
Effects of Adding Chitosan on Drug Entrapment Efficiency and Release Duration for Paclitaxel-Loaded Hydroxyapatite—Gelatin Composite Microspheres

Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Aqueous Solutions and Precipitation of HAp-Gel
2.2. Fourier Transform Infrared Spectroscopy Analysis (FTIR)
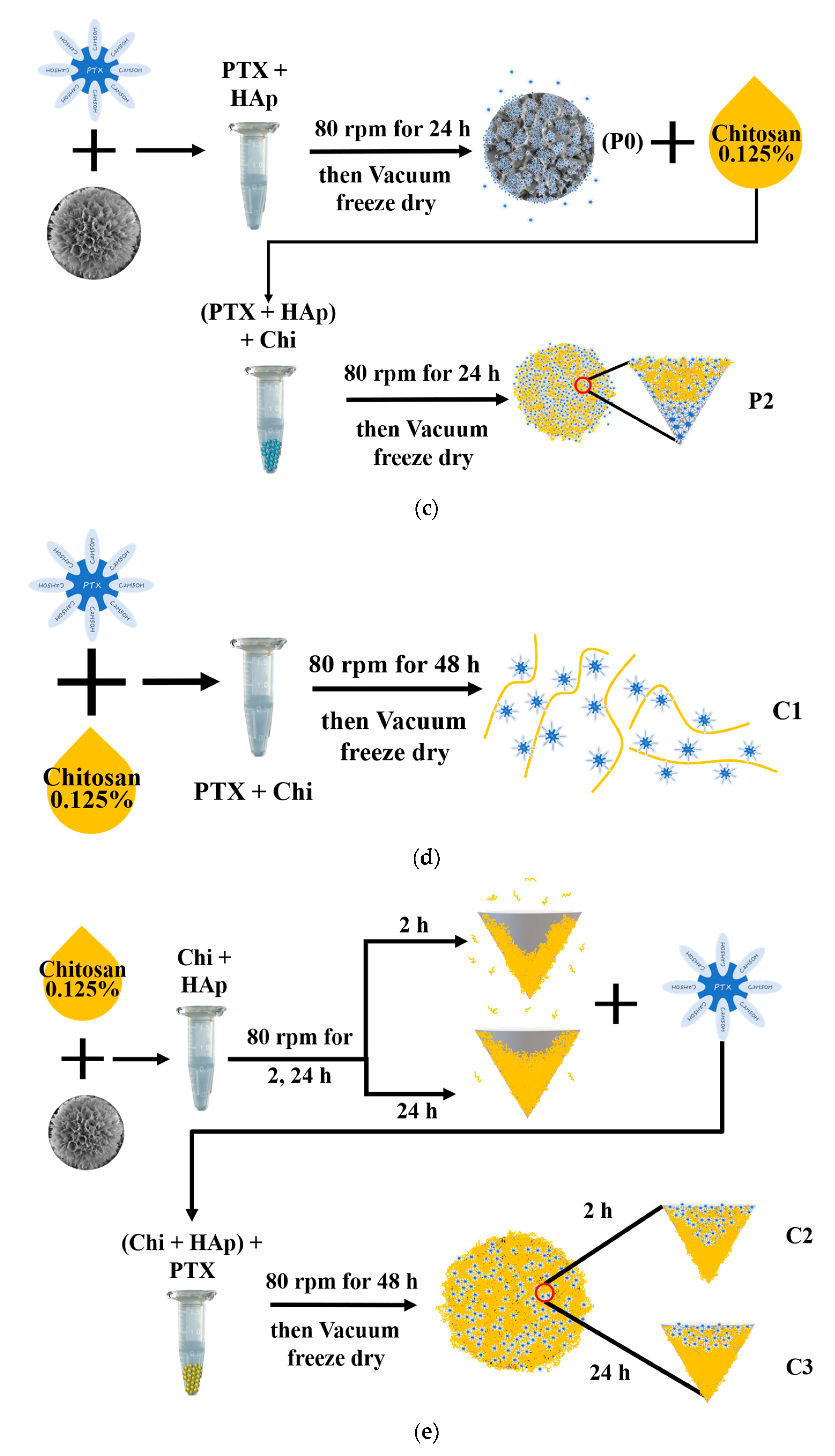
2.3. Loading of Drugs Onto HAp-Gel and/or Chitosan
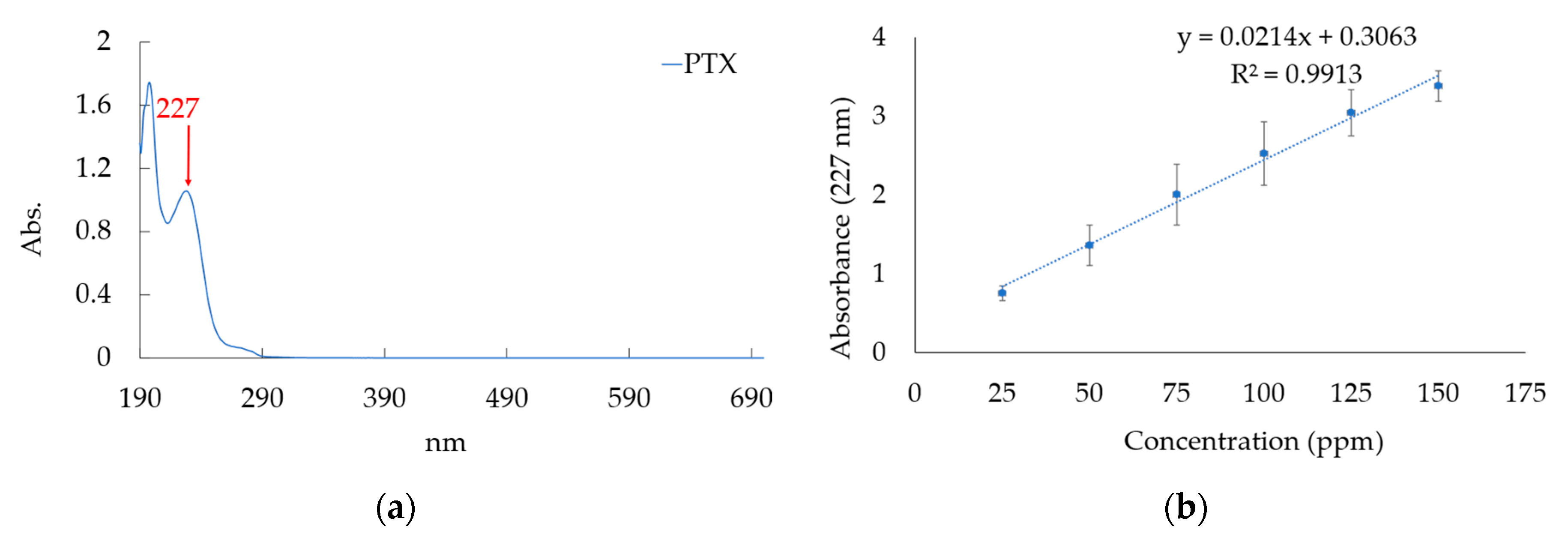
2.3.1. Creating Calibration Curve for PTX Concentration Analysis
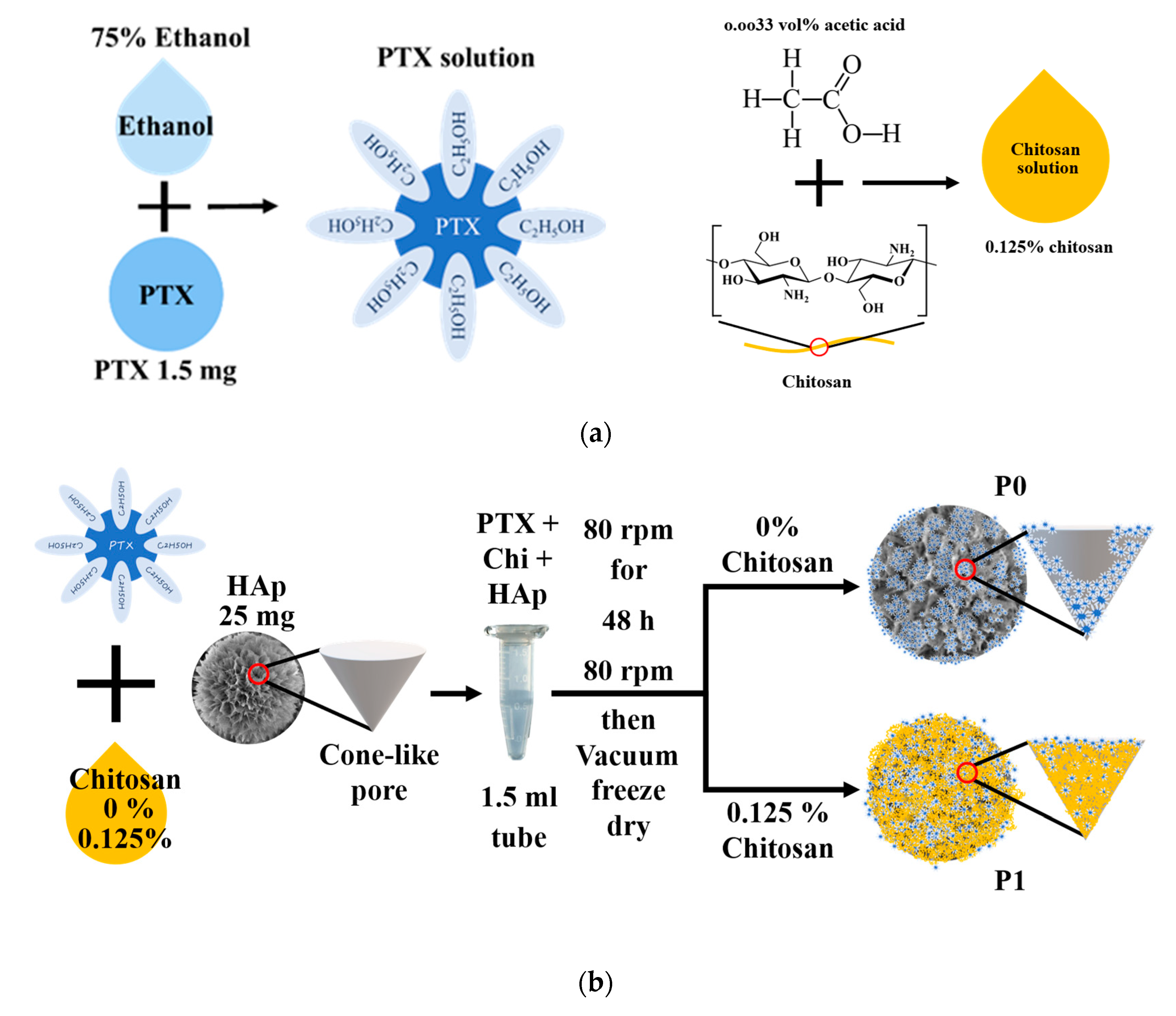
2.3.2. Drug Loading Process for PTX Divided into Two Categories
2.3.3. Bonding Capabilities of Chitosan with PTX and HAp-Gel
2.4. Surface Morphology of HAp-Gel, P0, P1, P2, C1, C2, and C3
2.5. Drug Release from PTX-HAp-Gel
2.6. Cell Experiments
2.6.1. Cultivation of Human Osteosarcoma Cells (G292, ATCC CRL-1423)
2.6.2. Direct Cell Toxicity Test
2.6.3. MTT Assay
2.7. Statistically Analysis
3. Results and Discussion
3.1. Fourier Transform Infrared Spectrograph Spectrometer
3.2. Surface Morphology of HAp-Gel

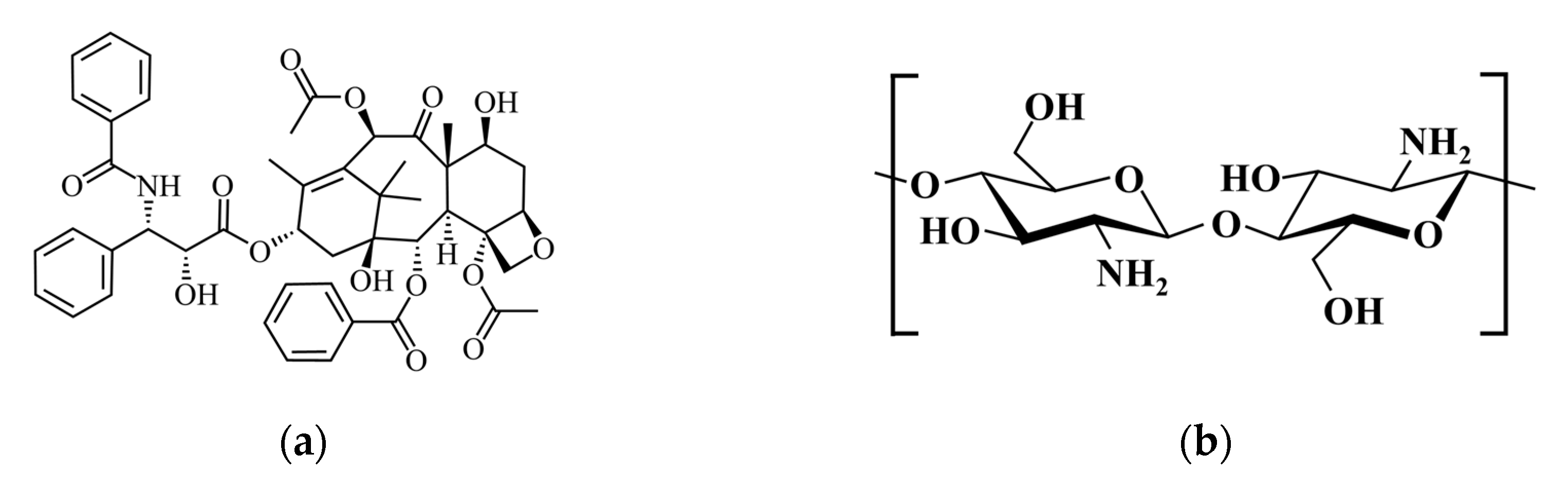
3.3. Loading of Drugs onto HAp-Gel
3.3.1. Creating Calibration Curve for PTX Concentration Analysis
3.3.2. Drug Loading Process for PTX

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | DEE (%) | DLC (%) |
|---|---|---|
| P0 | 58.91 ± 0.14 | 3.53 ± 0.01 |
| P1 | 57.27 ± 1.83 | 3.44 ± 0.1 |
