

Therapeutic Implications of PTEN in Non-Small Cell Lung Cancer
Abstract
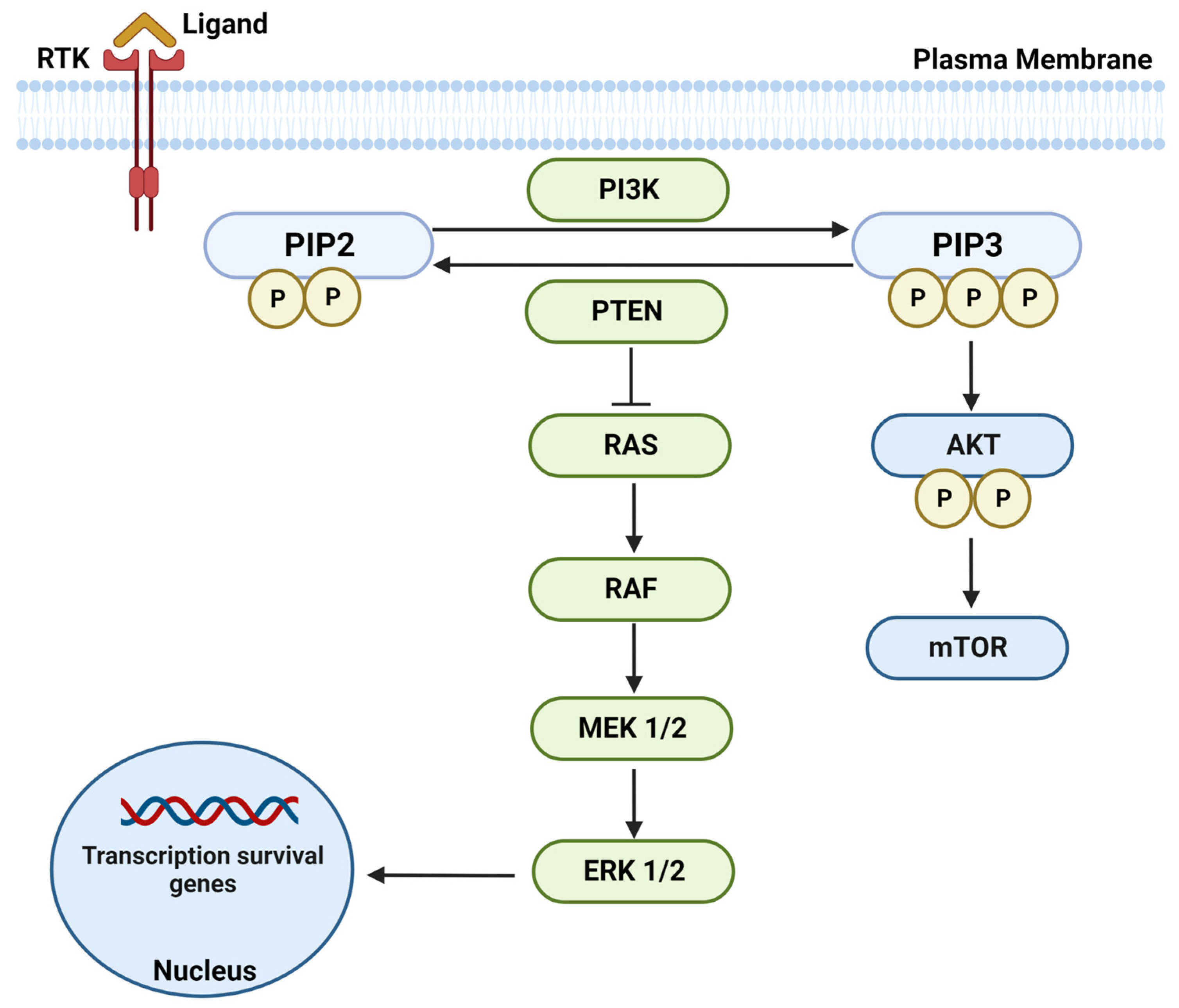
:1. Introduction
Biology and History of PTEN
2. Implications of PTEN in NSCLC
2.1. In Vitro and In Vivo Studies
2.2. Clinical Studies
3. Conclusions and Future Perspective
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PTEN | Phosphatase and tensin homolog |
| NSCLC | Non-small cell lung cancer |
| SCLC | Small cell lung cancer |
| COPD | Chronic obstructive pulmonary disease |
| TKI | Tyrosine kinase inhibitors |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| PI3K | Phosphatidylinositol 3-kinase |
| PIP3 | Phosphatidylinositol 3,4,5 trisphosphate |
| PIP2 | Phosphatidylinositol 4,5 bisphosphate |
| DUB | Deubiquitylates |
| HDACi | Histone deacetylase inhibitors |
| 5-AZA | 5-aza-2′deoxycytidine |
| HDAC | Histone deacetyl transferase |
| TSA | Trichostatin A |
| TGFβRII | Transforming growth factor β receptor type II |
| TIRF | Total internal reflection fluorescence |
| MTD | Maximum tolerated dose |
| DLT | Dose limiting toxicity |
| AE | Adverse events |
| TEAEs | Treatment-emergent adverse events |
| RP2D | Recommended Phase 2 dose |
| ORR | Overall response rate |
| TEAE | Treatment-emergent adverse event |
| SAE | Severe adverse events |
| Cmax | Maximum observed plasma concentration |
| Tmax | Time to maximum concentration |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Nasim, F.; Sabath, B.F.; Eapen, G.A. Lung Cancer. Med. Clin. N. Am. 2019, 103, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yin, Z.; Liu, L.; Gao, W.; Li, W.; Shu, Y.; Xu, J. Second Primary Lung Cancer After Breast Cancer: A Population-Based Study of 6,269 Women. Front. Oncol. 2018, 8, 427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, D.E.; Kazerooni, E.A.; Baum, S.L.; Eapen, G.A.; Ettinger, D.S.; Hou, L.; Jackman, D.M.; Klippenstein, D.; Kumar, R.; Lackner, R.P.; et al. Lung Cancer Screening, Version 3.2018, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2018, 16, 412–441. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, H.; Lee, A.; Eccles, L.; Yuan, Y.; Burlison, H.; Forshaw, C.; Varol, N. Treatment Patterns and Outcomes of Patients with Metastatic Non-Small Cell Lung Cancer in Five European Countries: A Real-World Evidence Survey. BMC Cancer 2023, 23, 603. [Google Scholar] [CrossRef]
- Travis, W.D.; Brambilla, E.; Burke, A.P.; Marx, A.; Nicholson, A.G. Introduction to the 2015 World Health Organization Classification of Tumors of the Lung, Pleura, Thymus, and Heart. J. Thorac. Oncol. 2015, 10, 1240–1242. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network. Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Lázaro, S.; Lorz, C.; Enguita, A.B.; Seller, I.; Paramio, J.M.; Santos, M. Pten and P53 Loss in the Mouse Lung Causes Adenocarcinoma and Sarcomatoid Carcinoma. Cancers 2022, 14, 3671. [Google Scholar] [CrossRef]
- O’Keeffe, L.M.; Taylor, G.; Huxley, R.R.; Mitchell, P.; Woodward, M.; Peters, S.A.E. Smoking as a Risk Factor for Lung Cancer in Women and Men: A Systematic Review and Meta-Analysis. BMJ Open 2018, 8, e021611. [Google Scholar] [CrossRef] [Green Version]
- Piñeros, M.; Laversanne, M.; Barrios, E.; de Camargo Cancela, M.; Cancela, C.; De Vries, E.; Pardo, C.; Bray, F. An Updated Profile of the Cancer Burden, Patterns and Trends in Latin America and the Caribbean. Lancet Reg. Health Am. 2022, 13, 100294. [Google Scholar] [CrossRef]
- Murphy, S.E. Biochemistry of Nicotine Metabolism and Its Relevance to Lung Cancer. J. Biol. Chem. 2021, 296, 100722. [Google Scholar] [CrossRef] [PubMed]
- Yousuf, H.; Hofstra, M.; Tijssen, J.; Leenen, B.; Lindemans, J.W.; van Rossum, A.; Narula, J.; Hofstra, L. Estimated Worldwide Mortality Attributed to Secondhand Tobacco Smoke Exposure, 1990–2016. JAMA Netw. Open 2020, 3, e201177. [Google Scholar] [CrossRef] [PubMed]
- Kutob, L.; Schneider, F. Lung Cancer Staging. Surg. Pathol. Clin. 2020, 13, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Woodard, G.A.; Jones, K.D.; Jablons, D.M. Lung Cancer Staging and Prognosis. Cancer Treat. Res. 2016, 170, 47–75. [Google Scholar] [CrossRef]
- Milovanovic, I.S.; Stjepanovic, M.; Mitrovic, D. Distribution Patterns of the Metastases of the Lung Carcinoma in Relation to Histological Type of the Primary Tumor: An Autopsy Study. Ann. Thorac. Med. 2017, 12, 191–198. [Google Scholar] [CrossRef]
- Tamura, T.; Kurishima, K.; Nakazawa, K.; Kagohashi, K.; Ishikawa, H.; Satoh, H.; Hizawa, N. Specific Organ Metastases and Survival in Metastatic Non-Small-Cell Lung Cancer. Mol. Clin. Oncol. 2015, 3, 217–221. [Google Scholar] [CrossRef] [Green Version]
- Leiter, A.; Veluswamy, R.R.; Wisnivesky, J.P. The Global Burden of Lung Cancer: Current Status and Future Trends. Nat. Rev. Clin. Oncol. 2023; ahead of print. [Google Scholar] [CrossRef]
- Alaswad, M. Locally Advanced Non-Small Cell Lung Cancer: Current Issues and Recent Trends. Rep. Pract. Oncol. Radiother. 2023, 28, 286–303. [Google Scholar] [CrossRef]
- Vansteenkiste, J.; Crinò, L.; Dooms, C.; Douillard, J.Y.; Faivre-Finn, C.; Lim, E.; Rocco, G.; Senan, S.; Van Schil, P.; Veronesi, G.; et al. 2nd ESMO Consensus Conference on Lung Cancer: Early-Stage Non-Small-Cell Lung Cancer Consensus on Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2014, 25, 1462–1474. [Google Scholar] [CrossRef] [PubMed]
