
Nanoparticle-Mediated Therapy with miR-198 Sensitizes Pancreatic Cancer to Gemcitabine Treatment through Downregulation of VCP-Mediated Autophagy
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Stable Cell Lines, Plasmids, Chemicals, and Antibodies
2.2. Preparation of LGA-PEI Polymer
2.3. Production and Characterization of Nucleic Acid-Containing Nanoparticles (LPNPs)
2.4. In Vitro Transfection Assay
2.5. Cell Viability Assay
2.6. Autophagy Characterization
2.7. Pancreatic Cancer Mouse Models
2.8. In Vivo Subchronic Toxicity Evaluation
2.9. MiRNA and mRNA Extraction and Reverse-Transcription
2.10. Western Blot Analysis
2.11. Hematoxylin-Eosin and Immunohistochemistry Staining
2.12. TUNEL Assay
2.13. Statistical Analysis
3. Results
3.1. LGA-PEI Nanoparticles (LPNPs) Efficiently Deliver Functional miR-198 into Pancreatic Cancer Cells In Vitro
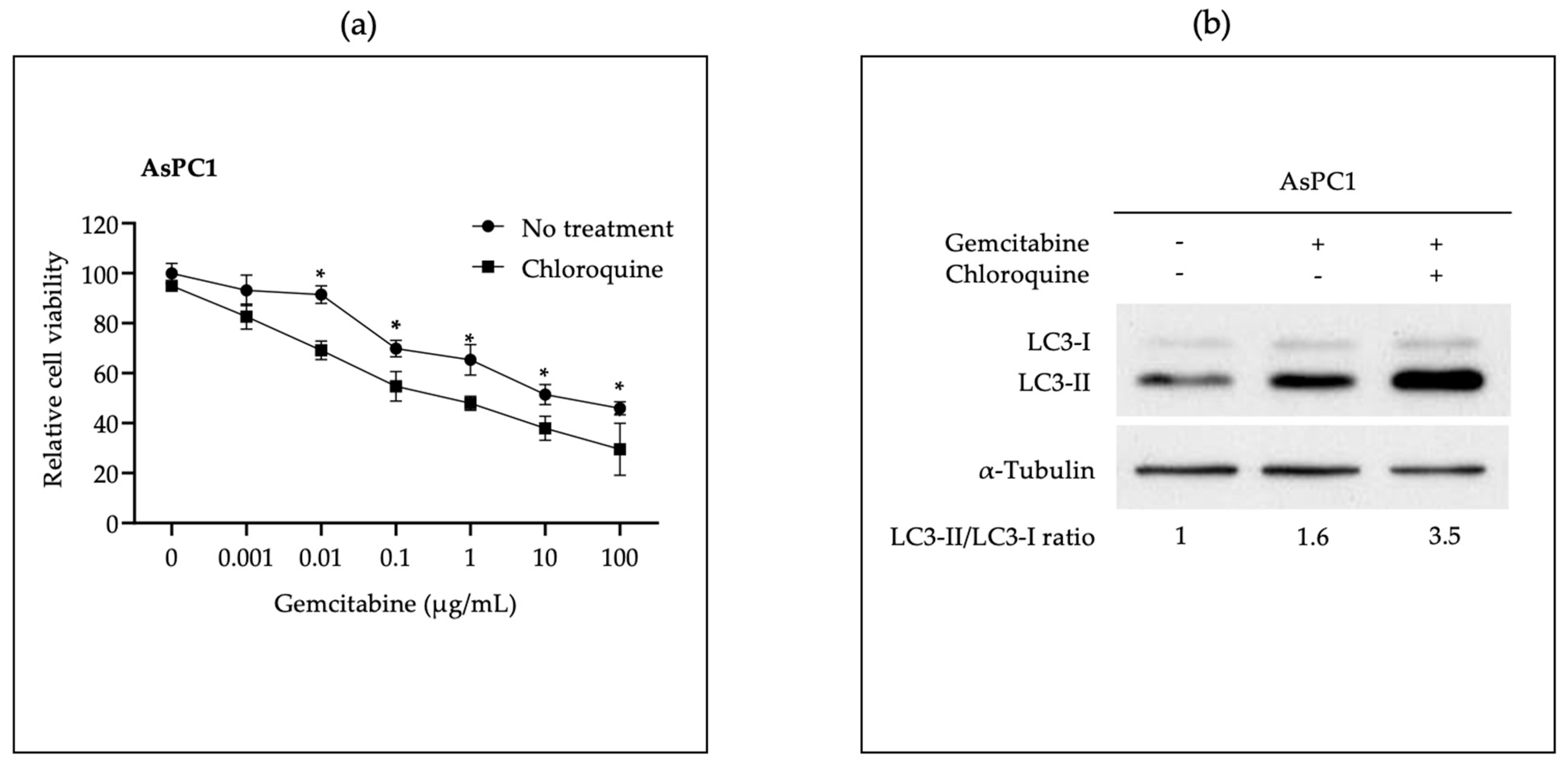
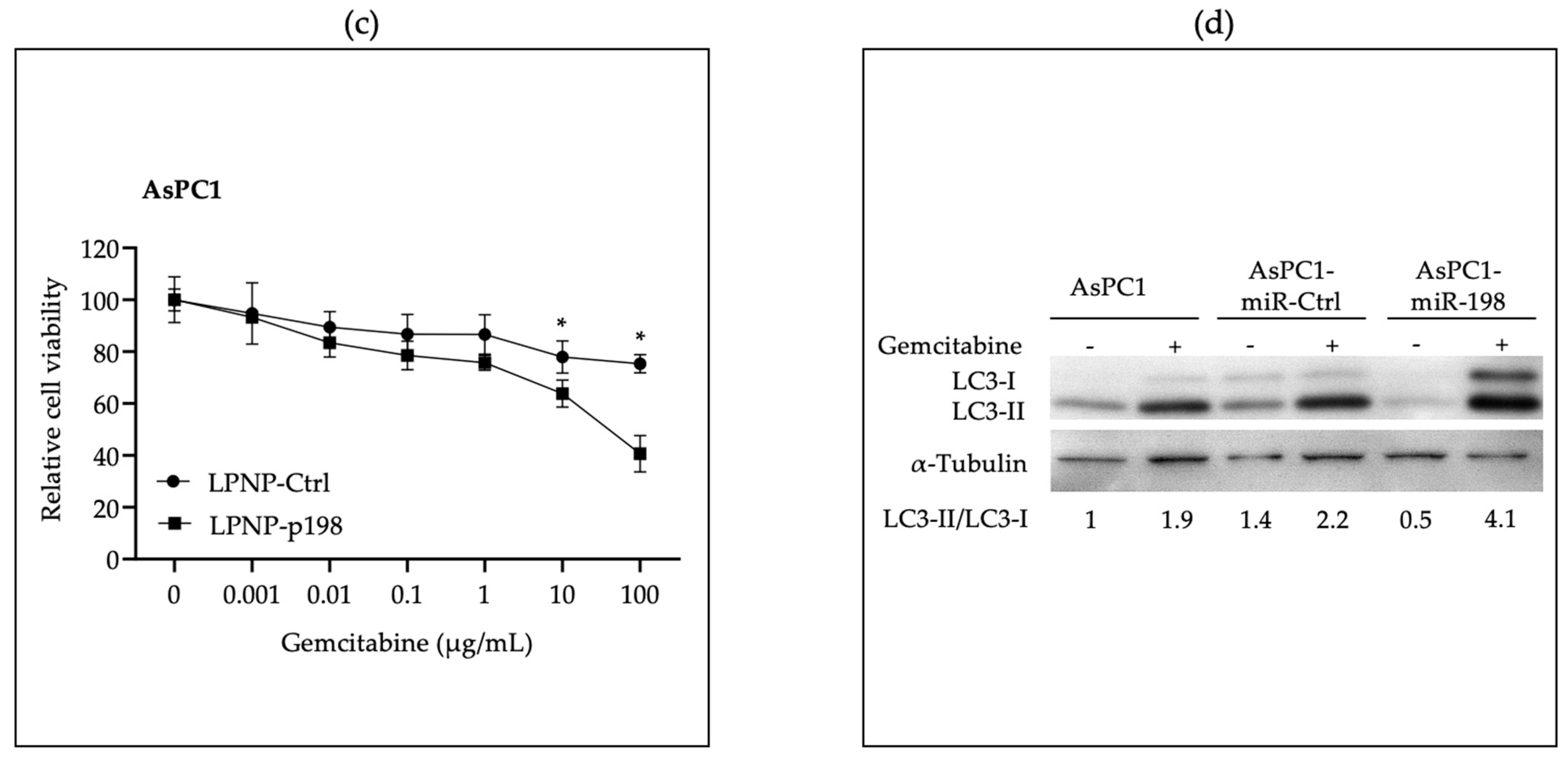
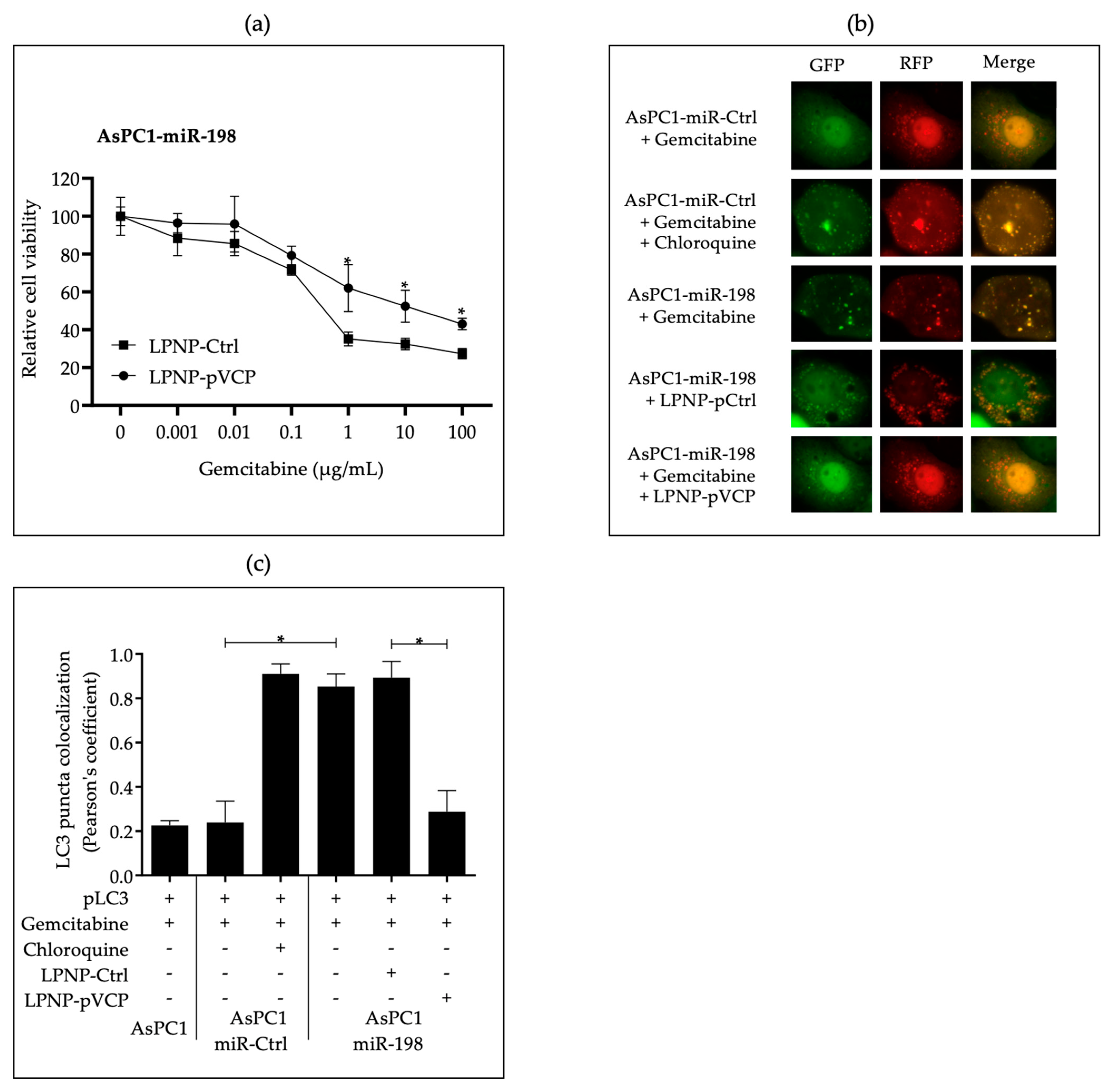
3.2. In Vitro Administration of miR-198 Sensitizes PDAC Cells to Gemcitabine Treatment through VCP Downregulation-Mediated Autophagy Maturation Process Inhibition
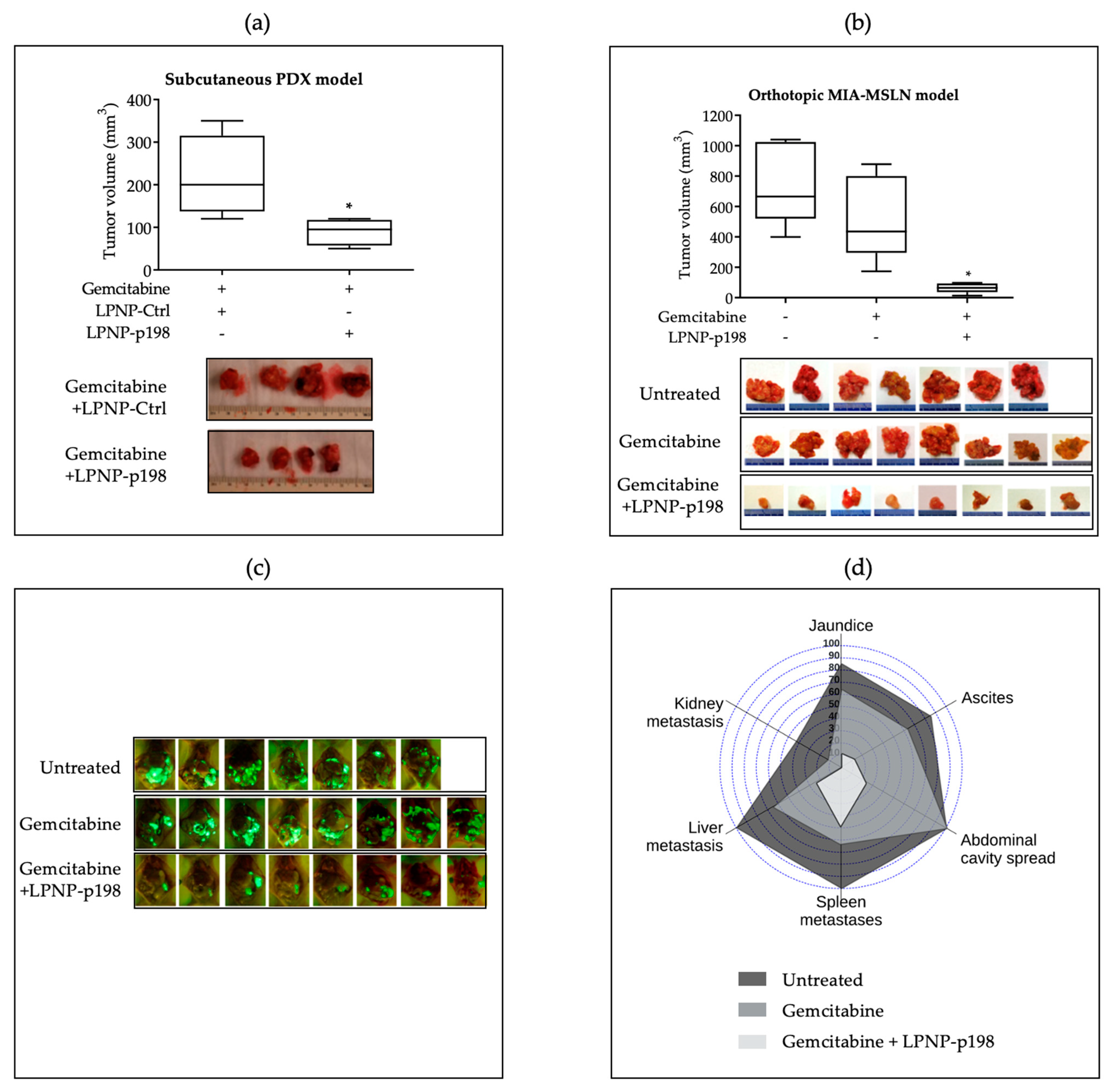
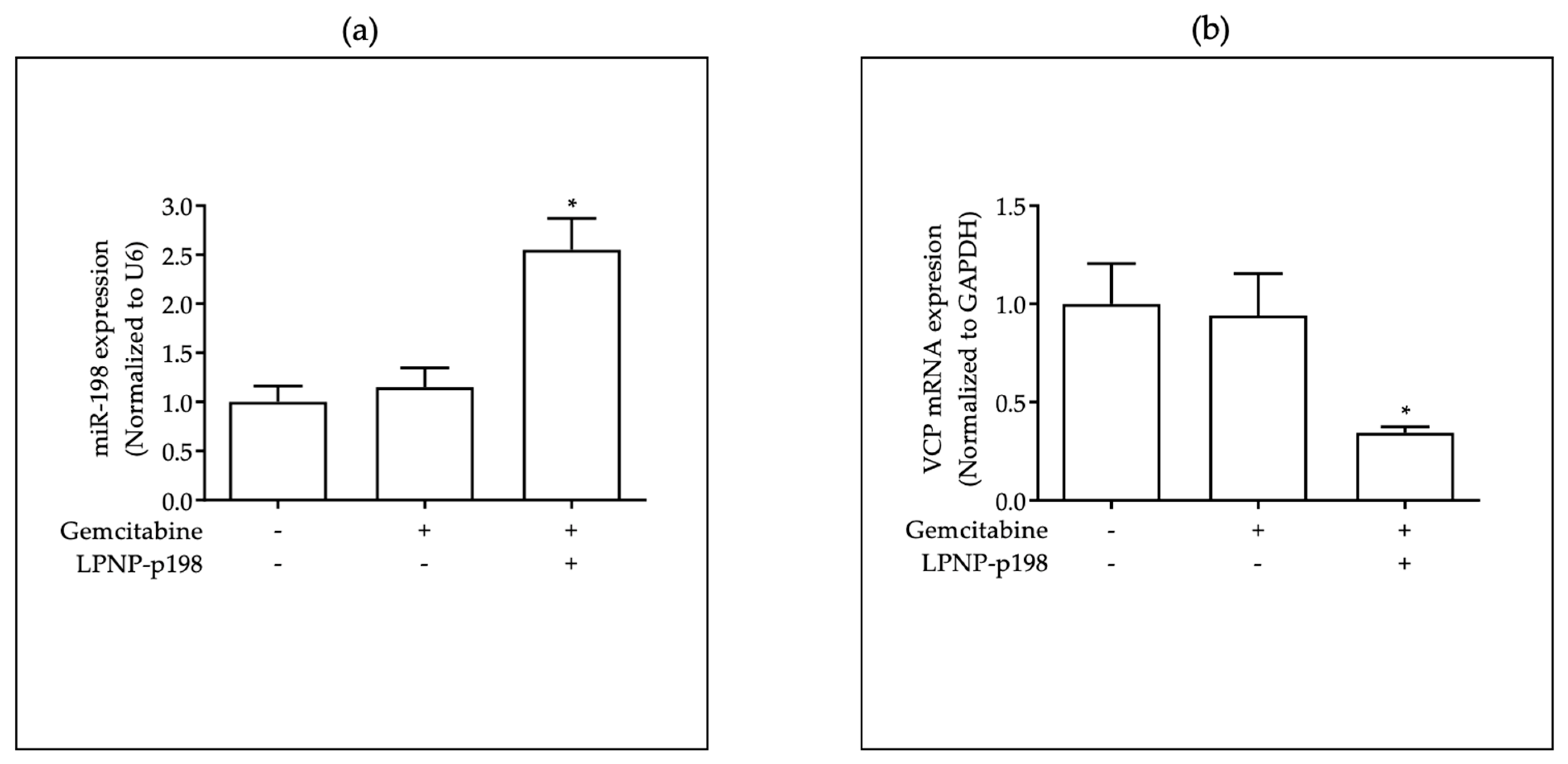
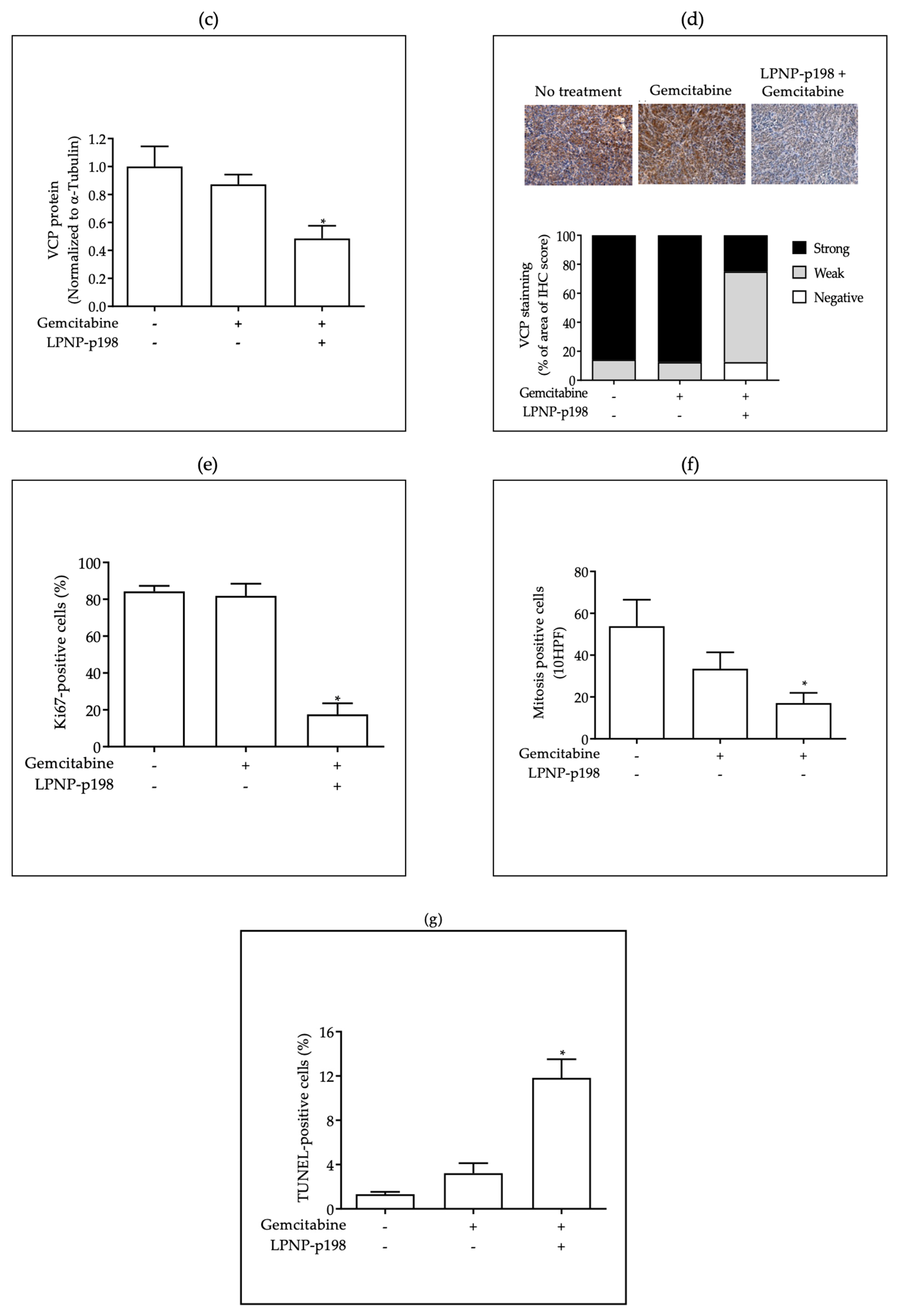
3.3. In Vivo Delivery of miR-198 Sensitizes PDAC Cells to Gemcitabine through Downregulation of VCP-Mediated Autophagosome Maturation, Leading to a Significant Reduction in Tumor Burden and Metastases
3.4. LPNPs Demonstrate a Favorable Safety Profile When Administered Systemically in a Repeated Dose Toxicity Study
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Capurso, G.; Sette, C. Drug Resistance in Pancreatic Cancer: New Player Caught in Act. eBioMedicine 2019, 40, 39–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Feng, W.; Wang, W.; Ye, X.; Chen, H.; Yu, C. Girdin Knockdown Increases Gemcitabine Chemosensitivity to Pancreatic Cancer by Modulating Autophagy. Front. Oncol. 2021, 11, 618764. [Google Scholar] [CrossRef] [PubMed]
- Amrutkar, M.; Gladhaug, I.P. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers 2017, 9, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ropolo, A.; Catrinacio, C.; Renna, F.J.; Boggio, V.; Orquera, T.; Gonzalez, C.D.; Vaccaro, M.I. A Novel E2F1-EP300-VMP1 Pathway Mediates Gemcitabine-Induced Autophagy in Pancreatic Cancer Cells Carrying Oncogenic KRAS. Front. Endocrinol. 2020, 11, 411. [Google Scholar] [CrossRef]
- Xi, Y.; Yuan, P.; Li, T.; Zhang, M.; Liu, M.-F.; Li, B. HENT1 Reverses Chemoresistance by Regulating Glycolysis in Pancreatic Cancer. Cancer Lett. 2020, 479, 112–122. [Google Scholar] [CrossRef]