| P2 | 94.21 ± 0.08 * | 5.65 ± 0.01 * |
| C1 | 41.94 ± 0.67 * | 251.64 ± 4.07 * |
| C2 | 58.71 ± 0.88 | 3.52 ± 0.05 |
| C3 | 27.57 ± 0.6 * | 1.65 ± 0.04 * |



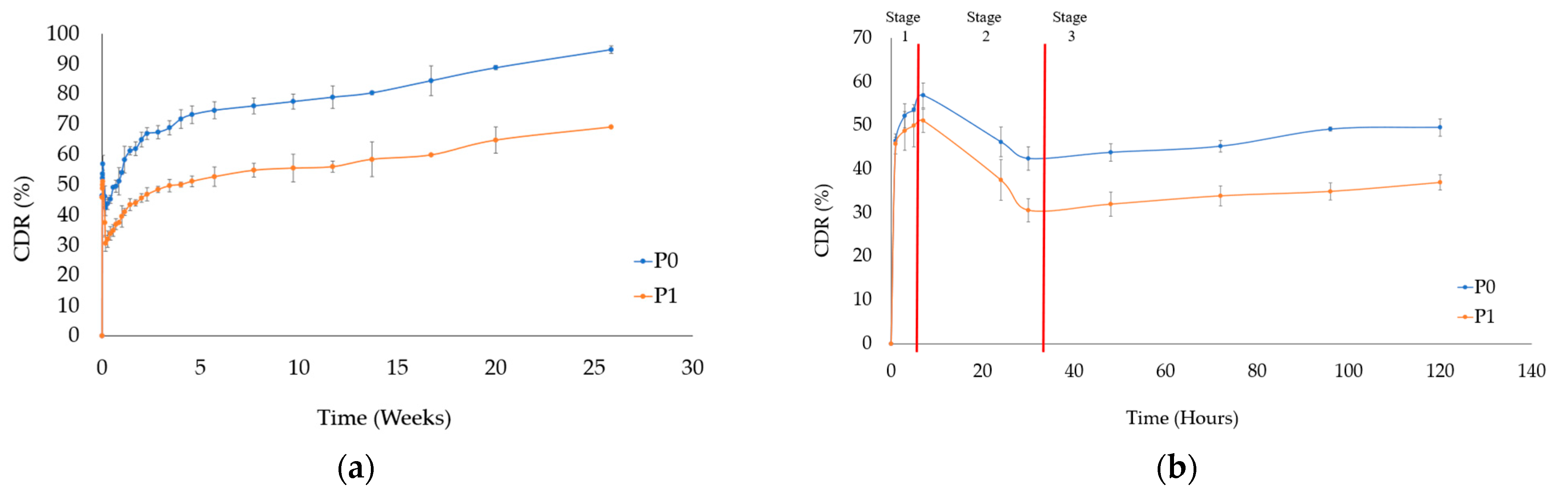
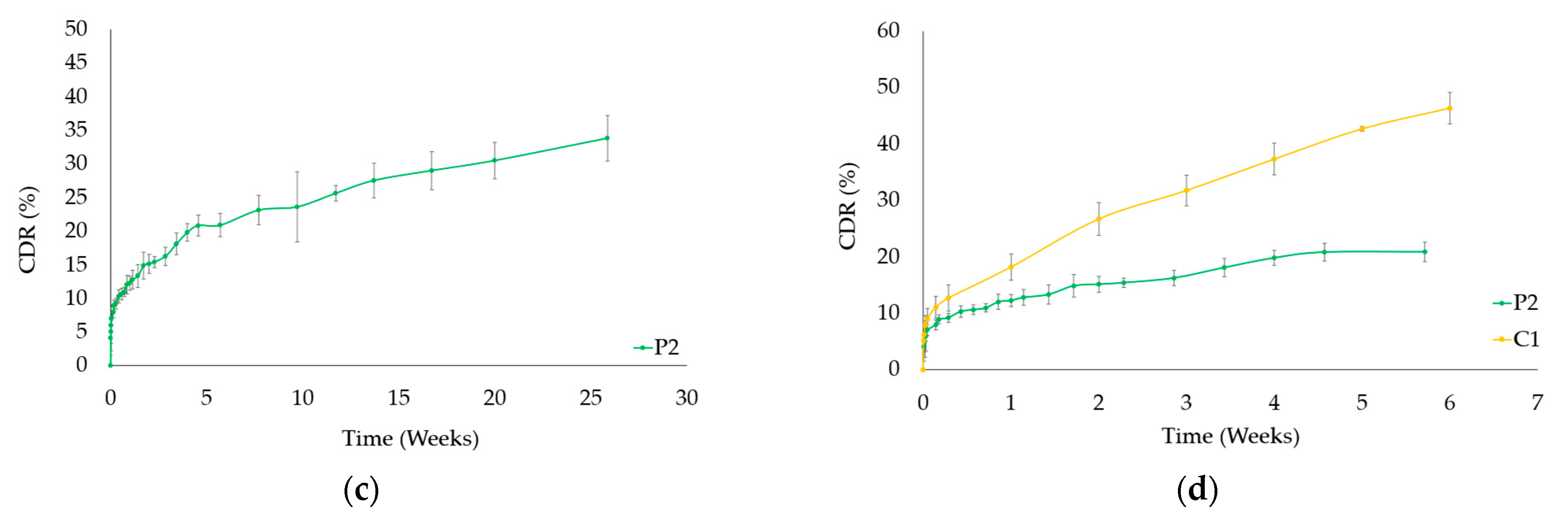
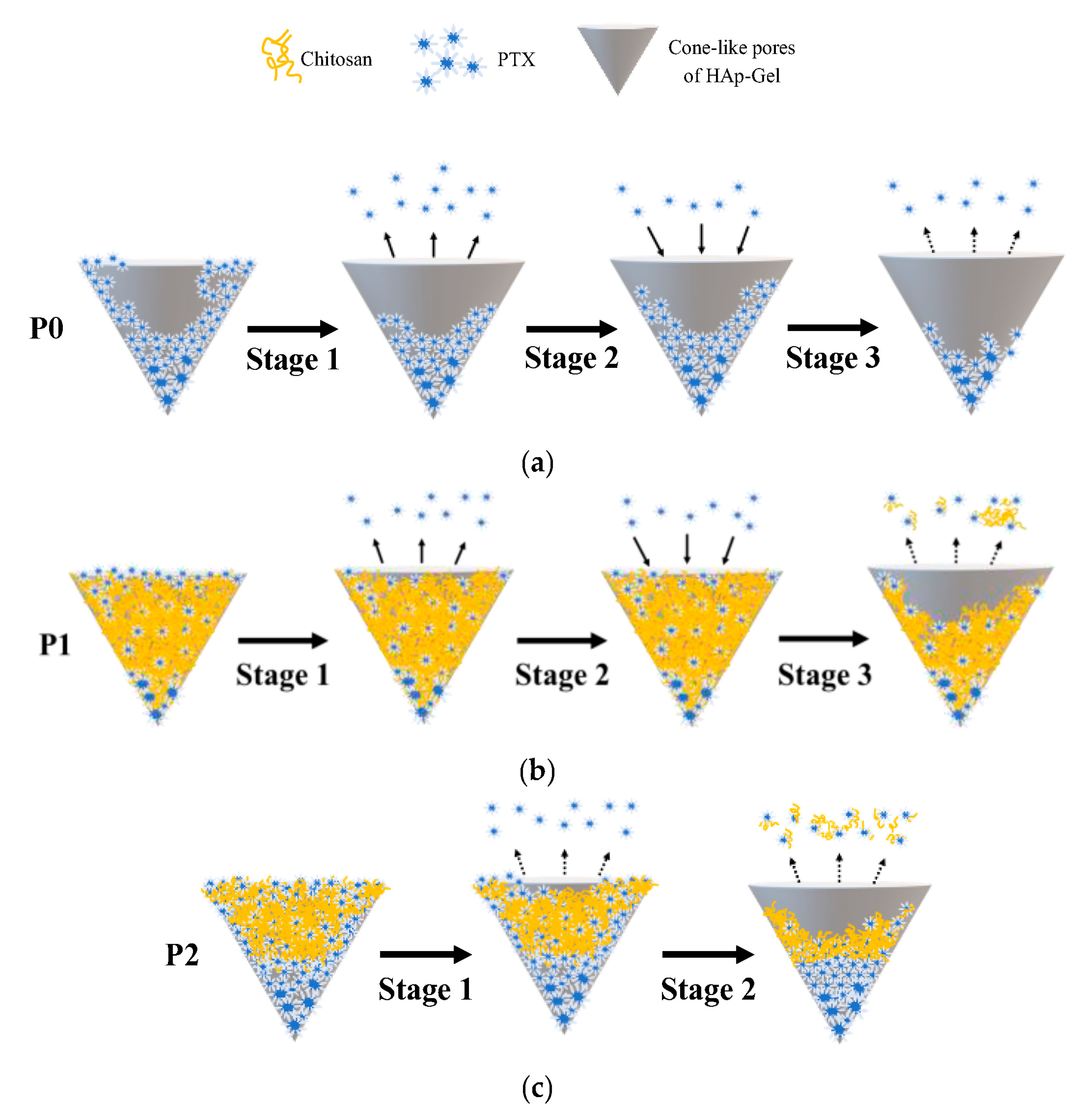
3.4. Drug Release from PTX-HAp-Gel



3.5. Cell Experiment
3.5.1. MTT Assay
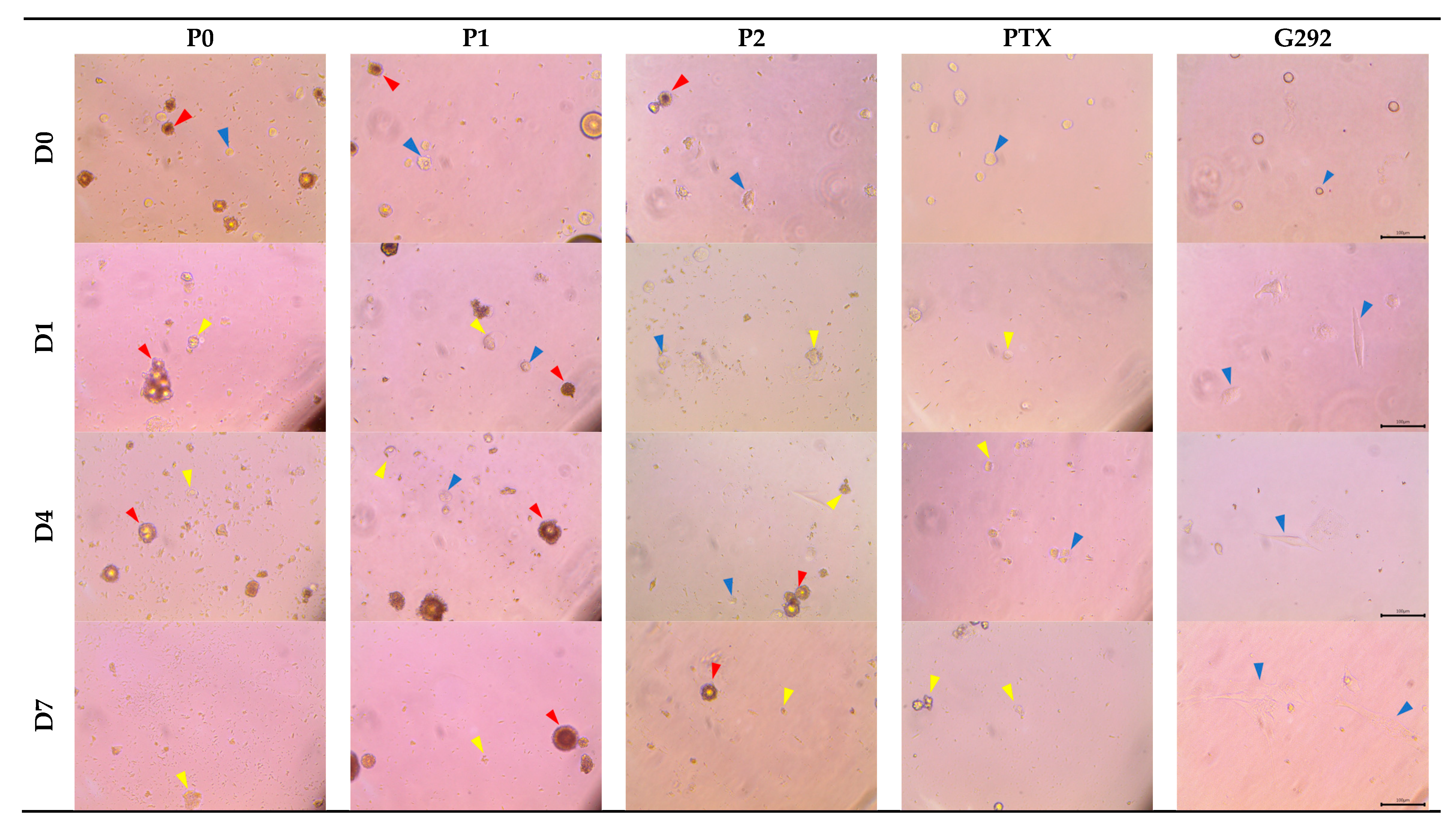
3.5.2. Observation of G292 Cell Morphology in the Cell Viability Assay

4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Daculsi, G.; Bouler, J.M.; LeGeros, R.Z. Adaptive Crystal Formation in Normal and Pathological Calcifications in Synthetic Calcium Phosphate and Related Biomaterials. In International Review of Cytology; Jeon, K.W., Ed.; Academic Press: Cambridge, MA, USA, 1997; Volume 172, pp. 129–191. [Google Scholar]
- Lebre, F.; Sridharan, R.; Sawkins, M.J.; Kelly, D.J.; O’Brien, F.J.; Lavelle, E.C. The shape and size of hydroxyapatite particles dictate inflammatory responses following implantation. Sci. Rep. 2017, 7, 2922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, C.; Peng, S.; Feng, P.; Shuai, C. Bone biomaterials and interactions with stem cells. Bone Res. 2017, 5, 17059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habibovic, P.; Barralet, J.E. Bioinorganics and biomaterials: Bone repair. Acta Biomater. 2011, 7, 3013–3026. [Google Scholar] [CrossRef] [PubMed]