- Icard, P.; Damotte, D.; Alifano, M. New Therapeutic Strategies for Lung Cancer. Cancers 2021, 13, 1937. [Google Scholar] [CrossRef] [PubMed]
- Oskarsdottir, G.N.; Halldorsson, H.; Sigurdsson, M.I.; Fridriksson, B.M.; Baldvinsson, K.; Orrason, A.W.; Jonsson, S.; Planck, M.; Gudbjartsson, T. Lobectomy for Non-Small Cell Lung Carcinoma: A Nationwide Study of Short- and Long-Term Survival. Acta Oncol. 2017, 56, 936–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raman, V.; Yang, C.F.J.; Deng, J.Z.; D’Amico, T.A. Surgical Treatment for Early Stage Non-Small Cell Lung Cancer. J. Thorac. Dis. 2018, 10, S898–S904. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.Y.; Zhou, Q. Lobectomy Should Remain the First Choice for Treating Early Stage Nonsmall Cell Lung Cancer. Eur. Respir. J. 2019, 54, 1900649. [Google Scholar] [CrossRef]
- Arriagada, R.; Auperin, A.; Burdett, S.; Higgins, J.P.; Johnson, D.H.; Le Chevalier, T.; Le Pechoux, C.; Parmar, M.K.B.; Pignon, J.P.; Souhami, R.L.; et al. Adjuvant Chemotherapy, with or without Postoperative Radiotherapy, in Operable Non-Small-Cell Lung Cancer: Two Meta-Analyses of Individual Patient Data. Lancet 2010, 375, 1267–1277. [Google Scholar] [CrossRef] [Green Version]
- Daly, M.E.; Singh, N.; Nofisat, I.; Antonoff, M.B.; Arenberg, D.A.; Bradley, J.; David, E.; Detterbeck, F.; Früh, M.; Früh, F.; et al. Management of Stage III Non-Small-Cell Lung Cancer: ASCO Guideline. J. Clin. Oncol. 2021, 40, 1356–1384. [Google Scholar] [CrossRef]
- Ettinger, D.S.; Wood, D.E.; Aisner, D.L.; Akerley, W.; Bauman, J.R.; Bharat, A.; Bruno, D.S.; Chang, J.Y.; Chirieac, L.R.; D’Amico, T.A.; et al. Non-Small Cell Lung Cancer, Version 3.2022. J. Natl. Compr. Cancer Netw. 2022, 20, 497–530. [Google Scholar] [CrossRef]
- Duma, N.; Santana-Davila, R.; Molina, J.R. Non-Small Cell Lung Cancer: Epidemiology, Screening, Diagnosis, and Treatment. Mayo Clin. Proc. 2019, 94, 1623–1640. [Google Scholar] [CrossRef]
- Naylor, E.C.; Desani, J.K.; Chung, P.K. Targeted Therapy and Immunotherapy for Lung Cancer. Surg. Oncol. Clin. N. Am. 2016, 25, 601–609. [Google Scholar] [CrossRef]
- Hosomi, Y.; Morita, S.; Sugawara, S.; Kato, T.; Fukuhara, T.; Gemma, A.; Takahashi, K.; Fujita, Y.; Harada, T.; Minato, K.; et al. Gefitinib Alone Versus Gefitinib Plus Chemotherapy for Non-Small-Cell Lung Cancer with Mutated Epidermal Growth Factor Receptor: NEJ009 Study. J. Clin. Oncol. 2020, 38, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.; Li, R.; Sun, J.; Lou, Y.; Zhang, W.; Bai, H.; Wang, H.; Shen, J.; Jing, B.; Shi, C.; et al. Erlotinib as Neoadjuvant Therapy in Stage IIIA (N2) EGFR Mutation-Positive Non-Small Cell Lung Cancer: A Prospective, Single-Arm, Phase II Study. Oncologist 2019, 24, 157-e64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara, M.G.; Martini, M.; D’Argento, E.; Forcella, C.; Vita, E.; Di Noia, V.; Sperduti, I.; Bilotta, M.; Ribelli, M.; Damiano, P.; et al. PTEN Loss as a Predictor of Tumor Heterogeneity and Poor Prognosis in Patients with EGFR-Mutant Advanced Non–Small-Cell Lung Cancer Receiving Tyrosine Kinase Inhibitors. Clin. Lung Cancer 2021, 22, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Marcar, L.; Bardhan, K.; Gheorghiu, L.; Dinkelborg, P.; Pfäffle, H.; Liu, Q.; Wang, M.; Piotrowska, Z.; Sequist, L.V.; Borgmann, K.; et al. Acquired Resistance of EGFR-Mutated Lung Cancer to Tyrosine Kinase Inhibitor Treatment Promotes PARP Inhibitor Sensitivity. Cell Rep. 2019, 27, 3422–3432.e4. [Google Scholar] [CrossRef] [Green Version]
- Krall, E.B.; Wang, B.; Munoz, D.M.; Ilic, N.; Raghavan, S.; Niederst, M.J.; Yu, K.; Ruddy, D.A.; Aguirre, A.J.; Kim, J.W.; et al. KEAP1 Loss Modulates Sensitivity to Kinase Targeted Therapy in Lung Cancer. Elife 2017, 6, e18970. [Google Scholar] [CrossRef] [Green Version]
- Salmena, L. PTEN: History of a Tumor Suppressor. Methods Mol. Biol. 2016, 1388, 3–11. [Google Scholar] [CrossRef]
- Parsons, R. Discovery of the PTEN Tumor Suppressor and Its Connection to the PI3K and AKT Oncogenes. Cold Spring Harb. Perspect. Med. 2020, 10, a036129. [Google Scholar] [CrossRef]
- Harris, H.; Miller, O.J.; Klein, G.; Worst, P.; Tachibana, T. Suppression of Malignancy by Cell Fusion. Nature 1969, 223, 363–368. [Google Scholar] [CrossRef]
- Knudson, A.G.J. Mutation and Cancer: Statistical Study of Retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef]
- Chen, L.; Liu, S.; Tao, Y. Regulating Tumor Suppressor Genes: Post-Translational Modifications. Signal Transduct. Target. Ther. 2020, 5, 90. [Google Scholar] [CrossRef]
- Yunis, J.J.; Ramsay, N. Retinoblastoma and Subband Deletion of Chromosome 13. Am. J. Dis. Child. 1978, 132, 161–163. [Google Scholar] [CrossRef] [PubMed]
- Cavenee, W.K.; Dryja, T.P.; Phillips, R.A.; Benedict, W.F.; Godbout, R.; Gallie, B.L.; Murphree, A.L.; Strong, L.C.; White, R.L. Expression of Recessive Alleles by Chromosomal Mechanisms in Retinoblastoma. Nature 1983, 305, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.H.; Bookstein, R.; Hong, F.; Young, L.J.; Shew, J.Y.; Lee, E.Y. Human Retinoblastoma Susceptibility Gene: Cloning, Identification, and Sequence. Science 1987, 235, 1394–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Álvarez-Garcia, V.; Tawil, Y.; Wise, H.M.; Leslie, N.R. Mechanisms of PTEN Loss in Cancer: It’s All about Diversity. Semin. Cancer Biol. 2019, 59, 66–79. [Google Scholar] [CrossRef]
