
Hacking the Immune Response to Solid Tumors: Harnessing the Anti-Cancer Capacities of Oncolytic Bacteria

 , , , , and
, , , , and 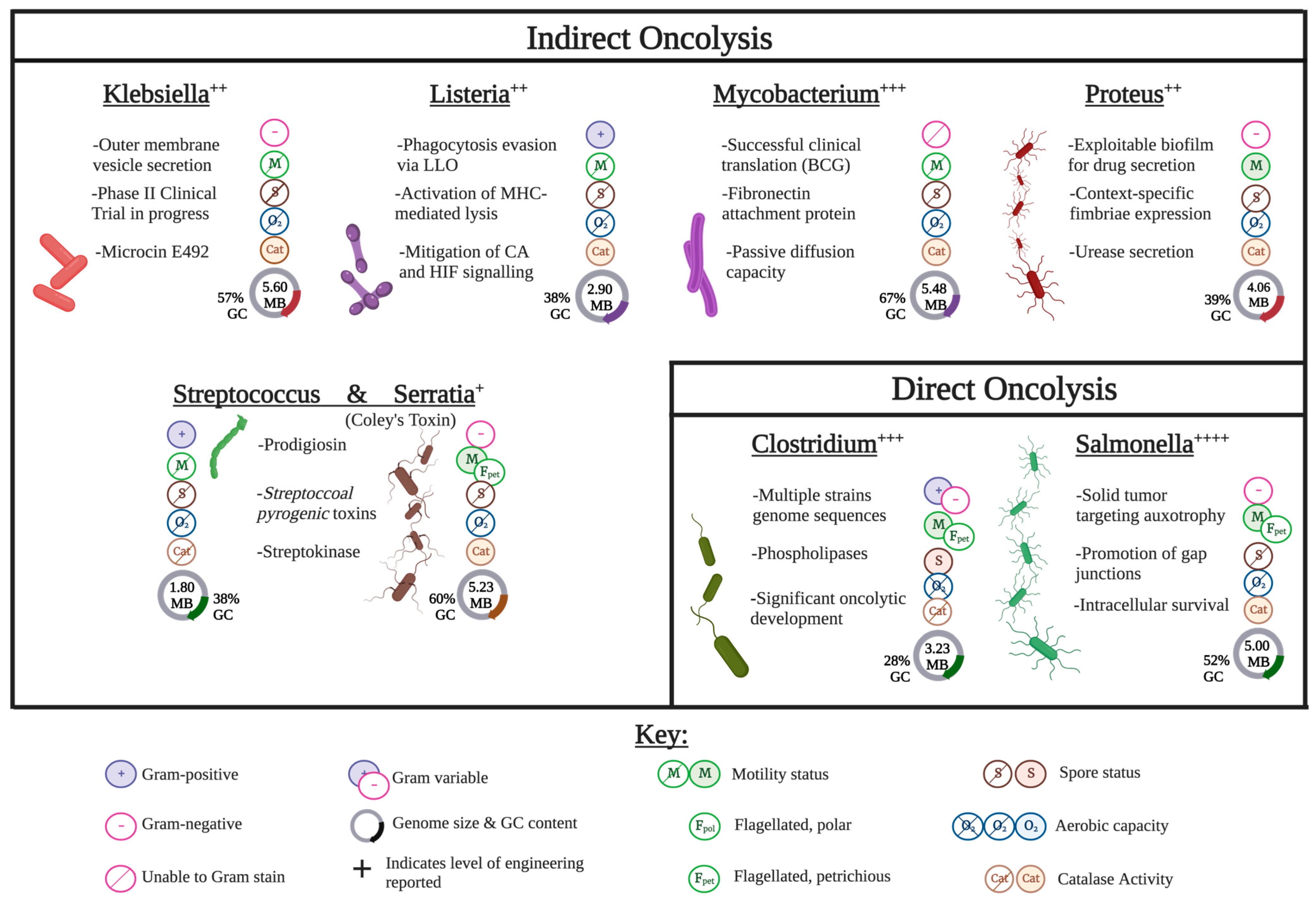{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction and Background
1.1. Essential Components of Host–Pathogen Interactions
1.2. Decades of Efficacy and Safety: The BCG Vaccine
2. Indirect Oncolytic Bacteria
2.1. Klebsiella
2.1.1. Klebsiella Basic Microbiology
2.1.2. Klebsiella Genome
2.1.3. Klebsiella Background and History
2.1.4. Klebsiella Cell Surface
2.1.5. Klebsiella Metabolism and Byproduct Secretion
2.1.6. Klebsiella Host–Pathogen Interactions
2.1.7. Klebsiella Oncolytic Development
2.2. Listeria monocytogenes
2.2.1. Listeria Basic Microbiology
2.2.2. Listeria Genome
2.2.3. Listeria Background and History
2.2.4. Listeria Cell Surface
2.2.5. Listeria Metabolism, Byproducts and Secretions
2.2.6. Listeria Host–Pathogen Interaction
2.2.7. Listeria Oncolytic Development
2.3. Mycobacterium
2.3.1. Mycobacterium Basic Microbiology
2.3.2. Mycobacterium Genome
2.3.3. Mycobacterium Background and History
2.3.4. Mycobacterium Cell Surface
Mycobacterium pargordonae (Pmg)
Mycobacterium indicus pranii (MIP)
Mycobacterium smegmatis
2.3.5. Mycobacterium Metabolism and Byproduct Secretion
2.3.6. Mycobacterium Host–Pathogen Interactions
2.3.7. Mycobacterium Oncolytic Development
2.4. Proteus mirabilis
2.4.1. Proteus mirabilis Basic Microbiology
2.4.2. Proteus mirabilis Genome
2.4.3. Proteus mirabilis Background and History
2.4.4. Proteus mirabilis Cell Surface
2.4.5. Proteus mirabilis Metabolism, Byproducts, and Secretions
2.4.6. Proteus mirabilis Host–Pathogen Interactions
2.4.7. Proteus mirabilis Oncolytic Development
2.5. Streptococcus/Serratia Mix
2.5.1. Streptococcus/Serratia Mix Background and History
2.5.2. Serratia marcescens
Serratia marcescens Basic Microbiology
Serratia marcescens Genome
Serratia marcescens Cell Surface
Serratia marcescens Metabolism and Byproduct Secretion
2.5.3. Streptococcus pyogenes
Streptococcus pyogenes Basic Microbiology
Streptococcus pyogenes Genome
Streptococcus pyogenes Cell Surface
Streptococcus pyogenes Metabolism and Byproduct Secretion
Streptococcus pyogenes Host–Pathogen Interactions
Streptococcus pyogenes Oncolytic Development
3. Direct Oncolysis
3.1. Clostridia
3.1.1. Clostridium acetobutylicum
Clostridium acetobutylicum Basic Microbiology
Clostridium acetobutylicum Genome
Clostridium acetobutylicum Background and History
Clostridium acetobutylicum Cell and Spore Surface
Clostridium acetobutylicum Metabolism, Byproducts, and Secretions
Clostridium acetobutylicum Host–Pathogen Interactions
Clostridium acetobutylicum Oncolytic Development
3.1.2. Hathewaya histolytica, Formerly Known as Clostridium histolyticum
Hathewaya histolytica Basic Microbiology
Hathewaya histolytica Genome
Hathewaya histolytica Background and History
Hathewaya histolytica Cell and Spore Surface
Hathewaya histolytica Metabolism, Byproducts, and Secretions
- Alpha-Toxin
- Beta-Toxin
- Gamma-, Delta-, and Epsilon Toxins
Hathewaya histolytica Host–Pathogen Interactions
Hathewaya histolytica Oncolytic Development
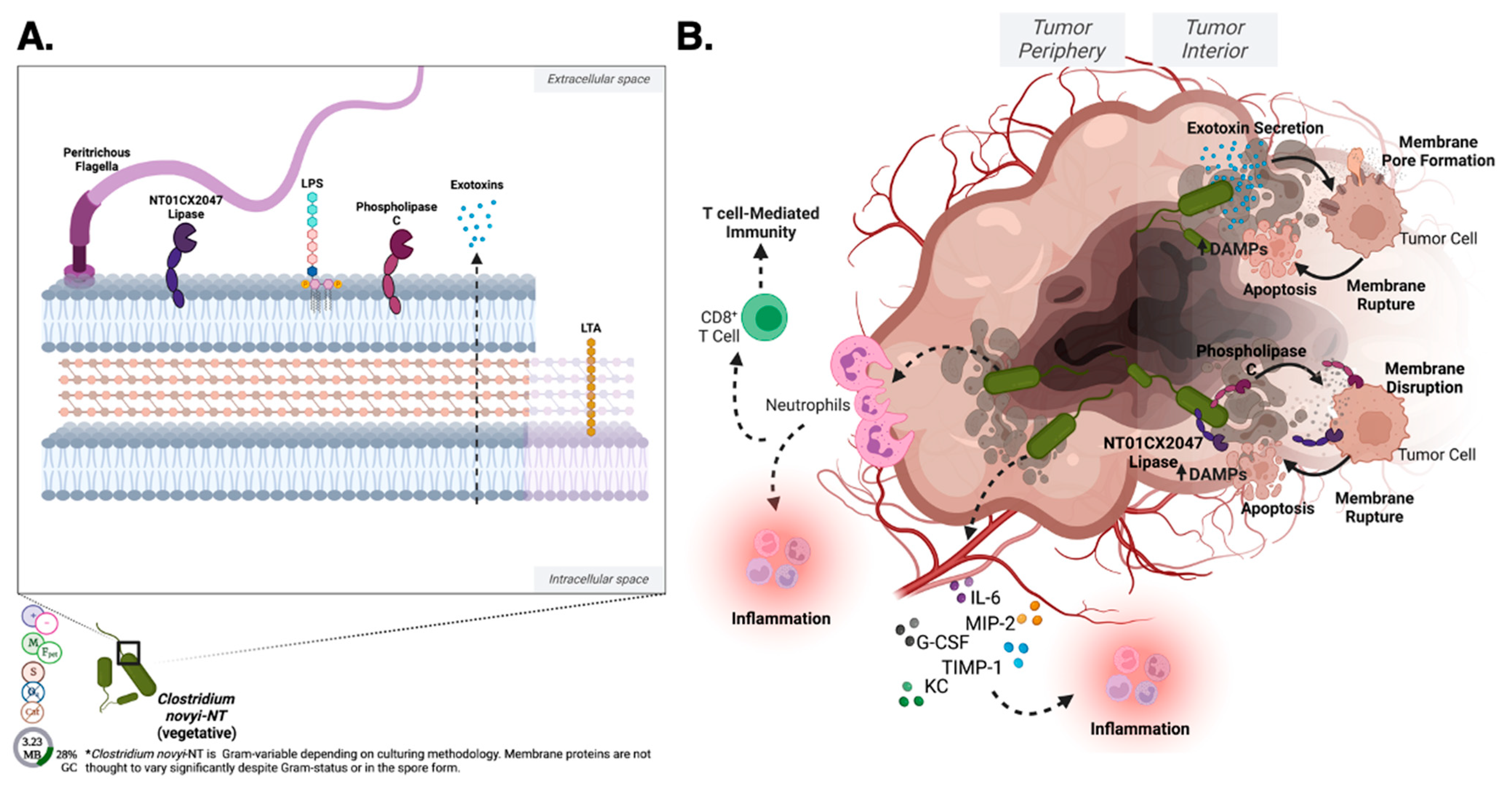
3.1.3. Clostridium novyi
Clostridium novyi Basic Microbiology
Clostridium novyi Genome
Clostridium novyi Background and History
Clostridium novyi Cell and Spore Surface
Clostridium novyi Metabolism, Byproducts, and Secretions
Clostridium novyi Host–Pathogen Interactions
Clostridium novyi Oncolytic Development
3.1.4. Clostridium sporogenes
Clostridium sporogenes Basic Microbiology
Clostridium sporogenes Genome
Clostridium sporogenes Background and History
Clostridium sporogenes Cell and Spore Surface
Clostridium sporogenes Metabolism, Byproducts, and Secretions
Clostridium sporogenes Host–Pathogen Interactions
Clostridium sporogenes Oncolytic Development
3.1.5. Clostridium tetani
Clostridium tetani Basic Microbiology
Clostridium tetani Genome
Clostridium tetani Background and History
Clostridium tetani Cell and Spore Surface
Clostridium tetani Metabolism, Byproducts, and Secretions
Clostridium tetani Host–Pathogen Interactions
Clostridium tetani Oncolytic Development
3.2. Salmonella
3.2.1. Salmonella Basic Microbiology
3.2.2. Salmonella Genome
3.2.3. Salmonella Background and History
3.2.4. Salmonella Cell Surface
3.2.5. Salmonella Metabolism, Byproducts, and Secretions
3.2.6. Salmonella Host–Pathogen Interactions
3.2.7. Salmonella Oncolytic Development
4. Prophylactic Bacteria
4.1. Bifidobacterium
4.1.1. Bifidobacterium Basic Microbiology
4.1.2. Bifidobacterium Genome
4.1.3. Bifidobacterium Background and History
4.1.4. Bifidobacterium Cell Surface
4.1.5. Bifidobacterium Metabolism and Byproduct Secretion
4.1.6. Bifidobacterium Host Interaction
4.1.7. Bifidobacterium Oncolytic Development
4.2. Caulobacter
4.2.1. Caulobacter Basic Microbiology
4.2.2. Caulobacter Genome
4.2.3. Caulobacter Background and History
4.2.4. Caulobacter Cell Surface
4.2.5. Caulobacter Metabolism and Byproduct Secretion
4.2.6. Caulobacter Oncolytic Development
4.3. Lactobacilli
4.3.1. Lactobacilli Basic Microbiology
4.3.2. Lactobacilli Genome
4.3.3. Lactobacilli Background and History
4.3.4. Lactobacilli Cell Surface
4.3.5. Lactobacilli Metabolism and Byproduct Secretion
4.3.6. Lactobacilli Oncolytic Development
5. Future Perspective
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC transporters | ATP Binding cassette transporters |
| ActA | Actin assembly-inducing surface protein |
| ADEPT | Antibody directed-enzyme-prodrug therapy |
| AFM | Atomic force microscopy |
| ATF | Ambient temperature fimbriae |
| ATP | Adenosine triphosphate |
| B-CLL | B cell lymphocytic leukemia |
| BCG | Bacille Calmette-Guerin |
| bETE | b-Exotoxin I exporter |
| C. novyi-NT | Clostridium novyi-Non Toxic |
| CCL-19 | Chemokine ligand 19 |
| CCR-7 | Chemokine receptor 7 |
| ClyA | Cytolysin A |
| COBALT | Combination bacteriolytic therapy |
| CPS | Capsular polysaccharides |
| Crp | Cyclic AMP receptor protein |
| CXCL-10 | Chemokine ligand 10 |
| CXCL-2 | Chemokine ligand 2 |
| DAMPs | Damage-associated molecular patterns |
| DEPT | Directed-enzyme-prodrug therapy |
| EPS | Exopolysaccaride |
| FAP | Fibronectin attachment protein |
| FEA | Flagella export apparatus |
| FPE | Fimbrilin Protein Exporter |
| GDEPT | Gene directed-enzyme-prodrug therapy |
| HIF-1α | Hypoxia Inducible Factor-1α |
| HMGB1 | High-mobility group box 1 protein |
| HMME | Hematoprophyrin monomethyl ether |
| IFN-γ | Interferon gamma |
| InlA | Internalin A |
| InlB | Internalin B |
| IPA | Indole-3-propionic acid |
| ISE | Insertion sequence elements |
| LLO | Listeriolysin O |
| LPS | Lipopolysaccharide |
| LTA | Lipoteichoic acid |
| LTP | Lipoprotein translocase |
| MHC | Major histocompatibility complex |
| MIF | Migration inhibitor factor |
| MIP | Mycobacterium indicus pranii |
| MMP-9 | Metalloproteinase 9 |
| MR/P | Mannose-resistant Proteus-like fimbriae |
| MscL | Mechanosensitive ion channel |
| mTNF-ɑ | Mouse tumor necrosis factor alpha |
| NETs | Neutrophil extracellular traps |
| NK cell | Natural Killer cell |
| NLRs | NOD-like receptors |
| NO | Nitric Oxide |
| Nrp | Non-ribosomal peptide |
| NTG | Nitroguanidine |
| ODN | Oligodexoynucleotides |
| OmpA | Outer membrane protein A |
| OMPs | Outer membrane proteins |
| OMV | Outer membrane vesicles |
| PAMPs | Pathogen-associated molecular patterns |
| Pbt | Proteobactin |
| PDX | Patient-derived xenograft |
| PE/PPE | Proline-glutamate or Proline-proline Glutamate |
| PEI | Polyethyleneimine |
| Pep1E | Peptide-1 exporter |
| Pep2E | Peptide-2 exporter |
| Pep4E | Peptide-4 exporter |
| Pep5E | Peptide-5 exporter |
| PMF | Proteus mirabilis fimbriae |
| Pmg | Mycobacterium pargordonae |
| PRRs | Pattern recognition receptors |
| Pta | Proteus-toxic agglutinin |
| rIL-2 | Rat interleukin-2 |
| RLRs | RIG-I-like receptors |
| SCID | Severe combined immunodeficiency |
| Sec | Secretory pathway |
| SLAP | Spacious Listeria-containing phagosomes |
| SLH | S-layer Homology domains |
| SPE | Streptococcal pyrogenic toxins |
| STING pathway | Stimulator interferon genes |
| Tad system | Tight adherence system |
| tat | Twin arginine translocation pathway |
| TB | Tuberculosis |
| TeTx | Tetanus toxin |
| TME | Tumor microenvironment |
| TNF-ɑ | Tumor necrosis factor-alpha |
| UCA | Uroepithelial cell adhesin |
| UTI | Urinary tract infection |
| WHO | World Health Organization |
References
- Rius-Rocabert, S.; Pinel, F.L.; Pozuelo, M.J.; Garcia, A.; Nistal-Villán, E. Oncolytic bacteria: Past, present and future. FEMS Microbiol. Lett. 2019, 366, fnz136. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, X.; Guo, Y.; Wang, J.; Zhang, D.; Mei, Y.; Shi, J.; Tan, W.; Zheng, J.H. Genetically engineered oncolytic bacteria as drug delivery systems for targeted cancer theranostics. Acta Biomater. 2021, 124, 72–87. [Google Scholar] [CrossRef]
- Iglesias-Garcia, J.; de la Iglesia-Garcia, D.; Olmos-Martinez, J.M.; Lariño-Noia, J.; Dominguez-Muñoz, J.E. Differential diagnosis of solid pancreatic masses. Minerva Gastroenterol. Dietol. 2020, 66, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Global Cancer Facts & Figures, 4th ed.; American Cancer Society, GLOBOCAN: 2018. Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/global-cancer-facts-and-figures/global-cancer-facts-and-figures-4th-edition.pdf (accessed on 17 July 2023).