- Holzwarth, J.M.; Ma, P.X. Biomimetic nanofibrous scaffolds for bone tissue engineering. Biomaterials 2011, 32, 9622–9629. [Google Scholar] [CrossRef] [Green Version]
- Palazzo, B.; Sidoti, M.C.; Roveri, N.; Tampieri, A.; Sandri, M.; Bertolazzi, L.; Galbusera, F.; Dubini, G.; Vena, P.; Contro, R. Controlled drug delivery from porous hydroxyapatite grafts: An experimental and theoretical approach. Mater. Sci. Eng. C 2005, 25, 207–213. [Google Scholar] [CrossRef]
- Watanabe, K.; Nishio, Y.; Makiura, R.; Nakahira, A.; Kojima, C. Paclitaxel-loaded hydroxyapatite/collagen hybrid gels as drug delivery systems for metastatic cancer cells. Int. J. Pharm. 2013, 446, 81–86. [Google Scholar] [CrossRef]
- Chao, S.C.; Wang, M.-J.; Pai, N.-S.; Yen, S.-K. Preparation and characterization of gelatin–hydroxyapatite composite microspheres for hard tissue repair. Mater. Sci. Eng. C 2015, 57, 113–122. [Google Scholar] [CrossRef]
- Lai, Y.-L.; Cheng, Y.-M.; Yen, S.-K. Doxorubicin-chitosan-hydroxyapatite composite coatings on titanium alloy for localized cancer therapy. Mater. Sci. Eng. C 2019, 104, 109953. [Google Scholar] [CrossRef]
- Kesharwani, P.; Tekade, R.; Jain, N. Spectrophotometric estimation of paclitaxel. Int. J. Adv. Pharm. Sci. 2011, 2, 29–32. [Google Scholar]
