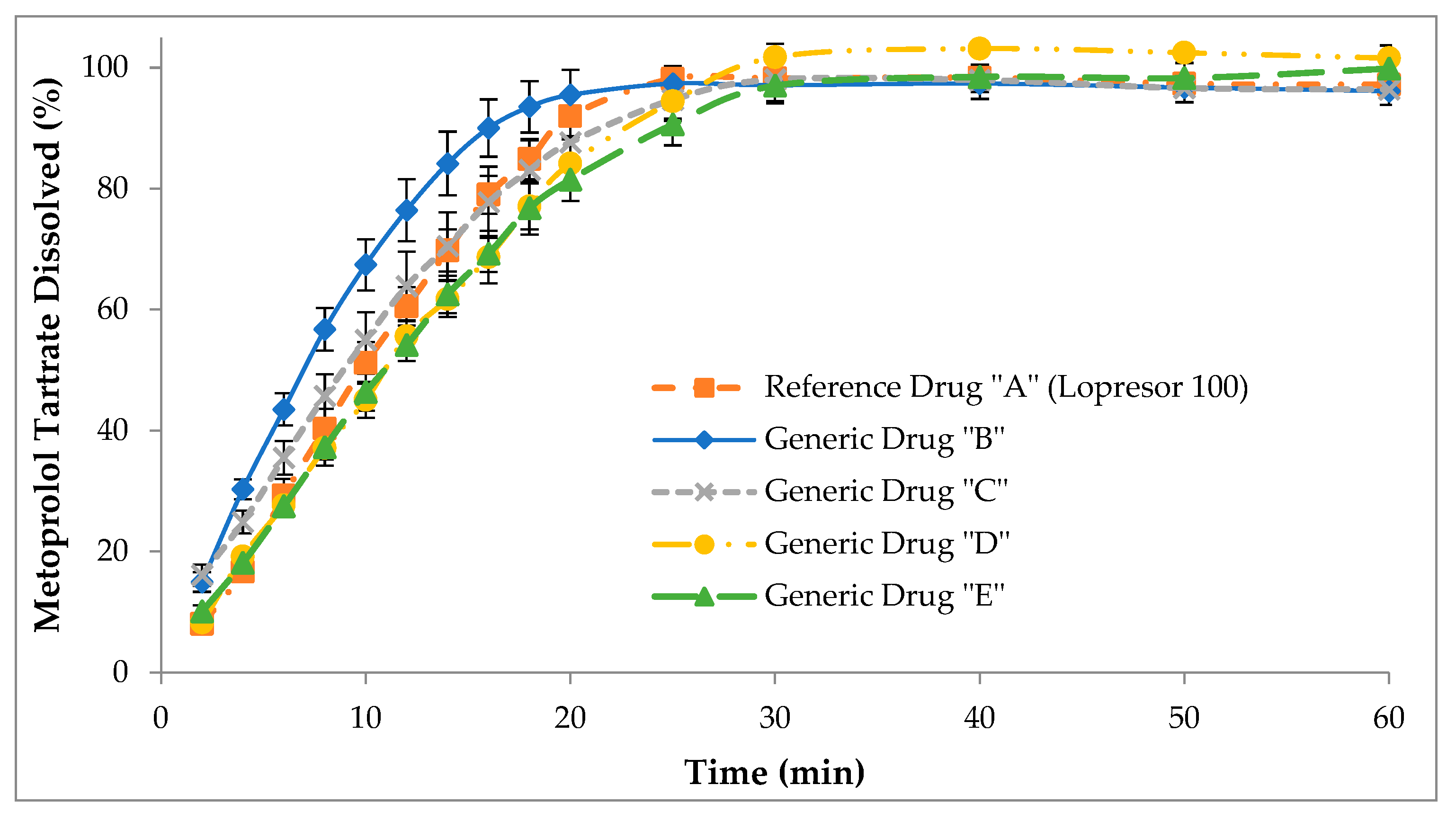
3.3. Dissolution Profiles Obtained in the USP IV Apparatus (Test 3)
3.4. Similarity Evaluation
3.4.1. f1, f2, and Bootstrap f2 Approaches
3.4.2. ANOVA-Based Method
3.4.3. Dissolution Efficiency
3.4.4. Multivariate Statistical Distance (MSD)
3.4.5. Time Series Approach
3.4.6. Dependent Models: Fit to Mathematical Models
3.4.7. Independent Model Comparison for Non-Accumulated Data (USP IV Apparatus)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Muselík, J.; Komersová, A.; Kubová, K.; Matzick, K.; Skalická, B. A critical overview of FDA and EMA statistical methods to compare in vitro drug dissolution profiles of pharmaceutical products. Pharmaceutics 2021, 13, 1703. [Google Scholar] [CrossRef]
- Anand, O.M.; Yu, L.X.; Conner, D.P.; Davit, B.M. Dissolution testing for generic drugs: An FDA perspective. AAPS J. 2011, 13, 328–335. [Google Scholar] [CrossRef]
- Mehtani, D.; Seth, A.; Sharma, P.; Maheshwari, R.; Abed, S.N.; Deb, P.K.; Chougule, M.B.; Tekade, R.K. Dissolution profile consideration in pharmaceutical product development. In Dosage Form Design Considerations; Elsevier: Amsterdam, The Netherlands, 2018; pp. 287–336. [Google Scholar]
- Yi, H.; Liu, F.; Zhang, G.; Cheng, Z. Evaluation of a Modified Flow-Through Method for Predictive Dissolution and In Vitro/In Vivo Correlations of Immediate Release and Extended Release Formulations. J. Nanomater. 2021, 2021, 9956962. [Google Scholar] [CrossRef]
- Zhang, G.; Sun, M.; Jiang, S.; Wang, L.; Tan, Y.; Cheng, Z. Investigating a Modified Apparatus to Discriminate the Dissolution Capacity In Vitro and Establish an IVIVC of Mycophenolate Mofetil Tablets in the Fed State. J. Pharm. Sci. 2021, 110, 1240–1247. [Google Scholar] [CrossRef]
- Singh, I.; Aboul-Enein, H.Y. Advantages of USP apparatus IV (flow-through cell apparatus) in dissolution studies. J. Iran. Chem. Soc. 2006, 3, 220–222. [Google Scholar] [CrossRef]
- Prabhu, N.B.; Marathe, A.S.; Jain, S.; Singh, P.P.; Sawant, K.; Rao, L.; Amin, P.D. Comparison of dissolution profiles for sustained release resinates of BCS class i drugs using USP apparatus 2 and 4: A technical note. AAPS PharmSciTech 2008, 9, 769–773. [Google Scholar] [CrossRef]
- Gao, Z. In vitro dissolution testing with flow-through method: A technical note. Aaps Pharmscitech 2009, 10, 1401. [Google Scholar] [CrossRef]
- Siew, A. Dissolution testing. Pharm. Technol. 2016, 40, 56–64. [Google Scholar]
- Yoshida, H.; Shibata, H.; Izutsu, K.; Goda, Y. Comparison of dissolution similarity assessment methods for products with large variations: F2 statistics and model-independent multivariate confidence region procedure for dissolution profiles of multiple oral products. Biol. Pharm. Bull. 2017, 40, 722–725. [Google Scholar] [CrossRef]
- Kollipara, S.; Boddu, R.; Ahmed, T.; Chachad, S. Simplified model-dependent and model-independent approaches for dissolution profile comparison for oral products: Regulatory perspective for generic product development. AAPS PharmSciTech 2022, 23, 53. [Google Scholar] [CrossRef]
- Index, M. An Encyclopedia of Chemicals and Drugs and Biologicals, 13th ed.; Merck and Co. Inc.: New Jersey, NJ, USA, 2001. [Google Scholar]
- Narendra, C.; Srinath, M.S.; Rao, B.P. Development of three layered buccal compact containing metoprolol tartrate by statistical optimization technique. Int. J. Pharm. 2005, 304, 102–114. [Google Scholar] [CrossRef]