- Li, J.; Hu, B.; Wang, T.; Huang, W.; Ma, C.; Zhao, Q.; Zhuo, L.; Zhang, T.; Jiang, Y. C-Src Confers Resistance to Mitotic Stress through Inhibition DMAP1/Bub3 Complex Formation in Pancreatic Cancer. Mol. Cancer 2018, 17, 174. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi, S.; Hosseini, M.; Shahidsales, S.; Maftouh, M.; A Ferns, G.; Ghayour-Mobarhan, M.; Mahdi Hassanian, S.; Avan, A. Targeting the Akt/PI3K Signaling Pathway as a Potential Therapeutic Strategy for the Treatment of Pancreatic Cancer. Curr. Med. Chem. 2017, 24, 1321–1331. [Google Scholar] [CrossRef]
- Sarvepalli, D.; Rashid, M.U.; Rahman, A.U.; Ullah, W.; Hussain, I.; Hasan, B.; Jehanzeb, S.; Khan, A.K.; Jain, A.G.; Khetpal, N. Gemcitabine: A Review of Chemoresistance in Pancreatic Cancer. Crit. Rev. Oncog. 2019, 24, 199–212. [Google Scholar] [CrossRef]
- Zeng, S.; Pöttler, M.; Lan, B.; Grützmann, R.; Pilarsky, C.; Yang, H. Chemoresistance in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 4504. [Google Scholar] [CrossRef] [Green Version]
- Ma, T.; Chen, W.; Zhi, X.; Liu, H.; Zhou, Y.; Chen, B.W.; Hu, L.; Shen, J.; Zheng, X.; Zhang, S. USP9X Inhibition Improves Gemcitabine Sensitivity in Pancreatic Cancer by Inhibiting Autophagy. Cancer Lett. 2018, 436, 129–138. [Google Scholar] [CrossRef]
- Zhang, X.; Kumstel, S.; Jiang, K.; Meng, S.; Gong, P.; Vollmar, B.; Zechner, D. LW6 Enhances Chemosensitivity to Gemcitabine and Inhibits Autophagic Flux in Pancreatic Cancer. J. Adv. Res. 2019, 20, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’Antonio, G. Pancreatic Cancers Require Autophagy for Tumor Growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.; Lee, J.H.; Kim, J.H.; Lee, S.; Yoo, S.; Jung, M.; Kim, S.J.; Yoo, H.J.; Pack, C.; Rho, J.K. Matrine Suppresses KRAS-driven Pancreatic Cancer Growth by Inhibiting Autophagy-mediated Energy Metabolism. Mol. Oncol. 2018, 12, 1203–1215. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Chen, Y.; Ji, Y.; Yu, Y.; Jin, Y.; Zhang, X.; Zhou, J. Gemcitabine Induces Apoptosis and Autophagy via the AMPK/MTOR Signaling Pathway in Pancreatic Cancer Cells. Biotechnol. Appl. Biochem. 2018, 65, 665–671. [Google Scholar] [CrossRef]
- Ertmer, A.; Huber, V.; Gilch, S.; Yoshimori, T.; Erfle, V.; Duyster, J.; Elsässer, H.P.; Schätzl, H.M. The Anticancer Drug Imatinib Induces Cellular Autophagy. Leukemia 2007, 21, 936–942. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Yu, D.; Lum, J.J.; Bui, T.; Christophorou, M.A.; Evan, G.I.; Thomas-Tikhonenko, A.; Thompson, C.B. Autophagy Inhibition Enhances Therapy-Induced Apoptosis in a Myc-Induced Model of Lymphoma. J. Clin. Investig. 2007, 117, 326–336. [Google Scholar] [CrossRef] [Green Version]
- Qadir, M.A.; Kwok, B.; Dragowska, W.H.; To, K.H.; Le, D.; Bally, M.B.; Gorski, S.M. Macroautophagy Inhibition Sensitizes Tamoxifen-Resistant Breast Cancer Cells and Enhances Mitochondrial Depolarization. Breast Cancer Res. Treat. 2008, 112, 389–403. [Google Scholar] [CrossRef]