- Mun, E.J.; Babiker, H.M.; Weinberg, U.; Kirson, E.D.; Von Hoff, D.D. Tumor-Treating Fields: A Fourth Modality in Cancer Treatment. Clin. Cancer Res. 2018, 24, 266–275. [Google Scholar] [CrossRef] [Green Version]
- Debela, D.T.; Muzazu, S.G.; Heraro, K.D.; Ndalama, M.T.; Mesele, B.W.; Haile, D.C.; Kitui, S.K.; Manyazewal, T. New approaches and procedures for cancer treatment: Current perspectives. SAGE Open Med. 2021, 9, 20503121211034370. [Google Scholar] [CrossRef]
- Zhu, S.; Zhang, T.; Zheng, L.; Liu, H.; Song, W.; Liu, D.; Li, Z.; Pan, C. Combination strategies to maximize the benefits of cancer immunotherapy. J. Hematol. Oncol. 2021, 14, 156. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.-G.; Li, Y.-M. Emerging Adjuvants for Cancer Immunotherapy. Front. Chem. 2020, 8, 601. [Google Scholar] [CrossRef]
- Ji, X.; Jiang, W.; Wang, J.; Zhou, B.; Ding, W.; Liu, S.; Huang, H.; Chen, G.; Sun, X. Application of individualized multimodal radiotherapy combined with immunotherapy in metastatic tumors. Front. Immunol. 2023, 13, 1106644. [Google Scholar] [CrossRef]
- Piechotta, V.; Adams, A.; Haque, M.; Scheckel, B.; Kreuzberger, N.; Monsef, I.; Jordan, K.; Kuhr, K.; Skoetz, N. Antiemetics for Adults for Prevention of Nausea and Vomiting Caused by Moderately or Highly Emetogenic Chemotherapy: A Network Meta-Analysis. Cochrane Database Syst. Rev. 2021, 11, CD012775. [Google Scholar]
- Yan, T.; Zhu, L.; Chen, J. Current advances and challenges in CAR T-Cell therapy for solid tumors: Tumor-associated antigens and the tumor microenvironment. Exp. Hematol. Oncol. 2023, 12, 14. [Google Scholar] [CrossRef]
- Shi, A.-M.; Tao, Z.-Q.; Li, H.; Wang, Y.-Q.; Zhao, J. Cancer stem cells targeting agents—A review. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 4064–4067. [Google Scholar] [PubMed]
- Huo, D.; Jiang, X.; Hu, Y. Recent Advances in Nanostrategies Capable of Overcoming Biological Barriers for Tumor Management. Adv. Mater. 2020, 32, e1904337. [Google Scholar] [CrossRef] [PubMed]
- Palanivelu, L.; Liu, C.-H.; Lin, L.-T. Immunogenic cell death: The cornerstone of oncolytic viro-immunotherapy. Front. Immunol. 2022, 13, 1038226. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef] [PubMed]
- Denning, N.-L.; Aziz, M.; Gurien, S.D.; Wang, P. DAMPs and NETs in Sepsis. Front. Immunol. 2019, 10, 2536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herwald, H.; Egesten, A. On PAMPs and DAMPs. J. Innate Immun. 2016, 8, 427–428. [Google Scholar] [CrossRef]
- Pallett, L.J.; Swadling, L.; Diniz, M.; Maini, A.A.; Schwabenland, M.; Gasull, A.D.; Davies, J.; Kucykowicz, S.; Skelton, J.K.; Thomas, N.; et al. Tissue CD14+CD8+ T cells reprogrammed by myeloid cells and modulated by LPS. Nature 2023, 614, 334–342. [Google Scholar] [CrossRef]
- Ciesielska, A.; Matyjek, M.; Kwiatkowska, K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell. Mol. Life Sci. 2021, 78, 1233–1261. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Seregin, S.S.; Yang, D.; Fukase, K.; Chamaillard, M.; Alnemri, E.S.; Inohara, N.; Chen, G.Y.; Núñez, G. The NLRP6 Inflammasome Recognizes Lipoteichoic Acid and Regulates Gram-Positive Pathogen Infection. Cell 2018, 175, 1651–1664.e14. [Google Scholar] [CrossRef] [Green Version]
- Kubicek-Sutherland, J.Z.; Vu, D.M.; Noormohamed, A.; Mendez, H.M.; Stromberg, L.R.; Pedersen, C.A.; Hengartner, A.C.; Klosterman, K.E.; Bridgewater, H.A.; Otieno, V.; et al. Direct detection of bacteremia by exploiting host-pathogen interactions of lipoteichoic acid and lipopolysaccharide. Sci. Rep. 2019, 9, 6203. [Google Scholar] [CrossRef] [Green Version]
- Su, S.-C.; Hua, K.-F.; Lee, H.; Chao, L.K.; Tan, S.-K.; Lee, H.; Yang, S.-F.; Hsu, H.-Y. LTA and LPS mediated activation of protein kinases in the regulation of inflammatory cytokines expression in macrophages. Clin. Chim. Acta 2006, 374, 106–115. [Google Scholar] [CrossRef]
- de Oliviera Nascimento, L.; Massari, P.; Wetzler, L. The Role of TLR2 in Infection and Immunity. Front. Immunol. 2012, 3, 79. [Google Scholar] [CrossRef] [Green Version]
- Liljeroos, M.; Vuolteenaho, R.; Morath, S.; Hartung, T.; Hallman, M.; Ojaniemi, M. Bruton’s tyrosine kinase together with PI 3-kinase are part of Toll-like receptor 2 multiprotein complex and mediate LTA induced Toll-like receptor 2 responses in macrophages. Cell. Signal. 2007, 19, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Gambhir, V.; Yildiz, C.; Mulder, R.; Siddiqui, S.; Guzzo, C.; Szewczuk, M.; Gee, K.; Basta, S. The TLR2 agonists lipoteichoic acid and Pam3CSK4 induce greater pro-inflammatory responses than inactivated Mycobacterium butyricum. Cell. Immunol. 2012, 280, 101–107. [Google Scholar] [CrossRef]
- Liu, M.; Mu, H.; Peng, W.; Zhao, L.; Hu, W.; Jiang, Z.; Gao, L.; Cao, X.; Li, N.; Han, J. Time-dependent C5a and C5aR expression in dental pulp cells following stimulation with LTA and LPS. Int. J. Mol. Med. 2019, 44, 823–834. [Google Scholar] [CrossRef] [Green Version]
- Jasenosky, L.D.; Nambu, A.; Tsytsykova, A.V.; Ranjbar, S.; Haridas, V.; Kruidenier, L.; Tough, D.F.; Goldfeld, A.E. Identification of a Distal Locus Enhancer Element That Controls Cell Type-Specific TNF and LTA Gene Expression in Human T Cells. J. Immunol. 2020, 205, 2479–2488. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K.K.; Genga, K.R.; Topchiy, E.; Cirstea, M.; Shimada, T.; Fjell, C.; Russell, J.A.; Boyd, J.H.; Walley, K.R. Reduced Proprotein convertase subtilisin/kexin 9 (PCSK9) function increases lipoteichoic acid clearance and improves outcomes in Gram positive septic shock patients. Sci. Rep. 2019, 9, 10588. [Google Scholar] [CrossRef] [Green Version]
- Mutig, N.; Geers-Knoerr, C.; Piep, B.; Pahuja, A.; Vogt, P.M.; Brenner, B.; Niederbichler, A.D.; Kraft, T. Lipoteichoic acid from Staphylococcus aureus directly affects cardiomyocyte contractility and calcium transients. Mol. Immunol. 2013, 56, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Kakihana, Y.; Ito, T.; Nakahara, M.; Yamaguchi, K.; Yasuda, T. Sepsis-induced myocardial dysfunction: Pathophysiology and management. J. Intensive Care 2016, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Grin, P.M.; Dwivedi, D.J.; Chathely, K.M.; Trigatti, B.L.; Prat, A.; Seidah, N.G.; Liaw, P.C.; Fox-Robichaud, A.E. Low-density lipoprotein (LDL)-dependent uptake of Gram-positive lipoteichoic acid and Gram-negative lipopolysaccharide occurs through LDL receptor. Sci. Rep. 2018, 8, 10496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.-H.; Hsu, C.-J.; Yang, W.-H.; Fong, Y.-C. Lipoteichoic acid enhances IL-6 production in human synovial fibroblasts via TLR2 receptor, PKCdelta and c-Src dependent pathways. Biochem. Pharmacol. 2010, 79, 1648–1657. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, E.C.; Bhatt, L.K.; Prabhavalkar, K.S. High Mobility Group Box-1 (HMGB1): A Potential Target in Therapeutics. Curr. Drug Targets 2019, 20, 1474–1485. [Google Scholar] [CrossRef]
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 proteins in cancer. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef] [Green Version]
- Murao, A.; Aziz, M.; Wang, H.; Brenner, M.; Wang, P. Release mechanisms of major DAMPs. Apoptosis 2021, 26, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Qiang, X.; Yang, W.-L.; Wu, R.; Zhou, M.; Jacob, A.; Dong, W.; Kuncewitch, M.; Ji, Y.; Yang, H.; Wang, H.W.; et al. Cold-Inducible RNA-Binding Protein (CIRP) Triggers Inflammatory Responses in Hemorrhagic Shock and Sepsis. Nat. Med. 2013, 19, 1489–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, C.M.; McRae, S.A.; Ranger, A.L.; Klegeris, A. Extracellular histones as damage-associated molecular patterns in neuroinflammatory responses. Rev. Neurosci. 2022, 34, 533–558. [Google Scholar] [CrossRef] [PubMed]
- Dubaniewicz, A. Microbial and human heat shock proteins as “danger signals” in sarcoidosis. Hum. Immunol. 2013, 74, 1550–1558. [Google Scholar] [CrossRef]
- Preissner, K.T.; Herwald, H. Extracellular nucleic acids in immunity and cardiovascular responses: Between alert and disease. Thromb. Haemost. 2017, 117, 1272–1282. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef]
- Yang, Y.; Li, H.; Fotopoulou, C.; Cunnea, P.; Zhao, X. Toll-like receptor-targeted anti-tumor therapies: Advances and challenges. Front. Immunol. 2022, 13, 1049340. [Google Scholar] [CrossRef]
- Liu, P.; Lu, Z.; Liu, L.; Li, R.; Liang, Z.; Shen, M.; Xu, H.; Ren, D.; Ji, M.; Yuan, S.; et al. NOD-like receptor signaling in inflammation-associated cancers: From functions to targeted therapies. Phytomedicine 2019, 64, 152925. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Masucci, M.T.; Minopoli, M.; Del Vecchio, S.; Carriero, M.V. The Emerging Role of Neutrophil Extracellular Traps (NETs) in Tumor Progression and Metastasis. Front. Immunol. 2020, 11, 1749. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Euler, M.; Hoffmann, M.H. The double-edged role of neutrophil extracellular traps in inflammation. Biochem. Soc. Trans. 2019, 47, 1921–1930. [Google Scholar] [CrossRef]
- van Horssen, R.; ten Hagen, T.L.M.; Eggermont, A.M.M. TNF-α in Cancer Treatment: Molecular Insights, Antitumor Effects, and Clinical Utility. Oncol. 2006, 11, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Montfort, A.; Colacios, C.; Levade, T.; Andrieu-Abadie, N.; Meyer, N.; Ségui, B. The TNF Paradox in Cancer Progression and Immunotherapy. Front. Immunol. 2019, 10, 1818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WANG, X.; LIN, Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol. Sin. 2008, 29, 1275–1288. [Google Scholar] [CrossRef] [Green Version]
- Hirano, T. IL-6 in inflammation, autoimmunity and cancer. Int. Immunol. 2021, 33, 127–148. [Google Scholar] [CrossRef] [PubMed]
- Rašková, M.; Lacina, L.; Kejík, Z.; Venhauerová, A.; Skaličková, M.; Kolář, M.; Jakubek, M.; Rosel, D.; Smetana, K.; Brábek, J. The Role of IL-6 in Cancer Cell Invasiveness and Metastasis—Overview and Therapeutic Opportunities. Cells 2022, 11, 3698. [Google Scholar] [CrossRef]
- Khan, F.H.; Dervan, E.; Bhattacharyya, D.D.; McAuliffe, J.D.; Miranda, K.M.; Glynn, S.A. The Role of Nitric Oxide in Cancer: Master Regulator or NOt? Int. J. Mol. Sci. 2020, 21, 9393. [Google Scholar] [CrossRef]
- Nakanishi, M.; Shimada, M.; Niida, H. Genetic instability in cancer cells by impaired cell cycle checkpoints. Cancer Sci. 2006, 97, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Ow, Y.-L.P.; Green, D.R.; Hao, Z.; Mak, T.W. Cytochrome c: Functions beyond respiration. Nat. Rev. Mol. Cell Biol. 2008, 9, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Morbidelli, L.; Donnini, S.; Ziche, M. Role of Nitric Oxide in the Modulation of Angiogenesis. Curr. Pharm. Des. 2023, 9, 521–530. [Google Scholar] [CrossRef]
- Dailey, K.M.; Allgood, J.E.; Johnson, P.R.; Ostlie, M.A.; Schaner, K.C.; Brooks, B.D.; Brooks, A.E. The next frontier of oncotherapy: Accomplishing clinical translation of oncolytic bacteria through genetic engineering. Future Microbiol 2021, 16, 341–368. [Google Scholar] [CrossRef]
- Pierce, K.M.; Miklavcic, W.R.; Cook, K.P.; Hennen, M.S.; Bayles, K.W.; Hollingsworth, M.A.; Brooks, A.E.; Pullan, J.E.; Dailey, K.M. The Evolution and Future of Targeted Cancer Therapy: From Nanoparticles, Oncolytic Viruses, and Oncolytic Bacteria to the Treatment of Solid Tumors. Nanomaterials 2021, 11, 3018. [Google Scholar] [CrossRef] [PubMed]
- Sára, M.; Sleytr, U.B. S-Layer Proteins. J. Bacteriol. 2000, 182, 859–868. [Google Scholar] [CrossRef] [Green Version]
- Rutherford, N.; Mourez, M. Surface display of proteins by Gram-negative bacterial autotransporters. Microb. Cell Factories 2006, 5, 22. [Google Scholar] [CrossRef] [Green Version]
- Genomic Analysis of the Protein Secretion Systems in Clostridium Acetobutylicum ATCC 824—ScienceDirect. Available online: https://www.sciencedirect.com/science/article/pii/S0167488905000777 (accessed on 25 January 2023).