- Peikova, L.; Pencheva, I.; Tzvetkova, B. Chemical stability-indicating HPLC study of fixed-dosage combination containing metoprolol tartrate and hydrochlorothiazide. J. Chem. Pharm. Res 2013, 5, 132–140. [Google Scholar]
- FDA Guidence for Industry. Dissolution Testing of Immediate Release Solid Oral Dosage Forms. Available online: chrome-extension://efaidnbmnnnibpcajpcglclefindmkaj/https://www.fda.gov/media/70936/download (accessed on 31 May 2023).
- European Medicines Agency. Guideline on the Investigation of Bioequivalence; European Medicines Agency: Amsterdam, The Netherlands, 2010.
- Diaz, D.A.; Colgan, S.T.; Langer, C.S.; Bandi, N.T.; Likar, M.D.; Van Alstine, L. Dissolution similarity requirements: How similar or dissimilar are the global regulatory expectations? AAPS J. 2016, 18, 15–22. [Google Scholar] [CrossRef]
- Khan, K.A. The concept of dissolution efficiency. J. Pharm. Pharmacol. 1975, 27, 48–49. [Google Scholar] [CrossRef]
- Ruiz, M.E.; Volonté, M.G. Biopharmaceutical relevance of dissolution profile comparison: Proposal of a combined approach. Dissolution Technol. 2014, 21, 32–43. [Google Scholar] [CrossRef]
- Tsong, Y.; Hammerstrom, T.; Sathe, P.; Shah, V.P. Statistical assessment of mean differences between two dissolution data sets. Drug Inf. J. 1996, 30, 1105–1112. [Google Scholar] [CrossRef]
- Paixão, P.; Gouveia, L.F.; Silva, N.; Morais, J.A.G. Evaluation of dissolution profile similarity–Comparison between the f2, the multivariate statistical distance and the f2 bootstrapping methods. Eur. J. Pharm. Biopharm. 2017, 112, 67–74. [Google Scholar] [CrossRef]
- Saranadasa, H. Defining the similarity of dissolution profiles using Hotelling’s [T. sup. 2] statistic. Pharm. Technol. Asia 2002, 15, 26–32. [Google Scholar]
- Gill, J.L. Design and analysis of experiments in the animal and medical sciences. Biometrics 1980, 36, 741. [Google Scholar]
- Chow, S.-C.; Fanny, Y.K. Statistical comparison between dissolution profiles of drug products. J. Biopharm. Stat. 1997, 7, 241–258. [Google Scholar] [CrossRef]
- Medina-López, R.; Guillén-Moedano, S.; Hurtado, M. In vitro release studies of furosemide reference tablets: Influence of agitation rate, USP apparatus and dissolution media. ADMET DMPK 2020, 8, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Juela, D.; Vera, M.; Cruzat, C.; Alvarez, X.; Vanegas, E. Mathematical modeling and numerical simulation of sulfamethoxazole adsorption onto sugarcane bagasse in a fixed-bed column. Chemosphere 2021, 280, 130687. [Google Scholar] [CrossRef] [PubMed]
- Sathe, P.M.; Tsong, Y.; Shah, V.P. In-vitro dissolution profile comparison: Statistics and analysis, model dependent approach. Pharm. Res. 1996, 13, 1799–1803. [Google Scholar] [CrossRef]
- Zhang, Y.; Huo, M.; Zhou, J.; Zou, A.; Li, W.; Yao, C.; Xie, S. DDSolver: An add-in program for modeling and comparison of drug dissolution profiles. AAPS J. 2010, 12, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.; Pateman, T. Pre-clinical pharmacokinetics. In A Handbook of Bioanalysis and Drug Metabolism; CRC Press: Boca Raton, FL, USA, 2021; pp. 113–131. ISBN 0203642538. [Google Scholar]
- Endashaw, E.; Tatiparthi, R.; Mohammed, T.; Tefera, Y.M.; Teshome, H.; Duguma, M. Dissolution Profile Evaluation of Seven Brands of Amoxicillin-Clavulanate Potassium 625 mg Tablets Retailed in Hawassa Town, Sidama Regional State, Ethiopia. 2022; preprint. [Google Scholar] [CrossRef]