- Gupta, U.; Sharma, S.; Khan, I.; Gothwal, A.; Sharma, A.K.; Singh, Y.; Chourasia, M.K.; Kumar, V. Enhanced apoptotic and anticancer potential of paclitaxel loaded biodegradable nanoparticles based on chitosan. Int. J. Biol. Macromol. 2017, 98, 810–819. [Google Scholar] [CrossRef]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [Green Version]
- Lei, Y.; Zhang, D.; Yu, J.; Dong, H.; Zhang, J.; Yang, S. Targeting autophagy in cancer stem cells as an anticancer therapy. Cancer Lett. 2017, 393, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Dang, Y.; Guan, J. Nanoparticle-based drug delivery systems for cancer therapy. Smart Mater. Med. 2020, 1, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Hauner, K.; Maisch, P.; Retz, M. Nebenwirkungen der Chemotherapie. Der Urol. 2017, 56, 472–479. [Google Scholar] [CrossRef]
- Mills, J.K.; Needham, D. Targeted drug delivery. Expert Opin. Ther. Pat. 1999, 9, 1499–1513. [Google Scholar] [CrossRef]
- Jhaveri, J.; Raichura, Z.; Khan, T.; Momin, M.; Omri, A. Chitosan Nanoparticles-Insight into Properties, Functionalization and Applications in Drug Delivery and Theranostics. Molecules 2021, 26, 272. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Guan, X.; Zhong, S.; Chen, J.; Zhu, H.; Li, Z.; Xu, F.; Chen, P.; Wang, H. Multi-stimuli responsive smart chitosan-based microcapsules for targeted drug delivery and triggered drug release. Ultrason. Sonochem. 