- Huggett, M.T.; Jermyn, M.; Gillams, A.; Illing, R.; Mosse, S.; Novelli, M.; Kent, E.; Bown, S.G.; Hasan, T.; Pogue, B.W. Phase I/II Study of Verteporfin Photodynamic Therapy in Locally Advanced Pancreatic Cancer. Br. J. Cancer 2014, 110, 1698–1704. [Google Scholar] [CrossRef] [Green Version]
- Zeh, H.J.; Bahary, N.; Boone, B.A.; Singhi, A.D.; Miller-Ocuin, J.L.; Normolle, D.P.; Zureikat, A.H.; Hogg, M.E.; Bartlett, D.L.; Lee, K.K. A Randomized Phase II Preoperative Study of Autophagy Inhibition with High-Dose Hydroxychloroquine and Gemcitabine/Nab-Paclitaxel in Pancreatic Cancer Patients. Clin. Cancer Res. 2020, 26, 3126–3134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, P.; Chakraborty, A.; Sarkar, D.; Langthasa, M.; Rahman, M.; Bari, M.; Singha, R.K.S.; Malakar, A.K.; Chakraborty, S. Interplay between MiRNAs and Human Diseases. J. Cell. Physiol. 2018, 233, 2007–2018. [Google Scholar] [CrossRef]
- Denzler, R.; McGeary, S.E.; Title, A.C.; Agarwal, V.; Bartel, D.P.; Stoffel, M. Impact of MicroRNA Levels, Target-Site Complementarity, and Cooperativity on Competing Endogenous RNA-Regulated Gene Expression. Mol. Cell 2016, 64, 565–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, B.; An, Y.; Lv, N.; Chen, J.; Tu, M.; Sun, J.; Wu, P.; Wei, J.; Jiang, K.; Miao, Y. MiRNA-181b Increases the Sensitivity of Pancreatic Ductal Adenocarcinoma Cells to Gemcitabine in Vitro and in Nude Mice by Targeting BCL-2. Oncol. Rep. 2013, 29, 1769–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.; Yu, X.-J.; Guo, X.-Z.; Sun, M.-H.; Wang, Z.; Song, Y.; Ni, Q.-X.; Li, H.-Y.; Mukaida, N.; Li, Y.-Y. MicroRNA-33a-Mediated Downregulation of Pim-3 Kinase Expression Renders Human Pancreatic Cancer Cells Sensitivity to Gemcitabine. Oncotarget 2015, 6, 14440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.; Wang, Z.; Li, Y.-Y.; Yu, B.-H.; Zhang, F.; Li, H.-Y. MiR-33a Suppresses the Nuclear Translocation of β-Catenin to Enhance Gemcitabine Sensitivity in Human Pancreatic Cancer Cells. Tumor Biol. 2015, 36, 9395–9403. [Google Scholar] [CrossRef]
- Yao, J.; Li, Z.; Wang, X.; Xu, P.; Zhao, L.; Qian, J. MiR-125a Regulates Chemo-Sensitivity to Gemcitabine in Human Pancreatic Cancer Cells through Targeting A20. Acta Biochim. Biophys. Sin. 2016, 48, 202–208. [Google Scholar] [CrossRef] [Green Version]
- Paik, W.H.; Kim, H.R.; Park, J.K.; Song, B.J.; Lee, S.H.; Hwang, J.-H. Chemosensitivity Induced by Down-Regulation of MicroRNA-21 in Gemcitabine-Resistant Pancreatic Cancer Cells by Indole-3-Carbinol. Anticancer Res. 2013, 33, 1473–1481. [Google Scholar] [PubMed]
- Zhang, X.; Zhao, P.; Wang, C.; Xin, B. SNHG14 Enhances Gemcitabine Resistance by Sponging MiR-101 to Stimulate Cell Autophagy in Pancreatic Cancer. Biochem. Biophys. Res. Commun. 2019, 510, 508–514. [Google Scholar] [CrossRef]