- Suarez-Sharp, S.; Delvadia, P.R.; Dorantes, A.; Duan, J.; Externbrink, A.; Gao, Z.; Ghosh, T.; Miksinski, S.P.; Seo, P. Regulatory perspectives on strength-dependent dissolution profiles and biowaiver approaches for Immediate Release (IR) oral tablets in new drug applications. AAPS J. 2016, 18, 578–588. [Google Scholar] [CrossRef]
- Hoffelder, T. Comparison of dissolution profiles: A statistician’s perspective. Ther. Innov. Regul. Sci. 2018, 52, 423–429. [Google Scholar] [CrossRef]
- Gray, V.A. Dissolution Testing in the Pharmaceutical Laboratory. Anal. Test. Pharm. GMP Lab. 2022, 7, 206–250. [Google Scholar]
- Speer, I.; Preis, M.; Breitkreutz, J. Dissolution testing of oral film preparations: Experimental comparison of compendial and non-compendial methods. Int. J. Pharm. 2019, 561, 124–134. [Google Scholar] [CrossRef]
- Fotaki, N.; Brown, W.; Kochling, J.; Chokshi, H.; Miao, H.; Tang, K.; Gray, V. Rationale for selection of dissolution media: Three case studies. Dissolut. Technol 2013, 20, 6–13. [Google Scholar] [CrossRef]
- European Medicines Agency. Reflection Paper on the Dissolution Specification for Generic Solid Oral Immediate Release Products with Systemic Action (EMA/CHMP/CVMP/QWP/336031/2017); European Medicines Agency: Amsterdam, The Netherlands, 2017.
- Mudie, D.M.; Samiei, N.; Marshall, D.J.; Amidon, G.E.; Bergström, C.A.S. Selection of in vivo predictive dissolution media using drug substance and physiological properties. AAPS J. 2020, 22, 1–13. [Google Scholar] [CrossRef]
- Rekhi, G.S.; Eddington, N.D.; Fossler, M.J.; Schwartz, P.; Lesko, L.J.; Augsburger, L.L. Evaluation of in vitro release rate and in vivo absorption characteristics of four metoprolol tartrate immediate-release tablet formulations. Pharm. Dev. Technol. 1997, 2, 11–24. [Google Scholar] [CrossRef]
- Roush, J.A. The role of the stomach in drug absorption as observed via absorption rate analysis. Int. J. Pharm. 2014, 471, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Holford, N. Absorption and half-life. Transl. Clin. Pharmacol. 2016, 24, 157–160. [Google Scholar] [CrossRef]
- Yska, J.P.; Wanders, J.T.M.; Odigie, B.; Apers, J.A.; Emous, M.; Totté, E.R.E.; Boerma, E.C.; Ubels, F.L.; Woerdenbag, H.J.; Frijlink, H.W. Effect of Roux-en-Y gastric bypass on the bioavailability of metoprolol from immediate and controlled release tablets: A single oral dose study before and after surgery. Eur. J. Hosp. Pharm. 2020, 27, e19–e24. [Google Scholar] [CrossRef] [PubMed]
- Beyssac, E.; Lavigne, J. Dissolution study of active pharmaceutical ingredients using the flow through apparatus USP 4. Dissolution Technol. 2005, 12, 23–25. [Google Scholar] [CrossRef]
- Medina-Lopez, J.R.; Dominguez-Reyes, A.; Hurtado, M. Comparison of Generic Furosemide Products by In Vitro Release Studies using USP Apparatus 2 and 4. Dissolution Technol. 2021, 28, 14–23. [Google Scholar] [CrossRef]
- Bai, G.E.; Armenante, P.M.; Plank, R.V.; Gentzler, M.; Ford, K.; Harmon, P. Hydrodynamic investigation of USP dissolution test apparatus II. J. Pharm. Sci. 2007, 96, 2327–2349. [Google Scholar] [CrossRef]
- Fotaki, N. Flow-through cell apparatus (USP apparatus 4): Operation and features. Dissolution Technol. 2011, 18, 46–49. [Google Scholar] [CrossRef]