- Dassa, E.; Bouige, P. The ABC of ABCs: A phylogenetic and functional classification of ABC systems in living organisms. Res. Microbiol. 2001, 152, 211–229. [Google Scholar] [CrossRef] [PubMed]
- Pivetti, C.D.; Yen, M.-R.; Miller, S.; Busch, W.; Tseng, Y.-H.; Booth, I.R.; Saier, J.M.H. Two Families of Mechanosensitive Channel Proteins|Microbiology and Molecular Biology Reviews. Available online: https://journals.asm.org/doi/full/10.1128/MMBR.67.1.66-85.2003 (accessed on 26 January 2023).
- Lange, C.; Aaby, P.; Behr, M.A.; Donald, P.R.; Kaufmann, S.H.E.; Netea, M.G.; Mandalakas, A.M. 100 years of Mycobacterium bovis bacille Calmette-Guérin. Lancet Infect. Dis. 2022, 22, e2–e12. [Google Scholar] [CrossRef] [PubMed]
- Kativhu, C.L.; Libraty, D.H. A Model to Explain How the Bacille Calmette Guérin (BCG) Vaccine Drives Interleukin-12 Production in Neonates. PLoS ONE 2016, 11, e0162148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkabani, M.; Greene, J.N.; Vincent, A.L.; Vanhook, S.; Sandin, R.L. Disseminated Mycobacterium Bovis after Intravesicular Bacillus Calmette-Guérin Treatments for Bladder Cancer. Cancer Control 2000, 7, 476–481. [Google Scholar] [CrossRef] [Green Version]
- Zlotta, A.R.; Van Vooren, J.P.; Denis, O.; Drowart, A.; Daffé, M.; Lefèvre, P.; Schandene, L.; De Cock, M.; De Bruyn, J.; Vandenbussche, P.; et al. What are the immunologically active components of bacille Calmette-Guérin in therapy of superficial bladder cancer? Int. J. Cancer 2000, 87, 844–852. [Google Scholar] [CrossRef]
- Klebsiella Pneumoniae (ID 815)—Genome—NCBI. Available online: https://www.ncbi.nlm.nih.gov/genome/?term=Klebsiella%20pneumoniae%5BOrganism%5D&cmd=DetailsSearch (accessed on 19 March 2023).
- Ashurst, J.V.; Dawson, A. Klebsiella Pneumonia. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Bazett, M.; Costa, A.M.; Bosiljcic, M.; Anderson, R.M.; Alexander, M.P.; Wong, S.W.Y.; Dhanji, S.; Chen, J.M.; Pankovich, J.; Lam, S.; et al. Harnessing innate lung anti-cancer effector functions with a novel bacterial-derived immunotherapy. Oncoimmunology 2017, 7, e1398875. [Google Scholar] [CrossRef]
- Wieland, C.W.; van Lieshout, M.H.P.; Hoogendijk, A.J.; van der Poll, T. Host defence during Klebsiella pneumonia relies on haematopoietic-expressed Toll-like receptors 4 and 2. Eur. Respir. J. 2011, 37, 848–857. [Google Scholar] [CrossRef] [Green Version]
- Josephs, S.F.; Ichim, T.E.; Prince, S.M.; Kesari, S.; Marincola, F.M.; Escobedo, A.R.; Jafri, A. Unleashing endogenous TNF-alpha as a cancer immunotherapeutic. J. Transl. Med. 2018, 16, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Elssen, C.H.M.J.; Vanderlocht, J.; Frings, P.W.H.; Senden-Gijsbers, B.L.M.G.; Schnijderberg, M.C.A.; van Gelder, M.; Meek, B.; Libon, C.; Ferlazzo, G.; Germeraad, W.T.V.; et al. Klebsiella pneumoniae-triggered DC recruit human NK cells in a CCR5-dependent manner leading to increased CCL19-responsiveness and activation of NK cells. Eur. J. Immunol. 2010, 40, 3138–3149. [Google Scholar] [CrossRef] [PubMed]
- Tugues, S.; Burkhard, S.H.; Ohs, I.; Vrohlings, M.; Nussbaum, K.; vom Berg, J.; Kulig, P.; Becher, B. New insights into IL-12-mediated tumor suppression. Cell Death Differ. 2015, 22, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Llobet, E.; March, C.; Giménez, P.; Bengoechea, J.A. Klebsiella pneumoniae OmpA Confers Resistance to Antimicrobial Peptides. Antimicrob. Agents Chemother. 2009, 53, 298–302. [Google Scholar] [CrossRef] [Green Version]
- Schroll, C.; Barken, K.B.; Krogfelt, K.A.; Struve, C. Role of type 1 and type 3 fimbriae in Klebsiella pneumoniae biofilm formation. BMC Microbiol. 2010, 10, 179. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Vuai, M.S.; Zhong, M. The role of bacteria in cancer therapy—Enemies in the past, but allies at present. Infect. Agents Cancer 2018, 13, 9. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Yang, Z.; Chen, Y.; Zhuang, W.; Niu, H.; Wu, J.; Ying, H. Clostridium acetobutylicum grows vegetatively in a biofilm rich in heteropolysaccharides and cytoplasmic proteins. Biotechnol. Biofuels 2018, 11, 315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunne, W.M. Bacterial Adhesion: Seen Any Good Biofilms Lately? Clin. Microbiol. Rev. 2002, 15, 155–166. [Google Scholar] [CrossRef] [Green Version]
- Mirzaei, R.; Sabokroo, N.; Ahmadyousefi, Y.; Motamedi, H.; Karampoor, S. Immunometabolism in biofilm infection: Lessons from cancer. Mol. Med. 2022, 28, 10. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hwang, I.; Lee, E.; Shin, S.J.; Lee, E.-J.; Rhee, J.H.; Yu, J.-W. Bacterial Outer Membrane Vesicle-Mediated Cytosolic Delivery of Flagellin Triggers Host NLRC4 Canonical Inflammasome Signaling. Front. Immunol. 2020, 11, 581165. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Lee, E.J.; Lee, J.H.; Jun, S.H.; Choi, C.W.; Kim, S.I.; Kang, S.S.; Hyun, S. Klebsiella pneumoniae secretes outer membrane vesicles that induce the innate immune response. FEMS Microbiol. Lett. 2012, 331, 17–24. [Google Scholar] [CrossRef]
- Hetz, C.; Bono, M.R.; Barros, L.F.; Lagos, R. Microcin E492, a channel-forming bacteriocin from Klebsiella pneumoniae, induces apoptosis in some human cell lines. Proc. Natl. Acad. Sci. USA 2002, 99, 2696–2701. [Google Scholar] [CrossRef]
- Darbandi, A.; Asadi, A.; Ari, M.M.; Ohadi, E.; Talebi, M.; Zadeh, M.H.; Emamie, A.D.; Ghanavati, R.; Kakanj, M. Bacteriocins: Properties and potential use as antimicrobials. J. Clin. Lab. Anal. 2022, 36, e24093. [Google Scholar] [CrossRef]
- Paczosa, M.K.; Mecsas, J. Klebsiella pneumoniae: Going on the Offense with a Strong Defense. Microbiol. Mol. Biol. Rev. 2016, 80, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Harimoto, T.; Hahn, J.; Chen, Y.-Y.; Im, J.; Zhang, J.; Hou, N.; Li, F.; Coker, C.; Gray, K.; Harr, N.; et al. A programmable encapsulation system improves delivery of therapeutic bacteria in mice. Nat. Biotechnol. 2022, 40, 1259–1269. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.-W.; Qiao, J.-Y.; Liu, X.-H.; Zhang, C.; Zhang, X.-Z. Customized materials-assisted microorganisms in tumor therapeutics. Chem. Soc. Rev. 2021, 50, 12576–12615. [Google Scholar] [CrossRef]
- Feldman, M.F.; Bridwell, A.E.M.; Scott, N.E.; Vinogradov, E.; McKee, S.R.; Chavez, S.M.; Twentyman, J.; Stallings, C.L.; Rosen, D.A.; Harding, C.M. A promising bioconjugate vaccine against hypervirulent Klebsiella pneumoniae. Proc. Natl. Acad. Sci. USA 2019, 116, 18655–18663. [Google Scholar] [CrossRef] [Green Version]
- Fang, C.-T.; Chuang, Y.-P.; Shun, C.-T.; Chang, S.-C.; Wang, J.-T. A Novel Virulence Gene in Klebsiella pneumoniae Strains Causing Primary Liver Abscess and Septic Metastatic Complications. J. Exp. Med. 2004, 199, 697–705. [Google Scholar] [CrossRef] [Green Version]
- Gröbner, S.; Lukowski, R.; Autenrieth, I.B.; Ruth, P. Lipopolysaccharide induces cell volume increase and migration of dendritic cells. Microbiol. Immunol. 2014, 58, 61–67. [Google Scholar] [CrossRef]
- Qu Biologics Inc. Open Label, Single Arm, Exploratory Study to Evaluate the Safety, Tolerability, Compliance and MOA, of QBKPN SSI in Subjects with 2 or More Second Primary Pre-invasive/Invasive Adenocarcinoma Following Surgical Resection of Stage I NSCLC; clinicaltrials.gov. 2016. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT02256852 (accessed on 17 July 2023).