- Li, C.; Zhao, Z.; Zhou, Z.; Liu, R. Linc-ROR Confers Gemcitabine Resistance to Pancreatic Cancer Cells via Inducing Autophagy and Modulating the MiR-124/PTBP1/PKM2 Axis. Cancer Chemother. Pharmacol. 2016, 78, 1199–1207. [Google Scholar] [CrossRef]
- Kwon, J.J.; Willy, J.A.; Quirin, K.A.; Wek, R.C.; Korc, M.; Yin, X.-M.; Kota, J. Novel Role of MiR-29a in Pancreatic Cancer Autophagy and Its Therapeutic Potential. Oncotarget 2016, 7, 71635. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Liu, G.; Cao, H.; Zhang, H.; Shao, F. Hsa_circ_0035483 Sponges Hsa-MiR-335 to Promote the Gemcitabine-Resistance of Human Renal Cancer Cells by Autophagy Regulation. Biochem. Biophys. Res. Commun. 2019, 519, 172–178. [Google Scholar] [CrossRef]
- Sun, Q.; Liu, T.; Yuan, Y.; Guo, Z.; Xie, G.; Du, S.; Lin, X.; Xu, Z.; Liu, M.; Wang, W. MiR-200c Inhibits Autophagy and Enhances Radiosensitivity in Breast Cancer Cells by Targeting UBQLN1. Int. J. Cancer 2015, 136, 1003–1012. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, N.; Liu, P.; Chen, Q.; Situ, H.; Xie, T.; Zhang, J.; Peng, C.; Lin, Y.; Chen, J. MicroRNA-25 Regulates Chemoresistance-Associated Autophagy in Breast Cancer Cells, a Process Modulated by the Natural Autophagy Inducer Isoliquiritigenin. Oncotarget 2014, 5, 7013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Bi, R.; Li, L.; Zhou, K.; Yin, H. LncRNA ANRIL Aggravates the Chemoresistance of Pancreatic Cancer Cells to Gemcitabine by Targeting Inhibition of MiR-181a and Targeting HMGB1-Induced Autophagy. Aging 2021, 13, 19272. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhu, Y.; Xie, Q. The Promising Role and Prognostic Value of MiR-198 in Human Diseases. Am. J. Transl. Res. 2022, 14, 2749. [Google Scholar] [PubMed]
- Kaushik, P.; Kumar, A. Emerging Role and Function of MiR-198 in Human Health and Diseases. Pathol.-Res. Pract. 2022, 229, 153741. [Google Scholar] [CrossRef]
- Marin-Muller, C.; Li, D.; Bharadwaj, U.; Li, M.; Chen, C.; Hodges, S.E.; Fisher, W.E.; Mo, Q.; Hung, M.C.; Yao, Q. A Tumorigenic Factor Interactome Connected through Tumor Suppressor MicroRNA-198 in Human Pancreatic Cancer. Clin. Cancer Res. 2013, 19, 5901–5913. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-A.; Yang, J.; Jo, Y.N.; Lee, C.W.; Lee, O.-J.; Choi, S.-Y.; Kang, J.S.; Yun, J. MicroRNA-198 Inhibits Non-Small Cell Lung Cancer Migration and Invasion through Targeting OTX1 and VCP. Cancer Res. 2014, 74, 4358. [Google Scholar] [CrossRef]
- Meyer, H.; Weihl, C.C. The VCP/P97 System at a Glance: Connecting Cellular Function to Disease Pathogenesis. J. Cell Sci. 2014, 127, 3877–3883. [Google Scholar] [CrossRef] [Green Version]
- Kaistha, B.P.; Schimanski, S.; Gress, T.M.; Buchholz, M. VCP/P97 Is Upregulated in Pancreatic Cancer and Promotes Proliferation and Cell Cycle Progression in Cancer Cells. Z. Gastroenterol. 2017, 55, KV-182. [Google Scholar]