- Suarez-Sharp, S.; Abend, A.; Hoffelder, T.; Leblond, D.; Delvadia, P.; Kovacs, E.; Diaz, D.A. In vitro dissolution profiles similarity assessment in support of drug product quality: What, how, when—Workshop summary report. AAPS J. 2020, 22, 1–12. [Google Scholar] [CrossRef]
- Liu, S.; Cai, X.; Shen, M.; Tsong, Y. In vitro dissolution profile comparison using bootstrap bias corrected similarity factor, f2. J. Biopharm. Stat. 2023; online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.B.; Robertson, V.K.; Rawat, A.; Flick, T.; Tang, Z.J.; Cauchon, N.S.; McElvain, J.S. Development and application of a biorelevant dissolution method using USP apparatus 4 in early phase formulation development. Mol. Pharm. 2010, 7, 1466–1477. [Google Scholar] [CrossRef] [PubMed]
- Mangas-Sanjuan, V.; Colon-Useche, S.; Gonzalez-Alvarez, I.; Bermejo, M.; Garcia-Arieta, A. Assessment of the regulatory methods for the comparison of highly variable dissolution profiles. AAPS J. 2016, 18, 1550–1561. [Google Scholar] [CrossRef]
- Simionato, L.D.; Petrone, L.; Baldut, M.; Bonafede, S.L.; Segall, A.I. Comparison between the dissolution profiles of nine meloxicam tablet brands commercially available in Buenos Aires, Argentina. Saudi Pharm. J. 2018, 26, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Jamil, R.; Polli, J.E. Sources of dissolution variability into biorelevant media. Int. J. Pharm. 2022, 620, 121745. [Google Scholar] [CrossRef]
- Polli, J.E.; Rekhi, G.S.; Augsburger, L.L.; Shah, V.P. Methods to compare dissolution profiles and a rationale for wide dissolution specifications for metoprolol tartrate tablets. J. Pharm. Sci. 1997, 86, 690–700. [Google Scholar] [CrossRef]
- O’hara, T.; Dunne, A.; Butler, J.; Devane, J.; Group, I.C.W. A review of methods used to compare dissolution profile data. Pharm. Sci. Technolo. Today 1998, 1, 214–223. [Google Scholar] [CrossRef]
- Shojaee, S.; Nokhodchi, A.; Maniruzzaman, M. Evaluation of the drug solubility and rush ageing on drug release performance of various model drugs from the modified release polyethylene oxide matrix tablets. Drug Deliv. Transl. Res. 2017, 7, 111–124. [Google Scholar] [CrossRef]
- Khan, K.A.; Rhodes, C.T. Effect of compaction pressure on the dissolution efficiency of some direct compression systems. Pharm. Acta Helv. 1972, 47, 594–607. [Google Scholar]
- Anderson, N.H.; Bauer, M.; Boussac, N.; Khan-Malek, R.; Munden, P.; Sardaro, M. An evaluation of fit factors and dissolution efficiency for the comparison of in vitro dissolution profiles. J. Pharm. Biomed. Anal. 1998, 17, 811–822. [Google Scholar] [CrossRef]
- Cardot, J.-M.; Roudier, B.; Schütz, H. Dissolution comparisons using a Multivariate Statistical Distance (MSD) test and a comparison of various approaches for calculating the measurements of dissolution profile comparison. AAPS J. 2017, 19, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Costa, P.; Lobo, J.M.S. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef] [PubMed]
- USP43–NF38 The United States pharmacopeia 43. The National formulary 38. USP Monogr. Metoprolol Tartrate Tablets. 2023, 42, 2926. [CrossRef]
- Hsu, J.-Y.; Hsu, M.-Y.; Liao, C.-C.; Hsu, H.-C. On the characteristics of the FDA’s similarity factor for comparison of drug dissolution. J. Food Drug Anal. 1998, 6, 5. [Google Scholar] [CrossRef]
- Ekenna, I.C.; Abali, S.O. Comparison of the Use of Kinetic Model Plots and DD Solver Software to Evaluate the Drug Release from Griseofulvin Tablets. J. Drug Deliv. Ther. 2022, 12, 5–13. [Google Scholar] [CrossRef]