- Greenberger, M.J.; Kunkel, S.L.; Strieter, R.M.; Lukacs, N.W.; Bramson, J.; Gauldie, J.; Graham, F.L.; Hitt, M.; Danforth, J.M.; Standiford, T.J. IL-12 gene therapy protects mice in lethal Klebsiella pneumonia. J. Immunol. 1996, 157, 3006–3012. [Google Scholar] [CrossRef]
- Vázquez-Boland, J.A.; Kuhn, M.; Berche, P.; Chakraborty, T.; Domínguez-Bernal, G.; Goebel, W.; González-Zorn, B.; Wehland, J.; Kreft, J. Listeria Pathogenesis and Molecular Virulence Determinants. Clin. Microbiol. Rev. 2001, 14, 584–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaillard, J.L.; Berche, P.; Frehel, C.; Gouin, E.; Cossart, P. Entry of L. monocytogenes into cells is mediated by internalin, a repeat protein reminiscent of surface antigens from gram-positive cocci. Cell 1991, 65, 1127–1141. [Google Scholar] [CrossRef]
- Domann, E.; Wehland, J.; Rohde, M.; Pistor, S.; Hartl, M.; Goebel, W.; Leimeister-Wächter, M.; Wuenscher, M.; Chakraborty, T. A novel bacterial virulence gene in Listeria monocytogenes required for host cell microfilament interaction with homology to the proline-rich region of vinculin. EMBO J. 1992, 11, 1981–1990. [Google Scholar] [CrossRef]
- Ireton, K.; Mortuza, R.; Gyanwali, G.C.; Gianfelice, A.; Hussain, M. Role of internalin proteins in the pathogenesis of Listeria monocytogenes. Mol. Microbiol. 2021, 116, 1407–1419. [Google Scholar] [CrossRef] [PubMed]
- Mengaud, J.; Ohayon, H.; Gounon, P.; Mege, R.-M.; Cossart, P. E-cadherin is the receptor for internalin, a surface protein required for entry of L. monocytogenes into epithelial cells. Cell 1996, 84, 923–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Naujokas, M.; Park, M.; Ireton, K. InIB-dependent internalization of Listeria is mediated by the Met receptor tyrosine kinase. Cell 2000, 103, 501–510. [Google Scholar] [CrossRef] [Green Version]
- Lecuit, M. Understanding how Listeria monocytogenes targets and crosses host barriers. Clin. Microbiol. Infect. 2005, 11, 430–436. [Google Scholar] [CrossRef] [Green Version]
- Mungunsukh, O.; Lee, Y.H.; Marquez, A.P.; Cecchi, F.; Bottaro, D.P.; Day, R.M. A tandem repeat of a fragment of Listeria monocytogenes internalin B protein induces cell survival and proliferation. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 299, L905–L914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birmingham, C.L.; Canadien, V.; Kaniuk, N.A.; Steinberg, B.E.; Higgins, D.E.; Brumell, J.H. Listeriolysin O allows Listeria monocytogenes replication in macrophage vacuoles. Nature 2008, 451, 350–354. [Google Scholar] [CrossRef]
- Glomski, I.J.; Decatur, A.L.; Portnoy, D.A. Listeria monocytogenes mutants that fail to compartmentalize listerolysin O activity are cytotoxic, avirulent, and unable to evade host extracellular defenses. Infect. Immun. 2003, 71, 6754–6765. [Google Scholar] [CrossRef] [Green Version]
- Quereda, J.J.; Morón-García, A.; Palacios-Gorba, C.; Dessaux, C.; García-Del Portillo, F.; Pucciarelli, M.G.; Ortega, A.D. Pathogenicity and virulence of Listeria monocytogenes: A trip from environmental to medical microbiology. Virulence 2021, 12, 2509–2545. [Google Scholar] [CrossRef]
- Repp, H.; Pamukci, Z.; Koschinski, A.; Domann, E.; Darji, A.; Birringer, J.; Brockmeier, D.; Chakraborty, T.; Dreyer, F. Listeriolysin of Listeria monocytogenes forms Ca2+-permeable pores leading to intracellular Ca2+ oscillations. Cell. Microbiol. 2002, 4, 483–491. [Google Scholar] [CrossRef]
- Föller, M.; Shumilina, E.; Lam, R.; Mohamed, W.; Kasinathan, R.; Huber, S.; Chakraborty, T.; Lang, F. Induction of suicidal erythrocyte death by listeriolysin from Listeria monocytogenes. Cell. Physiol. Biochem. 2007, 20, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Kohda, C.; Kawamura, I.; Baba, H.; Nomura, T.; Ito, Y.; Kimoto, T.; Watanabe, I.; Mitsuyama, M. Dissociated linkage of cytokine-inducing activity and cytotoxicity to different domains of listeriolysin O from Listeria monocytogenes. Infect. Immun. 2002, 70, 1334–1341. [Google Scholar] [CrossRef] [Green Version]
- Pamer, E.G.; Harty, J.T.; Bevan, M.J. Precise prediction of a dominant class I MHC-restricted epitope of Listeria monocytogenes. Nature 1991, 353, 852–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, F.; Smith, K.D.; Ozinsky, A.; Hawn, T.R.; Yi, E.C.; Goodlett, D.R.; Eng, J.K.; Akira, S.; Underhill, D.M.; Aderem, A. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 2001, 410, 1099–1103. [Google Scholar] [CrossRef] [PubMed]
- Way, S.S.; Thompson, L.J.; Lopes, J.E.; Hajjar, A.M.; Kollmann, T.R.; Freitag, N.E.; Wilson, C.B. Characterization of flagellin expression and its role in Listeria monocytogenes infection and immunity. Cell. Microbiol. 2004, 6, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Girardin, S.E.; Travassos, L.H.; Hervé, M.; Blanot, D.; Boneca, I.G.; Philpott, D.J.; Sansonetti, P.J.; Mengin-Lecreulx, D.; Bouma, B.; Kroon-Batenburg, L.M.J.; et al. Peptidoglycan molecular requirements allowing detection by Nod1 and Nod2. J. Biol. Chem. 2003, 278, 41702–41708. [Google Scholar] [CrossRef] [Green Version]
- Safley, S.A.; Jensen, P.E.; Reay, P.A.; Ziegler, H.K. Mechanisms of T cell epitope immunodominance analyzed in murine listeriosis. J. Immunol. 1995, 155, 4355–4366. [Google Scholar] [CrossRef]
- Oladejo, M.; Paterson, Y.; Wood, L.M. Clinical Experience and Recent Advances in the Development of Listeria-Based Tumor Immunotherapies. Front. Immunol. 2021, 12, 642316. [Google Scholar] [CrossRef] [PubMed]
- Jensen, E.R.; Glass, A.A.; Clark, W.R.; Wing, E.J.; Miller, J.F.; Gregory, S.H. Fas (CD95)-dependent cell-mediated immunity to Listeria monocytogenes. Infect. Immun. 1998, 66, 4143–4150. [Google Scholar] [CrossRef] [PubMed]
- White, D.W.; Harty, J.T. Perforin-deficient CD8+ T cells provide immunity to Listeria monocytogenes by a mechanism that is independent of CD95 and IFN-gamma but requires TNF-alpha. J. Immunol. 1998, 160, 898–905. [Google Scholar] [CrossRef]
- Kägi, D.; Ledermann, B.; Bürki, K.; Hengartner, H.; Zinkernagel, R.M. CD8+ T cell-mediated protection against an intracellular bacterium by perforin-dependent cytotoxicity. Eur. J. Immunol. 1994, 24, 3068–3072. [Google Scholar] [CrossRef]
- Barbuddhe, S.; Chakraborty, T. Biotechnological applications of Listeria’s sophisticated infection strategies. Microb. Biotechnol. 2008, 1, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Brunt, L.M.; Portnoy, D.A.; Unanue, E.R. Presentation of Listeria monocytogenes to CD8+ T cells requires secretion of hemolysin and intracellular bacterial growth. J. Immunol. 1990, 145, 3540–3546. [Google Scholar] [CrossRef]
- Stark, F.C.; Sad, S.; Krishnan, L. Intracellular bacterial vectors that induce CD8+ T cells with similar cytolytic abilities but disparate memory phenotypes provide contrasting tumor protection. Cancer Res. 2009, 69, 4327–4334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orme, I.M. Active and memory immunity to Listeria monocytogenes infection in mice is mediated by phenotypically distinct T-cell populations. Immunology 1989, 68, 93–95. [Google Scholar] [PubMed]
- Makino, M.; Kawai, M.; Kawamura, I.; Fujita, M.; Gejo, F.; Mitsuyama, M. Involvement of Reactive Oxygen Intermediate in the Enhanced Expression of Virulence-Associated Genes of Listeria monocytogenes inside Activated Macrophages. Microbiol. Immunol. 2005, 49, 805–811. [Google Scholar] [CrossRef]
- Kim, S.H.; Castro, F.; Paterson, Y.; Gravekamp, C. High efficacy of a Listeria-based vaccine against metastatic breast cancer reveals a dual mode of action. Cancer Res. 2009, 69, 5860–5866. [Google Scholar] [CrossRef] [Green Version]
- Mycobacterium (ID 13563)—Genome—NCBI. Available online: https://www.ncbi.nlm.nih.gov/genome/13563 (accessed on 19 March 2023).
- Harimoto, T.; Danino, T. Engineering bacteria for cancer therapy. Emerg. Top. Life Sci. 2019, 3, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Kresowik, T.P.; Griffith, T.S.; Shariat, S.F.; Lee, R.; Lowrance, W.T.; Bochner, B.H.; A Karam, J.; Raman, J.D.; I Karakiewicz, P.; Sun, M.; et al. Bacillus Calmette–Guerin immunotherapy for urothelial carcinoma of the bladder. Immunotherapy 2009, 1, 281–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, R.S.; Targoni, O.S.; Krieg, A.M.; Lehmann, P.V.; Harding, C.V. CpG Oligodeoxynucleotides Act as Adjuvants that Switch on T Helper 1 (Th1) Immunity. J. Exp. Med. 1997, 186, 1623–1631. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, S.; Egeter, O.; Hausmann, S.; Lipford, G.B.; Röcken, M.; Wagner, H.; Heeg, K. Cutting Edge: CpG Oligodeoxynucleotides Trigger Protective and Curative Th1 Responses in Lethal Murine Leishmaniasis. J. Immunol. 1998, 160, 3627–3630. [Google Scholar] [CrossRef] [PubMed]
- Häcker, G.; Redecke, V.; Häcker, H. Activation of the immune system by bacterial CpG-DNA. Immunology 2002, 105, 245–251. [Google Scholar] [CrossRef]
- Beatty, W.L.; Russell, D.G. Identification of Mycobacterial Surface Proteins Released into Subcellular Compartments of Infected Macrophages. Infect. Immun. 2000, 68, 6997–7002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Schorey, J.S.; Bong-Mastek, M.; Ritchey, J.; Brown, E.J.; Ratliff, T.L. Role of a bacillus calmette-guérin fibronectin attachment protein in BCG-induced antitumor activity. Int. J. Cancer 2000, 86, 83–88. [Google Scholar] [CrossRef]
- Middleton, A.M.; Chadwick, M.V.; Nicholson, A.G.; Dewar, A.; Groger, R.K.; Brown, E.J.; Wilson, R. The role of Mycobacterium avium complex fibronectin attachment protein in adherence to the human respiratory mucosa. Mol. Microbiol. 2000, 38, 381–391. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.P.; Hielscher, A. Fibronectin: How Its Aberrant Expression in Tumors May Improve Therapeutic Targeting. J. Cancer 2017, 8, 674–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efthymiou, G.; Saint, A.; Ruff, M.; Rekad, Z.; Ciais, D.; Van Obberghen-Schilling, E. Shaping Up the Tumor Microenvironment With Cellular Fibronectin. Front. Oncol. 2020, 10, 641. [Google Scholar] [CrossRef] [PubMed]
- Ates, L.S. New insights into the mycobacterial PE and PPE proteins provide a framework for future research. Mol. Microbiol. 2020, 113, 4–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampson, S.L. Mycobacterial PE/PPE Proteins at the Host-Pathogen Interface. Clin. Dev. Immunol. 2011, 2011, 497203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.-Y.; Yang, S.-B.; Choi, Y.-M.; Oh, S.-J.; Kim, B.-J.; Kook, Y.-H.; Kim, B.-J. Heat-killed Mycobacterium paragordonae therapy exerts an anti-cancer immune response via enhanced immune cell mediated oncolytic activity in xenograft mice model. Cancer Lett. 2020, 472, 142–150. [Google Scholar] [CrossRef]
- Podder, S.; Rakshit, S.; Ponnusamy, M.; Nandi, D. Efficacy of Bacteria in Cancer Immunotherapy: Special Emphasis on the Potential of Mycobacterial Species. Clin. Cancer Drugs 2016, 3, 100–108. [Google Scholar] [CrossRef]
- Rakshit, S.; Ponnusamy, M.; Papanna, S.; Saha, B.; Ahmed, A.; Nandi, D. Immunotherapeutic efficacy of Mycobacterium indicus pranii in eliciting anti-tumor T cell responses: Critical roles of IFNγ. Int. J. Cancer 2012, 130, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Gocher, A.M.; Workman, C.J.; Vignali, D.A.A. Interferon-γ: Teammate or opponent in the tumour microenvironment? Nat. Rev. Immunol. 2022, 22, 158–172. [Google Scholar] [CrossRef]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-γ in tumor progression and regression: A review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef]
- Eisenring, M.; vom Berg, J.; Kristiansen, G.; Saller, E.; Becher, B. IL-12 initiates tumor rejection via lymphoid tissue–inducer cells bearing the natural cytotoxicity receptor NKp46. Nat. Immunol. 2010, 11, 1030–1038. [Google Scholar] [CrossRef]
- Gerber, S.A.; Moran, J.P.; Frelinger, J.G.; Frelinger, J.A.; Fenton, B.M.; Lord, E.M. Mechanism of IL-12 mediated alterations in tumour blood vessel morphology: Analysis using whole-tissue mounts. Br. J. Cancer 2003, 88, 1453–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. Br. J. Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef]
- Kumar, P.; Tyagi, R.; Das, G.; Bhaskar, S. Mycobacterium indicus pranii and Mycobacterium bovis BCG lead to differential macrophage activation in Toll-like receptor-dependent manner. Immunology 2014, 143, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Lee, S.-Y.; Seo, H.; Kim, B.-J. Recombinant Mycobacterium smegmatis delivering a fusion protein of human macrophage migration inhibitory factor (MIF) and IL-7 exerts an anticancer effect by inducing an immune response against MIF in a tumor-bearing mouse model. J. Immunother. Cancer 2021, 9, e003180. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.S.; E de Bock, C.; Molloy, T.J.; Sadeqzadeh, E.; Geng, X.Y.; Hersey, P.; Zhang, X.D.; Thorne, R.F. Macrophage Migration Inhibitory Factor Engages PI3K/Akt Signalling and Is a Prognostic Factor in Metastatic Melanoma|BMC Cancer|Full Text. Available online: https://bmccancer.biomedcentral.com/articles/10.1186/1471-2407-14-630 (accessed on 22 December 2022).