- Wang, F.; Vij, K.; Li, L.; Dodhiawala, P.; Lim, K.-H.; Shao, J. Phospho-Ser784-VCP Drives Resistance of Pancreatic Ductal Adenocarcinoma to Genotoxic Chemotherapies and Predicts the Chemo-Sensitizing Effect of VCP Inhibitor. Cancers 2021, 13, 5076. [Google Scholar] [CrossRef]
- Kobayashi, H.; Shoji, K.; Kiyokawa, K.; Negishi, L.; Tomari, Y. VCP Machinery Mediates Autophagic Degradation of Empty Argonaute. Cell Rep. 2019, 28, 1144–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, V.; Cristofani, R.; Tedesco, B.; Crippa, V.; Chierichetti, M.; Casarotto, E.; Cozzi, M.; Mina, F.; Piccolella, M.; Galbiati, M. Valosin Containing Protein (VCP): A Multistep Regulator of Autophagy. Int. J. Mol. Sci. 2022, 23, 1939. [Google Scholar] [CrossRef] [PubMed]
- Huryn, D.M.; Kornfilt, D.J.P.; Wipf, P. P97: An Emerging Target for Cancer, Neurodegenerative Diseases, and Viral Infections. J. Med. Chem. 2019, 63, 1892–1907. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Doss, C.G.P.; Lee, S.S. Therapeutic MiRNA and SiRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol. Ther. Nucleic Acids 2017, 8, 132–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lü, J.M.; Liang, Z.; Wang, X.; Gu, J.; Yao, Q.; Chen, C. New Polymer of Lactic-Co-Glycolic Acid-Modified Polyethylenimine for Nucleic Acid Delivery. Nanomedicine 2016, 11, 1971–1991. [Google Scholar] [CrossRef] [Green Version]
- Lü, J.-M.; Liang, Z.; Liu, D.; Zhan, B.; Yao, Q.; Chen, C. Two Antibody-Guided Lactic-Co-Glycolic Acid-Polyethylenimine (LGA-PEI) Nanoparticle Delivery Systems for Therapeutic Nucleic Acids. Pharmaceuticals 2021, 14, 841. [Google Scholar] [CrossRef]
- Bharadwaj, U.; Marin-Muller, C.; Li, M.; Chen, C.; Yao, Q. Mesothelin Overexpression Promotes Autocrine IL-6/SIL-6R Trans-Signaling to Stimulate Pancreatic Cancer Cell Proliferation. Carcinogenesis 2011, 32, 1013–1024. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Bharadwaj, U.; Zhang, R.; Zhang, S.; Mu, H.; Fisher, W.E.; Brunicardi, F.C.; Chen, C.; Yao, Q. Mesothelin Is a Malignant Factor and Therapeutic Vaccine Target for Pancreatic Cancer. Mol. Cancer Ther. 2008, 7, 286–296. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Zhang, Y.; Liu, Z.; Bharadwaj, U.; Wang, H.; Wang, X.; Zhang, S.; Liuzzi, J.P.; Chang, S.-M.; Cousins, R.J. Aberrant Expression of Zinc Transporter ZIP4 (SLC39A4) Significantly Contributes to Human Pancreatic Cancer Pathogenesis and Progression. Proc. Natl. Acad. Sci. USA 2007, 104, 18636–18641. [Google Scholar] [CrossRef]
- Han, H.-J.; Russo, J.; Kohwi, Y.; Kohwi-Shigematsu, T. SATB1 Reprogrammes Gene Expression to Promote Breast Tumour Growth and Metastasis. Nature 2008, 452, 187–193. [Google Scholar] [CrossRef]