- Carter, B.S.; Ewing, C.M.; Ward, W.S.; Treiger, B.F.; Aalders, T.W.; Schalken, J.A.; Epstein, J.I.; Isaacs, W.B. Allelic Loss of Chromosomes 16q and 10q in Human Prostate Cancer. Proc. Natl. Acad. Sci. USA 1990, 87, 8751–8755. [Google Scholar] [CrossRef]
- Sonoda, Y.; Murakami, Y.; Tominaga, T.; Kayama, T.; Yoshimoto, T.; Sekiya, T. Deletion Mapping of Chromosome 10 in Human Glioma. Jpn. J. Cancer Res. 1996, 87, 363–367. [Google Scholar] [CrossRef]
- Murakami, Y.S.; Albertsen, H.; Brothman, A.R.; Leach, R.J.; White, R.L. Suppression of the Malignant Phenotype of Human Prostate Cancer Cell Line PPC-1 by Introduction of Normal Fragments of Human Chromosome 10. Cancer Res. 1996, 56, 2157–2160. [Google Scholar]
- Fujimoto, M.; Fults, D.W.; Thomas, G.A.; Nakamura, Y.; Heilbrun, M.P.; White, R.; Story, J.L.; Naylor, S.L.; Kagan-Hallet, K.S.; Sheridan, P.J. Loss of Heterozygosity on Chromosome 10 in Human Glioblastoma Multiforme. Genomics 1989, 4, 210–214. [Google Scholar] [CrossRef]
- Bigner, S.H.; Mark, J.; Mahaley, M.S.; Bigner, D.D. Patterns of the Early, Gross Chromosomal Changes in Malignant Human Gliomas. Hereditas 1984, 101, 103–113. [Google Scholar] [CrossRef]
- Lee, Y.-R.; Chen, M.; Pandolfi, P.P. The Functions and Regulation of the PTEN Tumour Suppressor: New Modes and Prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef]
- Worby, C.A.; Dixon, J.E. PTEN. Annu. Rev. Biochem. 2014, 83, 641–669. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a Putative Protein Tyrosine Phosphatase Gene Mutated in Human Brain, Breast, and Prostate Cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef] [PubMed]
- Steck, P.A.; Pershouse, M.A.; Jasser, S.A.; Yung, W.K.; Lin, H.; Ligon, A.H.; Langford, L.A.; Baumgard, M.L.; Hattier, T.; Davis, T.; et al. Identification of a Candidate Tumour Suppressor Gene, MMAC1, at Chromosome 10q23.3 That Is Mutated in Multiple Advanced Cancers. Nat. Genet. 1997, 15, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Liaw, D.; Marsh, D.J.; Li, J.; Dahia, P.L.; Wang, S.I.; Zheng, Z.; Bose, S.; Call, K.M.; Tsou, H.C.; Peacocke, M.; et al. Germline Mutations of the PTEN Gene in Cowden Disease, an Inherited Breast and Thyroid Cancer Syndrome. Nat. Genet. 1997, 16, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Fusco, N.; Sajjadi, E.; Venetis, K.; Gaudioso, G.; Lopez, G.; Corti, C.; Rocco, E.G.; Criscitiello, C.; Malapelle, U.; Invernizzi, M. Pten Alterations and Their Role in Cancer Management: Are We Making Headway on Precision Medicine? Genes 2020, 11, 719. [Google Scholar] [CrossRef] [PubMed]
- Gkountakos, A.; Sartori, G.; Falcone, I.; Piro, G.; Ciuffreda, L.; Carbone, C.; Tortora, G.; Scarpa, A.; Bria, E.; Milella, M.; et al. PTEN in Lung Cancer: Dealing with the Problem, Building on New Knowledge and Turning the Game Around. Cancers 2019, 11, 1141. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.; Zhu, Y.; Chen, W.; Merlino, G.; Yu, Y. PTEN Dual Lipid- and Protein-Phosphatase Function in Tumor Progression. Cancers 2022, 14, 3666. [Google Scholar] [CrossRef]
- Rossi, G.; Russo, A.; Tagliamento, M.; Tuzi, A.; Nigro, O.; Vallome, G.; Sini, C.; Grassi, M.; Dal Bello, M.G.; Coco, S.; et al. Precision Medicine for NSCLC in the Era of Immunotherapy: New Biomarkers to Select the Most Suitable Treatment or the Most Suitable Patient. Cancers 2020, 12, 1125. [Google Scholar] [CrossRef]
- Mukherjee, R.; Vanaja, K.G.; Boyer, J.A.; Gadal, S.; Solomon, H.; Chandarlapaty, S.; Levchenko, A.; Rosen, N. Regulation of PTEN Translation by PI3K Signaling Maintains Pathway Homeostasis. Mol. Cell 2021, 81, 708–723.e5. [Google Scholar] [CrossRef]
- Li, Q.; Li, Z.; Luo, T.; Shi, H. Targeting the PI3K/AKT/MTOR and RAF/MEK/ERK Pathways for Cancer Therapy. Mol. Biomed. 2022, 3, 47. [Google Scholar] [CrossRef]
- Vidotto, T.; Melo, C.M.; Castelli, E.; Koti, M.; dos Reis, R.B.; Squire, J.A. Emerging Role of PTEN Loss in Evasion of the Immune Response to Tumours. Br. J. Cancer 2020, 122, 1732–1743. [Google Scholar] [CrossRef] [PubMed]
- Luongo, F.; Colonna, F.; Calapà, F.; Vitale, S.; Fiori, M.E.; De Maria, R. Pten Tumor-Suppressor: The Dam of Stemness in Cancer. Cancers 2019, 11, 1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanaei, M.J.; Razi, S.; Pourbagheri-Sigaroodi, A.; Bashash, D. The PI3K/Akt/MTOR Pathway in Lung Cancer; Oncogenic Alterations, Therapeutic Opportunities, Challenges, and a Glance at the Application of Nanoparticles. Transl. Oncol. 2022, 18, 101364. [Google Scholar] [CrossRef]
- Hopkins, B.D.; Fine, B.; Steinbach, N.; Dendy, M.; Rapp, Z.; Shaw, J.; Pappas, K.; Yu, J.S.; Hodakoski, C.; Mense, S.; et al. A Secreted PTEN Phosphatase That Enters Cells to Alter Signaling and Survival. Science 2013, 341, 399–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngeow, J.; Eng, C. PTEN in Hereditary and Sporadic Cancer. Cold Spring Harb. Perspect. Med. 2020, 10, a036087. [Google Scholar] [CrossRef] [Green Version]
- Fischer, T.; Hartmann, O.; Reissland, M.; Prieto-Garcia, C.; Klann, K.; Pahor, N.; Schülein-Völk, C.; Baluapuri, A.; Polat, B.; Abazari, A.; et al. PTEN Mutant Non-Small Cell Lung Cancer Require ATM to Suppress pro-Apoptotic Signalling and Evade Radiotherapy. Cell Biosci. 2022, 12, 50. [Google Scholar] [CrossRef]