- Bruschi, M.L. (Ed.) 5–Mathematical models of drug release. In Strategies to Modify the Drug Release from Pharmaceutical Systems; Woodhead Publishing: Cambridge, UK, 2015; pp. 63–86. ISBN 978-0-08-100092-2. [Google Scholar]
- Danyuo, Y.; Ani, C.J.; Salifu, A.A.; Obayemi, J.D.; Dozie-Nwachukwu, S.; Obanawu, V.O.; Akpan, U.M.; Odusanya, O.S.; Abade-Abugre, M.; McBagonluri, F. Anomalous release kinetics of prodigiosin from poly-N-isopropyl-acrylamid based hydrogels for the treatment of triple negative breast cancer. Sci. Rep. 2019, 9, 3862. [Google Scholar] [CrossRef]
- Ahsan, W.; Alam, S.; Javed, S.; Alhazmi, H.A.; Albratty, M.; Najmi, A.; Sultan, M.H. Study of Drug Release Kinetics of Rosuvastatin Calcium Immediate-Release Tablets Marketed in Saudi Arabia. Dissolution Technol 2022, 29, GC1. [Google Scholar] [CrossRef]
- Tsong, Y.; Sathe, P.M.; Shah, V.P. In vitro dissolution profile comparison. In Encyclipedia Biopharmaceutical Statistics, 2nd ed.; Marcel Dekker, Inc.: New York, NY, USA, 2003; p. 456J462. [Google Scholar]
- LeBlond, D.; Altan, S.; Novick, S.; Peterson, J.; Shen, Y.; Yang, H. In vitro dissolution curve comparisons: A critique of current practice. Dissolution Technol 2016, 23, 14–23. [Google Scholar] [CrossRef]
- FDA, U.S. Bioavailability Studies Submitted in NDAs or INDs—General Considerations and Guidance for Industry; Silver Spring: Rockville, MD, USA, 2022. [Google Scholar]
- Han, Y.R.; Lee, P.I.; Pang, K.S. Finding Tmax and Cmax in multicompartmental models. Drug Metab. Dispos. 2018, 46, 1796–1804. [Google Scholar] [CrossRef]
- Gray, V.A. Power of the dissolution test in distinguishing a change in dosage form critical quality attributes. AAPS PharmSciTech 2018, 19, 3328–3332. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Equation |
|---|---|
| Zero-order | |
| First-order | |
| Higuchi | |
| Hixon–Crowell | |
| Korsmeyer–Peppas | |
| Weibull | |
| Gompertz | |
| Logistic |
| Drug | Difference Factor (f1) | Similarity Factor (f2) | Bootstrap f2 | |||
|---|---|---|---|---|---|---|
| USP Dissolution Apparatus | ||||||
| II | IV | II | IV | II | IV | |
| Generic Drug “B” | 30.65 * | 58.78 | 42.76 | 36.58 | 39.84 | 34.60 |
| Generic Drug “C” | 8.13 | 18.32 | 64.60 | 53.78 | 59.96 | 50.81 |
| Generic Drug “D” | 9.89 | 10.99 | 60.39 | 65.30 | 55.25 | 61.05 |
| Generic Drug “E” | 10.40 | 26.99 | 59.47 | 46.46 | 54.76 | 44.09 |
| Drug | USP II Apparatus | USP IV Apparatus | ||||
|---|---|---|---|---|---|---|
| Two-Factor p Value | 95% CI | Decision | Two-Factor p Value | 95% CI | Decision | |
| Generic Drug “B” | <0.01 | Significant differences at 4–16 min | Non-similar | <0.01 | Significant differences at 4–20 min | Non-similar |
| Generic Drug “C” | 0.10 | No significant differences | Similar | <0.01 | Significant differences at 16–35 min | Non-similar |
| Generic Drug “D” | <0.01 | Significant differences at 16 min | Non-similar | <0.01 | Significant differences at 35 and 30 min | Non-similar |
| Generic Drug “E” | <0.01 | Significant differences at 16 and 20 min | Non-similar | <0.01 | Significant differences at 10 and 35 min | Non-similar |
| Drug | USP II Apparatus | USP IV Apparatus | ||
|---|---|---|---|---|
| DE % 90% CI for Mean Ratio G/R | Decision 1 | DE % 90% CI for Mean Ratio G/R | Decision | |
