
Anticholinesterase and Serotoninergic Evaluation of Benzimidazole–Carboxamides as Potential Multifunctional Agents for the Treatment of Alzheimer’s Disease
Abstract
:1. Introduction
2. Materials and Methods
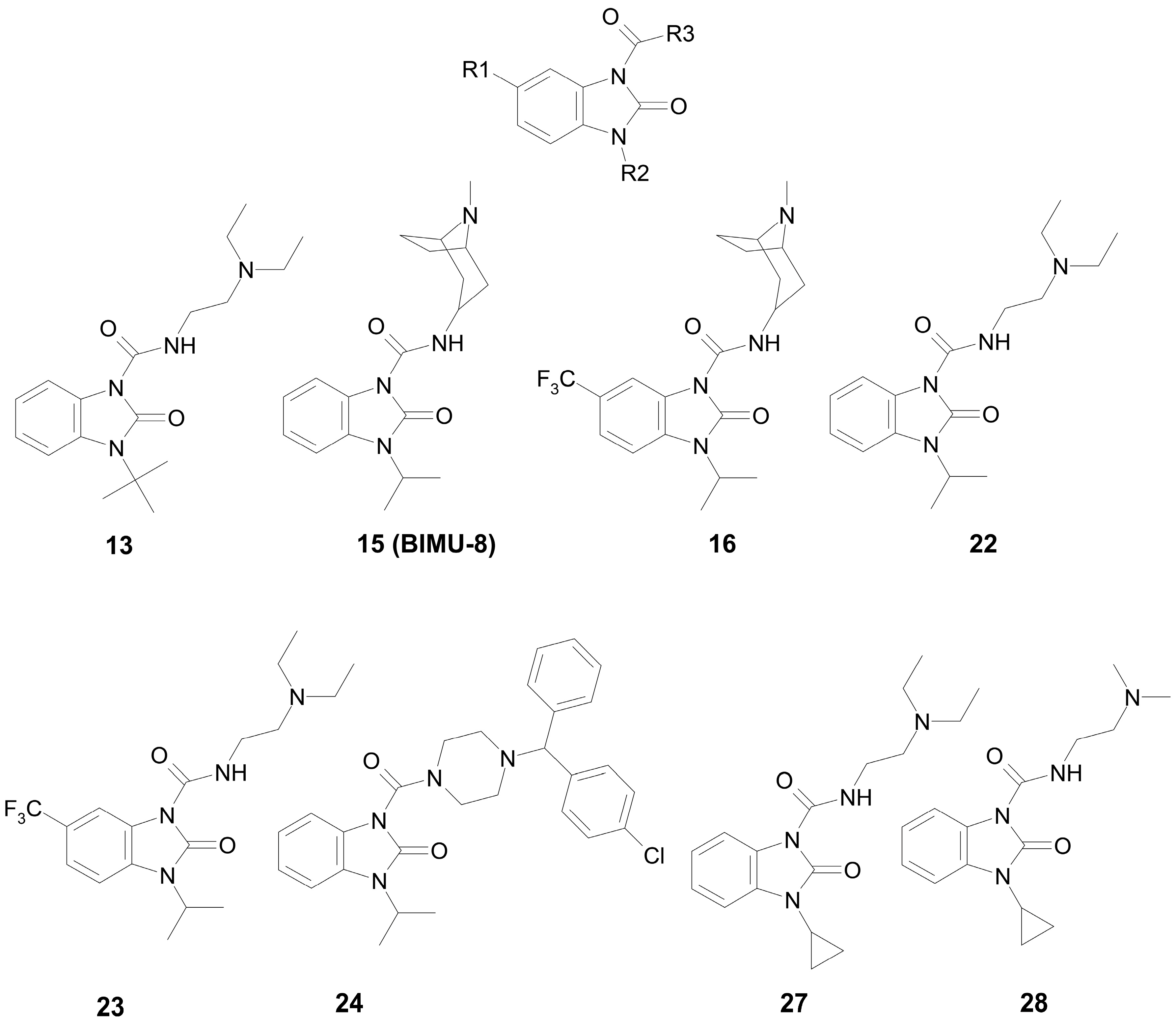
2.1. Choice and Synthesis of the Benzimidazole–Carboxamides
2.2. Anticholinesterase Activity of Benzimidazole–Carboxamides
2.3. Building of 3D Models for Molecular Modeling
2.4. Molecular Docking
2.5. Molecular Dynamics of Drug-ChE Complexes in Water
2.6. Molecular Dynamics of the Drug Complexes with 5-HT4R in Lipid Bilayer
3. Results
3.1. Anticholinesterase Activity of Benzimidazole–Carboxamides In Vitro
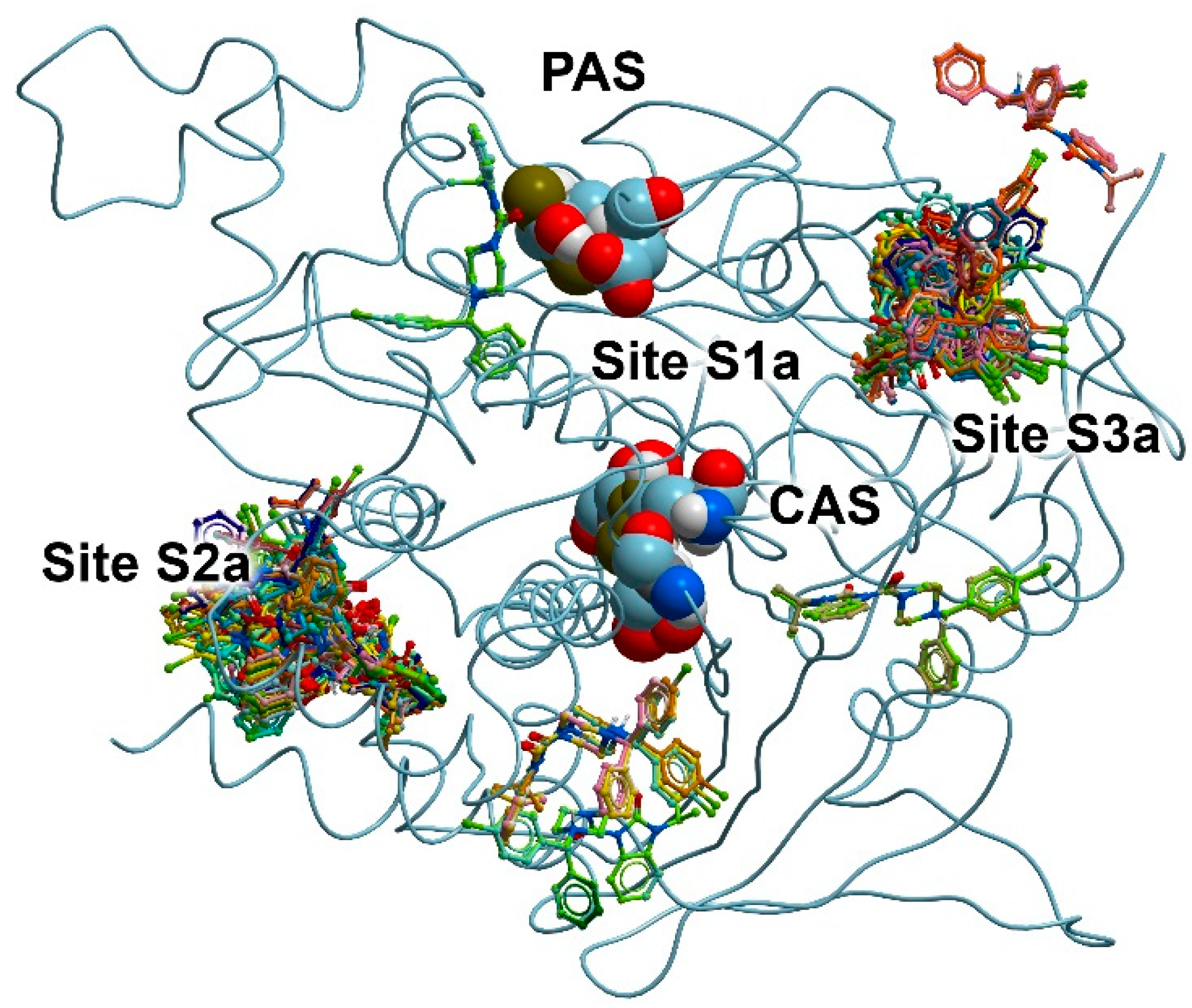
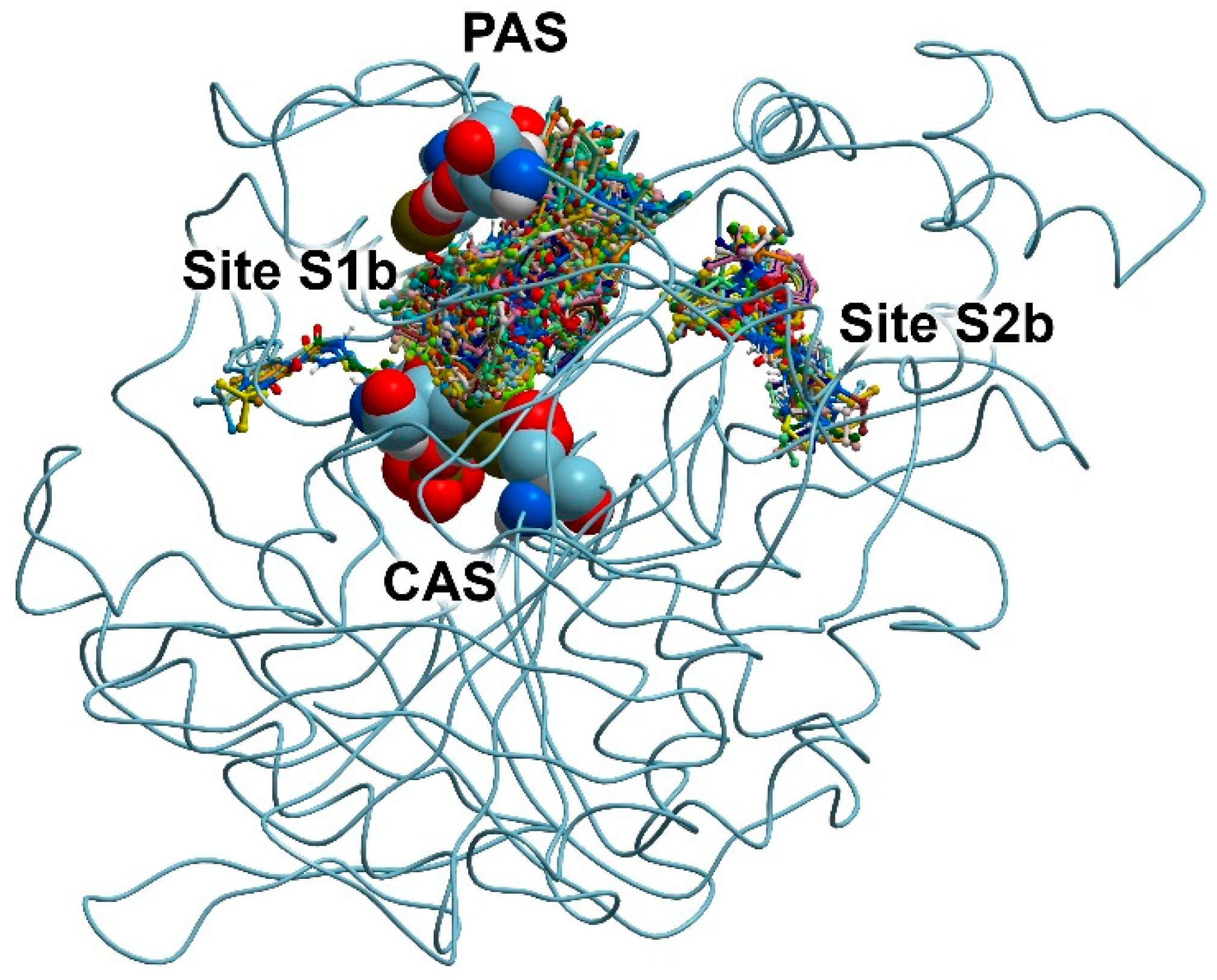
3.2. Search for Binding Sites on the Surface of AChE and BChE
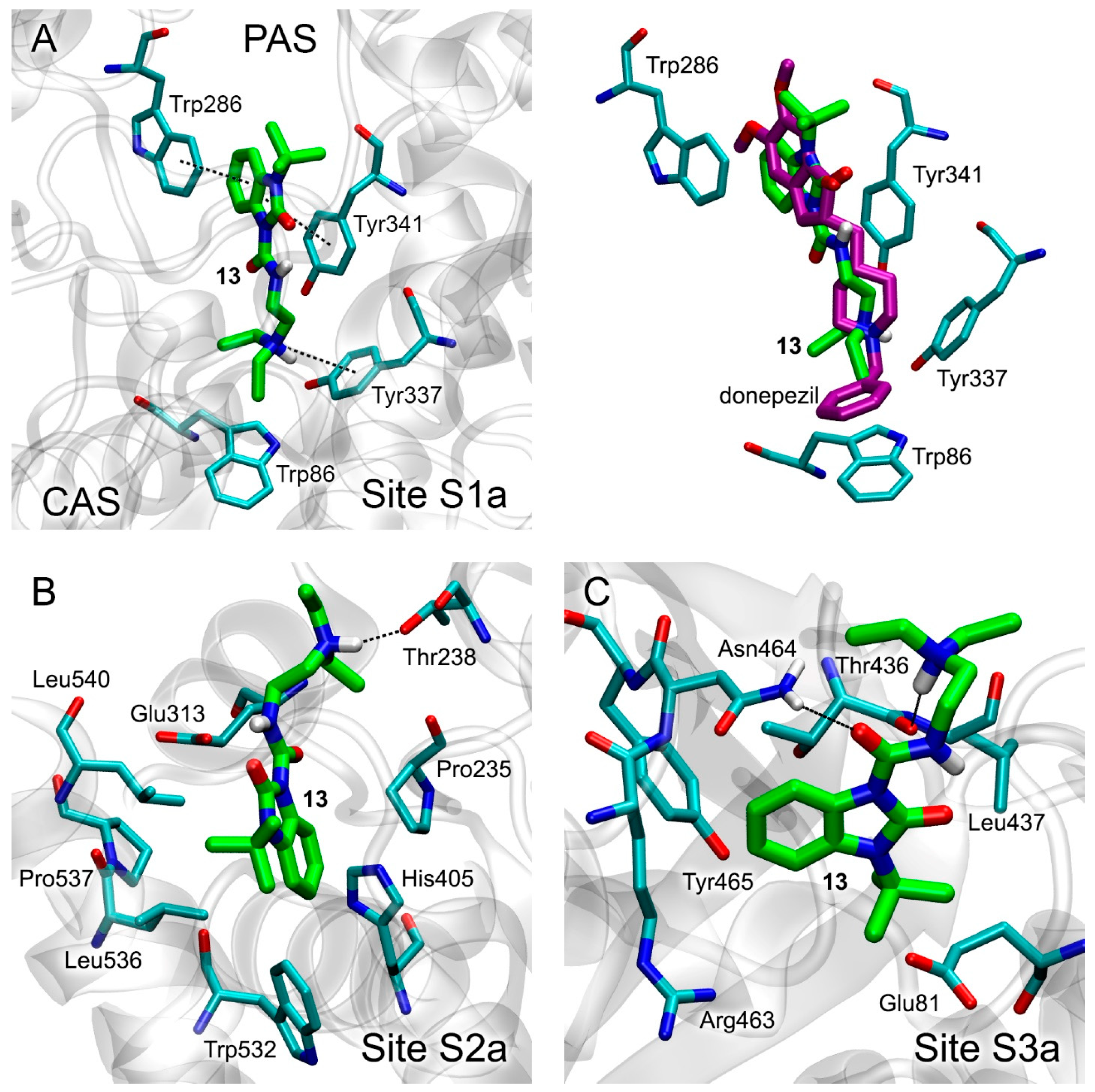
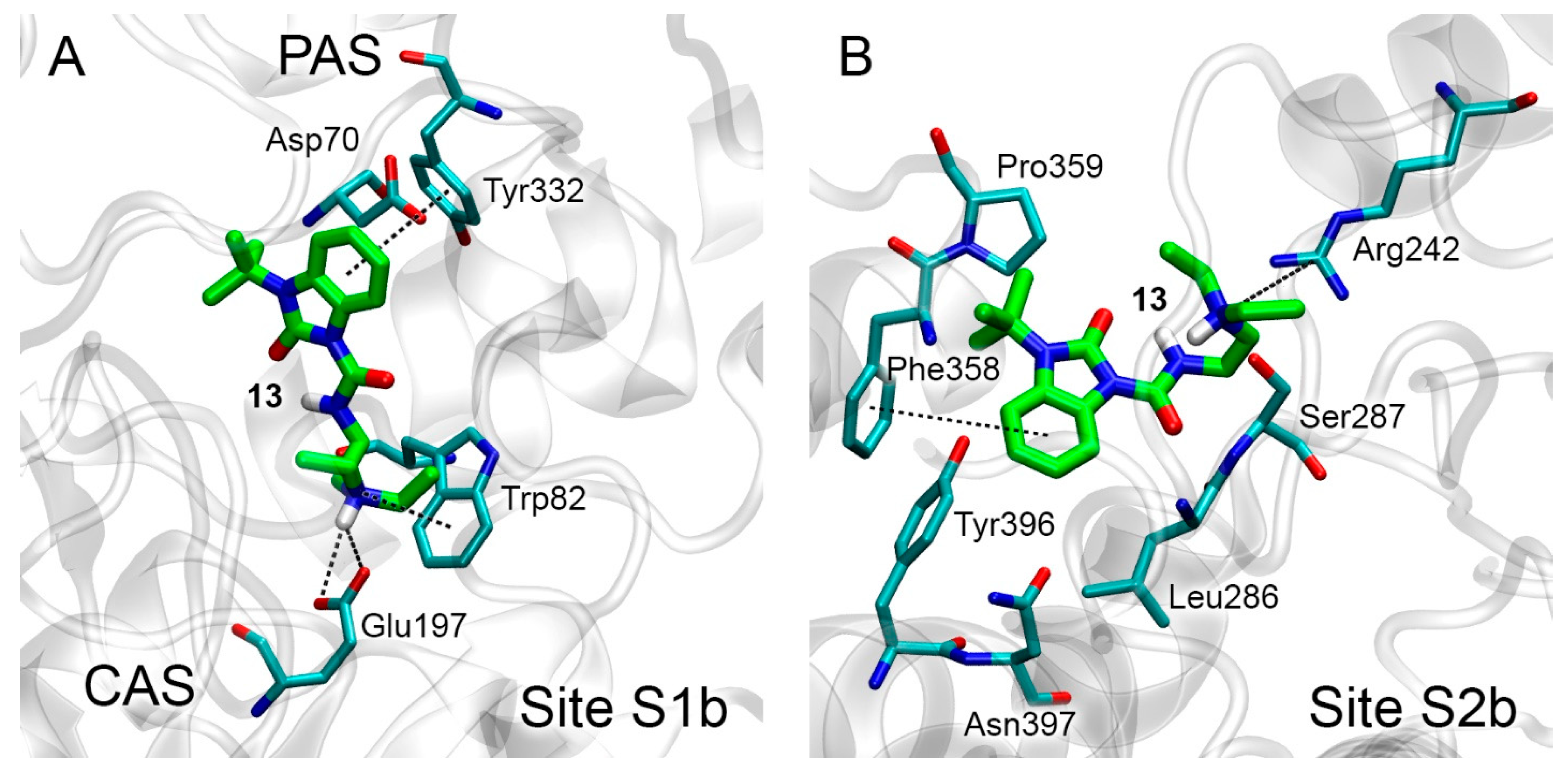
3.3. Docking of Benzimidazolones into the Binding Sites of AChE
3.4. Docking of Benzimidazolones into the Binding Sites of BChE
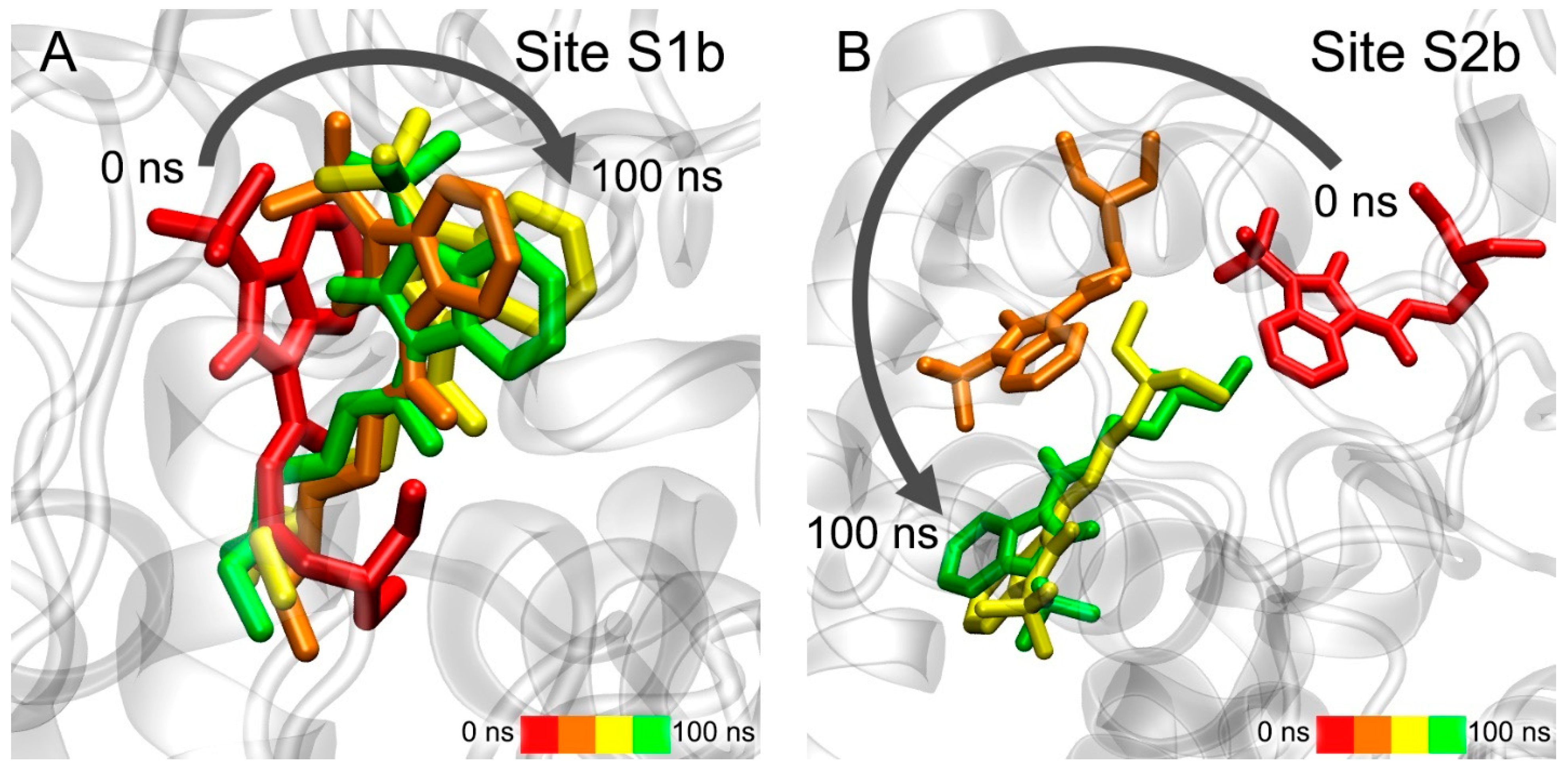
3.5. Interaction of Benzimidazolones with AChE and BChE According to MD Simulation
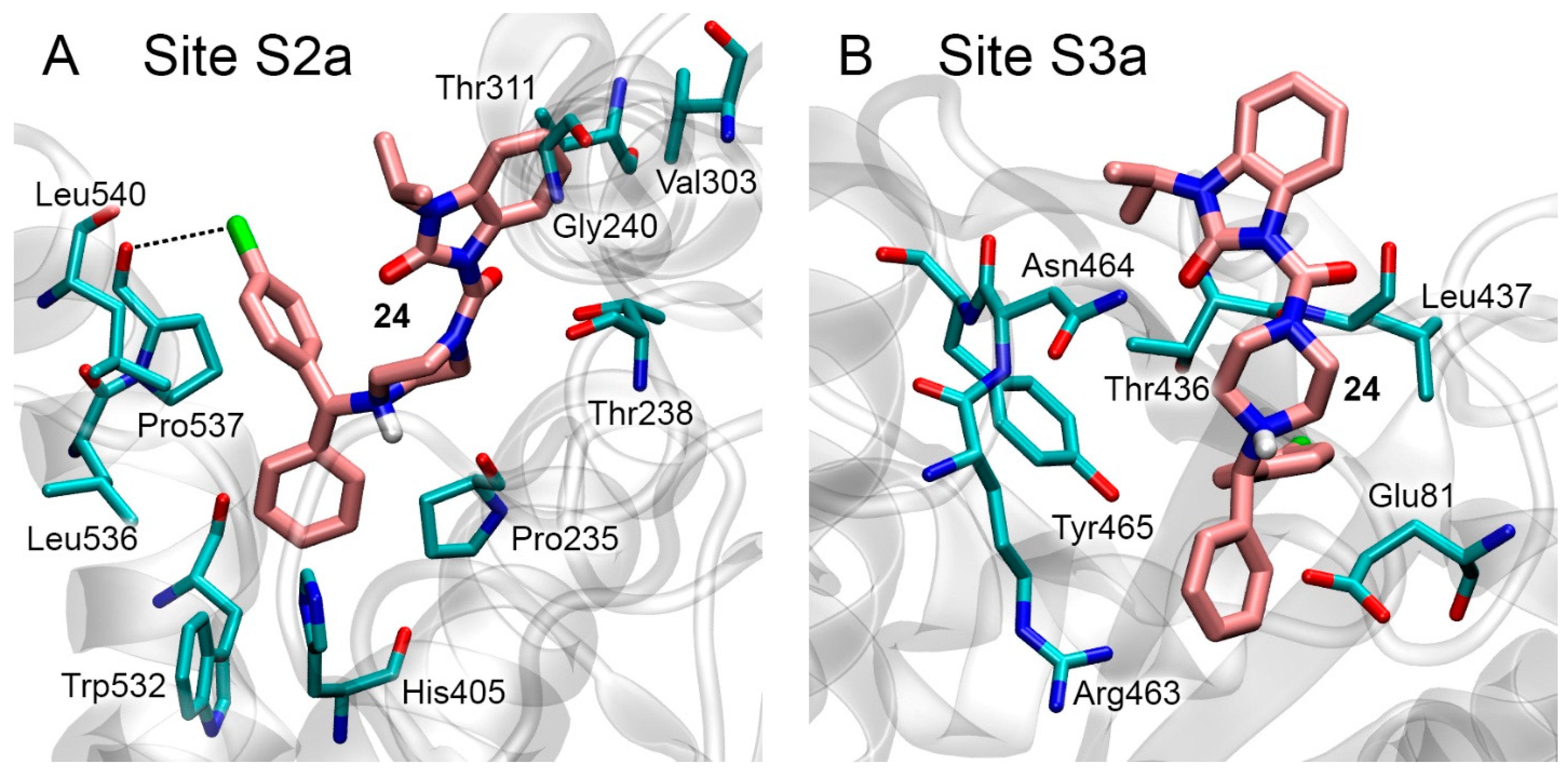
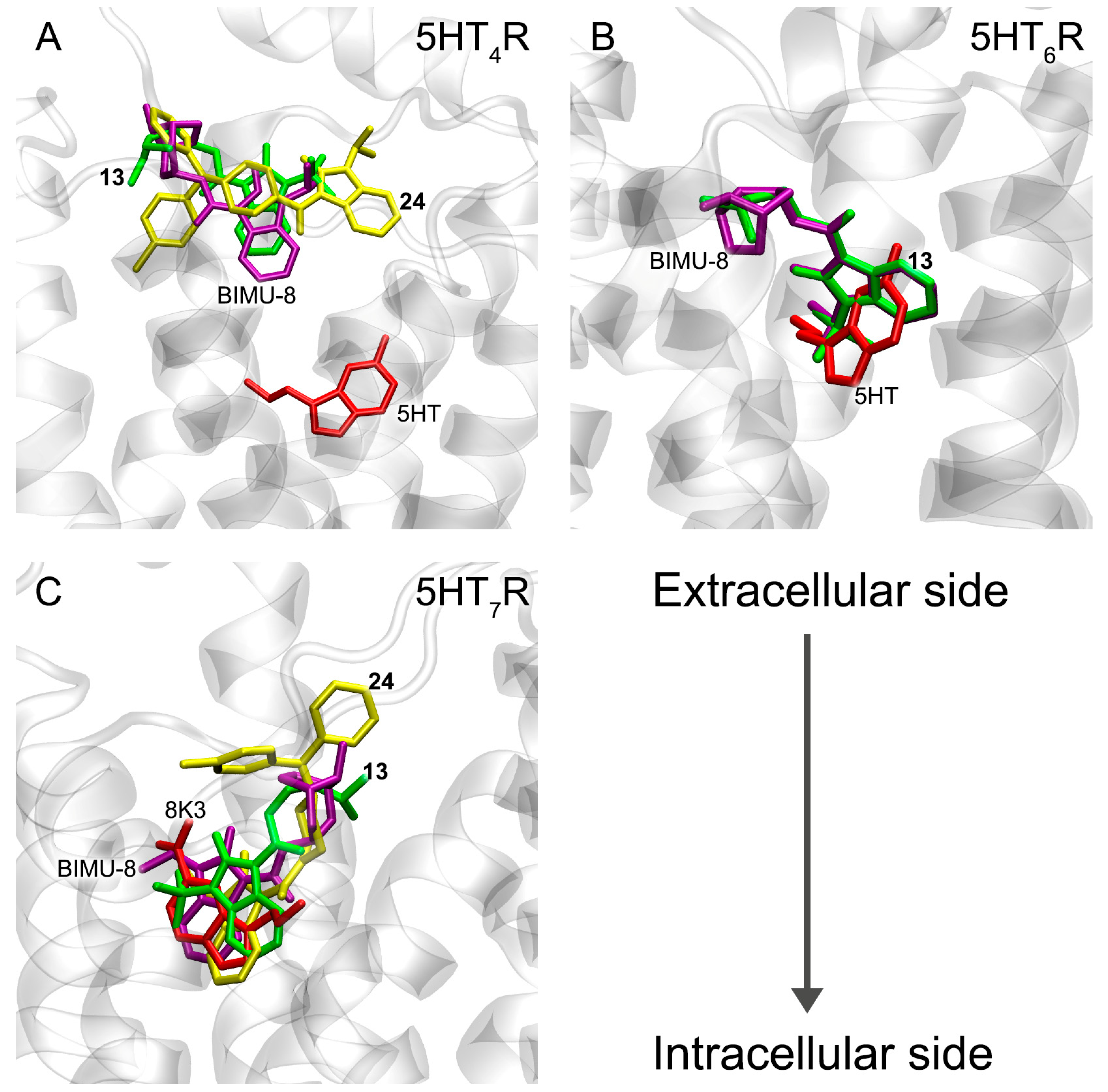
3.6. Interaction of Compounds 13, 15 and 24 with 5-HT4R, 5-HT6R and 5-HT7R According to Molecular Docking Data
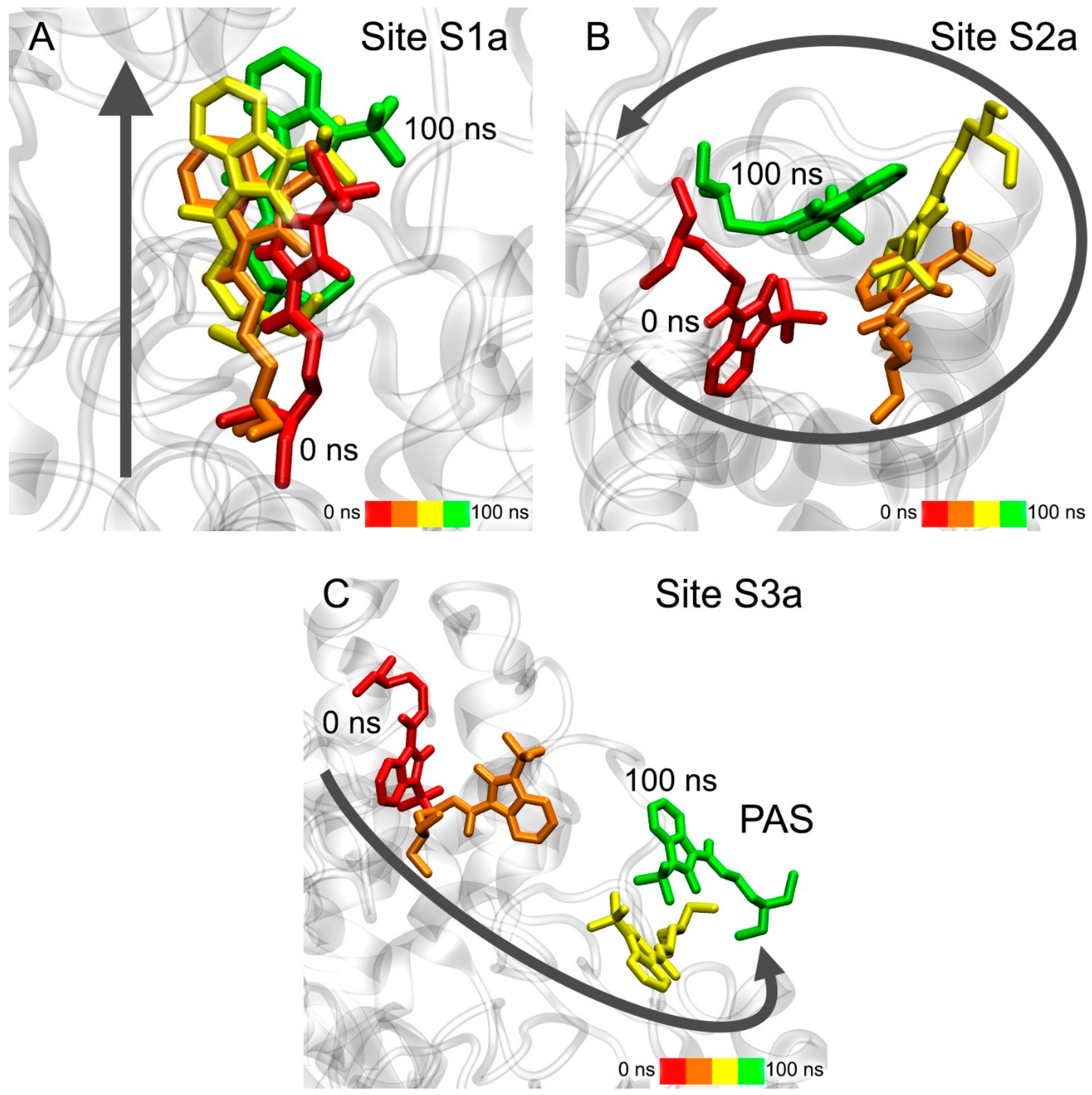
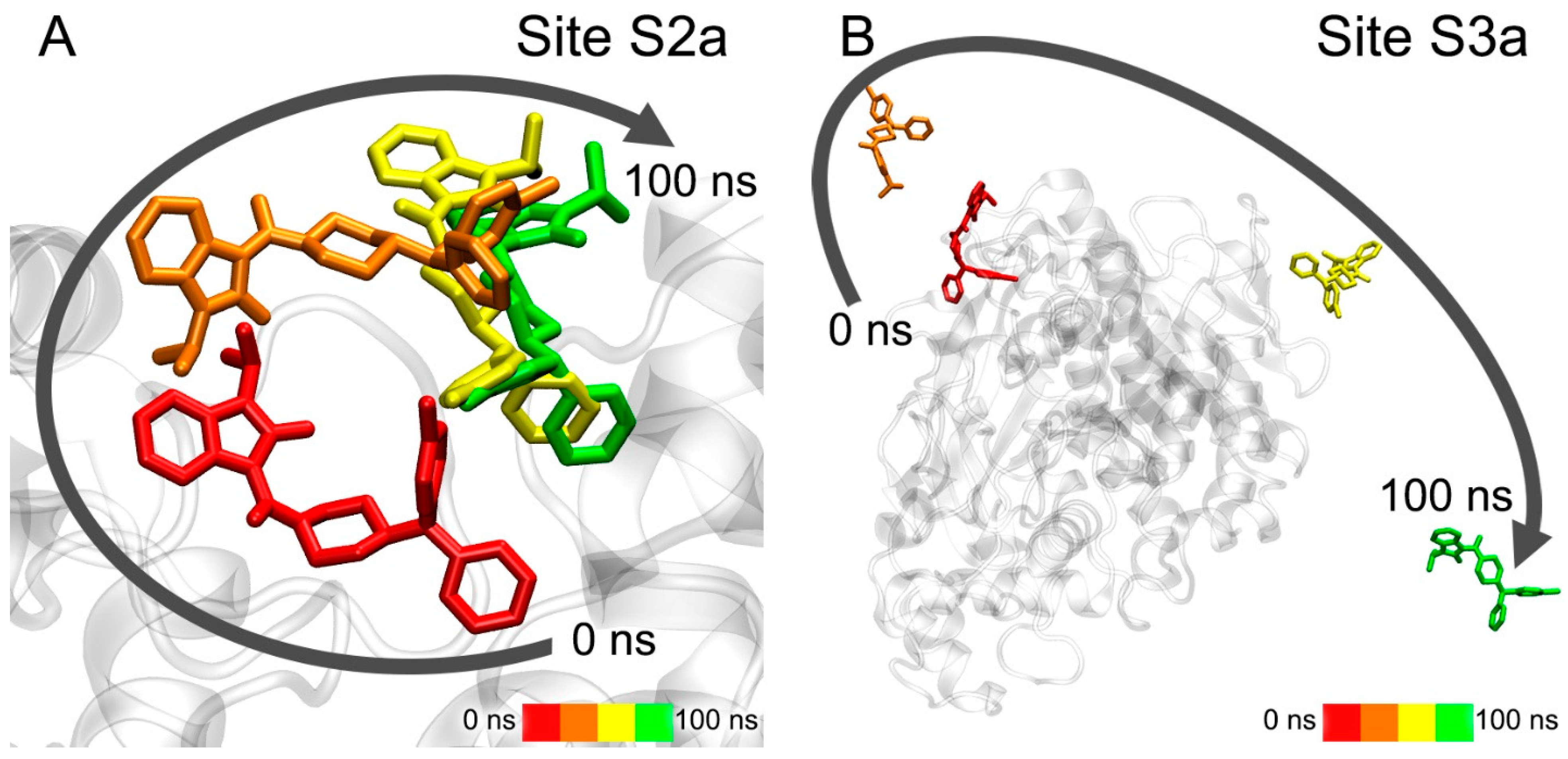
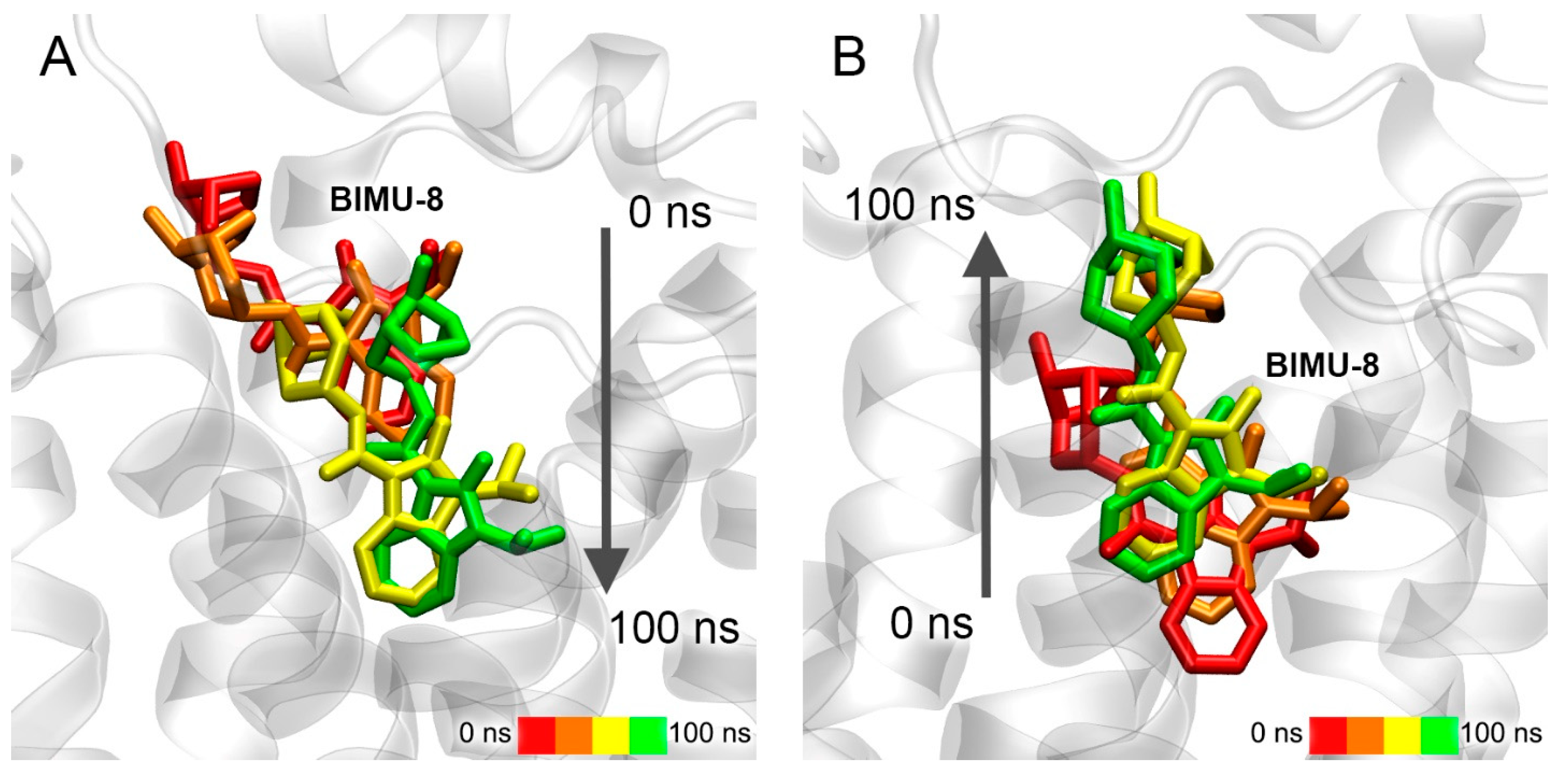
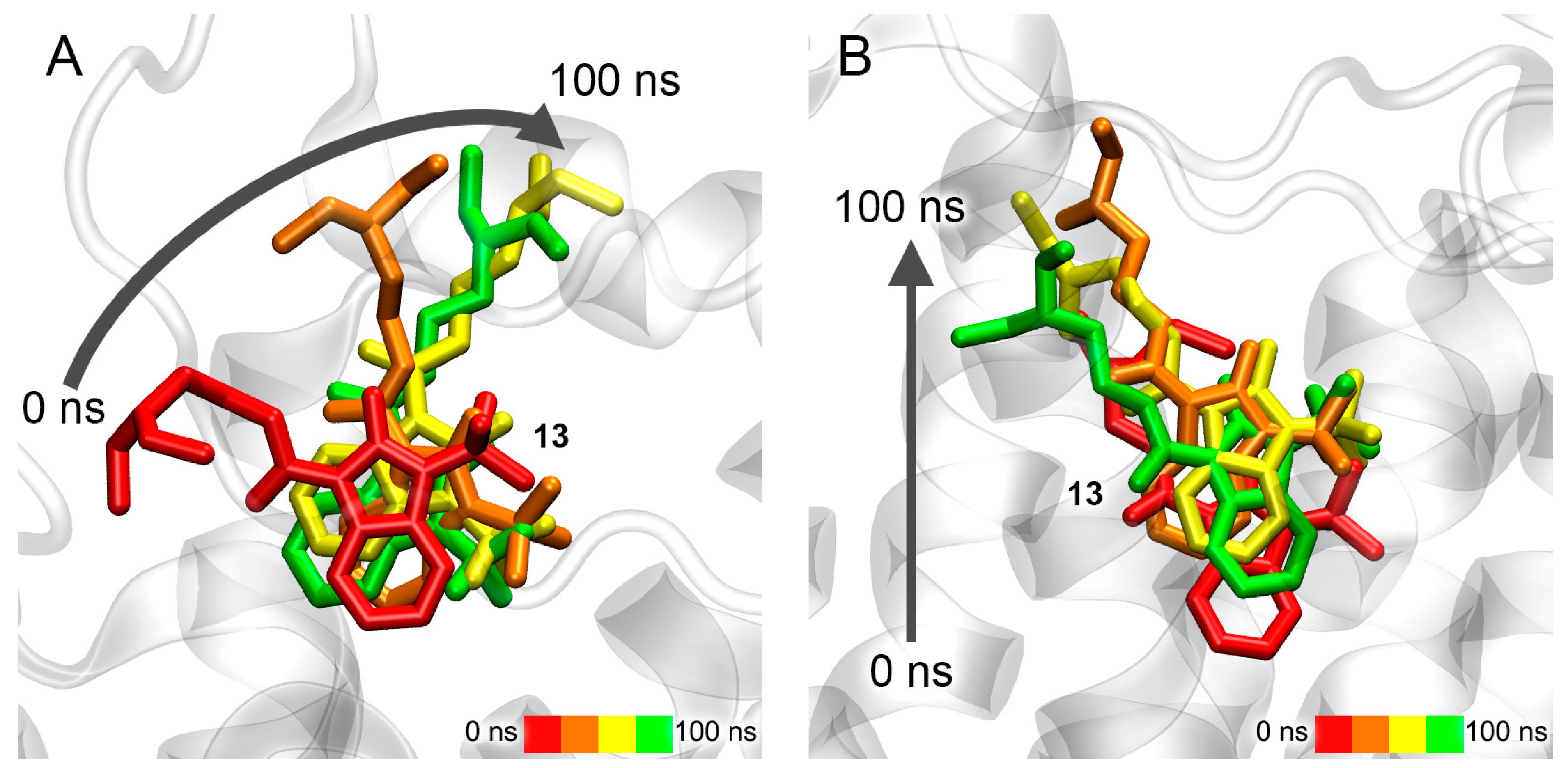
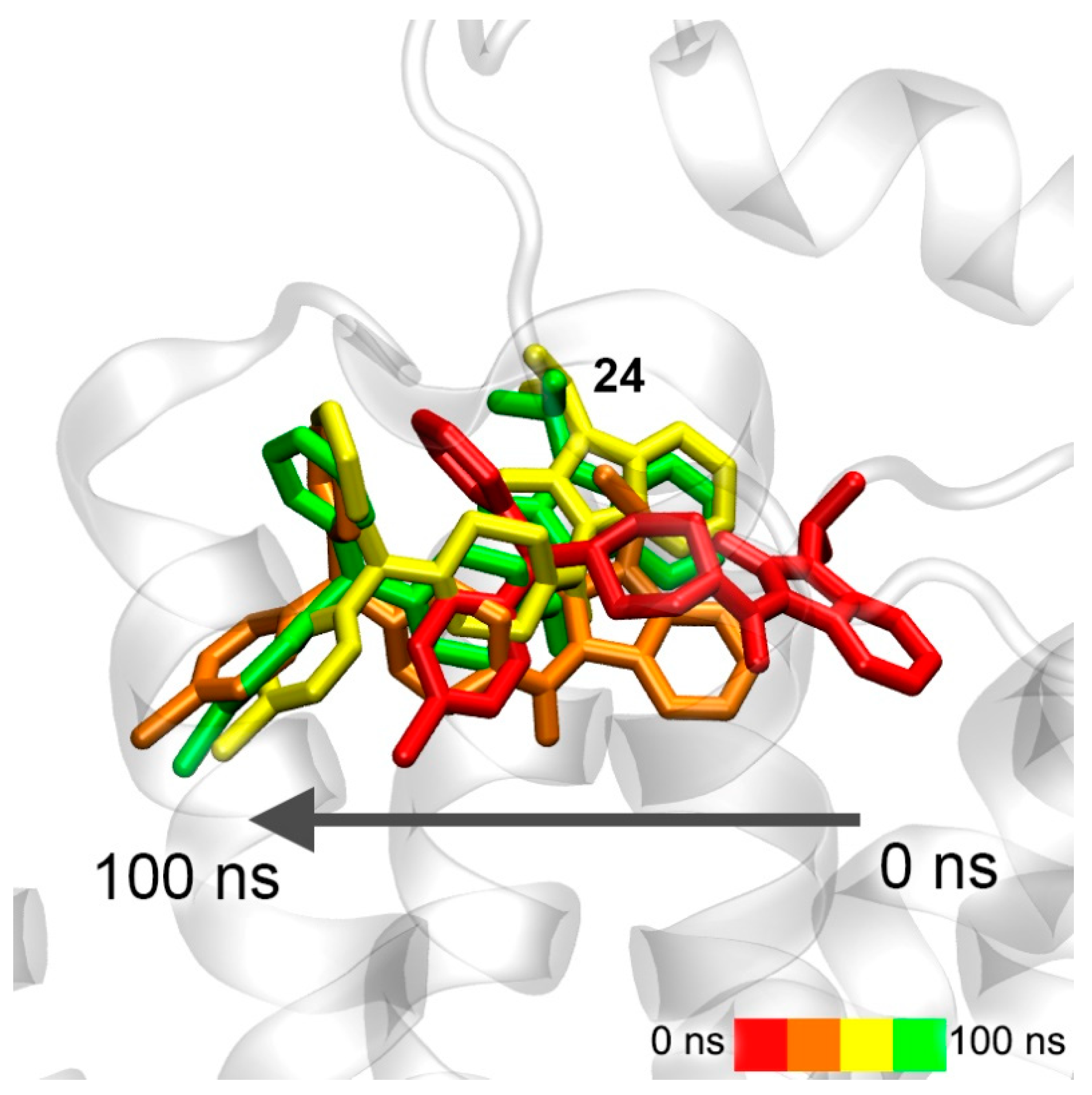
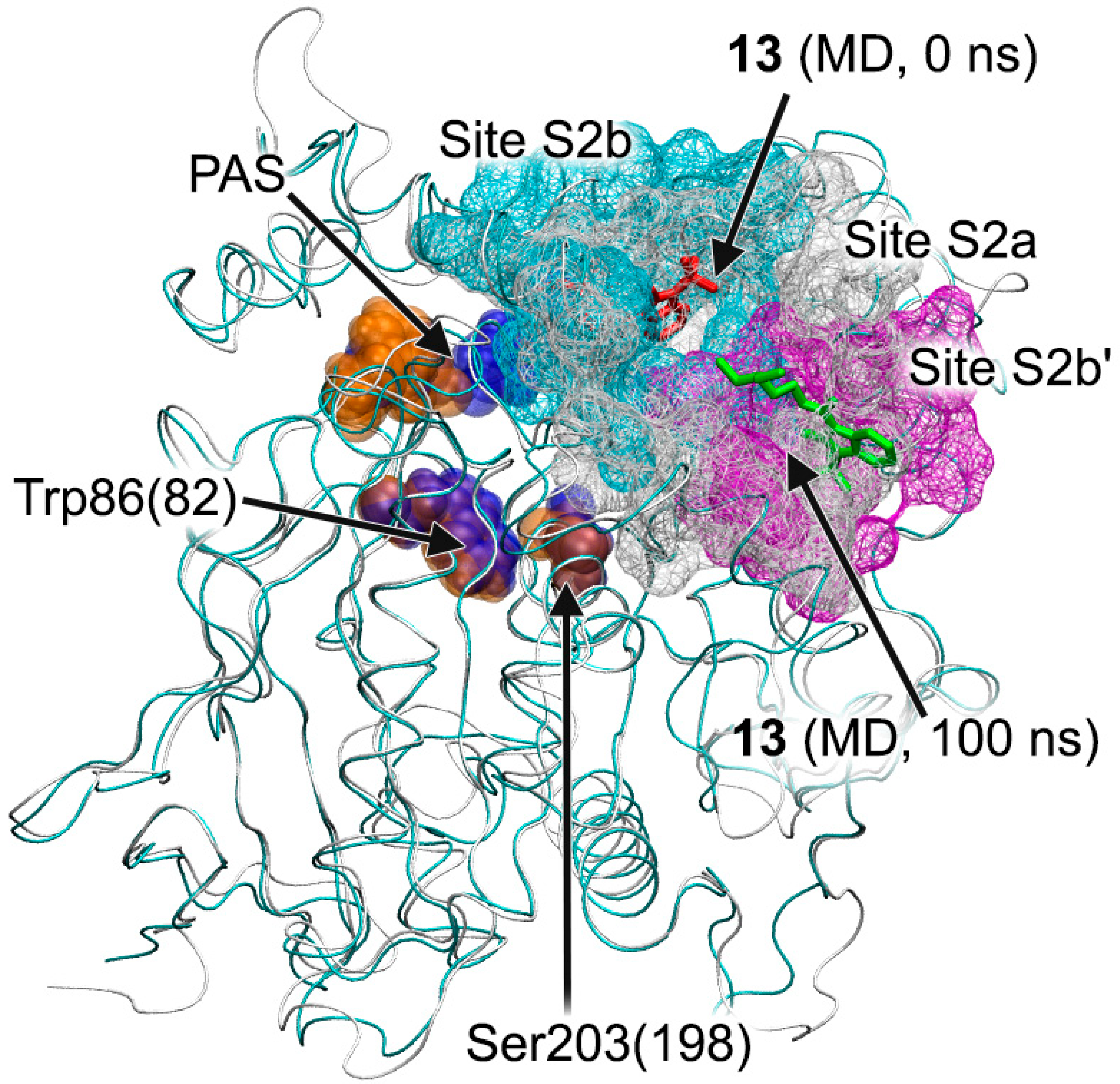
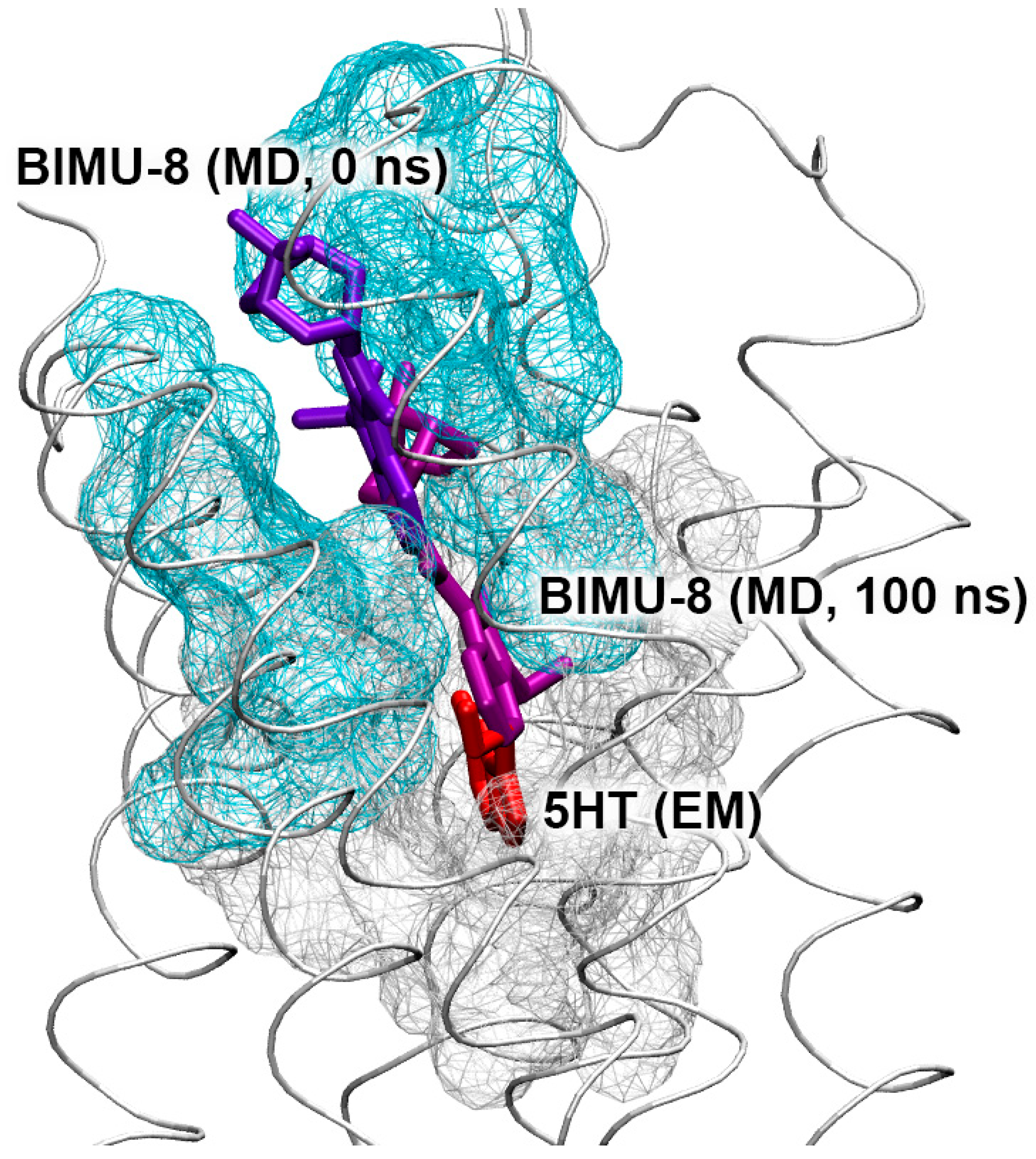
3.7. Interaction of Compounds 13, 15 and 24 with 5-HT4R According to MD Simulation
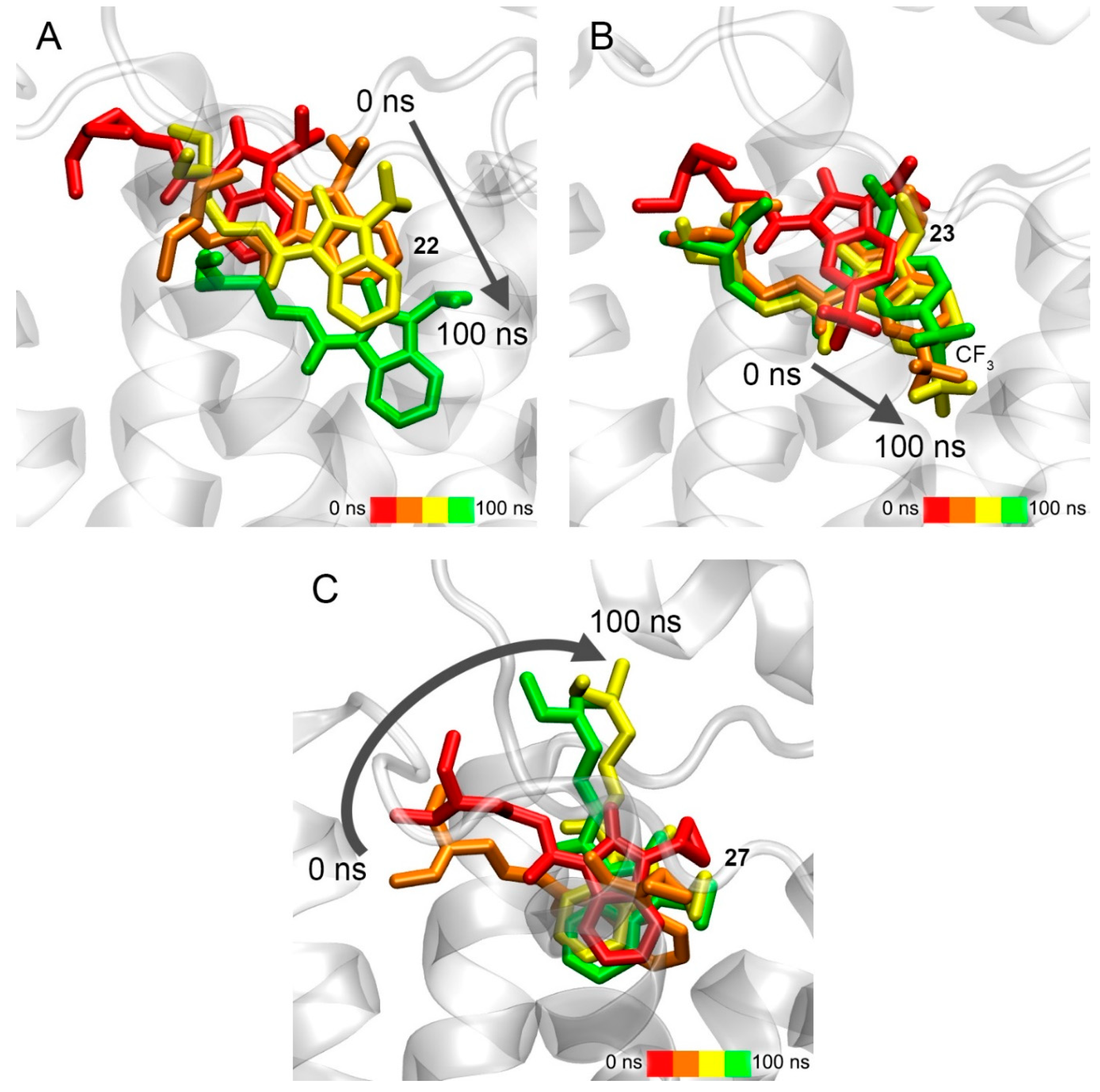
3.8. Interaction of Compounds 22, 23 and 27 with 5-HT4R According to Molecular Modeling Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 5HT | serotonin |
| 5-HTR | serotonin receptor |
| 8K3 | 3-(2-azanylethyl)-1H-indole-5-carboxamide |
| ACh | acetylcholine |
| AChE | acetylcholinesterase |
| AD | Alzheimer’s disease |
| ATCh | acetylthiocholine |
| BBB | blood–brain barrier |
| BChE | butyrylcholinesterase |
| BTCh | butyrylthiocholine |
| CAS | catalytic active site |
| ChE | cholinesterases |
| DMSO | dimethyl sulfoxide |
| DTNB | 5,5′-dithio-bis-2-nitrobenzoic acid |
| GPCR | G-protein-coupled receptor |
| hCB2R | cannabinoid receptors type 2 |
| hH3R | histamine H3 receptors |
| LD50 | median lethal dose |
| LINCS | linear constraint solver for molecular simulations |
| MD | molecular dynamics |
| NPT | constant number of particles N, pressure P and temperature T |
| NVT | constant number of particles N, volume V and temperature T |