- Cook, G.M.; Berney, M.; Gebhard, S.; Heinemann, M.; Cox, R.A.; Danilchanka, O.; Niederweis, M. Physiology of Mycobacteria. Adv. Microb. Physiol. 2009, 55, 81–319. [Google Scholar] [CrossRef] [Green Version]
- Sousa, S.; Borges, V.; Joao, I.; Gomes, J.P.; Jordao, L. Nontuberculous Mycobacteria Persistence in a Cell Model Mimicking Alveolar Macrophages. Microorganisms 2019, 7, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M.M.; Odell, J.A. Nontuberculous mycobacterial pulmonary infections. J. Thorac. Dis. 2014, 6, 210–220. [Google Scholar] [CrossRef]
- Zhang, J.; Jiang, R.; Takayama, H.; Tanaka, Y. Survival of Virulent Mycobacterium tuberculosis Involves Preventing Apoptosis Induced by Bcl-2 Upregulation and Release Resulting from Necrosis in J774 Macrophages. Microbiol. Immunol. 2005, 49, 845–852. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Zhou, S.; Gravekamp, C.; Bermudes, D.; Liu, K. Tumour-targeting bacteria engineered to fight cancer. Nat. Rev. Cancer 2018, 18, 727–743. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.J.; Tait, S.W.G. Targeting BCL-2 regulated apoptosis in cancer. Open Biol. 2018, 8, 180002. [Google Scholar] [CrossRef] [Green Version]
- Noguera-Ortega, E.; Guallar-Garrido, S.; Julián, E. Mycobacteria-Based Vaccines as Immunotherapy for Non-urological Cancers. Cancers 2020, 12, 1802. [Google Scholar] [CrossRef] [PubMed]
- De Leon, J.; Jiang, G.; Ma, Y.; Rubin, E.; Fortune, S.; Sun, J. Mycobacterium tuberculosis ESAT-6 Exhibits a Unique Membrane-interacting Activity That Is Not Found in Its Ortholog from Non-pathogenic Mycobacterium smegmatis. J. Biol. Chem. 2012, 287, 44184–44191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Wang, J.; Zhao, F.; Yu, F.; Chen, D.; Cai, K.; Yang, C.; Chen, J.; Dou, J. Antitumor efficacy of viable tumor vaccine modified by heterogenetic ESAT-6 antigen and cytokine IL-21 in melanomatous mouse. Immunol. Res. 2012, 52, 240–249. [Google Scholar] [CrossRef]
- Ushigusa, T.; Koyama, Y.; Ito, T.; Watanabe, K.; Chambers, J.K.; Hasegawa, A.; Uchida, K.; Kanegi, R.; Hatoya, S.; Inaba, T.; et al. Innate immunity mediated by dendritic cells/macrophages plays a central role in the early period in tumor treatment using gene of Mycobacterium tuberculosis antigen. J. Vet. Med. Sci. 2018, 80, 190–196. [Google Scholar] [CrossRef] [Green Version]
- Murata, T.; Arakawa, M.; Sugiya, Y.; Inazu, Y.; Hattori, Z.; Suzuki, Y.; Minakami, H.; Nakahara, M.; Okazaki, H. Oncolytic effect of Proteus mirabilis upon tumor bearing animal. Life Sci. 1965, 4, 1055–1067. [Google Scholar] [CrossRef]
- Pearson, M.M.; Sebaihia, M.; Churcher, C.; Quail, M.A.; Seshasayee, A.S.; Luscombe, N.M.; Abdellah, Z.; Arrosmith, C.; Atkin, B.; Chillingworth, T.; et al. Complete Genome Sequence of Uropathogenic Proteus mirabilis, a Master of both Adherence and Motility. J. Bacteriol. 2008, 190, 4027–4037. [Google Scholar] [CrossRef] [Green Version]
- Scavone, P.; Iribarnegaray, V.; Caetano, A.L.; Schlapp, G.; Härtel, S.; Zunino, P. Fimbriae have distinguishable roles in Proteus mirabilis biofilm formation. Pathog. Dis. 2016, 74, ftw033. [Google Scholar] [CrossRef] [Green Version]
- Bahrani, F.K.; Johnson, D.E.; Robbins, D.; Mobley, H.L. Proteus mirabilis flagella and MR/P fimbriae: Isolation, purification, N-terminal analysis, and serum antibody response following experimental urinary tract infection. Infect. Immun. 1991, 59, 3574–3580. [Google Scholar] [CrossRef]
- Zhang, H.; Diao, H.; Jia, L.; Yuan, Y.; Thamm, D.H.; Wang, H.; Jin, Y.; Pei, S.; Zhou, B.; Yu, F.; et al. Proteus mirabilis inhibits cancer growth and pulmonary metastasis in a mouse breast cancer model. PLoS ONE 2017, 12, e0188960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arakawa, M.; Sugiura, K.; Reilly, H.C.; Stock, C.C. Oncolytic effect of Proteus mirabilis upon tumor-bearing animals. II. Effect on transplantable mouse and rat tumors. Gan 1968, 59, 117–122. [Google Scholar]
- Braun, V.; Focareta, T. Pore-forming bacterial protein hemolysins (cytolysins). Crit. Rev. Microbiol. 1991, 18, 115–158. [Google Scholar] [CrossRef] [PubMed]
- Williams, F.D.; Anderson, D.M.; Hoffman, P.S.; Schwarzhoff, R.H.; Leonard, S. Evidence against the involvement of chemotaxis in swarming of Proteus mirabilis. J. Bacteriol. 1976, 127, 237–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, Y.-L.; Chien, H.-F.; Huang, K.-T.; Lin, W.-Y.; Liaw, S.-J. cAMP receptor protein regulates mouse colonization, motility, fimbria-mediated adhesion, and stress tolerance in uropathogenic Proteus mirabilis. Sci. Rep. 2017, 7, 7282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaffer, J.N.; Norsworthy, A.N.; Sun, T.T.; Pearson, M.M. Proteus Mirabilis Fimbriae- and Urease-Dependent Clusters Assemble in an Extracellular Niche to Initiate Bladder Stone Formation. Proc. Natl. Acad. Sci. USA 2016, 113, 4494–4499. [Google Scholar] [CrossRef] [PubMed]
- Jansen, A.M.; Lockatell, V.; Johnson, D.E.; Mobley, H.L.T. Mannose-Resistant Proteus-Like Fimbriae Are Produced by Most Proteus mirabilis Strains Infecting the Urinary Tract, Dictate the In Vivo Localization of Bacteria, and Contribute to Biofilm Formation. Infect. Immun. 2004, 72, 7294–7305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.K.; Harrison, B.A.; Latta, R.; Altman, E. The binding of Proteus mirabilis nonagglutinating fimbriae to ganglio-series asialoglycolipids and lactosyl ceramide. Can. J. Microbiol. 2000, 46, 961–966. [Google Scholar] [CrossRef]
- Massad, G.; Lockatell, C.V.; Johnson, D.E.; Mobley, H.L. Proteus mirabilis fimbriae: Construction of an isogenic pmfA mutant and analysis of virulence in a CBA mouse model of ascending urinary tract infection. Infect. Immun. 1994, 62, 536–542. [Google Scholar] [CrossRef] [Green Version]
- Zunino, P.; Sosa, V.; Allen, A.G.; Preston, A.; Schlapp, G.; Maskell, D.J. Proteus mirabilis fimbriae (PMF) are important for both bladder and kidney colonization in mice. Microbiology 2003, 149, 3231–3237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massad, G.; Bahrani, F.K.; Mobley, H.L. Proteus Mirabilis Fimbriae: Identification, Isolation, and Characterization of a New Ambient-Temperature Fimbria. Infect. Immun. 1994, 62, 1989–1994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zunino, P.; Geymonat, L.; Allen, A.G.; Legnani-Fajardo, C.; Maskell, D.J. Virulence of a Proteus mirabilis ATF isogenic mutant is not impaired in a mouse model of ascending urinary tract infection. FEMS Immunol. Med. Microbiol. 2000, 29, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Armbruster, C.E.; Mobley, H.L.T.; Pearson, M.M. Pathogenesis of Proteus mirabilis Infection. EcoSal. Plus 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Jones, B.D.; Lockatell, C.V.; Johnson, D.E.; Warren, J.W.; Mobley, H.L. Construction of a urease-negative mutant of Proteus mirabilis: Analysis of virulence in a mouse model of ascending urinary tract infection. Infect. Immun. 1990, 58, 1120–1123. [Google Scholar] [CrossRef]
- Himpsl, S.D.; Pearson, M.M.; Arewång, C.J.; Nusca, T.D.; Sherman, D.H.; Mobley, H.L.T. Proteobactin and a yersiniabactin-related siderophore mediate iron acquisition in Proteus mirabilis. Mol. Microbiol. 2010, 78, 138–157. [Google Scholar] [CrossRef] [Green Version]
- Gaisser, S.; Hughes, C. A Locus Coding for Putative Non-Ribosomal Peptide/Polyketide Synthase Functions Is Mutated in a Swarming-Defective Proteus Mirabilis Strain. Mol. Gen. Genet. MGG 1997, 250, 415–427. [Google Scholar] [CrossRef]
- Drechsel, H.; Thieken, A.; Reissbrodt, R.; Jung, G.; Winkelmann, G. Alpha-keto acids are novel siderophores in the genera Proteus, Providencia, and Morganella and are produced by amino acid deaminases. J. Bacteriol. 1993, 175, 2727–2733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaca, W.; Rózalski, A. Characterization of cell-bound and cell-free hemolytic activity of Proteus strains. Eur. J. Epidemiol. 1991, 7, 159–165. [Google Scholar] [CrossRef]
- Swihart, K.G.; Welch, R.A. Cytotoxic activity of the Proteus hemolysin HpmA. Infect. Immun. 1990, 58, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Welch, R.A. Identification of two different hemolysin determinants in uropathogenic Proteus isolates. Infect. Immun. 1987, 55, 2183–2190. [Google Scholar] [CrossRef] [Green Version]
- Alamuri, P.; Mobley, H.L.T. A novel autotransporter of uropathogenic Proteus mirabilis is both a cytotoxin and an agglutinin. Mol. Microbiol. 2008, 68, 997–1017. [Google Scholar] [CrossRef] [Green Version]
- Alamuri, P.; Eaton, K.A.; Himpsl, S.D.; Smith, S.N.; Mobley, H.L.T. Vaccination with Proteus Toxic Agglutinin, a Hemolysin-Independent Cytotoxin In Vivo, Protects against Proteus mirabilis Urinary Tract Infection. Infect. Immun. 2009, 77, 632–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senior, B.W.; Albrechtsen, M.; Kerr, M.A. A survey of IgA protease production among clinical isolates of Proteeae. J. Med. Microbiol. 1988, 25, 27–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wassif, C.; Cheek, D.; Belas, R. Molecular analysis of a metalloprotease from Proteus mirabilis. J. Bacteriol. 1995, 177, 5790–5798. [Google Scholar] [CrossRef] [Green Version]
- GlpC Gene is Responsible for Biofilm Formation and Defense against Phagocytes and Imparts Tolerance to pH and Organic Solvents in Proteus Vulgaris. Available online: https://www.geneticsmr.com/articles/5025 (accessed on 21 January 2023).
- O’Toole, G.; Kaplan, H.B.; Kolter, R. Biofilm formation as microbial development. Annu. Rev. Microbiol. 2000, 54, 49–79. [Google Scholar] [CrossRef] [PubMed]
- Armbruster, C.E.; Smith, S.N.; Johnson, A.O.; DeOrnellas, V.; Eaton, K.A.; Yep, A.; Mody, L.; Wu, W.; Mobley, H.L.T. The Pathogenic Potential of Proteus mirabilis Is Enhanced by Other Uropathogens during Polymicrobial Urinary Tract Infection. Infect. Immun. 2017, 85, e00808–e00816. [Google Scholar] [CrossRef] [Green Version]
- Seo, S.-U.; Kamada, N.; Muñoz-Planillo, R.; Kim, Y.-G.; Kim, D.; Koizumi, Y.; Hasegawa, M.; Himpsl, S.D.; Browne, H.P.; Lawley, T.D.; et al. Distinct commensals induce interleukin-1β via NLRP3 inflammasome in inflammatory monocytes to promote intestinal inflammation in response to injury. Immunity 2015, 42, 744–755. [Google Scholar] [CrossRef] [Green Version]
- Steiner, T.S. How flagellin and toll-like receptor 5 contribute to enteric infection. Infect. Immun. 2007, 75, 545–552. [Google Scholar] [CrossRef] [Green Version]
- Umpiérrez, A.; Scavone, P.; Romanin, D.; Marqués, J.M.; Chabalgoity, J.A.; Rumbo, M.; Zunino, P. Innate immune responses to Proteus mirabilis flagellin in the urinary tract. Microbes Infect. 2013, 15, 688–696. [Google Scholar] [CrossRef] [PubMed]
- Okumura, R.; Kurakawa, T.; Nakano, T.; Kayama, H.; Kinoshita, M.; Motooka, D.; Gotoh, K.; Kimura, T.; Kamiyama, N.; Kusu, T.; et al. Lypd8 promotes the segregation of flagellated microbiota and colonic epithelia. Nature 2016, 532, 117–121. [Google Scholar] [CrossRef]
- Xue, Y.; Li, Q.; Park, C.G.; Klena, J.D.; Anisimov, A.P.; Sun, Z.; Wei, X.; Chen, T. Proteus mirabilis Targets Atherosclerosis Plaques in Human Coronary Arteries via DC-SIGN (CD209). Front. Immunol. 2021, 11, 579010. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hoedt, E.C.; Liu, Q.; Berendsen, E.; Teh, J.J.; Hamilton, A.; O’ Brien, A.W.; Ching, J.Y.L.; Wei, H.; Yang, K.; et al. Elucidation of Proteus mirabilis as a Key Bacterium in Crohn’s Disease Inflammation. Gastroenterology 2021, 160, 317–330.e11. [Google Scholar] [CrossRef] [PubMed]
- Hypoxia-Inducible Factors and Cancer—PMC. Available online: https://www-ncbi-nlm-nih-gov.proxy.rvu.edu/pmc/articles/PMC5607450/ (accessed on 27 January 2023).
- Walzer, T.; Bléry, M.; Chaix, J.; Fuseri, N.; Chasson, L.; Robbins, S.H.; Jaeger, S.; André, P.; Gauthier, L.; Daniel, L.; et al. Identification, activation, and selective in vivo ablation of mouse NK cells via NKp46. Proc. Natl. Acad. Sci. USA 2007, 104, 3384–3389. [Google Scholar] [CrossRef] [PubMed]
- Gerosa, F.; Baldani-Guerra, B.; Nisii, C.; Marchesini, V.; Carra, G.; Trinchieri, G. Reciprocal Activating Interaction between Natural Killer Cells and Dendritic Cells. J. Exp. Med. 2002, 195, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, N.C.; Lozier, A.; Flament, C.; Ricciardi-Castagnoli, P.; Bellet, D.; Suter, M.; Perricaudet, M.; Tursz, T.; Maraskovsky, E.; Zitvogel, L. Dendritic cells directly trigger NK cell functions: Cross-talk relevant in innate anti-tumor immune responses in vivo. Nat. Med. 1999, 5, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Cann, S.A.H.; Netten, J.P.V.; Netten, C. van Dr William Coley and tumour regression: A place in history or in the future. Postgrad. Med. J. 2003, 79, 672–680. [Google Scholar] [CrossRef]
- McCarthy, E.F. The Toxins of William B. Coley and the Treatment of Bone and Soft-Tissue Sarcomas. Iowa Orthop. J. 2006, 26, 154–158. [Google Scholar]
- Karbach, J.; Neumann, A.; Brand, K.; Wahle, C.; Siegel, E.; Maeurer, M.; Ritter, E.; Tsuji, T.; Gnjatic, S.; Old, L.J.; et al. Phase I Clinical Trial of Mixed Bacterial Vaccine (Coley’s Toxins) in Patients with NY-ESO-1 Expressing Cancers: Immunological Effects and Clinical Activity. Clin. Cancer Res. 2012, 18, 5449–5459. [Google Scholar] [CrossRef] [Green Version]
- Coley, W.B. The Treatment of Inoperable Sarcoma by Bacterial Toxins (the Mixed Toxins of the Streptococcus erysipelas and the Bacillus prodigiosus). Proc. R. Soc. Med. 1910, 3, 1–48. [Google Scholar] [CrossRef] [PubMed]
- Maletzki, C.; Klier, U.; Obst, W.; Kreikemeyer, B.; Linnebacher, M. Reevaluating the Concept of Treating Experimental Tumors with a Mixed Bacterial Vaccine: Coley’s Toxin. J. Immunol. Res. 2012, 2012, e230625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herra, C.; Falkiner, F.R. Serratia Marcescens—Infectious Disease and Antimicrobial Agents. Available online: http://www.antimicrobe.org/b26.asp (accessed on 9 February 2023).
- O’Rear, J.; Alberti, L.; Harshey, R.M. Mutations that impair swarming motility in Serratia marcescens 274 include but are not limited to those affecting chemotaxis or flagellar function. J. Bacteriol. 1992, 174, 6125–6137. [Google Scholar] [CrossRef] [Green Version]
- Serratia Marcescens (ID 1112)—Genome—NCBI. Available online: https://www.ncbi.nlm.nih.gov/genome/?term=serratia+marcescens%5BOrganism%5D (accessed on 19 March 2023).