| Generic Drug “B” | 117.91 114.68–121.24 | Non-similar | 117.71 115.39–120.07 | Non-similar |
| Generic Drug “C” | 106.58 103.57–109.67 | Similar | 90.19 88.93–91.47 | Non-similar |
| Generic Drug “D” | 95.72 93.02–98.51 | Similar | 94.54 92.92–96.20 | Similar |
| Generic Drug “E” | 98.56 95.88–101.31 | Similar | 83.40 82.07–84.76 | Non-similar |
| Sampling Time (min) | 90% Confidence Intervals (90% CI) | |||
|---|---|---|---|---|
| USP II Apparatus | ||||
| Generic Drug B | Generic Drug C | Generic Drug D | Generic Drug E | |
| 2 | [5.65, 8.23] | [7.13, 9.03] | [−0.73, 1.23] | [1.02, 3.16] |
| 4 | [12.44, 14.81] | [7.16, 9.24] | [1.41, 3.64] | [−0.07, 2.96] |
| 6 | [12.74, 15.80] | [4.60, 7.91] | [−3.42, 0.20] | [−3.83, 0.34] |
| 8 | [14.42, 18.35] | [3.46, 7.26] | [−5.18, −1.29] | [−5.67, −0.58] |
| 10 | [13.94, 18.52] | [1.21, 6.42] | [−8.09, −4.11] | [−7.82, −1.82] |
| 12 | [13.65, 18.20] | [−0.03, 6.82] | [−6.70, −3.12] | [−9.15, −3,62] |
| 14 | [11.82, 16.93] | [−2.65, 4.11] | [−10.29, −5.73] | [−10.37, −4.16] |
| 16 | [8.83, 13.19] | [−4.01, 1.52] | [−12.91, −7.70] | [−12.55, −7.03] |
| 18 | [6.80, 10.45] | [−4.21, 0.57] | [−10.54, −5.12] | [−11.77, −4.42] |
| 20 | [1.99, 4.99] | [−6.50, −2.19] | [−10.56, −5.11] | [−13.81, −7.25] |
| 25 | [−2.56, 0.95] | [−5.50, −1.41] | [−5.68, −1.79] | [−9.55, −5.53] |
| Conclusion | Non-similar | Similar | Non-similar | Non-similar |
| Sampling Time (min) | 90% Confidence Intervals (90% CI) | |||
|---|---|---|---|---|
| USP IV Apparatus | ||||
| Generic Drug B | Generic Drug C | Generic Drug D | Generic Drug E | |
| 1 | [1.66, 2.24] | [0.67, 1.05] | [0.75, 1.09] | [0.33, 068] |
| 2 | [4.15, 5.35] | [1.90, 2.49] | [1.95, 2.49] | [0.51,1.16] |
| 3 | [6.61, 8.62] | [2.35, 3.39] | [2.54, 3.45] | [−0.17, 1.01] |
| 4 | [9.38, 12.08] | [2.28, 3.78] | [2.53, 3.77] | [−1.19, 0.43] |
| 5 | [12.01, 15.24] | [1.96, 3.47] | [2.11, 3.42] | [−2.52, −0.75] |
| 6 | [14.38, 18.38] | [1.25, 2.86] | [1.33, 2.78] | [−4.27, −2.25] |
| 7 | [16.27, 21.05] | [−0.01, 1.76] | 0.28, 1.92] | [−6.28, −3.90] |
| 8 | [18.08, 23.58] | [−1.33, 0.63] | [−0.43, 1.45] | [−8.02, −5.21] |
| 10 | [20.70, 27.41] | [−4.03, −1.68] | [−1.92, 0.44] | [−11.00, −7.51] |
| 12 | [22.05, 30.09] | [−6.65, −3.87] | [−3.29, −046] | [−13.41, −9.41] |
| 14 | [21.88, 30.81] | [−9.50, −6.17] | [−4.76, −057] | [−15.97, −11.39] |
| 16 | [20.66, 29.09] | [−12.11, −8.34] | [−6.28, −2.93] | [−18.28, −13.44] |
| 18 | [18.27, 26.07] | [−14.63, −10.50] | [−7.99, −4.37] | [−20.47, −15.37] |
| 20 | [13.77, 21.26] | [−17.07, −12.57] | [−9.88, −6.02] | [−22.68, −17.27] |
| 25 | [2.06, 9.49] | [−23.87, −17.56] | [−15.86, −10.73] | [−27.94, −21.25] |
| 30 | [−0.72, 6.25] | [−19.95, −13.46] | [−11.69, −6.39] | [−22.10, −15.81] |
| 35 | [−1.73, 5.04] | [−14.47, −8.15] | [−7.06, −2.26] | [−14.20, −8.02] |
| 40 | [−2.14, 4.67] | [−9.10, −2.56] | [−4.70, 0.43] | [−7.97, −2.26] |
| Decision | Non-similar | No-similar | Non-similar | Non-similar |
| Drug | USP II Apparatus | USP IV Apparatus | ||||
|---|---|---|---|---|---|---|
| 95% CI (Global Similarity) | 95% CI (Local Similarity) | Decision | 95% CI (Global Similarity) | 95% CI (Local Similarity) | Decision | |
| Generic Drug “B” | 123.01–144.84 | Significant differences before 18 min | Non-similar | 157.67–215.20 | Significant differences at all times of the profile | Non-similar |
| Generic Drug “C” | 108.50–126.72 | Significant differences before 12 min | Non-similar | 97.10–129.22 | Non-similar | |