| PAS | peripheral anionic site |
| PBS | phosphate-buffered saline |
| PDB | protein data bank |
| TIP3P | transferable intermolecular potential with 3 points (water model) |
| VMD | Visual Molecular Dynamics |
References
- Li, X.; Feng, X.; Sun, X.; Hou, N.; Han, F.; Liu, Y. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2019. Front. Aging Neurosci. 2022, 14, 937486. [Google Scholar] [CrossRef] [PubMed]
- García-Morales, V.; González-Acedo, A.; Melguizo-Rodríguez, L.; Pardo-Moreno, T.; Costela-Ruiz, V.J.; Montiel-Troya, M.; Ramos-Rodríguez, J.J. Current Understanding of the Physiopathology, Diagnosis and Therapeutic Approach to Alzheimer’s Disease. Biomedicines 2021, 9, 1910. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.P.; Xie, Y.; Meng, X.Y.; Kang, J.S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Nasb, M.; Tao, W.; Chen, N. Alzheimer’s Disease Puzzle: Delving into Pathogenesis Hypotheses. Aging Dis. 2023; online ahead of print. [Google Scholar] [CrossRef]
- Mantzavinos, V.; Alexiou, A. Biomarkers for Alzheimer’s Disease Diagnosis. Curr. Alzheimer Res. 2017, 14, 1149–1154. [Google Scholar] [CrossRef]
- Talevi, A. Multi-target pharmacology: Possibilities and limitations of the “skeleton key approach” from a medicinal chemist perspective. Front. Pharmacol. 2015, 6, 205. [Google Scholar] [CrossRef]
- Nepovimova, E.; Korabecny, J.; Hepnarova, V.; Jun, D.; Dolezal, R.; Muckova, L.; Jost, P.; Soukup, O.; Janockova, J.; Pham, N.L.; et al. N-alkylated Tacrine Derivatives as Potential Agents in Alzheimer’s Disease Therapy. Curr. Alzheimer Res. 2019, 16, 333–343. [Google Scholar] [CrossRef]
- Chalupova, K.; Korabecny, J.; Bartolini, M.; Monti, B.; Lamba, D.; Caliandro, R.; Pesaresi, A.; Brazzolotto, X.; Gastellier, A.J.; Nachon, F.; et al. Novel tacrine-tryptophan hybrids: Multi-target directed ligands as potential treatment for Alzheimer’s disease. Eur. J. Med. Chem. 2019, 168, 491–514. [Google Scholar] [CrossRef]
- Hamulakova, S.; Janovec, L.; Soukup, O.; Jun, D.; Janockova, J.; Hrabinova, M.; Sepsova, V.; Kuca, K. Tacrine-coumarin and Tacrine-7-chloroquinoline Hybrids with Thiourea Linkers: Cholinesterase Inhibition Properties, Kinetic Study, Molecular Docking and Permeability Assay for Blood-brain Barrier. Curr. Alzheimer Res. 2018, 15, 1096–1105. [Google Scholar] [CrossRef]
- Hamulakova, S.; Kudlickova, Z.; Janovec, L.; Mezencev, R.; Deckner, Z.J.; Chernoff, Y.O.; Janockova, J.; Ihnatova, V.; Bzonek, P.; Novakova, N.; et al. Design and synthesis of novel tacrine-indole hybrids as potential multitarget-directed ligands for the treatment of Alzheimer’s disease. Future Med. Chem. 2021, 13, 785–804. [Google Scholar] [CrossRef]