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escobar-Díaz, E.; López-Martín, E.M.; del Cerro, M.H.; Puig-Kroger, A.; Soto-Cerrato, V.; Montaner, B.; Giralt, E.; García-Marco, J.A.; Pérez-Tomás, R.; Garcia-Pardo, A. AT514, a cyclic depsipeptide from Serratia marcescens, induces apoptosis of B-chronic lymphocytic leukemia cells: Interference with the Akt/NF-κB survival pathway. Leukemia 2005, 19, 572–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Du, M.; Chen, Z.; Yuan, Z. Recent Advances in Bacteria-Based Cancer Treatment. Cancers 2022, 14, 4945. [Google Scholar] [CrossRef] [PubMed]
- Soto-Cerrato, V.; Montaner, B.; Martinell, M.; Vilaseca, M.; Giralt, E.; Pérez-Tomás, R. Cell cycle arrest and proapoptotic effects of the anticancer cyclodepsipeptide serratamolide (AT514) are independent of p53 status in breast cancer cells. Biochem. Pharmacol. 2005, 71, 32–41. [Google Scholar] [CrossRef]
- Montaner, B.; Navarro, S.; Piqué, M.; Vilaseca, M.; Martinell, M.; Giralt, E.; Gil, J.; Pérez-Tomás, R. Prodigiosin from the supernatant of Serratia marcescens induces apoptosis in haematopoietic cancer cell lines. Br. J. Pharmacol. 2000, 131, 585–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darshan, N.; Manonmani, H.K. Prodigiosin and its potential applications. J. Food Sci. Technol. 2015, 52, 5393–5407. [Google Scholar] [CrossRef]
- Ishii, K.; Adachi, T.; Imamura, K.; Takano, S.; Usui, K.; Suzuki, K.; Hamamoto, H.; Watanabe, T.; Sekimizu, K. Serratia marcescens Induces Apoptotic Cell Death in Host Immune Cells via a Lipopolysaccharide- and Flagella-dependent Mechanism. J. Biol. Chem. 2012, 287, 36582–36592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandinova, A.; Lee, S.W. The p53 Pathway as a Target in Cancer Therapeutics: Obstacles and Promise. Sci. Transl. Med. 2011, 3, 64rv1. [Google Scholar] [CrossRef] [Green Version]
- Effects of the proapoptotic drug prodigiosin on cell cycle-related proteins in Jurkat T cells. Histol. Histopathol. 2003, 18, 379–385. [CrossRef]
- Wang, Z.; Li, B.; Zhou, L.; Yu, S.; Su, Z.; Song, J.; Sun, Q.; Sha, O.; Wang, X.; Jiang, W.; et al. Prodigiosin inhibits Wnt/β-catenin signaling and exerts anticancer activity in breast cancer cells. Proc. Natl. Acad. Sci. USA 2016, 113, 13150–13155. [Google Scholar] [CrossRef]
- Streptococcus Pyogenes (ID 175)—Genome—NCBI. Available online: https://www.ncbi.nlm.nih.gov/genome/?term=streptococcus+pyogenes%5BOrganism%5D (accessed on 21 March 2023).
- Sela, S.; Marouni, M.J.; Perry, R.; Barzilai, A. Effect of lipoteichoic acid on the uptake of Streptococcus pyogenes by HEp-2 cells. FEMS Microbiol. Lett. 2000, 193, 187–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, P.A.; Juncosa, B. Group A Streptococcal Adherence. In Streptococcus Pyogenes: Basic Biology to Clinical Manifestations; Ferretti, J.J., Stevens, D.L., Fischetti, V.A., Eds.; University of Oklahoma Health Sciences Center: Oklahoma City, OK, USA, 2016. [Google Scholar]
- Walker, M.J.; Barnett, T.C.; McArthur, J.D.; Cole, J.N.; Gillen, C.M.; Henningham, A.; Sriprakash, K.S.; Sanderson-Smith, M.L.; Nizet, V. Disease Manifestations and Pathogenic Mechanisms of Group A Streptococcus. Clin. Microbiol. Rev. 2014, 27, 264–301. [Google Scholar] [CrossRef] [Green Version]
- Metzgar, D.; Zampolli, A. The M Protein of Group A Streptococcus is a Key Virulence Factor and a Clinically Relevant Strain Identification Marker. Virulence 2011, 2, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Golińska, E.; van der Linden, M.; Więcek, G.; Mikołajczyk, D.; Machul, A.; Samet, A.; Piórkowska, A.; Dorycka, M.; Heczko, P.B.; Strus, M. Virulence factors of Streptococcus pyogenes strains from women in peri-labor with invasive infections. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the role of CD4+ T cells in cancer immunotherapy—New insights into old paradigms. Cancer Gene Ther. 2021, 28, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Peuhkurinen, K.J.; Risteli, L.; Melkko, J.T.; Linnaluoto, M.; Jounela, A.; Risteli, J. Thrombolytic therapy with streptokinase stimulates collagen breakdown. Circulation 1991, 83, 1969–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, W.; Lameda, V.; Olivar, L.C.; Navarro, C.; Fuenmayor, J.; Pérez, A.; Mindiola, A.; Rojas, M.; Martínez, M.S.; Velasco, M.; et al. Bacteria in cancer therapy: Beyond immunostimulation. J. Cancer Metastasis Treat. 2018, 4, 4. [Google Scholar] [CrossRef]
- Bobek, V.; Pinterova, D.; Kolostova, K.; Boubelik, M.; Douglas, J.; Teyssler, P.; Pavlasek, J.; Kovarik, J. Streptokinase increases the sensitivity of colon cancer cells to chemotherapy by gemcitabine and cis-platine in vitro. Cancer Lett. 2006, 237, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Babashamsi, M.; Razavian, M.H.; Nejadmoghaddam, M.R. Production and Purification of Streptokinase by Protected Affinity Chromatography. Avicenna J. Med. Biotechnol. 2009, 1, 47–51. [Google Scholar] [PubMed]
- Khanna, A.; Khanna, M.; Aggarwal, A. Serratia Marcescens- A Rare Opportunistic Nosocomial Pathogen and Measures to Limit its Spread in Hospitalized Patients. J. Clin. Diagn. Res. 2013, 7, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Yang, M.; Shi, L.; Yao, Y.; Jiang, Q.; Li, X.; Tang, L.-H.; Zheng, B.-J.; Yuen, K.-Y.; Smith, D.K.; et al. Explicit hypoxia targeting with tumor suppression by creating an “obligate” anaerobic Salmonella Typhimurium strain. Sci. Rep. 2012, 2, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, M.Q.; Ellem, K.A.O.; Dunn, P.; West, M.J.; Bai, C.X.; Vogelstein, B. Facultative or obligate anaerobic bacteria have the potential for multimodality therapy of solid tumours. Eur. J. Cancer 2007, 43, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Mowday, A.M.; Guise, C.P.; Ackerley, D.F.; Minton, N.P.; Lambin, P.; Dubois, L.J.; Theys, J.; Smaill, J.B.; Patterson, A.V. Advancing Clostridia to Clinical Trial: Past Lessons and Recent Progress. Cancers 2016, 8, 63. [Google Scholar] [CrossRef] [Green Version]
- Torrey, J.; Kahn, M.C. The Treatment of Flexner-Jobling Rat Carcinomas with Bacterial Proteolytic Ferments. J. Cancer Res. 1927, 11, 334–376. [Google Scholar]
- Dang, L.H.; Bettegowda, C.; Huso, D.L.; Kinzler, K.W.; Vogelstein, B. Combination bacteriolytic therapy for the treatment of experimental tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 15155–15160. [Google Scholar] [CrossRef] [PubMed]
- Hara-Kudo, Y.; Ogura, A.; Noguchi, Y.; Kumagai, S. Characteristics of toxicity and haemorrhagic toxin produced by Clostridium sporogenes in various animals and cultured cells. J. Med. Microbiol. 1997, 46, 270–275. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-S.; Toth, J.; Kasap, M. Nitrogen-fixation genes and nitrogenase activity in Clostridium acetobutylicum and Clostridium beijerinckii. J. Ind. Microbiol. Biotechnol. 2001, 27, 281–286. [Google Scholar] [CrossRef]
- Wei, M.Q.; Mengesha, A.; Good, D.; Anné, J. Bacterial targeted tumour therapy-dawn of a new era. Cancer Lett. 2008, 259, 16–27. [Google Scholar] [CrossRef]
- Janku, F.; Zhang, H.H.; Pezeshki, A.M.; Goel, S.; Murthy, R.; Wang-Gillam, A.; Shepard, D.R.; Helgason, T.; Masters, T.; Hong, D.S.; et al. Intratumoral Injection of Clostridium novyi-NT Spores in Patients with Treatment-Refractory Advanced Solid Tumors. Clin. Cancer Res. 2020, 27, 96–106. [Google Scholar] [CrossRef]
- Nolling, J.; Breton, G.; Omelchenko, M.V.; Makarova, K.S.; Zeng, Q.; Gibson, R.; Lee, H.M.; Dubois, J.; Qiu, D.; Hitti, J.; et al. Genome Sequence and Comparative Analysis of the Solvent-Producing Bacterium Clostridium Acetobutylicum. Available online: https://journals.asm.org/doi/epub/10.1128/JB.183.16.4823-4838.2001 (accessed on 25 January 2023).
- Cornillot, E.; Nair, R.V.; Papoutsakis, E.T.; Soucaille, P. The genes for butanol and acetone formation in Clostridium acetobutylicum ATCC 824 reside on a large plasmid whose loss leads to degeneration of the strain. J. Bacteriol. 1997, 179, 5442–5447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fierobe, H.P.; Mingardon, F.; Chanal, A. Engineering Cellulase Activity into Clostridium acetobutylicum. Methods Enzymol. 2012, 510, 301–316. [Google Scholar]
- Cornillot, E.; Croux, C.; Soucaille, P. Physical and genetic map of the Clostridium acetobutylicum ATCC 824 chromosome. J. Bacteriol. 1997, 179, 7426–7434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lütke-Eversloh, T.; Bahl, H. Metabolic engineering of Clostridium acetobutylicum: Recent advances to improve butanol production. Curr. Opin. Biotechnol. 2011, 22, 634–647. [Google Scholar] [CrossRef]
- Jones, D.T.; Woods, D.R. Acetone-butanol fermentation revisited. Microbiol. Rev. 1986, 50, 484–524. [Google Scholar] [CrossRef] [PubMed]
- Giménez, J.A.; Sugiyama, H. Comparison of toxins of Clostridium butyricum and Clostridium botulinum type E. Infect. Immun. 1988, 56, 926–929. [Google Scholar] [CrossRef] [Green Version]
- Summary of Clostridium Acetobutylicum ATCC 824, Version 26.5. Available online: https://biocyc.org/organism-summary?object=GCF_000008765 (accessed on 26 January 2023).
- Bayles, K.W. Are the molecular strategies that control apoptosis conserved in bacteria? Trends Microbiol. 2003, 11, 306–311. [Google Scholar] [CrossRef]
- Pallen, M.J. The ESAT-6/WXG100 Superfamily—And a New GRAM-Positive Secretion System? Trends Microbiol. 2002, 10, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, A.L.; Nagai, S.; Houen, G.; Andersen, P.; Andersen, A.B. Purification and characterization of a low-molecular-mass T-cell antigen secreted by Mycobacterium tuberculosis. Infect. Immun. 1995, 63, 1710–1717. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.A. Clostridial Toxins as Therapeutic Agents: Benefits of Nature’s Most Toxic Proteins | Annual Review of Microbiology. Annu. Rev. Microbiol. 1999, 53, 551–575. [Google Scholar] [CrossRef]
- Ghelardi, E.; Celandroni, F.; Salvetti, S.; Beecher, D.J.; Gominet, M.; Lereclus, D.; Wong, A.C.L.; Senesi, S. Requirement of flhA for Swarming Differentiation, Flagellin Export, and Secretion of Virulence-Associated Proteins in Bacillus thuringiensis | Journal of Bacteriology. J. Bacteriol. 2002, 184, 6424–6433. [Google Scholar] [CrossRef] [Green Version]
- Lyristis, M.; Boynton, Z.L.; Petersen, D.; Kan, Z.; Bennett, G.N.; Rudolph, F.B. Cloning, Sequencing, and Characterization of the Gene Encoding Flagellin, flaC, and the Post-translational Modification of Flagellin, FlaC, from Clostridium acetobutylicum ATCC824. Anaerobe 2000, 6, 69–79. [Google Scholar] [CrossRef]
- Boedtkjer, E.; Pedersen, S.F. The Acidic Tumor Microenvironment as a Driver of Cancer. Annu. Rev. Physiol. 2020, 82, 103–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biebl, H. Acetobutylicum. In Encyclopedia of Food Microbiology; Robinson, R.K., Ed.; Elsevier: Oxford, UK, 1999; pp. 445–451. ISBN 978-0-12-227070-3. [Google Scholar]
- Gutierrez, N.A.; Maddox, I.S. Role of Chemotaxis in Solvent Production by Clostridium acetobutylicum. Appl. Environ. Microbiol. 1987, 53, 1924–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dürre, P.; Hollergschwandner, C. Initiation of endospore formation in Clostridium acetobutylicum. Anaerobe 2004, 10, 69–74. [Google Scholar] [CrossRef]
- Brüggemann, H.; Bäumer, S.; Fricke, W.F.; Wiezer, A.; Liesegang, H.; Decker, I.; Herzberg, C.; Martínez-Arias, R.; Merkl, R.; Henne, A.; et al. The Genome Sequence of Clostridium Tetani, the Causative Agent of Tetanus Disease. Proc. Natl. Acad. Sci. USA 2003, 100, 1316–1321. [Google Scholar] [CrossRef] [PubMed]
- Gill, D.M. Bacterial toxins: A table of lethal amounts. Microbiol. Rev. 1982, 46, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, N.A.; Maddox, I.S. The effect of some culture maintenance and inoculum development techniques on solvent production by Clostridium acetobutylicum. Can. J. Microbiol. 1987, 33, 82–84. [Google Scholar] [CrossRef]
- Theys, J.; Landuyt, A.W.; Nuyts, S.; Van Mellaert, L.; Lambin, P.; Anné, J. Clostridium as a tumor-specific delivery system of therapeutic proteins. Cancer Detect. Prev. 2001, 25, 548–557. [Google Scholar]
- Theys, J.; Nuyts, S.; Landuyt, W.; Van Mellaert, L.; Dillen, C.; Böhringer, M.; Dürre, P.; Lambin, P.; Anné, J. Stable Escherichia coli-Clostridium acetobutylicum shuttle vector for secretion of murine tumor necrosis factor alpha. Appl. Environ. Microbiol. 1999, 65, 4295–4300. [Google Scholar] [CrossRef] [PubMed]
- Barbé, S.; Van Mellaert, L.; Theys, J.; Geukens, N.; Lammertyn, E.; Lambin, P.; Anné, J. Secretory production of biologically active rat interleukin-2 by Clostridium acetobutylicum DSM792 as a tool for anti-tumor treatment. FEMS Microbiol. Lett. 2005, 246, 67–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, R.C.; Plummer, H.C.; Siebenmann, C.O.; Chapman, M.G. Effect of Histolyticus Infection and Toxin on Transplantable Mouse Tumors. Proc. Soc. Exp. Biol. Med. 1947, 66, 461–467. [Google Scholar] [CrossRef]
- (Proteome:UP000308489) in UniProtKB Search (2563)|UniProt. Available online: https://www.uniprot.org/uniprotkb?query= (accessed on 26 January 2023).