| Generic Drug “D” | 82.11–107.90 | Significant differences at 4 min and between 8–20 min | Non-similar | 103.95–133.50 | Non-similar | |
| Generic Drug “E” | 73.50–119.10 | Significant differences at all times, except at 6 min | Non-similar | 63.57–116.63 | Non-similar | |
| Limit of similarity: 87.50–114.29% 1 | ||||||
| Model | Statistics 1 | USP II Apparatus | USP IV Apparatus | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference “A” | Generic “B” | Generic “C” | Generic “D” | Generic “E” | Reference “A” | Generic “B” | Generic “C” | Generic “D” | Generic “E” | ||
| Zero-order | r2 | 0.9922 | 0.9166 | 0.9502 | 0.9898 | 0.9929 | 0.9749 | 0.9657 | 0.9919 | 0.9953 | 0.9883 |
| k0 (%Dis ∗ min−1) | 4.9024 | 5.9298 | 5.0479 | 4.4087 | 4.4161 | 3.3844 | 4.9917 | 2.8718 | 3.1668 | 2.4538 | |
| MSE | 5.8329 | 63.9021 | 28.3103 | 5.4318 | 3.7852 | 14.3348 | 32.9669 | 1.8453 | 1.9696 | 3.1387 | |
| AIC | 36.2512 | 57.9025 | 50.2955 | 33.8004 | 31.2852 | 74.3746 | 85.3320 | 48.9331 | 46.5559 | 53.3278 | |
| First-order | r2 | 0.9286 | 0.9612 | 0.9777 | 0.9602 | 96.3200 | 0.9144 | 0.9403 | 0.9842 | 0.9654 | 0.9583 |
| k1 (min−1) | 0.0770 | 0.1136 | 0.0844 | 0.0660 | 0.0663 | 0.0447 | 0.0827 | 0.0370 | 0.0417 | 0.0300 | |
| MSE | 53.1229 | 31.1351 | 13.0971 | 22.6939 | 20.2464 | 48.8620 | 58.3842 | 5.1922 | 14.4517 | 11.2917 | |
| AIC | 56.4263 | 50.4021 | 42.4009 | 47.7845 | 47.2864 | 91.9112 | 92.9871 | 58.6913 | 74.7632 | 71.5421 | |
| Higuchi | r2 | 0.8133 | 0.9128 | 0.9075 | 0.8409 | 0.8451 | 0.7250 | 0.8315 | 0.8205 | 0.7920 | 0.7554 |
| kH (%Dis ∗ min−1/2) | 17.1046 | 21.0966 | 17.9065 | 15.4558 | 15.4901 | 11.5370 | 0.1756 | 10.0318 | 10.9832 | 8.4341 | |
| MSE | 138.6870 | 67.5427 | 51.8659 | 88.2738 | 83.5642 | 155.4990 | 156.9909 | 56.9645 | 85.8074 | 65.9340 | |
| AIC | 65.0834 | 58.3930 | 56.1195 | 60.8167 | 60.4724 | 108.4083 | 108.3192 | 94.2654 | 100.1338 | 96.4672 | |
| Hixson–Crowell | r2 | 0.9621 | 0.9908 | 0.9942 | 0.9814 | 0.9848 | 0.9377 | 0.9688 | 0.9940 | 0.9800 | 0.9704 |
| ks (%Dis ∗ min−1/3) | 0.0223 | 0.0313 | 0.0240 | 0.0194 | 0.0194 | 0.0137 | 0.0239 | 0.0114 | 0.0127 | 0.0094 | |
| MSE | 28.1260 | 7.5387 | 3.4102 | 10.7124 | 8.4516 | 35.5652 | 30.6325 | 2.6745 | 8.3986 | 7.9981 | |
| AIC | 50.6311 | 36.0721 | 30.1136 | 40.2526 | 38.9975 | 87.3931 | 83.4847 | 45.8812 | 67.0090 | 66.6547 | |
| Korsmeyer–Peppas | r2 | 0.9933 | 0.9847 | 0.9961 | 0.9945 | 0.9974 | 0.9936 | 0.9746 | 0.9954 | 0.9963 | 0.9975 |
| kk (%Dis ∗ min−n) | 5.0637 | 11.8948 | 9.1967 | 5.3606 | 5.4351 | 1.8844 | 6.6770 | 3.1345 | 2.8404 | 1.6715 | |
| n | 0.9920 | 0.7322 | 0.7703 | 0.9264 | 0.9217 | 1.2229 | 0.8935 | 0.9679 | 1.0423 | 1.1469 | |
| MSE | 5.6997 | 13.8821 | 2.6908 | 3.5050 | 1.6278 | 3.9871 | 26.6685 | 1.5489 | 1.6252 | 0.7375 | |
| AIC | 36.9205 | 43.8078 | 27.3260 | 28.0736 | 24.7320 | 56.6951 | 83.1922 | 44.7129 | 44.7520 | 30.4641 | |
| Weibull Parameters | Ln Differences | USP II Apparatus | |||
|---|---|---|---|---|---|
| Generic Drug “B” | Generic Drug “C” | Generic Drug “D” | Generic Drug “E” | ||
| 1 | 90% CI | −0.125 to −0.090 | 0.403 to 0.454 | 0.078 to 0.184 | 0.157 to 0.248 |
| 2 STD Similarity region 3 | −0.045–0.045 | ||||
| 2 | 90% CI | −0.189 to −0.072 | −0.122 to −0.107 | −0.071 to −0.037 | −0.077 to −0.055 |
| 2 STD Similarity region | −0.008–0.008 | ||||