- Nepovimova, E.; Svobodova, L.; Dolezal, R.; Hepnarova, V.; Junova, L.; Jun, D.; Korabecny, J.; Kucera, T.; Gazova, Z.; Motykova, K.; et al. Tacrine—Benzothiazoles: Novel class of potential multitarget anti-Alzheimeŕs drugs dealing with cholinergic, amyloid and mitochondrial systems. Bioorg. Chem. 2021, 107, 104596. [Google Scholar] [CrossRef] [PubMed]
- Bachurin, S.O.; Shevtsova, E.F.; Makhaeva, G.F.; Grigoriev, V.V.; Boltneva, N.P.; Kovaleva, N.V.; Lushchekina, S.V.; Shevtsov, P.N.; Neganova, M.E.; Redkozubova, O.M.; et al. Novel conjugates of aminoadamantanes with carbazole derivatives as potential multitarget agents for AD treatment. Sci. Rep. 2017, 7, 45627. [Google Scholar] [CrossRef] [PubMed]
- Bachurin, S.O.; Makhaeva, G.F.; Shevtsova, E.F.; Aksinenko, A.Y.; Grigoriev, V.V.; Shevtsov, P.N.; Goreva, T.V.; Epishina, T.A.; Kovaleva, N.V.; Pushkareva, E.A.; et al. Conjugation of Aminoadamantane and γ-Carboline Pharmacophores Gives Rise to Unexpected Properties of Multifunctional Ligands. Molecules 2021, 26, 5527. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Lushchekina, S.V.; Boltneva, N.P.; Sokolov, V.B.; Grigoriev, V.V.; Serebryakova, O.G.; Vikhareva, E.A.; Aksinenko, A.Y.; Barreto, G.E.; Aliev, G.; et al. Conjugates of γ-Carbolines and Phenothiazine as new selective inhibitors of butyrylcholinesterase and blockers of NMDA receptors for Alzheimer Disease. Sci. Rep. 2015, 5, 13164. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Shevtsova, E.F.; Boltneva, N.P.; Lushchekina, S.V.; Kovaleva, N.V.; Rudakova, E.V.; Bachurin, S.O.; Richardson, R.J. Overview of novel multifunctional agents based on conjugates of γ-carbolines, carbazoles, tetrahydrocarbazoles, phenothiazines, and aminoadamantanes for treatment of Alzheimer’s disease. Chem. Biol. Interact. 2019, 308, 224–234. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Lushchekina, S.V.; Kovaleva, N.V.; Astakhova, T.Y.; Boltneva, N.P.; Rudakova, E.V.; Serebryakova, O.G.; Proshin, A.N.; Serkov, I.V.; Trofimova, T.P.; et al. Amiridine-piperazine hybrids as cholinesterase inhibitors and potential multitarget agents for Alzheimer’s disease treatment. Bioorg. Chem. 2021, 112, 104974. [Google Scholar] [CrossRef] [PubMed]
- Waly, O.M.; Saad, K.M.; El-Subbagh, H.I.; Bayomi, S.M.; Ghaly, M.A. Synthesis, biological evaluation, and molecular modeling simulations of new heterocyclic hybrids as multi-targeted anti-Alzheimer’s agents. Eur. J. Med. Chem. 2022, 231, 114152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, Z.; Mou, C.; Zou, J.; Xie, Y.; Liu, Z.; Benjamin Naman, C.; Mao, Y.; Wei, J.; Huang, X.; et al. Design and synthesis of novel tacrine-dipicolylamine dimers that are multiple-target-directed ligands with potential to treat Alzheimer’s disease. Bioorg. Chem. 2021, 116, 105387. [Google Scholar] [CrossRef]