- Li, X.; Xie, W.; Xie, C.; Huang, C.; Zhu, J.; Liang, Z.; Deng, F.; Zhu, M.; Zhu, W.; Wu, R.; et al. Curcumin Modulates MiR-19/PTEN/AKT/P53 Axis to Suppress Bisphenol A-Induced MCF-7 Breast Cancer Cell Proliferation. Phytother. Res. 2014, 28, 1553–1560. [Google Scholar] [CrossRef]
- Zhang, W.; Bai, W.; Zhang, W. MiR-21 Suppresses the Anticancer Activities of Curcumin by Targeting PTEN Gene in Human Non-Small Cell Lung Cancer A549 Cells. Clin. Transl. Oncol. 2014, 16, 708–713. [Google Scholar] [CrossRef]
- Wang, X.; He, H.; Lu, Y.; Ren, W.; Teng, K.-Y.; Chiang, C.-L.; Yang, Z.; Yu, B.; Hsu, S.; Jacob, S.T.; et al. Indole-3-Carbinol Inhibits Tumorigenicity of Hepatocellular Carcinoma Cells via Suppression of MicroRNA-21 and Upregulation of Phosphatase and Tensin Homolog. Biochim. Biophys. Acta 2015, 1853, 244–253. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Tian, X.; Lu, M.; Zhang, X.; Ma, K.; Lv, Z.; Wang, Z.; Hu, Y.; Xun, C.; Zhang, Z.; et al. Genistein Exerts Growth Inhibition on Human Osteosarcoma MG-63 Cells via PPARγ Pathway. Int. J. Oncol. 2015, 46, 1131–1140. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Shao, T.; Zheng, W.; Ding, J. Curcumin Suppresses Tumor Growth of Gemcitabine-Resistant Non-Small Cell Lung Cancer by Regulating LncRNA-MEG3 and PTEN Signaling. Clin. Transl. Oncol. 2021, 23, 1386–1393. [Google Scholar] [CrossRef]
- Ranjan, A.; Ramachandran, S.; Gupta, N.; Kaushik, I.; Wright, S.; Srivastava, S.; Das, H.; Srivastava, S.; Prasad, S.; Srivastava, S.K. Role of Phytochemicals in Cancer Prevention. Int. J. Mol. Sci. 2019, 20, 4981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.A.; Hannan, M.A.; Dash, R.; Rahman, M.H.; Islam, R.; Uddin, M.J.; Sohag, A.A.M.; Rahman, M.H.; Rhim, H. Phytochemicals as a Complement to Cancer Chemotherapy: Pharmacological Modulation of the Autophagy-Apoptosis Pathway. Front. Pharmacol. 2021, 12, 639628. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Fang, T.; Pang, T.; Chen, Z.; Cheng, L.; Ma, D.; Xi, Z. The Potential Application of Branch-PCR Assembled PTEN Gene Nanovector in Lung Cancer Gene Therapy. ChemBioChem 2022, 23, e202200387. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.-X.; Cao, L.-Y.; Chen, X.; Xiao, J.; Zou, Y.; Chen, Q. PTEN Inhibits Cell Proliferation, Promotes Cell Apoptosis, and Induces Cell Cycle Arrest via Downregulating the PI3K/AKT/HTERT Pathway in Lung Adenocarcinoma A549 Cells. BioMed Res. Int. 2016, 2016, 2476842. [Google Scholar] [CrossRef] [Green Version]
- Sacco, J.J.; Yau, T.Y.; Darling, S.; Patel, V.; Liu, H.; Urbé, S.; Clague, M.J.; Coulson, J.M. The Deubiquitylase Ataxin-3 Restricts PTEN Transcription in Lung Cancer Cells. Oncogene 2014, 33, 4265–4272. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Zhang, J.; Su, Y.; Hou, Y.; Wang, Z.; Zhao, L.; Sun, S.; Fu, H. Overexpression of PTEN May Increase the Effect of Pemetrexed on A549 Cells via Inhibition of the PI3K/AKT/MTOR Pathway and Carbohydrate Metabolism. Mol. Med. Rep. 2019, 20, 3793–3801. [Google Scholar] [CrossRef] [Green Version]
- Noro, R.; Gemma, A.; Miyanaga, A.; Kosaihira, S.; Minegishi, Y.; Nara, M.; Kokubo, Y.; Seike, M.; Kataoka, K.; Matsuda, K.; et al. PTEN Inactivation in Lung Cancer Cells and the Effect of Its Recovery on Treatment with Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors. Int. J. Oncol. 2007, 31, 1157–1163. [Google Scholar]
- Yu, Y.X.; Wang, Y.; Liu, H. Overexpression of PTEN Suppresses Non-Small-Cell Lung Carcinoma Metastasis through Inhibition of Integrin AVβ6 Signaling. Am. J. Transl. Res. 2017, 9, 3304–3314. [Google Scholar]
- Malkoski, S.P.; Cleaver, T.G.; Thompson, J.J.; Sutton, W.P.; Haeger, S.M.; Rodriguez, K.J.; Lu, S.-L.; Merrick, D.; Wang, X.-J. Role of PTEN in Basal Cell Derived Lung Carcinogenesis. Mol. Carcinog. 2014, 53, 841–846. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Jiang, S.; Mao, C.; Zheng, H.; Cao, B.; Zhang, Z.; Zhao, J.; Zeng, Y.; Mao, X. The Deubiquitinase USP10 Restores PTEN Activity and Inhibits Non-Small Cell Lung Cancer Cell Proliferation. J. Biol. Chem. 2021, 297, 101088. [Google Scholar] [CrossRef] [PubMed]
- Cong, S.; Di, X.; Li, R.; Cao, Y.; Jin, X.; Tian, C.; Zhao, M.; Wang, K. RBM10 Regulates Alternative Splicing of LncRNA Neat1 to Inhibit the Invasion and Metastasis of NSCLC. Cancer Cell Int. 2022, 22, 338. [Google Scholar] [CrossRef]
- Cai, J.; Li, R.; Xu, X.; Zhang, L.; Lian, R.; Fang, L.; Huang, Y.; Feng, X.; Liu, X.; Li, X.; et al. CK1α Suppresses Lung Tumour Growth by Stabilizing PTEN and Inducing Autophagy. Nat. Cell Biol. 2018, 20, 465–478. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Wang, X.; Chen, W.; Zhang, J.; Xiong, Y.; Cao, L.; Zhang, L.; Zhao, N.; Yang, Y.; Wang, L. PTPIP51 Inhibits Non-Small-Cell Lung Cancer by Promoting PTEN-Mediated EGFR Degradation. Life Sci. 2022, 297, 120293. [Google Scholar] [CrossRef] [PubMed]
- He, Y.-M.; Zhou, X.-M.; Jiang, S.-Y.; Zhang, Z.-B.; Cao, B.-Y.; Liu, J.-B.; Zeng, Y.-Y.; Zhao, J.; Mao, X.-L. TRIM25 Activates AKT/MTOR by Inhibiting PTEN via K63-Linked Polyubiquitination in Non-Small Cell Lung Cancer. Acta Pharmacol. Sin. 2022, 43, 681–691. [Google Scholar] [CrossRef]