| Decision | Non-similar | Non-similar | Non-similar | Non-similar | |
| Weibull Parameters | Ln Differences | USP IV Apparatus | |||
|---|---|---|---|---|---|
| Generic Drug “B” | Generic Drug “C” | Generic Drug “D” | Generic Drug “E” | ||
| 1 | 90% CI | 0.484 to −0.654 | 0.374 to 0.521 | 0.333 to 0.461 | 0.164 to 0.319 |
| 2 STD Similarity region 3 | −0.093–0.0933 | ||||
| 2 | 90% CI | −0.084 to −0.048 | −0.147 to −0.114 | −0.118 to −0.092 | −0.117 to −0.085 |
| 2 STD Similarity region | −0.019–0.019 | ||||
| Decision | Non-similar | Non-similar | Non-similar | Non-similar | |
| Parameter | Geometric Mean ± SE | Geometric Point Estimate Ratio | |||||||
|---|---|---|---|---|---|---|---|---|---|
| (90% CI) | (90% CI) | ||||||||
| Reference (A) | Generic (B) | Generic (C) | Generic (D) | Generic (E) | B/A | C/A | D/A | E/A | |
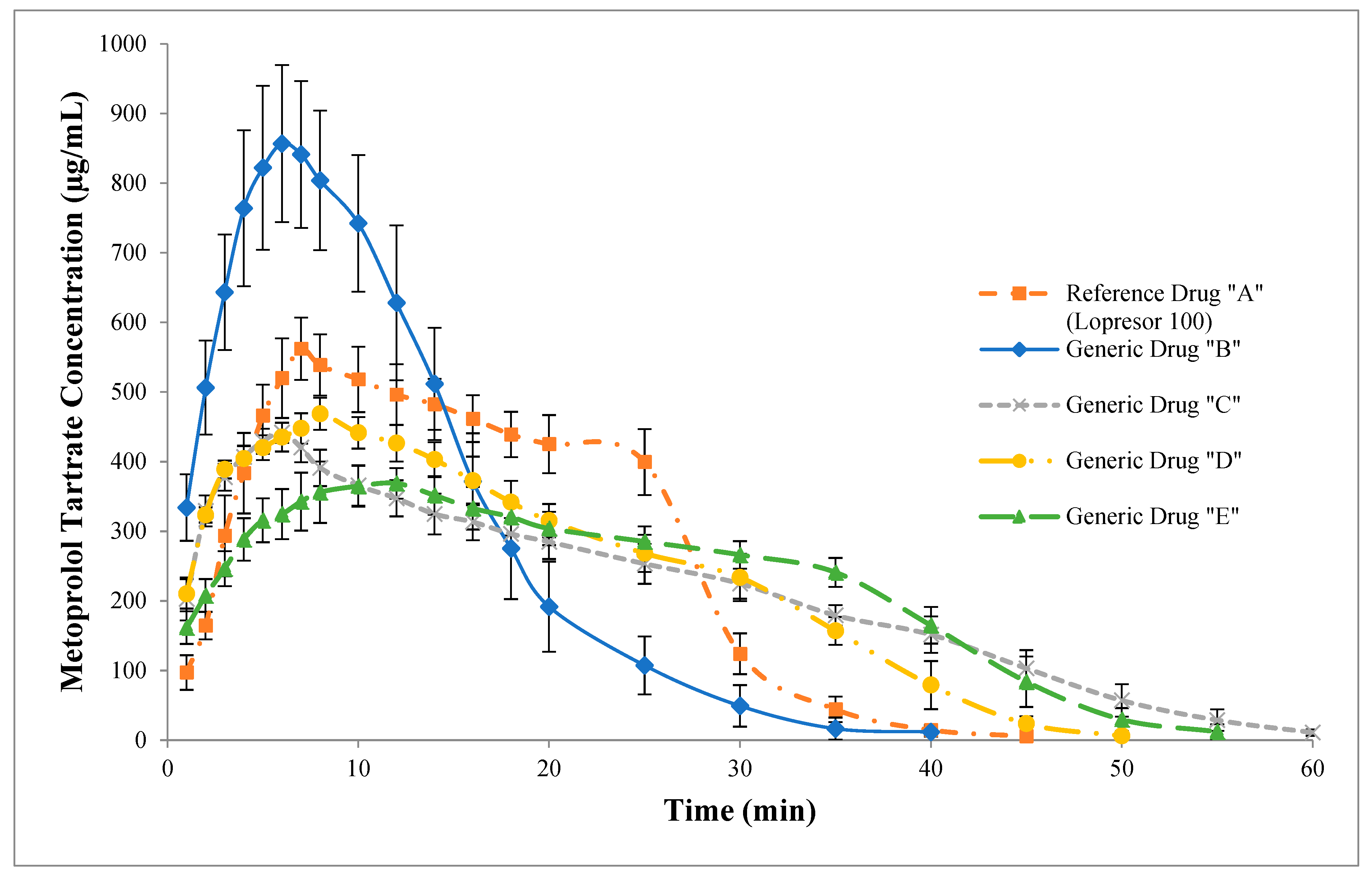
| Cmax (µg/mL) | 565.21 ± 13.39 | 869.69 ± 31.73 | 441.77 ± 4.16 | 468.73 ± 6.65 | 384.54 ± 7.95 | 153.29 | 78.36 | 83.10 | 68.09 |
| (541.17–589.26) | (812.72–926.67) | (434.30–449.25) | (456.78–480.68) | (370.27–398.81) | (142.55–164.84) | (74.91–81.98) | (79.23–87.15) | (64.48–71.89) | |
| AUC0∞ (µg·min/mL) | 12,507.10 ± 189.35 | 12,580.20 ± 135.68 | 12,507.20 ± 151.82 | 12,234.30 ± 157.75 | 12,352.10 ± 115.31 | 101.57 | 99.19 | 97.86 | 98.84 |
| (12,167.10–12,847.10) | (12,336.50–12,823.90) | (12,234.50–12,779.80) | (11,951.00–12,517.60) | (12,145.00–12,559.20) | (98.61–104.63) | (96.12–102.36) | (97.61–98.11) | (95.21–102.62) | |
| AUC0Cmax (µg·min/mL) | 2203.29 ± 57.32 | 3857.76 ± 185.18 | 1966.84 ± 20.74 | 2864.59 ± 22.51 | 2896.96 ± 188.61 | 173.57 | 89.52 | 131.09 | 128.99 |
| (2099.41–2307.18) | (3525.20–4190.33) | (1929.58–2004.09) | (2824.16–2905.02) | (2558.25–3235.68) | (157.88–190.82) | (85.11–94.17) | (124.82–137.68) | (114.23–145.67) | |
| Tmax (min) | 7.25 ± 0.25 | 6.42 ± 0.19 | 6.00 ± 0.00 | 8.00 ± 0.00 | 10.33 ± 0.54 | 88.57 | 83.20 | 110.94 | 141.17 |
| (6.80–7.70) | (6.07–6.76) | (6.00–6.00) | (8.00–8.00) | (9.36–11.31) | (82.53–95.06) | (78.88–87.77) | (105.17–117.02) | (127.35–156.49) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solis-Cruz, B.; Hernandez-Patlan, D.; Morales Hipólito, E.A.; Tellez-Isaias, G.; Alcántara Pineda, A.; López-Arellano, R. Discriminative Dissolution Method Using the Open-Loop Configuration of the USP IV Apparatus to Compare Dissolution Profiles of Metoprolol Tartrate Immediate-Release Tablets: Use of Kinetic Parameters. Pharmaceutics 2023, 15, 2191. https://doi.org/10.3390/pharmaceutics15092191
Solis-Cruz B, Hernandez-Patlan D, Morales Hipólito EA, Tellez-Isaias G, Alcántara Pineda A, López-Arellano R. Discriminative Dissolution Method Using the Open-Loop Configuration of the USP IV Apparatus to Compare Dissolution Profiles of Metoprolol Tartrate Immediate-Release Tablets: Use of Kinetic Parameters. Pharmaceutics. 2023; 15(9):2191. https://doi.org/10.3390/pharmaceutics15092191
Chicago/Turabian StyleSolis-Cruz, Bruno, Daniel Hernandez-Patlan, Elvia A. Morales Hipólito, Guillermo Tellez-Isaias, Alejandro Alcántara Pineda, and Raquel López-Arellano. 2023. "Discriminative Dissolution Method Using the Open-Loop Configuration of the USP IV Apparatus to Compare Dissolution Profiles of Metoprolol Tartrate Immediate-Release Tablets: Use of Kinetic Parameters" Pharmaceutics 15, no. 9: 2191. https://doi.org/10.3390/pharmaceutics15092191