- Liu, C.; Kou, X.; Wang, X.; Wu, J.; Yang, A.; Shen, R. Novel chrysin derivatives as hidden multifunctional agents for anti-Alzheimer’s disease: Design, synthesis and in vitro evaluation. Eur. J. Pharm. Sci. 2021, 166, 105976. [Google Scholar] [CrossRef]
- Wu, J.; Kou, X.; Ju, H.; Zhang, H.; Yang, A.; Shen, R. Design, synthesis and biological evaluation of naringenin carbamate derivatives as potential multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2021, 49, 128316. [Google Scholar] [CrossRef]
- Darras, F.H.; Pockes, S.; Huang, G.; Wehle, S.; Strasser, A.; Wittmann, H.J.; Nimczick, M.; Sotriffer, C.A.; Decker, M. Synthesis, biological evaluation, and computational studies of Tri- and tetracyclic nitrogen-bridgehead compounds as potent dual-acting AChE inhibitors and hH3 receptor antagonists. ACS Chem. Neurosci. 2014, 5, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Dolles, D.; Nimczick, M.; Scheiner, M.; Ramler, J.; Stadtmüller, P.; Sawatzky, E.; Drakopoulos, A.; Sotriffer, C.; Wittmann, H.J.; Strasser, A.; et al. Aminobenzimidazoles and Structural Isomers as Templates for Dual-Acting Butyrylcholinesterase Inhibitors and hCB2 R Ligands to Combat Neurodegenerative Disorders. ChemMedChem. 2016, 11, 1270–1283. [Google Scholar] [CrossRef]
- Dolles, D.; Hoffmann, M.; Gunesch, S.; Marinelli, O.; Möller, J.; Santoni, G.; Chatonnet, A.; Lohse, M.J.; Wittmann, H.J.; Strasser, A.; et al. Structure-activity relationships and computational investigations into the development of potent and balanced dual-acting butyrylcholinesterase inhibitors and human cannabinoid receptor 2 agonists with pro-cognitive activity in vivo. J. Med. Chem. 2018, 61, 1646–1663. [Google Scholar] [CrossRef] [PubMed]
- Lecoutey, C.; Hedou, D.; Freret, T.; Giannoni, P.; Gaven, F.; Since, M.; Bouet, V.; Ballandonne, C.; Corvaisier, S.; Malzert Fréon, A.; et al. Design of donecopride, a dual serotonin subtype 4 receptor agonist/acetylcholinesterase inhibitor with potential interest for Alzheimer’s disease treatment. Proc. Natl. Acad. Sci. USA 2014, 111, E3825–E3830. [Google Scholar] [CrossRef]
- Rochais, C.; Lecoutey, C.; Gaven, F.; Giannoni, P.; Hamidouche, K.; Hedou, D.; Dubost, E.; Genest, D.; Yahiaoui, S.; Freret, T.; et al. Novel multitarget-directed ligands (MTDLs) with acetylcholinesterase (AChE) inhibitory and serotonergic subtype 4 receptor (5-HT4R) agonist activities as potential agents against Alzheimer’s disease: The design of donecopride. J. Med. Chem. 2015, 58, 3172–3187. [Google Scholar] [CrossRef] [PubMed]