- Jin, G.; Kim, M.J.; Jeon, H.-S.; Choi, J.E.; Kim, D.S.; Lee, E.B.; Cha, S.I.; Yoon, G.S.; Kim, C.H.; Jung, T.H.; et al. PTEN Mutations and Relationship to EGFR, ERBB2, KRAS, and TP53 Mutations in Non-Small Cell Lung Cancers. Lung Cancer 2010, 69, 279–283. [Google Scholar] [CrossRef]
- Sos, M.L.; Koker, M.; Weir, B.A.; Heynck, S.; Rabinovsky, R.; Zander, T.; Seeger, J.M.; Weiss, J.; Fischer, F.; Frommolt, P.; et al. PTEN Loss Contributes to Erlotinib Resistance in EGFR-Mutant Lung Cancer by Activation of Akt and EGFR. Cancer Res. 2009, 69, 3256–3261. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.B.; Xu, X.; Lee, H.J.; Jheon, S.; Lee, C.T.; Choe, G.; Chung, J.H. Loss of PTEN Expression Is an Independent Poor Prognostic Factor in Non-Small Cell Lung Cancer. J. Pathol. Transl. Med. 2011, 45, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Cumberbatch, M.; Tang, X.; Beran, G.; Eckersley, S.; Wang, X.; Ellston, R.P.A.; Dearden, S.; Cosulich, S.; Smith, P.D.; Behrens, C.; et al. Identification of a Subset of Human Non-Small Cell Lung Cancer Patients with High PI3Kβ and Low PTEN Expression, More Prevalent in Squamous Cell Carcinoma. Clin. Cancer Res. 2014, 20, 595–603. [Google Scholar] [CrossRef] [Green Version]
- Panagiotou, I.; Tsiambas, E.; Lazaris, A.C.; Kavantzas, N.; Konstantinou, M.; Kalkandi, P.; Ragkos, V.; Metaxas, G.E.; Roukas, D.K.; Vilaras, G.; et al. PTEN Expression in Non Small Cell Lung Carcinoma Based on Digitized Image Analysis. J. BUON 2012, 17, 719–723. [Google Scholar]
- City of Hope Medical Center. PI3K Inhibitor BKM120, Carboplatin, and Pemetrexed Disodium in Treating Patients with Stage IV Non-Small Cell Lung Cancer. In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT01723800. Available online: https://clinicaltrials.gov/study/NCT01723800 (accessed on 24 July 2023).
- University of Oxford. Palliative Thoracic Radiotherapy Plus BKM120 (BKM120). In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT02128724. Available online: https://clinicaltrials.gov/study/NCT02128724 (accessed on 24 July 2023).
- National Cancer Centre, Singapore. A Trial of Gefitinib in Combination with BKM120 in Patients with Advanced Non-Small Cell Lung Cancer, with Enrichment for Patients Whose Tumours Harbour Molecular Alterations of PI3K Pathway and Known to Overexpress EGFR (BKM120). In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT01570296. Available online: https://clinicaltrials.gov/study/NCT01570296 (accessed on 24 July 2023).
- EMD Serono. Trial of MEK Inhibitor and PI3K/mTOR Inhibitor in Subjects with Locally Advanced or Metastatic Solid Tumors. In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT01390818. Available online: https://clinicaltrials.gov/study/NCT01390818 (accessed on 24 July 2023).
- Genentech, Inc. A Study of the Safety and Pharmacology of PI3-Kinase Inhibitor GDC-0941 in Combination with Either Paclitaxel And Carboplatin (with or without Bevacizumab) or Pemetrexed, Cisplatin, and Bevacizumab in Patients with Advanced Non-Small Cell Lung Cancer. In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT00974584. Available online: https://clinicaltrials.gov/study/NCT00974584 (accessed on 24 July 2023).
- BeiGene. Study of BGB-10188 as Monotherapy, and in Combination with Zanubrutinib, and Tislelizumab. In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT04282018. Available online: https://clinicaltrials.gov/study/NCT04282018 (accessed on 24 July 2023).
- Totus Medicines. A Study to Evaluate the Safety and Tolerability of TOS-358 in Adults with Select Solid Tumors. In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT05683418. Available online: https://clinicaltrials.gov/study/NCT05683418 (accessed on 24 July 2023).
- Chia Tai Tianqing Pharmaceutical Group. TQ-B3525 Tablets Combined with Osimertinib Mesylate Tablets in the Treatment of Advanced Non-Small Cell Lung Cancer. In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT05284994. Available online: https://clinicaltrials.gov/study/NCT05284994 (accessed on 24 July 2023).
- Hao, Z. Pembrolizumab + Idelalisib for Lung Cancer Study (PIL). In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT03257722. Available online: https://clinicaltrials.gov/study/NCT03257722 (accessed on 24 July 2023).
- National Cancer Institute (NCI). MK2206 and Erlotinib Hydrochloride in Treating Patients with Advanced Non-Small Cell Lung Cancer Who Have Progressed After Previous Response to Erlotinib Hydrochloride Therapy. In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT01294306. Available online: https://clinicaltrials.gov/study/NCT01294306 (accessed on 24 July 2023).
- Celgene. Study to Assess Safety, Pharmacokinetics, and Efficacy of Oral CC-223 for Patients with Advanced Solid Tumors, Non-Hodgkin Lymphoma or Multiple Myeloma. In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT01177397. Available online: https://clinicaltrials.gov/study/NCT01177397 (accessed on 24 July 2023).
- Celgene. Study Assessing Safety, Pharmacokinetics and Efficacy of CC-223 with Either Erlotinib or Oral Azacitidine in Advanced Non-Small Cell Lung Cancer. In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT01545947. Available online: https://clinicaltrials.gov/study/NCT01545947 (accessed on 24 July 2023).