2017, 38, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Dong, F.; Dong, X.; Zhou, L.; Xiao, H.-H.; Ho, P.-Y.; Wong, M.-S.; Wang, Y. Doxorubicin-loaded biodegradable self-assembly zein nanoparticle and its anti-cancer effect: Preparation, in vitro evaluation, and cellular uptake. Colloids Surf. B Biointerfaces 2015, 140, 324–331. [Google Scholar] [CrossRef]
- Feng, S.-S.; Chien, S. Chemotherapeutic engineering: Application and further development of chemical engineering principles for chemotherapy of cancer and other diseases. Chem. Eng. Sci. 2003, 58, 4087–4114. [Google Scholar] [CrossRef]
- Wu, H.; Li, Z.; Tang, J.; Yang, X.; Zhou, Y.; Guo, B.; Wang, L.; Zhu, X.; Tu, C.; Zhang, X. The in vitro and in vivo anti-melanoma effects of hydroxyapatite nanoparticles: Influences of material factors. Int. J. Nanomed. 2019, 14, 1177–1191. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Ng, S.; Heng, B.C.; Guo, J.; Ma, L.; Tan, T.T.Y.; Ng, K.W.; Loo, S.C.J. Cytotoxicity of hydroxyapatite nanoparticles is shape and cell dependent. Arch. Toxicol. 2013, 87, 1037–1052. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Liu, C.; Qian, J.; Wang, J.; Zhang, Y. Size-mediated cytotoxicity and apoptosis of hydroxyapatite nanoparticles in human hepatoma HepG2 cells. Biomaterials 2010, 31, 730–740. [Google Scholar] [CrossRef]