- De Cates, A.N.; Wright, L.C.; Martens, M.A.G.; Gibson, D.; Türkmen, C.; Filippini, N.; Cowen, P.J.; Harmer, C.J.; Murphy, S.E. Déjà-vu? Neural and behavioural effects of the 5-HT4 receptor agonist, prucalopride, in a hippocampal-dependent memory task. Transl. Psychiatry 2021, 11, 497. [Google Scholar] [CrossRef]
- De Cates, A.N.; Martens, M.A.G.; Wright, L.C.; Gould van Praag, C.D.; Capitão, L.P.; Gibson, D.; Cowen, P.J.; Harmer, C.J.; Murphy, S.E. The Effect of the 5-HT4 Agonist, Prucalopride, on a Functional Magnetic Resonance Imaging Faces Task in the Healthy Human Brain. Front. Psychiatry 2022, 13, 859123. [Google Scholar] [CrossRef]
- Nirogi, R.; Grandhi, V.R.; Medapati, R.; Ganuga, N.; Abraham, R.; Thentu, J.B.; Palacharla, V.R.C.; Petlu, S.; Srirangavaram, M.; Subramanian, R.; et al. Usmarapride (SUVN-D4010), a 5-HT4 receptor partial agonist for the potential treatment of Alzheimer’s disease: Behavioural, neurochemical and pharmacological profiling. Eur. J. Pharmacol. 2023, 947, 175625. [Google Scholar] [CrossRef]
- Khoury, R.; Grysman, N.; Gold, J.; Patel, K.; Grossberg, G.T. The role of 5 HT6-receptor antagonists in Alzheimer’s disease: An update. Expert Opin. Investig. Drugs 2018, 27, 523–533. [Google Scholar] [CrossRef]
- Szałaj, N.; Godyń, J.; Jończyk, J.; Pasieka, A.; Panek, D.; Wichur, T.; Więckowski, K.; Zaręba, P.; Bajda, M.; Pislar, A.; et al. Multidirectional in vitro and in cellulo studies as a tool for identification of multi-target-directed ligands aiming at symptoms and causes of Alzheimer’s disease. J. Enzyme Inhib. Med. Chem. 2020, 35, 1944–1952. [Google Scholar] [CrossRef]
- Czarnota-Łydka, K.; Kucwaj-Brysz, K.; Pyka, P.; Haberek, W.; Podlewska, S.; Handzlik, J. Multitargeting the Action of 5-HT6 Serotonin Receptor Ligands by Additional Modulation of Kinases in the Search for a New Therapy for Alzheimer’s Disease: Can It Work from a Molecular Point of View? Int. J. Mol. Sci. 2022, 23, 8768. [Google Scholar] [CrossRef] [PubMed]
- Nirogi, R.; Jayarajan, P.; Shinde, A.; Mohammed, A.R.; Grandhi, V.R.; Benade, V.; Goyal, V.K.; Abraham, R.; Jasti, V.; Cummings, J. Progress in Investigational Agents Targeting Serotonin-6 Receptors for the Treatment of Brain Disorders. Biomolecules 2023, 13, 309. [Google Scholar] [CrossRef] [PubMed]
- Quintero-Villegas, A.; Valdés-Ferrer, S.I. Central nervous system effects of 5-HT7 receptors: A potential target for neurodegenerative diseases. Mol. Med. 2022, 28, 70. [Google Scholar] [CrossRef] [PubMed]
- Villegas, A.Q.; Manzo, H.S.A.; Almada, M.O.V.; Modragon, C.B.; Guzman, R.G. Procognitive and neuroprotective effect of 5-ht7 agonist in an animal model by icv amyloid-b injection (p1.1-005). Neurology 2019, 92 (Suppl. 15), p1.1-005. [Google Scholar]