- Novartis Pharmaceuticals. Combination of RAD001 with Carboplatin, Paclitaxel and Bevacizumab in Non-small-cell Lung Cancer (NSCLC) Patients. In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT00457119. Available online: https://clinicaltrials.gov/study/NCT00457119 (accessed on 24 July 2023).
- Novartis Pharmaceuticals. Combination of RAD001 and Erlotinib in Patients with Advanced Non Small Cell Lung Cancer Previously Treated Only with Chemotherapy. In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT00456833. Available online: https://clinicaltrials.gov/study/NCT00456833 (accessed on 24 July 2023).
- Washington University School of Medicine. A Phase I Study of SUNITINIB and Rapamycin in Advanced Non-Small Cell Lung Cancer (NSCLC) (SU/Rapamycin). In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT00555256. Available online: https://clinicaltrials.gov/study/NCT00555256 (accessed on 24 July 2023).
- Cancer Research and Biostatistics Clinical Trials Consortium. Study of Everolimus, Pemetrexed, Carboplatin, and Bevacizumab to Treat Stage IV Lung Cancer. In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT01700400. Available online: https://clinicaltrials.gov/study/NCT01700400 (accessed on 24 July 2023).
- Memorial Sloan Kettering Cancer Center. Gefitinib and Everolimus in Treating Patients with Stage IIIB or Stage IV or Recurrent Non-Small Cell Lung Cancer. In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT00096486. Available online: https://clinicaltrials.gov/study/NCT00096486 (accessed on 24 July 2023).
- Puma Biotechnology. Neratinib with and without Temsirolimus for Patients with HER2 Activating Mutations in Non-Small Cell Lung Cancer. In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT01827267. Available online: https://clinicaltrials.gov/study/NCT01827267 (accessed on 24 July 2023).
- Boehringer Ingelheim. Trial of Continuous Once Daily Oral Treatment Using BIBW 2992 (Afatinib) Plus Sirolimus (Rapamune®) in Patients with Non-small Cell Lung Cancer Harbouring an EGFR Mutation and/or Disease Progression Following Prior Erlotinib or Gefitinib. In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT00993499. Available online: https://clinicaltrials.gov/study/NCT00993499 (accessed on 24 July 2023).
- Novartis Pharmaceuticals. A Study of PDR001 in Combination with LCL161, Everolimus or Panobinostat. In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT02890069. Available online: https://clinicaltrials.gov/study/NCT02890069 (accessed on 24 July 2023).
- Infinity Pharmaceuticals. Phase 1b/2 Study of Retaspimycin HCl (IPI-504) in Combination with Everolimus in KRAS Mutant Non-small Cell Lung Cancer. In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT01427946. Available online: https://clinicaltrials.gov/study/NCT01427946 (accessed on 24 July 2023).
- Mayo Clinic. Sirolimus and Auranofin in Treating Patients with Advanced or Recurrent Non-Small Cell Lung Cancer or Small Cell Lung Cancer. In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT01737502. Available online: https://clinicaltrials.gov/study/NCT01737502 (accessed on 24 July 2023).
- Washington University School of Medicine. Temsirolimus and Radiation for Non-Small Cell Lung Cancer. In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT00796796. Available online: https://clinicaltrials.gov/study/NCT00796796 (accessed on 24 July 2023).
- Clarke, J. A Trial of AMG 479, Everolimus (RAD001) and Panitumumab in Patients with Advanced Cancer—QUILT-3.007 (RAP). In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT01061788. Available online: https://clinicaltrials.gov/study/NCT01061788 (accessed on 24 July 2023).
- National Cancer Institute (NCI). CCI-779 in Treating Patients with Stage IIIB (with Pleural Effusion) or Stage IV Non-Small Cell Lung Cancer. In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT00079235. Available online: https://clinicaltrials.gov/study/NCT00079235 (accessed on 24 July 2023).
- Emory University. Trial of RAD001 in Patients with Operable Non-Small Cell Lung Cancer (NSCLC). In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT00401778. Available online: https://clinicaltrials.gov/study/NCT00401778 (accessed on 24 July 2023).
- Novartis Pharmaceuticals. Safety of Everolimus and Pemetrexed in Lung Cancer Patients. In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT00434174. Available online: https://clinicaltrials.gov/study/NCT00434174 (accessed on 24 July 2023).
- Novartis Pharmaceuticals. Study Investigating the Effect of Everolimus Monotherapy in Patients with Advanced Non-small Cell Lung Cancer (NSCLC). In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT00124280. Available online: https://clinicaltrials.gov/study/NCT00124280 (accessed on 24 July 2023).
- A&G Pharmaceutical Inc First in Human Phase 1 Study of AG01 Anti-Progranulin/GP88 Antibody in Advanced Solid Tumor Malignancies. In ClinicalTrials.gov; National Library of Medicine: Bethesda, MD, USA; ClinicalTrials.gov Identifier: NCT05627960. Available online: https://clinicaltrials.gov/study/NCT05627960 (accessed on 24 July 2023).


{kind=link}
| Cell Lines | Therapies | Target (s) | Findings | Refs. |
|---|---|---|---|---|
| A549 cells | - | PI3K/AKT/hTERT signaling pathway | PTEN downregulates PI3K/AKT/hTERT pathway to suppress the growth of A549 cells. | [75] |
| A549 cells | - | PI3K/AKT signaling pathway | ATXN3 provides an autonomous, complementary therapeutic target in cancers with epigenetic downregulation of PTEN. | [76] |
| A549 cells | Pemetrexed | PI3K/AKT/mTOR signaling pathway | Pemetrexed inhibits the proliferation of lung cancer cells via inhibiting the PI3K/AKT/mTOR pathway and through carbohydrate metabolism. | [77] |
| Twelve adenocarcinoma cell lines (ABC-1, A549, PC3, PC7, RERF-LCMS, RERF- LCKJ, VMRC-LCD, RERF-LCOK, PC14, PC9, PC9/f9, and PC9/f14). Eight squamous cell carcinoma cell lines (QG56, EBC-1, LK-2, LC-1/sq, PC1, RERF-LCAI, PC10, and SQ5). Five small cell carcinoma cell lines (NCI-H69, SBC3, NCI-N231, and Lu135). | 5-AZA, HDAC inhibitor, TSA, and gefitinib | PI3K/AKT signaling pathway | The combination of gefitinib with 5-AZA and TSA exerts synergistic effects in the inhibition of cell survival via targeting the PI3K/AKT pathway and, thus, could be explored for treating lung cancer. | [78] |
| H1975, A549, HCC827, and H1650 cell lines and female BALB/c nu/nu mouse model. | - | The integrin αVβ6 signaling pathway | Overexpression of PTEN resulted in the suppression of cancer progression by the inhibition of integrin αVβ6. | [79] |
| Transgenic mouse harboring TGFβRII conditional deletion allele, PTEN conditional deletion allele, lox-stop-lox-(LSL) KrasG12D knock-in allele, K5CrePR, and K14CrePR1 transgenes. | - | - | PTEN deletion in airway basal cells can initiate both lung adenocarcinoma and squamous cell carcinoma formation, and these tumors were relatively larger and highly malignant compared to the tumors initiated via KrasG12D mutation. | [80] |