- Usman, R.M.; Razzaq, F.; Akbar, A.; Farooqui, A.A.; Iftikhar, A.; Latif, A.; Hassan, H.; Zhao, J.; Carew, J.S.; Nawrocki, S.T. Role and Mechanism of Autophagy-regulating Factors in Tumorigenesis and Drug Resistance. Asia Pac. J. Clin. Oncol. 2021, 17, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, P.M.P.; de Sousa, R.W.R.; de Oliveira Ferreira, J.R.; Militão, G.C.G.; Bezerra, D.P. Chloroquine and Hydroxychloroquine in Antitumor Therapies Based on Autophagy-Related Mechanisms. Pharmacol. Res. 2021, 168, 105582. [Google Scholar] [CrossRef] [PubMed]
- Le, K.; Wang, J.; Zhang, T.; Guo, Y.; Chang, H.; Wang, S.; Zhu, B. Overexpression of Mesothelin in Pancreatic Ductal Adenocarcinoma (PDAC). Int. J. Med. Sci. 2020, 17, 422. [Google Scholar] [CrossRef] [Green Version]
- Bharadwaj, U.; Li, M.; Chen, C.; Yao, Q. Mesothelin-Induced Pancreatic Cancer Cell Proliferation Involves Alteration of Cyclin E via Activation of Signal Transducer and Activator of Transcription Protein 3. Mol. Cancer Res. 2008, 6, 1755–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raufi, A.G.; Liguori, N.R.; Carlsen, L.; Parker, C.; Hernandez Borrero, L.; Zhang, S.; Tian, X.; Louie, A.; Zhou, L.; Seyhan, A.A. Therapeutic Targeting of Autophagy in Pancreatic Ductal Adenocarcinoma. Front. Pharmacol. 2021, 12, 751568. [Google Scholar] [CrossRef]
- Tempero, M.A.; Reni, M.; Riess, H. APACT: Phase III, Multicenter, International, Open-Label, Randomized Trial of Adjuvant Nab-Paclitaxel plus Gemcitabine vs Gemcitabine for Surgically Resected Pancreatic Adenocarcinoma. J. Clin. Oncol. 2019, 37, 4000. [Google Scholar] [CrossRef]
- Howe, E.N.; Cochrane, D.R.; Richer, J.K. The MiR-200 and MiR-221/222 MicroRNA Families: Opposing Effects on Epithelial Identity. J. Mammary Gland Biol. Neoplasia 2012, 17, 65–77. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Fang, B.; Zeng, F.; Ma, C.; Pang, H.; Cheng, L.; Shi, Y.; Wang, H.; Yin, B.; Xia, J. Down-Regulation of MiR-223 Reverses Epithelial-Mesenchymal Transition in Gemcitabine-Resistant Pancreatic Cancer Cells. Oncotarget 2015, 6, 1740. [Google Scholar] [CrossRef] [Green Version]
- Moriyama, T.; Ohuchida, K.; Mizumoto, K.; Yu, J.; Sato, N.; Nabae, T.; Takahata, S.; Toma, H.; Nagai, E.; Tanaka, M. MicroRNA-21 Modulates Biological Functions of Pancreatic Cancer Cells Including Their Proliferation, Invasion, and ChemoresistancemiR-21 Elicits Malignant Progression in Pancreatic Cancer. Mol. Cancer Ther. 2009, 8, 1067–1074. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-F.; Luo, D.; Li, X.; Li, Z.-Q.; Yu, X.; Zhu, H.