- Hashemi-Firouzi, N.; Komaki, A.; Soleimani, A.S.; Shahidi, S. The effects of the 5-HT7 receptor on hippocampal long-term potentiation and apoptosis in a rat model of Alzheimer’s disease. Brain Res. Bull. 2017, 135, 85–91. [Google Scholar] [CrossRef]
- Chelusnova, Y.V.; Voronina, P.A.; Belinskaia, D.A.; Goncharov, N.V. Benzimidazole-carboxamides as potential therapeutics for Alzheimer’s disease: Primary in silico and in vitro analyses. Bull. Exp. Biol. Med. 2023, 175, 326–334. [Google Scholar] [CrossRef]
- Dumuis, A.; Sebben, M.; Monferini, E.; Nicola, M.; Turconi, M.; Ladinsky, H.; Bockaert, J. Azabicycloalkyl benzimidazolone derivatives as a novel class of potent agonists at the 5-HT4 receptor positively coupled to adenylate cyclase in brain. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1991, 343, 245–251. [Google Scholar] [CrossRef]
- Prokofieva, D.S.; Jenkins, R.O.; Goncharov, N.V. Microplate biochemical determination of Russian VX: Influence of admixtures and avoidance of false negative results. Anal. Biochem. 2012, 424, 108–113. [Google Scholar] [CrossRef]
- Froimowitz, M. HyperChem: A software package for computational chemistry and molecular modeling. Biotechniques 1993, 14, 1010–1013. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef] [PubMed]
- Carletti, E.; Li, H.; Li, B.; Ekström, F.; Nicolet, Y.; Loiodice, M.; Gillon, E.; Froment, M.T.; Lockridge, O.; Schopfer, L.M.; et al. Aging of cholinesterases phosphylated by tabun proceeds through O-dealkylation. J. Am. Chem. Soc. 2008, 130, 16011–16020. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Xu, P.; Shen, D.D.; Simon, I.A.; Mao, C.; Tan, Y.; Zhang, H.; Harpsøe, K.; Li, H.; Zhang, Y.; et al. GPCRs steer Gi and Gs selectivity via TM5-TM6 switches as revealed by structures of serotonin receptors. Mol. Cell. 2022, 82, 2681–2695.e6. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Dassault Systèmes BIOVIA. Discovery Studio Modeling Environment, Release 2020; Dassault Systèmes: San Diego, CA, USA, 2020. [Google Scholar]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Jorgensen, W.L. Quantum and statistical mechanical studies of liquids. 10. Transferable intermolecular potential functions for water, alcohols, and ethers. Application to liquid water. J. Am. Chem. Soc. 1981, 103, 335–340. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Foloppe, N.; MacKerell, A.D., Jr. All-atom empirical force field for nucleic acids: I. Parameter optimization based on small molecule and condensed phase macromolecular target data. J. Comput. Chem. 2000, 21, 86–104. [Google Scholar] [CrossRef]