- Ali, A.; Ahmed, S. A review on chitosan and its nanocomposites in drug delivery. Int. J. Biol. Macromol. 2018, 109, 273–286. [Google Scholar] [CrossRef]
- Ganji, F.; Abdekhodaie, M.J.; Ramazani S.A., A. Gelation time and degradation rate of chitosan-based injectable hydrogel. J. Sol-Gel Sci. Technol. 2007, 42, 47–53. [Google Scholar] [CrossRef]
- Shim, S.; Yoo, H.S. The Application of Mucoadhesive Chitosan Nanoparticles in Nasal Drug Delivery. Mar. Drugs 2020, 18, 605. [Google Scholar] [CrossRef]
- Ravi Kumar, M.N.V. A review of chitin and chitosan applications. React. Funct. Polym. 2000, 46, 1–27. [Google Scholar] [CrossRef]
- Islam, N.; Dmour, I.; Taha, M.O. Degradability of chitosan micro/nanoparticles for pulmonary drug delivery. Heliyon 2019, 5, e01684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Zhang, Y.; Zhou, Y. Application Progress of Modified Chitosan and Its Composite Biomaterials for Bone Tissue Engineering. Int. J. Mol. Sci. 2022, 23, 6574. [Google Scholar] [CrossRef]
- Celesti, C.; Iannazzo, D.; Espro, C.; Visco, A.; Legnani, L.; Veltri, L.; Visalli, G.; Di Pietro, A.; Bottino, P.; Chiacchio, M.A. Chitosan/POSS Hybrid Hydrogels for Bone Tissue Engineering. Materials 2022, 15, 8208. [Google Scholar] [CrossRef]
- DNA Structure, Replication, and Technology. Available online: http://www.shmoop.com/dna/dna-replication.html (accessed on 14 September 2022).