-W. PVT1 Knockdown Inhibits Autophagy and Improves Gemcitabine Sensitivity by Regulating the MiR-143/HIF-1α/VMP1 Axis in Pancreatic Cancer. Pancreas 2021, 50, 227–234. [Google Scholar] [CrossRef]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T. Protective Autophagy Elicited by RAF→ MEK→ ERK Inhibition Suggests a Treatment Strategy for RAS-Driven Cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D. Combination of ERK and Autophagy Inhibition as a Treatment Approach for Pancreatic Cancer. Nat Med 2019, 25, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Koltai, T.; Reshkin, S.J.; Carvalho, T.M.A.; Di Molfetta, D.; Greco, M.R.; Alfarouk, K.O.; Cardone, R.A. Resistance to Gemcitabine in Pancreatic Ductal Adenocarcinoma: A Physiopathologic and Pharmacologic Review. Cancers 2022, 14, 2486. [Google Scholar] [CrossRef]
- Hansen, T.E.; Johansen, T. Following Autophagy Step by Step. BMC Biol. 2011, 9, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, S.M.; Wrobel, L.; Ashkenazi, A.; Fernandez-Estevez, M.; Tan, K.; Bürli, R.W.; Rubinsztein, D.C. VCP/P97 Regulates Beclin-1-Dependent Autophagy Initiation. Nat. Chem. Biol. 2021, 17, 448–455. [Google Scholar] [CrossRef]
- Herzog, L.K.; Kevei, É.; Marchante, R.; Böttcher, C.; Bindesbøll, C.; Lystad, A.H.; Pfeiffer, A.; Gierisch, M.E.; Salomons, F.A.; Simonsen, A. The Machado–Joseph Disease Deubiquitylase Ataxin-3 Interacts with LC3C/GABARAP and Promotes Autophagy. Aging Cell 2020, 19, e13051. [Google Scholar] [CrossRef]
- Johnson, A.E.; Shu, H.; Hauswirth, A.G.; Tong, A.; Davis, G.W. VCP-Dependent Muscle Degeneration Is Linked to Defects in a Dynamic Tubular Lysosomal Network in Vivo. eLife 2015, 4, e07366. [Google Scholar] [CrossRef]
- Ju, J.-S.; Fuentealba, R.A.; Miller, S.E.; Jackson, E.; Piwnica-Worms, D.; Baloh, R.H.; Weihl, C.C. Valosin-Containing Protein (VCP) Is Required for Autophagy and Is Disrupted in VCP Disease. J. Cell Biol. 2009, 187, 875–888. [Google Scholar] [CrossRef]
- Kilgas, S.; Ramadan, K. Inhibitors of the ATPase P97/VCP: From Basic Research to Clinical Applications. Cell Chem. Biol. 2023. [Google Scholar] [CrossRef]
- Sundaram, G.M.; Common, J.E.A.; Gopal, F.E.; Srikanta, S.; Lakshman, K.; Lunny, D.P.; Lim, T.C.; Tanavde, V.; Lane, E.B.; Sampath, P. ‘See-Saw’ Expression of MicroRNA-198 and FSTL1 from a Single Transcript in Wound Healing. Nature 2013, 495, 103–106. [Google Scholar] [CrossRef]
- Yu, X.; Eischeid-Scholz, H.; Meder, L.; Kondylis, V.; Büttner, R.; Odenthal, M. SQSTM1/P62 Promotes MiR-198 Loading into Extracellular Vesicles and Its Autophagy-Related Secretion. Hum. Cell 2022, 35, 1766–1784. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T. How to Interpret LC3 Immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Gillson, J.; Abd El-Aziz, Y.S.; Leck, L.Y.W.; Jansson, P.J.; Pavlakis, N.; Samra, J.S.; Mittal, A.; Sahni, S. Autophagy: A Key Player in Pancreatic Cancer Progression and a Potential Drug Target. Cancers 2022, 14, 3528. [Google Scholar] [CrossRef] [PubMed]