- Bussi, G.; Zykova-Timan, T.; Parrinello, M. Isothermal-isobaric molecular dynamics using stochastic velocity rescaling. J. Chem. Phys. 2009, 130, 074101. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 3, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1473. [Google Scholar] [CrossRef]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.J.; Holian, B.L. The Nose-Hoover Thermostat. J. Chem. Phys. 1985, 83, 4069–4074. [Google Scholar] [CrossRef]
- Dileep, K.V.; Ihara, K.; Mishima-Tsumagari, C.; Kukimoto-Niino, M.; Yonemochi, M.; Hanada, K.; Shirouzu, M.; Zhang, K.Y.J. Crystal structure of human acetylcholinesterase in complex with tacrine: Implications for drug discovery. Int. J. Biol. Macromol. 2022, 210, 172–181. [Google Scholar] [CrossRef]
- Roca, C.; Requena, C.; Sebastián-Pérez, V.; Malhotra, S.; Radoux, C.; Pérez, C.; Martinez, A.; Antonio Páez, J.; Blundell, T.L.; Campillo, N.E. Identification of new allosteric sites and modulators of AChE through computational and experimental tools. J. Enzyme Inhib. Med. Chem. 2018, 33, 1034–1047. [Google Scholar] [CrossRef]
- Yu, P.; Chen, Z.; Liu, Y.; Gu, Z.; Wang, X.; Zhang, Y.; Ma, Y.; Dong, M.; Tian, Z. Bioactivity-Guided Separation of Anti-Cholinesterase Alkaloids from Uncaria rhynchophlly (Miq.) Miq. Ex Havil Based on HSCCC Coupled with Molecular Docking. Molecules 2022, 27, 2013. [Google Scholar] [CrossRef]
- Selvakumar, R.; Geib, S.J.; Sankar, A.M.; Premkumar, T.; Govindarajan, S. The chemistry of aminoguanidine derivatives—Preparation, crystal structure, thermal properties, and molecular docking studies of aminoguanidinium salts of several carboxylic acids. J. Phys. Chem. Solids 2015, 86, 49–56. [Google Scholar] [CrossRef]
- Zhou, J.; Cunningham, K.A. Positive-allosteric modulation of the 5-HT2C receptor: Implications for neuropsychopharmacology and neurotherapeutics. Neuropsychopharmacology 2019, 44, 230–231. [Google Scholar] [CrossRef] [PubMed]
- Galeotti, N.; Ghelardini, C.; Bartolini, A. Role of 5-HT4 receptors in the mouse passive avoidance test. J. Pharmacol. Exp. Ther. 1998, 286, 1115–1121. [Google Scholar] [PubMed]
- Berque-Bestel, I.; Lezoualc’h, F. Serotonin 5-HT4 receptors as pharmacological targets for the treatment of Alzheimer’s disease. In Emerging Drugs and Targets for Alzheimer’s Disease: V2: Neuronal Plasticity; Martinez, A., Ed.; RSC Publishing: London, UK, 2010; pp. 169–185. [Google Scholar]
- Pellissier, L.P.; Sallander, J.; Campillo, M.; Gaven, F.; Queffeulou, E.; Pillot, M.; Dumuis, A.; Claeysen, S.; Bockaert, J.; Pardo, L. Conformational toggle switches implicated in basal constitutive and agonist-induced activated states of 5-hydroxytryptamine-4 receptors. Mol. Pharmacol. 2009, 75, 982–990. [Google Scholar] [CrossRef] [PubMed]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | Ki, μM | Type of Inhibition |