- Paclitaxel (Taxol®). Available online: https://www.oncolink.org/cancer-treatment/oncolink-rx/paclitaxel-taxol-r (accessed on 11 October 2022).
- Zhao, Y.; Cai, C.; Liu, M.; Zhao, Y.; Wu, Y.; Fan, Z.; Ding, Z.; Zhang, H.; Wang, Z.; Han, J. Drug-binding albumins forming stabilized nanoparticles for co-delivery of paclitaxel and resveratrol: In vitro/in vivo evaluation and binding properties investigation. Int. J. Biol. Macromol. 2020, 153, 873–882. [Google Scholar] [CrossRef]
- Gaitanis, A.; Staal, S. Liposomal Doxorubicin and nab-Paclitaxel: Nanoparticle Cancer Chemotherapy in Current Clinical Use. In Cancer Nanotechnology: Methods and Protocols; Grobmyer, S.R., Moudgil, B.M., Eds.; Humana Press: Totowa, NJ, USA, 2010; pp. 385–392. [Google Scholar]
- Kampan, N.C.; Madondo, M.T.; McNally, O.M.; Quinn, M.; Plebanski, M. Paclitaxel and Its Evolving Role in the Management of Ovarian Cancer. BioMed Res. Int. 2015, 2015, 413076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham-Gurysh, E.G.; Moore, K.M.; Schorzman, A.N.; Lee, T.; Zamboni, W.C.; Hingtgen, S.D.; Bachelder, E.M.; Ainslie, K.M. Tumor Responsive and Tunable Polymeric Platform for Optimized Delivery of Paclitaxel to Treat Glioblastoma. ACS Appl. Mater. Interfaces 2020, 12, 19345–19356. [Google Scholar] [CrossRef] [PubMed]
- Paclitaxel. Available online: https://chemocare.com/chemotherapy/drug-info/paclitaxel.aspx (accessed on 29 September 2022).