| A549 and H1299 cell lines and NSCLC tissues. | - | AKT/mTOT | USP10 inhibits NSCLC proliferation by preventing PTEN from K63-linked polyubiquitination and inhibiting the activation of AKT/mTOR pathway. | [81] |
| A549 and H1299 NSCLC cell lines and NSCLC tissues. | - | PTEN/PI3K/AKT/ mTOR/Neat1 | RBM10 targets PTEN/PI3K/AKT/mTOR/Neat1 axis to regulate the growth of NSCLC cells. | [82] |
| A549 cells, xenograft model, and NSCLC tissues. | - | CK1α/PTEN/FOXO3a/Atg7 | CK1α/PTEN/FOXO3a/Atg7 axis inhibits the growth of A549 NSCLC cells and A549 lung tumor xenografts via inducing autophagy. | [83] |
| PC9 and A549 NSCLC cell lines, PDX mouse model, and NSCLC tissues. | Gefitinib | EGFR | PTPIP51 inhibits NSCLC proliferation, induces apoptosis, decreases tumor growth, and increases gefitinib sensitivity via PTEN activation and EGFR degradation. | [84] |
| Study Design | Therapy | Target (s) | Findings | Refs. |
|---|---|---|---|---|
| To define somatic mutations in PTEN in small number of NSCLC cases. | - | - | A subset of PTEN mutation-positive patients exhibited a positive correlation with p53 and EGFR genes, and none of these patients had the KRAS gene mutation. | [86] |
| To determine the role of homozygous deletion of PTEN as a candidate for EGFR inhibitor resistance. | Erlotinib | AKT signaling pathway | The results were centrally related to the co-occurrence of PTEN loss and EFGR mutations. In EGFR-mutant NSCLC, PTEN loss acts as a mechanism of initiation or acquired resistance to erlotinib-induced apoptosis. | [87] |
| To analyze PTEN expression and the correlation between PTEN expression and clinicopathological parameters in NSCLC. | - | - | 42.4% (122/288) of NSCLC patients had a loss of PTEN expression. Squamous cell carcinoma, male gender, and smoking exhibited a correlation with the loss of PTEN expression. | [88] |
| To identify a subset of NSCLC based on PI3Kβ and PTEN expression. | - | PI3Kβ pathway | In NSCLC patients, SCC was more prevalent with elevated expression of PI3Kβ accompanied by a reduction/loss of PTEN, for whom selective PI3Kβ inhibitors may be predicted to achieve more significant clinical benefit. | [89] |
| To analyze PTEN expression in NSCLC. | - | - | In NSCLC, PTEN deregulation is associated with an aggressive phenotype, and this is an essential factor in determining tailored targeted therapy regimens, especially when combined with HER2/PI3K inhibitors. | [90] |
| Study Title | Study Phase/ Status | Conditions | Intervention | Primary Outcome Measures | NCT Number | Refs. |
|---|---|---|---|---|---|---|
| PI3K inhibitor BKM120, carboplatin, and pemetrexed disodium in treating patients with stage IV non-small cell lung cancer. | Phase I (Completed) | Recurrent NSCLC, Stage IV NSCLC | PI3K inhibitor BKM120, pemetrexed disodium, carboplatin | Maximum tolerated dose (MTD), defined as the highest dose in which fewer than 33% of study participants experience a dose limiting toxicity (DLT), toxicity profile of PI3K inhibitor BKM120. | NCT01723800 | [91] |
| Palliative thoracic radiotherapy, plus BKM120. | Phase I (Completed) | NSCLC | BKM120 | Dose escalation analysis: number of DLTs observed in evaluable patients; MTD was defined as the highest dose at which no more than 1 of 6 evaluable patients or 0 of 3 evaluable patients experience DLT. | NCT02128724 | [92] |
| A trial of gefitinib in combination with BKM120 in patients with advanced non-small cell lung cancer, with enrichment for patients whose tumors harbor molecular alterations of PI3K pathway and are known to overexpress EGFR. | Phase I (Completed) | NSCLC, solid tumors | Gefitinib and BKM120 | Recommended Phase 2 dose for gefitinib and BKM120 combination therapy. | NCT01570296 | [93] |
| Trial of MEK inhibitor and PI3K/mTOR inhibitor in subjects with locally advanced or metastatic solid tumors. | Phase I (Completed) | Locally advanced solid tumor, metastatic solid tumor, breast cancer, NSCLC, melanoma, colorectal cancer | MSC1936369B (pimasertib), SAR245409 (PI3K), and mTOR inhibitor. | Number of subjects with DLT. | NCT01390818 | [94] |
| A study of the safety and pharmacology of PI3-kinase inhibitor, GDC-0941, in combination with either paclitaxel and carboplatin (with or without bevacizumab) or pemetrexed, cisplatin, and bevacizumab in patients with advanced NSCLC. | Phase I (Completed) | Non-squamous NSCLC | GDC-0941, bevacizumab, carboplatin, cisplatin, paclitaxel, pemetrexed | Number of participants with DLTs; percentage of participants with adverse events (AEs). | NCT00974584 | [95] |
| Study of BGB-10188 as monotherapy and in combination with zanubrutinib and tislelizumab. | Phase I, Phase II (Ongoing) | Chronic lymphocytic leukemia, small lymphocytic lymphoma, follicular lymphoma, marginal zone lymphoma, mantle cell lymphoma, diffuse large B-cell lymphoma, advanced solid tumor, NSCLC, small cell lung cancer, metastatic melanoma | BGB-10188, Zanubrutinib, tislelizumab | The recommended Phase 2 dose (RP2D) of BGB-10188 monotherapy, RP2D of BGB-10188 in combination with Zanubrutinib, RP2D of BGB-10188 in combination with tislelizumab, overall response rate (ORR), number of participants experiencing treatment-emergent adverse events (TEAEs), number of participants experiencing severe adverse events (SAEs), number of participants experiencing AEs leading to discontinuation. | NCT04282018 | [96] |
| A study to evaluate the safety and tolerability of TOS-358 in adults with select solid tumors. | Phase I (Ongoing) | Colorectal cancer, gastric cancer, HER2-negative breast cancer, NSCLC, squamous cell carcinoma of head and neck, urothelial carcinoma, cervical cancer, ovarian cancer, endometrial cancer | TOS-358 | Determine the rate of DLTs, incidence, and severity of AEs, and specific laboratory abnormalities. | NCT05683418 | [97] |
| TQ-B3525 tablets combined with osimertinib mesylate tablets in the treatment of advanced NSCLC. | Phase I, Phase II (Ongoing) | NSCLC | TQ-B3525 tablets, osimertinib mesylate tablets | DLT, baseline time to increase chemotherapy drug dose to toxicity intolerance, objective response rate (ORR). | NCT05284994 | [98] |
| Pembrolizumab + idelalisib for lung cancer study. | Phase I, Phase II (Unknown) | NSCLC, metastasis, recurrence | Pembrolizumab, idelalisib | Number of participants with DLT. | NCT03257722 | [99] |
| MK2206 and erlotinib hydrochloride in treating patients with advanced NSCLC who have progressed after previous response to erlotinib hydrochloride therapy. | Phase II (Completed) | Adenosquamous lung carcinoma, bronchioloalveolar carcinoma, large cell lung carcinoma, lung adenocarcinoma, recurrent NSCLC, squamous cell lung carcinoma | Akt inhibitor MK2206, erlotinib hydrochloride | Disease-control rate, response. | NCT01294306 | [100] |