| 13 | H | t-Bu | N,N-(Diethylamino)ethyl | 36.1 | competitive |
| 15 (BIMU-8) | H | i-Pr | 8-methyl-8-azabicyclo[3.2.1]octan-6-yl | 126.9 | mixed (α = 6.6) |
| 16 | CF3 | i-Pr | 8-methyl-8-azabicyclo[3.2.1]octan-6-yl | 31.0 | mixed (α = 5.1) |
| 22 | H | i-Pr | N,N-(diethylamino)ethyl | 97.1 | competitive |
| 23 | CF3 | i-Pr | N,N-(diethylamino)ethyl | 60.5 | mixed (α = 3.1) |
| 24 | H | i-Pr | [(4-chlorophenyl)(phenyl)methyl]-piperazin-1-yl | 0.13 | non-competitive |
| 27 | H | c-Pr | N,N-(diethylamino)ethyl | 131.8 | mixed (α = 10.9) |
| 28 | H | c-Pr | N,N-(dimethylamino)ethyl | 137.9 | mixed (α = 12.6) |
 | |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | Ki, μM | Type of Inhibition |
| 13 | H | t-Bu | N,N-(Diethylamino)ethyl | 1.7 | non-competitive |
| 15 (BIMU-8) | H | i-Pr | 8-methyl-8-azabicyclo[3.2.1]octan-6-yl | 9.7 | competitive |
| 16 | CF3 | i-Pr | 8-methyl-8-azabicyclo[3.2.1]octan-6-yl | 21.3 | competitive |
| 22 | H | i-Pr | N,N-(diethylamino)ethyl | 15.2 | competitive |
| 23 | CF3 | i-Pr | N,N-(diethylamino)ethyl | 4.5 | non-competitive |
| 24 | H | i-Pr | [(4-chlorophenyl)(phenyl)methyl]-piperazin-1-yl | no inhibition | |
| 27 | H | c-Pr | N,N-(diethylamino)ethyl | 16.2 | competitive |
| 28 | H | c-Pr | N,N-(dimethylamino)ethyl | 28.1 | non-competitive |
| Compound | AChE | BChE | |||
|---|---|---|---|---|---|
| S1a | S2a | S3a | S1b | S2b | |
| 13 | −7.5 | −6.7 | −5.7 | −6.8 | −6.6 |
| 24 | No Int | −7.9 | −7.4 | −0.3 | No Int |
| ACh | −4.7 | −4.3 | |||
| Compound | 5-HT4R | 5-HT6R | 5-HT7R |
|---|---|---|---|
| 13 | −7.8 | −5.2 | −7.5 |
| 24 | −10.7 | No Int | −8.9 |
| 15 (BIMU-8) | −8.5 | −4.6 | −8.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belinskaia, D.A.; Voronina, P.A.; Krivorotov, D.V.; Jenkins, R.O.; Goncharov, N.V. Anticholinesterase and Serotoninergic Evaluation of Benzimidazole–Carboxamides as Potential Multifunctional Agents for the Treatment of Alzheimer’s Disease. Pharmaceutics 2023, 15, 2159. https://doi.org/10.3390/pharmaceutics15082159
Belinskaia DA, Voronina PA, Krivorotov DV, Jenkins RO, Goncharov NV. Anticholinesterase and Serotoninergic Evaluation of Benzimidazole–Carboxamides as Potential Multifunctional Agents for the Treatment of Alzheimer’s Disease. Pharmaceutics. 2023; 15(8):2159. https://doi.org/10.3390/pharmaceutics15082159
Chicago/Turabian StyleBelinskaia, Daria A., Polina A. Voronina, Denis V. Krivorotov, Richard O. Jenkins, and Nikolay V. Goncharov. 2023. "Anticholinesterase and Serotoninergic Evaluation of Benzimidazole–Carboxamides as Potential Multifunctional Agents for the Treatment of Alzheimer’s Disease" Pharmaceutics 15, no. 8: 2159. https://doi.org/10.3390/pharmaceutics15082159