- Teow, H.M.; Zhou, Z.; Najlah, M.; Yusof, S.R.; Abbott, N.J.; D’Emanuele, A. Delivery of paclitaxel across cellular barriers using a dendrimer-based nanocarrier. Int. J. Pharm. 2013, 441, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Ho, E.A.; Vassileva, V.; Allen, C.; Piquette-Miller, M. In vitro and in vivo characterization of a novel biocompatible polymer–lipid implant system for the sustained delivery of paclitaxel. J. Control. Release 2005, 104, 181–191. [Google Scholar] [CrossRef]
- Vigata, M.; Meinert, C.; Pahoff, S.; Bock, N.; Hutmacher, D.W. Gelatin Methacryloyl Hydrogels Control the Localized Delivery of Albumin-Bound Paclitaxel. Polymers 2020, 12, 501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nsereko, S.; Amiji, M. Localized delivery of paclitaxel in solid tumors from biodegradable chitin microparticle formulations. Biomaterials 2002, 23, 2723–2731. [Google Scholar] [CrossRef]
- Cabanes, A.; Briggs, K.E.; Gokhale, P.C.; Treat, J.A.; Rahman, A. Comparative in vivo studies with paclitaxel and liposome-encapsulated paclitaxel. Int. J. Oncol. 1998, 12, 1035–1075. [Google Scholar] [CrossRef]
- Crosasso, P.; Ceruti, M.; Brusa, P.; Arpicco, S.; Dosio, F.; Cattel, L. Preparation, characterization and properties of sterically stabilized paclitaxel-containing liposomes. J. Control. Release 2000, 63, 19–30. [Google Scholar] [CrossRef]
- Wu, M.-Y.; Liang, Y.-H.; Yen, S.-K. Effects of Chitosan on Loading and Releasing for Doxorubicin Loaded Porous Hydroxyapatite–Gelatin Composite Microspheres. Polymers 2022, 14, 4276. [Google Scholar]
- Lai, Y.-L.; Lai, S.-B.; Yen, S.-K. Paclitaxel/hydroxyapatite composite coatings on titanium alloy for biomedical applications. Mater. Sci. Eng. C 2017, 79, 622–628. [Google Scholar] [CrossRef]
- Szebeni, J.; Alving, C.R.; Muggia, F.M. Complement Activation by Cremophor EL as a Possible Contributor to Hypersensitivity to Paclitaxel: An In Vitro Study. JNCI J. Natl. Cancer Inst. 1998, 90, 300–306. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.N.; Kim, J.Y.; Kim, J.W.; Kim, J.H.; Kim, H.I.; Yune, S.; Choi, D.-C.; Lee, B.-J. The Hidden Culprit: A Case of Repeated Anaphylaxis to Cremophor. Allergy Asthma Immunol. Res. 2016, 8, 174–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, M.; Hosoya, O.; Taoka, S.; Seki, T.; Kawaguchi, T.; Sugibayashi, K.; Juni, K.; Morimoto, Y. Relationship between Solubility of Chitosan in Alcoholic Solution and Its Gelation. Chem. Pharm. Bull. 1999, 47, 1044–1046. [Google Scholar] [CrossRef] [Green Version]
- Arora, M. Cell culture media: A review. Mater. Methods 2013, 3, 24. [Google Scholar] [CrossRef]
- van Meerloo, J.; Kaspers, G.J.L.; Cloos, J. Cell Sensitivity Assays: The MTT Assay. In Cancer Cell Culture: Methods and Protocols; Cree, I.A., Ed.; Humana Press: Totowa, NJ, USA, 2011; pp. 237–245. [Google Scholar]
- Martins, M.A.; Santos, C.; Almeida, M.M.; Costa, M.E.V. Hydroxyapatite micro- and nanoparticles: Nucleation and growth mechanisms in the presence of citrate species. J. Colloid Interface Sci. 2008, 318, 210–216. [Google Scholar] [CrossRef]
- Ashok, M.; Meenakshi Sundaram, N.; Narayana Kalkura, S. Crystallization of hydroxyapatite at physiological temperature. Mater. Lett. 2003, 57, 2066–2070. [Google Scholar] [CrossRef]
- Mousia, Z.; Farhat, I.A.; Pearson, M.; Chesters, M.A.; Mitchell, J.R. FTIR microspectroscopy study of composition fluctuations in extruded amylopectin–gelatin blends. Biopolymers 2001, 62, 208–218. [Google Scholar] [CrossRef]
- Jeong, J.; Kim, J.H.; Shim, J.H.; Hwang, N.S.; Heo, C.Y. Bioactive calcium phosphate materials and applications in bone regeneration. Biomater. Res. 2019, 23, 4. [Google Scholar] [CrossRef] [Green Version]
- Manna, S.; Donnell, A.M.; Kaval, N.; Al-Rjoub, M.F.; Augsburger, J.J.; Banerjee, R.K. Improved design and characterization of PLGA/PLA-coated Chitosan based micro-implants for controlled release of hydrophilic drugs. Int. J. Pharm. 2018, 547, 122–132. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, M.-Y.; Kao, I.-F.; Fu, C.-Y.; Yen, S.-K. Effects of Adding Chitosan on Drug Entrapment Efficiency and Release Duration for Paclitaxel-Loaded Hydroxyapatite—Gelatin Composite Microspheres. Pharmaceutics 2023, 15, 2025. https://doi.org/10.3390/pharmaceutics15082025
Wu M-Y, Kao I-F, Fu C-Y, Yen S-K. Effects of Adding Chitosan on Drug Entrapment Efficiency and Release Duration for Paclitaxel-Loaded Hydroxyapatite—Gelatin Composite Microspheres. Pharmaceutics. 2023; 15(8):2025. https://doi.org/10.3390/pharmaceutics15082025
Chicago/Turabian StyleWu, Meng-Ying, I-Fang Kao, Chien-Yao Fu, and Shiow-Kang Yen. 2023. "Effects of Adding Chitosan on Drug Entrapment Efficiency and Release Duration for Paclitaxel-Loaded Hydroxyapatite—Gelatin Composite Microspheres" Pharmaceutics 15, no. 8: 2025. https://doi.org/10.3390/pharmaceutics15082025