- Stevens, D.L.; Bisno, A.L.; Chambers, H.F.; Everett, E.D.; Dellinger, P.; Goldstein, E.J.C.; Gorbach, S.L.; Hirschmann, J.V.; Kaplan, E.L.; Montoya, J.G.; et al. Practice Guidelines for the Diagnosis and Management of Skin and Soft-Tissue Infections. Clin. Infect. Dis. 2005, 41, 1373–1406. [Google Scholar] [CrossRef] [PubMed]
- Hatheway, C.L. Toxigenic clostridia. Clin. Microbiol. Rev. 1990, 3, 66–98. [Google Scholar] [CrossRef]
- Brazier, J.S.; Gal, M.; Hall, V.; Morris, T.E. Outbreak of Clostridium histolyticum infections in injecting drug users in England and Scotland. Euro Surveill. 2004, 9, 15–16. [Google Scholar] [CrossRef]
- Takagi, A.; Kawata, T.; Yamamoto, S.; Kubo, T.; Okita, S. Electron Microscopic Studies on Ultrathin Sections of Spores of Clostridium Tetani and Clostridium Histolyticum, with Special Reference to Sporulation and Spore Germination Process. Jpn. J. Microbiol. 1960, 4, 137–155. [Google Scholar] [CrossRef]
- Yoshihara, K.; Matsushita, O.; Minami, J.; Okabe, A. Cloning and Nucleotide Sequence Analysis of the colH Gene from Clostridium Histolyticum Encoding a Collagenase and a Gelatinase. J. Bacteriol. 1994, 176, 6489–6496. [Google Scholar] [CrossRef] [Green Version]
- Moore, R.J.; Lacey, J.A. Genomics of the Pathogenic Clostridia. Microbiol. Spectr. 2019, 7, 10–1128. [Google Scholar] [CrossRef]
- Diehm, Y.F.; Marstaller, K.; Seckler, A.-M.; Berger, M.R.; Zepp, M.; Gaida, M.M.; Thomé, J.; Kotsougiani-Fischer, D.; Kneser, U.; Fischer, S. The collagenase of the bacterium Clostridium histolyticum does not favor metastasis of breast cancer. Breast Cancer 2022, 29, 599–609. [Google Scholar] [CrossRef]
- Thomas, A.; Bayat, A. The emerging role of Clostridium histolyticum collagenase in the treatment of Dupuytren disease. Ther. Clin. Risk Manag. 2010, 6, 557–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jóźwiak, J.; Komar, A.; Jankowska, E.; Martirosian, G. Determination of the cytotoxic effect of Clostridium histolyticum culture supernatant on HeLa cells in the presence of protease inhibitors. FEMS Immunol. Med. Microbiol. 2005, 45, 137–142. [Google Scholar] [CrossRef] [Green Version]
- Dargatz, H.; Diefenthal, T.; Witte, V.; Reipen, G.; von Wettstein, D. The heterodimeric protease clostripain from Clostridium histolyticum is encoded by a single gene. Mol. Gen. Genet. 1993, 240, 140–145. [Google Scholar] [CrossRef]
- Plomp, M.; McCaffery, J.M.; Cheong, I.; Huang, X.; Bettegowda, C.; Kinzler, K.W.; Zhou, S.; Vogelstein, B.; Malkin, A.J. Spore Coat Architecture of Clostridium novyi NT Spores. J. Bacteriol. 2007, 189, 6457–6468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dailey, K.M.; Jacobson, R.I.; Johnson, P.R.; Woolery, T.J.; Kim, J.; Jansen, R.J.; Mallik, S.; Brooks, A.E. The genome and transcriptomes of the anti-tumor agent Clostridium novyi-NT. Nat. Biotechnol. 2007, 24, 1573–1580. [Google Scholar] [CrossRef]
- Agrawal, N.; Bettegowda, C.; Cheong, I.; Geschwind, J.-F.; Drake, C.G.; Hipkiss, E.L.; Tatsumi, M.; Dang, L.H.; Diaz, L.A.; Pomper, M.; et al. Bacteriolytic therapy can generate a potent immune response against experimental tumors. Proc. Natl. Acad. Sci. USA 2004, 101, 15172–15177. [Google Scholar] [CrossRef] [PubMed]
- Dailey, K.M.; Jacobson, R.I.; Johnson, P.E.; Woolery, T.J.; Kim, J.; Mallik, S.; Brooks, A.E. Methods and Techniques to Facilitate the Development of Clostridium novyi-NT as an Effective, Therapeutic Oncolytic Bacteria. Front. Microbiol. 2021, 12, 624618. [Google Scholar] [CrossRef]
- Sebaihia, M.; Peck, M.W.; Minton, N.P.; Thomson, N.R.; Holden, M.T.G.; Mitchell, W.J.; Carter, A.T.; Bentley, S.D.; Mason, D.R.; Crossman, L.; et al. Genome sequence of a proteolytic (Group I) Clostridium botulinum strain Hall A and comparative analysis of the clostridial genomes. Genome Res. 2007, 17, 1082–1092. [Google Scholar] [CrossRef] [Green Version]
- Sebaihia, M.; Wren, B.W.; Mullany, P.; Fairweather, N.F.; Minton, N.; Stabler, R.; Thomson, N.R.; Roberts, A.P.; Cerdeño-Tárraga, A.M.; Wang, H.; et al. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat. Genet. 2006, 38, 779–786. [Google Scholar] [CrossRef]
- Shimizu, T.; Ohtani, K.; Hirakawa, H.; Ohshima, K.; Yamashita, A.; Shiba, T.; Ogasawara, N.; Hattori, M.; Kuhara, S.; Hayashi, H. Complete genome sequence of Clostridium perfringens, an anaerobic flesh-eater. Proc. Natl. Acad. Sci. USA 2002, 99, 996–1001. [Google Scholar] [CrossRef]
- Bettegowda, C.; Dang, L.H.; Abrams, R.; Huso, D.L.; Dillehay, L.; Cheong, I.; Agrawal, N.; Borzillary, S.; McCaffery, J.M.; Watson, E.L.; et al. Overcoming the hypoxic barrier to radiation therapy with anaerobic bacteria. Proc. Natl. Acad. Sci. USA 2003, 100, 15083–15088. [Google Scholar] [CrossRef]
- Cheong, I.; Huang, X.; Bettegowda, C.; Diaz, L.A.; Kinzler, K.W.; Zhou, S.; Vogelstein, B. A bacterial protein enhances the release and efficacy of liposomal cancer drugs. Science 2006, 314, 1308–1311. [Google Scholar] [CrossRef] [Green Version]
- Staedtke, V.; Bai, R.-Y.; Sun, W.; Huang, J.; Kibler, K.K.; Tyler, B.M.; Gallia, G.L.; Kinzler, K.; Vogelstein, B.; Zhou, S.; et al. Clostridium novyi-NT can cause regression of orthotopically implanted glioblastomas in rats. Oncotarget 2015, 6, 5536–5546. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.A. Shedding Light on Immunotherapy for Cancer. N. Engl. J. Med. 2004, 350, 1461–1463. [Google Scholar] [CrossRef] [Green Version]
- Maeda, H.; Nakagawa, K.; Murayama, K.; Goto, M.; Watanabe, K.; Takeuchi, M.; Yamagata, Y. Cloning a neutral protease of Clostridium histolyticum, determining its substrate specificity, and designing a specific substrate. Appl. Microbiol. Biotechnol. 2015, 99, 10489–10499. [Google Scholar] [CrossRef]
- Dang, L.H.; Bettegowda, C.; Agrawal, N.; Cheong, I.; Huso, D.; Frost, P.; Loganzo, F.; Greenberger, L.; Barkoczy, J.; Pettit, G.R.; et al. Targeting Vascular and Avascular Compartments of Tumors with C. novyi-NT and Anti-microtubule Agents. Cancer Biol. Ther. 2004, 3, 326–337. [Google Scholar] [CrossRef] [Green Version]
- Ji, J.; Park, W.R.; Cho, S.; Yang, Y.; Li, W.; Harris, K.; Huang, X.; Gu, S.; Kim, D.-H.; Zhang, Z.; et al. Iron-Oxide Nanocluster Labeling of Clostridium novyi-NT Spores for MR Imaging–Monitored Locoregional Delivery to Liver Tumors in Rat and Rabbit Models. J. Vasc. Interv. Radiol. 2019, 30, 1106–1115.e1. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Zhang, Z.; Khazaie, K.; Saha, S.; Lewandowski, R.J.; Zhang, G.; Larson, A.C. MRI-Monitored Intra-Tumoral Injection of Iron-Oxide Labeled Clostridium novyi-NT Anaerobes in Pancreatic Carcinoma Mouse Model. PLoS ONE 2014, 9, e116204. [Google Scholar] [CrossRef]
- Weickert, M.J.; Chambliss, G.H.; Sugiyama, H. Production of toxin by Clostridium botulinum type A strains cured by plasmids. Appl. Environ. Microbiol. 1986, 51, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Mose, J.; Mose, G. Oncolysis by clostridia. I. Activity of clostridium butyricum (m-55) and other nonpathogenic clostridia against the ehrlich carcinoma. Cancer Res. 1964, 24, 212–216. [Google Scholar]
- Dietmargericke, K.E. Oncolysis by Clostridia V. Transplanted Tumors of the Hamster. Cancer Res. 1964, 24, 239–243. [Google Scholar]
- Effect of Acidic pH on the Ability of Clostridium Sporogenes MD1 to Take up and Retain Intracellular Potassium|FEMS Microbiology Letters|Oxford Academic. Available online: https://academic.oup.com/femsle/article/267/1/46/644876 (accessed on 22 February 2023).
- Betz, J.V.; Anderson, K.E. Isolation and Characterization of Bacteriophages Active on Clostridium Sporogenes. J. Bacteriol. 1964, 87, 408–415. [Google Scholar] [CrossRef]
- Konopelski, P.; Mogilnicka, I. Biological Effects of Indole-3-Propionic Acid, a Gut Microbiota-Derived Metabolite, and Its Precursor Tryptophan in Mammals’ Health and Disease. Int. J. Mol. Sci. 2022, 23, 1222. [Google Scholar] [CrossRef]
- Guo, C.-J.; Allen, B.M.; Hiam, K.J.; Dodd, D.; Van Treuren, W.; Higginbottom, S.; Nagashima, K.; Fischer, C.R.; Sonnenburg, J.L.; Spitzer, M.H.; et al. Depletion of microbiome-derived molecules in the host using Clostridium genetics. Science 2019, 366, eaav1282. [Google Scholar] [CrossRef]
- Bendheim, P.E.; Poeggeler, B.; Neria, E.; Ziv, V.; Pappolla, M.A.; Chain, D.G. Development of indole-3-propionic acid (OXIGONTM) for alzheimer’s disease. J. Mol. Neurosci. 2002, 19, 213–217. [Google Scholar] [CrossRef]
- Karbownik, M.; Reiter, R.J.; Garcia, J.J.; Cabrera, J.; Burkhardt, S.; Osuna, C.; Lewiński, A. Indole-3-propionic acid, a melatonin-related molecule, protects hepatic microsomal membranes from iron-induced oxidative damage: Relevance to cancer reduction. J. Cell. Biochem. 2001, 81, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Bhave, M.S.; Hassanbhai, A.M.; Anand, P.; Luo, K.Q.; Teoh, S.H. Effect of Heat-Inactivated Clostridium sporogenes and Its Conditioned Media on 3-Dimensional Colorectal Cancer Cell Models. Sci. Rep. 2015, 5, 15681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heppner, F.; Möse, J.R. The liquefaction (oncolysis) of malignant gliomas by a non pathogenic Clostridium. Acta Neurochir. 1978, 42, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Theys, J.; Landuyt, W.; Nuyts, S.; Van Mellaert, L.; Bosmans, E.; Rijnders, A.; Van Den Bogaert, W.; van Oosterom, A.; Anné, J.; Lambin, P. Improvement of Clostridium tumour targeting vectors evaluated in rat rhabdomyosarcomas. FEMS Immunol. Med. Microbiol. 2001, 30, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Nuyts, S.; Van Mellaert, L.; Theys, J.; Landuyt, W.; Lambin, P.; Anné, J. Clostridium spores for tumor-specific drug delivery. Anticancer. Drugs 2002, 13, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Minton, N.P. Clostridia in cancer therapy. Nat. Rev. Microbiol. 2003, 1, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Malmgren, R.A.; Flanigan, C.C. Localization of the Vegetative Form of Clostridium tetani in Mouse Tumors Following Intravenous Spore Administration. Cancer Res. 1955, 15, 473–478. [Google Scholar]
- Garrigues, L.; Do, T.D.; Bideaux, C.; Guillouet, S.E.; Meynial-Salles, I. Insights into Clostridium tetani: From genome to bioreactors. Biotechnol. Adv. 2022, 54, 107781. [Google Scholar] [CrossRef]
- Schiavo, G.G.; Benfenati, F.; Poulain, B.; Rossetto, O.; de Laureto, P.P.; DasGupta, B.R.; Montecucco, C. Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature 1992, 359, 832–835. [Google Scholar] [CrossRef] [PubMed]
- Variable Factors Influencing the Production of Tetanus Toxin. Available online: https://journals.asm.org/doi/epdf/10.1128/jb.67.3.271-277.1954?src=getftr (accessed on 20 March 2023).