| Study to assess safety, pharmacokinetics, and efficacy of oral CC-223 for patients with advanced solid tumors, non-Hodgkin lymphoma, or multiple myeloma. | Phase I, Phase II (Completed) | Multiple myeloma, diffuse large B-Cell lymphoma, glioblastoma multiforme, hepatocellular carcinoma, NSCLC, neuroendocrine tumors of non-pancreatic origin, hormone receptor-positive breast cancer | CC-223 | Number of participants with DLT, maximum observed plasma concentration (Cmax) of CC-223, time to maximum concentration (Tmax) of CC-223, apparent volume of distribution (Vz/F) of CC-223. | NCT01177397 | [101] |
| Study assessing safety, pharmacokinetics, and efficacy of CC-223 with either erlotinib or oral azacitidine in advanced NSCLC. | Phase I (Completed) | NSCLC | CC-223, erlotinib, CC-223, oral azacitidine | Adverse events, number of participants with adverse events, MTD, PK-Cmax, Pk-Maximum observed concentration in plasma (Cmax), PK-AUC, PK-Tmax. | NCT01545947 | [102] |
| Combination of RAD001 with carboplatin, paclitaxel, and bevacizumab in NSCLC patients. | Phase I (Completed) | NSCLC | RAD001 | Establish the feasible doses/regimens of RAD001 in combination with chemotherapy. Primary endpoint is the end-of-cycle DLT rate every 3 months or once critical DLT occurs. | NCT00457119 | [103] |
| Combination of RAD001 and erlotinib in patients with advanced NSCLC previously treated only with chemotherapy. | Phase I (Completed) | NSCLC | RAD001, erlotinib | Dose limiting toxicities (DLTs) and PK drug–drug interaction (DDI). Phase 2: Tumor response assessed by CT scans. | NCT00456833 | [104] |
| A Phase I study of SUNITINIB and rapamycin in advanced NSCLC. | Phase I (Completed) | NSCLC | Sunitinib and rapamycin | To define the optimal dose of sunitinib when given in combination with rapamycin 2mg daily, determine the dose limiting toxicity of sunitinib and rapamycin. | NCT00555256 | [105] |
| Study of everolimus, pemetrexed, carboplatin, and bevacizumab to treat stage IV lung cancer | Phase I (Completed) | NSCLC | Everolimus, pemetrexed, carboplatin, bevacizumab | Define the MTD and RP2TD of the combination of everolimus with pemetrexed, carboplatin, and bevacizumab in patients with Stage IV non-squamous NSCLC. | NCT01700400 | [106] |
| Gefitinib and everolimus in treating patients with Stage IIIB, Stage IV, or recurrent NSCLC. | Phase I (Completed) | NSCLC | Everolimus, gefitinib | Overall objective response, efficacy of the combination of daily oral gefitinib and daily oral RAD001 in patients with advanced NSCLC. | NCT00096486 | [107] |
| Neratinib with and without temsirolimus for patients with HER2-activating mutations in NSCLC. | Phase II (Completed) | HER2-mutant NSCLC | Neratinib, temsirolimus | ORR | NCT01827267 | [108] |
| Trial of continuous once-daily oral treatment using BIBW 2992 (afatinib) plus sirolimus (Rapamune®) in patients with NSCLC harboring an EGFR mutation and/or disease progression following prior erlotinib or gefitinib. | Phase I (Completed) | NSCLC | BIBW 2992, sirolimus (rapamycin) | Occurrence of DLT, number of participants with DLT. | NCT00993499 | [109] |
| A study of PDR001 in combination with LCL161, everolimus, or panobinostat. | Phase I (Completed) | Colorectal cancer, non-small cell lung carcinoma (adenocarcinoma), triple-negative breast cancer, renal cell carcinoma | PDR001, LCL161, everolimus, panobinostat, QBM076, HDM201 | Incidence of DLTs, frequency of dose interruptions and reductions, frequency and severity of treatment-emergent AEs and SAEs. | NCT02890069 | [110] |
| Phase 1b/2 study of retaspimycin HCl (IPI-504) in combination with everolimus in KRAS-mutant NSCLC. | Phase I, Phase II (Completed) | NSCLC | IPI-504, everolimus | ORR | NCT01427946 | [111] |
| Sirolimus and auranofin in treating patients with advanced or recurrent NSCLC or small cell lung cancer. | Phase I, Phase II (Completed) | Extensive stage small cell lung carcinoma, lung adenocarcinoma, recurrent NSCLC, recurrent small cell lung carcinoma, squamous cell lung carcinoma, Stage IIIA NSCLC, Stage IIIB NSCLC, Stage IV NSCLC | Auranofin, sirolimus | MTD of auranofin, the number and severity of all AEs and PFS. | NCT01737502 | [112] |
| Temsirolimus and radiation for NSCLC. | Phase I (Completed) | NSCLC | Temsirolimus, radiation therapy | Determine the MTD of temsirolimus given with radiation. | NCT00796796 | [113] |
| A Trial of AMG 479, everolimus (RAD001), and panitumumab in patients with advanced cancer—QUILT-3.007. | Phase I (Completed) | Advanced solid tumors, NSCLC | AMG 479, everolimus, panitumumab | To define the MTD and/or recommended RP2TD for the doublet AMG 479 in combination with everolimus in subjects with advanced solid tumors, to define the MTD and/or recommended RP2TD for the triplet AMG 479 in combination with everolimus and panitumumab in subjects with advanced solid tumors. | NCT01061788 | [114] |
| CCI-779 in treating patients with Stage IIIB (with pleural effusion) or stage IV NSCLC. | Phase II (Completed) | Unspecified adult solid tumor, NSCLC | Combination of sorafenib and everolimus | To confirm tumor response. | NCT00079235 | [115] |
| Trial of RAD001 in patients with operable NSCLC. | Phase I (Completed) | Lung cancer | RAD001 | Clinical response, effects of RAD001 on the regulation of key proteins involved with the mTOR axis in tumor specimens and buccal mucosa in patients with operable NSCLC, inhibition of proliferation (Ki67) and induction of apoptosis (TUNEL assay) in tumor specimens and buccal mucosa. | NCT00401778 | [116] |
| Safety of everolimus and pemetrexed in lung cancer patients. | Phase I (Completed) | NSCLC | Everolimus, pemetrexed | Establish feasible dose levels/regimens of everolimus combined with pemetrexed chemotherapy. | NCT00434174 | [117] |
| Study investigating the effect of everolimus monotherapy in patients with advanced NSCLC. | Phase II (Completed) | NSCLC | RAD001 | Clinical efficacy based on the evaluation of objective tumor response rate. | NCT00124280 | [118] |
| First in human Phase 1 study of AG01 anti-progranulin/GP88 antibody in advanced solid tumor malignancies. | Phase I (Ongoing) | Triple-negative breast cancer, hormone-resistant breast cancer, NSCLC, mesothelioma | AG-01 compound | MTD and/or maximum administered dose (MAD). | NCT05627960 | [119] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sirhan, Z.; Alojair, R.; Thyagarajan, A.; Sahu, R.P. Therapeutic Implications of PTEN in Non-Small Cell Lung Cancer. Pharmaceutics 2023, 15, 2090. https://doi.org/10.3390/pharmaceutics15082090
Sirhan Z, Alojair R, Thyagarajan A, Sahu RP. Therapeutic Implications of PTEN in Non-Small Cell Lung Cancer. Pharmaceutics. 2023; 15(8):2090. https://doi.org/10.3390/pharmaceutics15082090
Chicago/Turabian StyleSirhan, Zaid, Rawan Alojair, Anita Thyagarajan, and Ravi P. Sahu. 2023. "Therapeutic Implications of PTEN in Non-Small Cell Lung Cancer" Pharmaceutics 15, no. 8: 2090. https://doi.org/10.3390/pharmaceutics15082090