- Kulshrestha, A.; Katara, G.K.; Ibrahim, S.A.; Riehl, V.; Sahoo, M.; Dolan, J.; Meinke, K.W.; Pins, M.R.; Beaman, K.D. Targeting V-ATPase Isoform Restores Cisplatin Activity in Resistant Ovarian Cancer: Inhibition of Autophagy, Endosome Function, and ERK/MEK Pathway. J. Oncol. 2019, 2019, 2343876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ré, A.E.L.; Fernández-Barrena, M.G.; Almada, L.L.; Mills, L.D.; Elsawa, S.F.; Lund, G.; Ropolo, A.; Molejon, M.I.; Vaccaro, M.I.; Fernandez-Zapico, M.E. Novel AKT1-GLI3-VMP1 Pathway Mediates KRAS Oncogene-Induced Autophagy in Cancer Cells. J. Biol. Chem. 2012, 287, 25325–25334. [Google Scholar] [PubMed] [Green Version]
- Yamamoto, K.; Iwadate, D.; Kato, H.; Nakai, Y.; Tateishi, K.; Fujishiro, M. Targeting Autophagy as a Therapeutic Strategy against Pancreatic Cancer. J. Gastroenterol. 2022, 57, 603–618. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter (Units) | Group (Expressed in Mean ± S.D., n = 4) | ||
|---|---|---|---|
| Untreated (Saline Solution) | LPNP-pCtrl (2.5 mg/kg) | LPNP-p198 (2.5 mg/kg) | |
| Albumin (g/dL) | 2.4 ± 0.1 | 2.2 ± 0.2 | 2.4 ± 0.2 |
| Globulin (g/dL) | 2.6 ± 0.3 | 2.5 ± 0.1 | 2.6 ± 0.2 |
| Albumin/Globulin (ratio) | 0.94 ± 0.07 | 0.88 ± 0.12 | 0.91 ± 0.04 |
| Total protein (g/dL) | 5.0 ± 0.5 | 4.6 ± 0.1 | 5.1 ± 0.2 |
| Alkaline phosphatase (U/L) | 42 ± 7 | 49 ± 10 | 39 ± 8 |
| Alanine transaminase (U/L) | 56 ± 21 | 53 ± 8 | 45 ± 5 |
| Lipase (U/L) | 848 ± 103 | 845 ± 81 | 826 ± 48 |
| Amylase (U/L) | 1565 ± 164 | 1557 ± 77 | 1500 ± 83 |
| Creatine (mg/dL) | 0.08 ± 0.1 | 0.10 ± 0.08 | 0.13 ± 0.1 |
| Urea nitrogen (mg/dL) | 0.20 ± 0.14 | 0.18 ± 0.1 | 0.23 ± 0.15 |
| Total bilirubin (mg/dL) | 21 ± 2 | 20 ± 1 | 19 ± 2 |
| Glucose (mg/dL) | 171.3 ± 29.3 | 156.8 ± 7.7 | 157.0 ± 17.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marin-Muller, C.; Li, D.; Lü, J.-M.; Liang, Z.; Vega-Martínez, O.; Crawford, S.E.; Estes, M.K.; Fisher, W.E.; Chen, C.; Yao, Q. Nanoparticle-Mediated Therapy with miR-198 Sensitizes Pancreatic Cancer to Gemcitabine Treatment through Downregulation of VCP-Mediated Autophagy. Pharmaceutics 2023, 15, 2038. https://doi.org/10.3390/pharmaceutics15082038
Marin-Muller C, Li D, Lü J-M, Liang Z, Vega-Martínez O, Crawford SE, Estes MK, Fisher WE, Chen C, Yao Q. Nanoparticle-Mediated Therapy with miR-198 Sensitizes Pancreatic Cancer to Gemcitabine Treatment through Downregulation of VCP-Mediated Autophagy. Pharmaceutics. 2023; 15(8):2038. https://doi.org/10.3390/pharmaceutics15082038
Chicago/Turabian StyleMarin-Muller, Christian, Dali Li, Jian-Ming Lü, Zhengdong Liang, Osvaldo Vega-Martínez, Sue E. Crawford, Mary K. Estes, William E. Fisher, Changyi Chen, and Qizhi Yao. 2023. "Nanoparticle-Mediated Therapy with miR-198 Sensitizes Pancreatic Cancer to Gemcitabine Treatment through Downregulation of VCP-Mediated Autophagy" Pharmaceutics 15, no. 8: 2038. https://doi.org/10.3390/pharmaceutics15082038