- Bowden, G.H.W. Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996; ISBN 978-0-9631172-1-2. [Google Scholar]
- Parry, C.M.; Hien, T.T.; Dougan, G.; White, N.J.; Farrar, J.J. Typhoid fever. N. Engl. J. Med. 2002, 347, 1770–1782. [Google Scholar] [CrossRef] [Green Version]
- Crump, J.A.; Sjölund-Karlsson, M.; Gordon, M.A.; Parry, C.M. Epidemiology, Clinical Presentation, Laboratory Diagnosis, Antimicrobial Resistance, and Antimicrobial Management of Invasive Salmonella Infections. Clin. Microbiol. Rev. 2015, 28, 901–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, S.; Dougan, G. The genome of Salmonella enterica serovar Typhi. Clin. Infect. Dis. 2007, 45 (Suppl. S1), S29–S33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, Z.; Feng, Z.-C.; Li, C.; Yang, X.; Ma, M.-T.; Rong, P.-F. Salmonella-Mediated Cancer Therapy: An Innovative Therapeutic Strategy. J. Cancer 2019, 10, 4765–4776. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Chen, Y.; Liu, X.; Min, J.-J.; Tan, W.; Zheng, J.H. Targeted cancer immunotherapy with genetically engineered oncolytic Salmonella typhimurium. Cancer Lett. 2020, 469, 102–110. [Google Scholar] [CrossRef]
- Badie, F.; Ghandali, M.; Tabatabaei, S.A.; Safari, M.; Khorshidi, A.; Shayestehpour, M.; Mahjoubin-Tehran, M.; Morshedi, K.; Jalili, A.; Tajiknia, V.; et al. Use of Salmonella Bacteria in Cancer Therapy: Direct, Drug Delivery and Combination Approaches. Front. Oncol. 2021, 11, 624759. [Google Scholar] [CrossRef]
- Kim, B.-J.; Forbes, N.S. Single-cell analysis demonstrates how nutrient deprivation creates apoptotic and quiescent cell populations in tumor cylindroids. Biotechnol. Bioeng. 2008, 101, 797–810. [Google Scholar] [CrossRef] [Green Version]
- Kasinskas, R.W.; Forbes, N.S. Salmonella typhimurium lacking ribose chemoreceptors localize in tumor quiescence and induce apoptosis. Cancer Res. 2007, 67, 3201–3209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.-Z.; Kazmierczak, R.A.; Eisenstark, A. Strains, Mechanism, and Perspective: Salmonella-Based Cancer Therapy. Int. J. Microbiol. 2016, 2016, 5678702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toso, J.F.; Gill, V.J.; Hwu, P.; Marincola, F.M.; Restifo, N.P.; Schwartzentruber, D.J.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Stock, F.; et al. Phase I study of the intravenous administration of attenuated Salmonella typhimurium to patients with metastatic melanoma. J. Clin. Oncol. 2002, 20, 142–152. [Google Scholar] [CrossRef]
- Uchugonova, A.; Zhao, M.; Zhang, Y.; Weinigel, M.; König, K.; Hoffman, R.M. Cancer-cell killing by engineered Salmonella imaged by multiphoton tomography in live mice. Anticancer. Res. 2012, 32, 4331–4337. [Google Scholar] [PubMed]
- Saccheri, F.; Pozzi, C.; Avogadri, F.; Barozzi, S.; Faretta, M.; Fusi, P.; Rescigno, M. Bacteria-induced gap junctions in tumors favor antigen cross-presentation and antitumor immunity. Sci. Transl. Med. 2010, 2, 44ra57. [Google Scholar] [CrossRef]
- Oviedo-Orta, E.; Evans, W.H. Gap junctions and connexin-mediated communication in the immune system. Biochim. Biophys. Acta 2004, 1662, 102–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Yang, M.; Li, X.-M.; Jiang, P.; Baranov, E.; Li, S.; Xu, M.; Penman, S.; Hoffman, R.M. Tumor-targeting bacterial therapy with amino acid auxotrophs of GFP-expressing Salmonella typhimurium. Proc. Natl. Acad. Sci. USA 2005, 102, 755–760. [Google Scholar] [CrossRef]
- Murakami, T.; Igarashi, K.; Kawaguchi, K.; Kiyuna, T.; Zhang, Y.; Zhao, M.; Hiroshima, Y.; Nelson, S.D.; Dry, S.M.; Li, Y.; et al. Tumor-targeting Salmonella typhimurium A1-R regresses an osteosarcoma in a patient-derived xenograft model resistant to a molecular-targeting drug. Oncotarget 2017, 8, 8035–8042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, J.; Wang, S.; Wang, Y.; Huang, Y.; Chen, X. Isolation and Molecular Identification of Auxotrophic Mutants to Develop a Genetic Manipulation System for the Haloarchaeon Natrinema sp. J7-2. Archaea 2015, 2015, 483194. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Yang, M.; Ma, H.; Li, X.; Tan, X.; Li, S.; Yang, Z.; Hoffman, R.M. Targeted therapy with a Salmonella typhimurium leucine-arginine auxotroph cures orthotopic human breast tumors in nude mice. Cancer Res. 2006, 66, 7647–7652. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Geller, J.; Ma, H.; Yang, M.; Penman, S.; Hoffman, R.M. Monotherapy with a tumor-targeting mutant of Salmonella typhimurium cures orthotopic metastatic mouse models of human prostate cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 10170–10174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tome, Y.; Suetsugu, A.; Zhang, L.; Zhang, N.; Hoffman, R.M.; Zhao, M. Determination of the optimal route of administration of Salmonella typhimurium A1-R to target breast cancer in nude mice. Anticancer. Res. 2012, 32, 2501–2508. [Google Scholar] [PubMed]
- Zhang, Y.; Miwa, S.; Zhang, N.; Hoffman, R.M.; Zhao, M. Tumor-targeting Salmonella typhimurium A1-R arrests growth of breast-cancer brain metastasis. Oncotarget 2015, 6, 2615–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Zhang, L.; Hoffman, R.M.; Zhao, M. Vessel destruction by tumor-targeting Salmonella typhimurium A1-R is enhanced by high tumor vascularity. Cell Cycle 2010, 9, 4518–4524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagakura, C.; Hayashi, K.; Zhao, M.; Yamauchi, K.; Yamamoto, N.; Tsuchiya, H.; Tomita, K.; Bouvet, M.; Hoffman, R.M. Efficacy of a genetically-modified Salmonella typhimurium in an orthotopic human pancreatic cancer in nude mice. Anticancer Res. 2009, 29, 1873–1878. [Google Scholar] [PubMed]
- Yam, C.; Zhao, M.; Hayashi, K.; Ma, H.; Kishimoto, H.; McElroy, M.; Bouvet, M.; Hoffman, R.M. Monotherapy with a tumor-targeting mutant of S. typhimurium inhibits liver metastasis in a mouse model of pancreatic cancer. J. Surg. Res. 2010, 164, 248–255. [Google Scholar] [CrossRef] [Green Version]
- Hiroshima, Y.; Zhao, M.; Zhang, Y.; Maawy, A.; Hassanein, M.K.; Uehara, F.; Miwa, S.; Yano, S.; Momiyama, M.; Suetsugu, A.; et al. Comparison of efficacy of Salmonella typhimurium A1-R and chemotherapy on stem-like and non-stem human pancreatic cancer cells. Cell Cycle 2013, 12, 2774–2780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, Y.; Miwa, S.; Zhang, Y.; Hiroshima, Y.; Yano, S.; Uehara, F.; Yamamoto, M.; Toneri, M.; Bouvet, M.; Matsubara, H.; et al. Efficacy of tumor-targeting Salmonella typhimurium A1-R on nude mouse models of metastatic and disseminated human ovarian cancer. J. Cell Biochem. 2014, 115, 1996–2003. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Miwa, S.; Zhang, Y.; Zhao, M.; Yano, S.; Uehara, F.; Yamamoto, M.; Hiroshima, Y.; Toneri, M.; Bouvet, M.; et al. Intraperitoneal administration of tumor-targeting Salmonella typhimurium A1-R inhibits disseminated human ovarian cancer and extends survival in nude mice. Oncotarget 2015, 6, 11369–11377. [Google Scholar] [CrossRef] [Green Version]
- Yano, S.; Zhang, Y.; Zhao, M.; Hiroshima, Y.; Miwa, S.; Uehara, F.; Kishimoto, H.; Tazawa, H.; Bouvet, M.; Fujiwara, T.; et al. Tumor-targeting Salmonella typhimurium A1-R decoys quiescent cancer cells to cycle as visualized by FUCCI imaging and become sensitive to chemotherapy. Cell Cycle 2014, 13, 3958–3963. [Google Scholar] [CrossRef] [Green Version]
- Hiroshima, Y.; Zhang, Y.; Zhao, M.; Zhang, N.; Murakami, T.; Maawy, A.; Mii, S.; Uehara, F.; Yamamoto, M.; Miwa, S.; et al. Tumor-Targeting Salmonella typhimurium A1-R in Combination with Trastuzumab Eradicates HER-2-Positive Cervical Cancer Cells in Patient-Derived Mouse Models. PLoS ONE 2015, 10, e0120358. [Google Scholar] [CrossRef]
- Murakami, T.; DeLong, J.; Eilber, F.C.; Zhao, M.; Zhang, Y.; Zhang, N.; Singh, A.; Russell, T.; Deng, S.; Reynoso, J.; et al. Tumor-targeting Salmonella typhimurium A1-R in combination with doxorubicin eradicate soft tissue sarcoma in a patient-derived orthotopic xenograft (PDOX) model. Oncotarget 2016, 7, 12783–12790. [Google Scholar] [CrossRef] [Green Version]
- Hiroshima, Y.; Zhao, M.; Zhang, Y.; Zhang, N.; Maawy, A.; Murakami, T.; Mii, S.; Uehara, F.; Yamamoto, M.; Miwa, S.; et al. Tumor-Targeting Salmonella typhimurium A1-R Arrests a Chemo-Resistant Patient Soft-Tissue Sarcoma in Nude Mice. PLoS ONE 2015, 10, e0134324. [Google Scholar] [CrossRef]
- Kiyuna, T.; Murakami, T.; Tome, Y.; Kawaguchi, K.; Igarashi, K.; Zhang, Y.; Zhao, M.; Li, Y.; Bouvet, M.; Kanaya, F.; et al. High efficacy of tumor-targeting Salmonella typhimurium A1-R on a doxorubicin- and dactolisib-resistant follicular dendritic-cell sarcoma in a patient-derived orthotopic xenograft PDOX nude mouse model. Oncotarget 2016, 7, 33046–33054. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Zhao, M.; Hiroshima, Y.; Zhang, Y.; Shurell, E.; Eilber, F.C.; Bouvet, M.; Noda, M.; Hoffman, R.M. Efficacy of Tumor-Targeting Salmonella A1-R on a Melanoma Patient-Derived Orthotopic Xenograft (PDOX) Nude-Mouse Model. PLoS ONE 2016, 11, e0160882. [Google Scholar] [CrossRef] [Green Version]
- Momiyama, M.; Zhao, M.; Kimura, H.; Tran, B.; Chishima, T.; Bouvet, M.; Endo, I.; Hoffman, R.M. Inhibition and eradication of human glioma with tumor-targeting Salmonella typhimurium in an orthotopic nude-mouse model. Cell Cycle 2012, 11, 628–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-E.; Phan, T.X.; Nguyen, V.H.; Dinh-Vu, H.-V.; Zheng, J.H.; Yun, M.; Park, S.-G.; Hong, Y.; Choy, H.E.; Szardenings, M.; et al. Salmonella typhimurium Suppresses Tumor Growth via the Pro-Inflammatory Cytokine Interleukin-1β. Theranostics 2015, 5, 1328–1342. [Google Scholar] [CrossRef] [Green Version]
- Na, H.S.; Kim, H.J.; Lee, H.-C.; Hong, Y.; Rhee, J.H.; Choy, H.E. Immune response induced by Salmonella typhimurium defective in ppGpp synthesis. Vaccine 2006, 24, 2027–2034. [Google Scholar] [CrossRef]
- Jiang, S.-N.; Park, S.-H.; Lee, H.J.; Zheng, J.H.; Kim, H.-S.; Bom, H.-S.; Hong, Y.; Szardenings, M.; Shin, M.G.; Kim, S.-C.; et al. Engineering of bacteria for the visualization of targeted delivery of a cytolytic anticancer agent. Mol. Ther. 2013, 21, 1985–1995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phan, T.X.; Nguyen, V.H.; Duong, M.T.-Q.; Hong, Y.; Choy, H.E.; Min, J.-J. Activation of inflammasome by attenuated Salmonella typhimurium in bacteria-mediated cancer therapy. Microbiol. Immunol. 2015, 59, 664–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, W.; Duong, M.T.-Q.; Zuo, C.; Qin, Y.; Zhang, Y.; Guo, Y.; Hong, Y.; Zheng, J.H.; Min, J.-J. Targeting of pancreatic cancer cells and stromal cells using engineered oncolytic Salmonella typhimurium. Mol. Ther. 2022, 30, 662–671. [Google Scholar] [CrossRef]
- Nguyen, V.H.; Kim, H.-S.; Ha, J.-M.; Hong, Y.; Choy, H.E.; Min, J.-J. Genetically engineered Salmonella typhimurium as an imageable therapeutic probe for cancer. Cancer Res. 2010, 70, 18–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.H.; Nguyen, V.H.; Jiang, S.-N.; Park, S.-H.; Tan, W.; Hong, S.H.; Shin, M.G.; Chung, I.-J.; Hong, Y.; Bom, H.-S.; et al. Two-step enhanced cancer immunotherapy with engineered Salmonella typhimurium secreting heterologous flagellin. Sci. Transl. Med. 2017, 9, eaak9537. [Google Scholar] [CrossRef] [PubMed]
- Kaimala, S.; Mohamed, Y.A.; Nader, N.; Issac, J.; Elkord, E.; Chouaib, S.; Fernandez-Cabezudo, M.J.; Al-Ramadi, B.K. Salmonella-mediated tumor regression involves targeting of tumor myeloid suppressor cells causing a shift to M1-like phenotype and reduction in suppressive capacity. Cancer Immunol. Immunother. 2014, 63, 587–599. [Google Scholar] [CrossRef]
- Saltzman, D.A.; Katsanis, E.; Heise, C.P.; Hasz, D.E.; Vigdorovich, V.; Kelly, S.M.; Curtiss, R.; Leonard, A.S.; Anderson, P.M. Antitumor mechanisms of attenuated Salmonella typhimurium containing the gene for human interleukin-2: A novel antitumor agent? J. Pediatr. Surg. 1997, 32, 301–306. [Google Scholar] [CrossRef]
- Bartee, M.Y.; Dunlap, K.M.; Bartee, E. Tumor-Localized Secretion of Soluble PD1 Enhances Oncolytic Virotherapy. Cancer Res. 2017, 77, 2952–2963. [Google Scholar] [CrossRef] [Green Version]
- Leschner, S.; Westphal, K.; Dietrich, N.; Viegas, N.; Jablonska, J.; Lyszkiewicz, M.; Lienenklaus, S.; Falk, W.; Gekara, N.; Loessner, H.; et al. Tumor invasion of Salmonella enterica serovar Typhimurium is accompanied by strong hemorrhage promoted by TNF-alpha. PLoS ONE 2009, 4, e6692. [Google Scholar] [CrossRef] [Green Version]
- Tu, D.-G.; Chang, W.-W.; Lin, S.-T.; Kuo, C.-Y.; Tsao, Y.-T.; Lee, C.-H. Salmonella inhibits tumor angiogenesis by downregulation of vascular endothelial growth factor. Oncotarget 2016, 7, 37513–37523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocijancic, D.; Felgner, S.; Schauer, T.; Frahm, M.; Heise, U.; Zimmermann, K.; Erhardt, M.; Weiss, S. Local application of bacteria improves safety of Salmonella-mediated tumor therapy and retains advantages of systemic infection. Oncotarget 2017, 8, 49988–50001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godinez, I.; Raffatellu, M.; Chu, H.; Paixão, T.A.; Haneda, T.; Santos, R.L.; Bevins, C.L.; Tsolis, R.M.; Bäumler, A.J. Interleukin-23 orchestrates mucosal responses to Salmonella enterica serotype Typhimurium in the intestine. Infect. Immun. 2009, 77, 387–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, H.; Pham, O.H.; Li, L.; Atif, S.M.; Lee, S.-J.; Ravesloot, M.M.; Stolfi, J.L.; Nuccio, S.-P.; Broz, P.; Monack, D.M.; et al. Toll-like receptor and inflammasome signals converge to amplify the innate bactericidal capacity of T helper 1 cells. Immunity 2014, 40, 213–224. [Google Scholar] [CrossRef]
- Raffatellu, M.; Santos, R.L.; Verhoeven, D.E.; George, M.D.; Wilson, R.P.; Winter, S.E.; Godinez, I.; Sankaran, S.; Paixao, T.A.; Gordon, M.A.; et al. Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes Salmonella dissemination from the gut. Nat. Med. 2008, 14, 421–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godinez, I.; Haneda, T.; Raffatellu, M.; George, M.D.; Paixão, T.A.; Rolán, H.G.; Santos, R.L.; Dandekar, S.; Tsolis, R.M.; Bäumler, A.J. T cells help to amplify inflammatory responses induced by Salmonella enterica serotype Typhimurium in the intestinal mucosa. Infect. Immun. 2008, 76, 2008–2017. [Google Scholar] [CrossRef] [Green Version]
- Kocijancic, D.; Leschner, S.; Felgner, S.; Komoll, R.-M.; Frahm, M.; Pawar, V.; Weiss, S. Therapeutic benefit of Salmonella attributed to LPS and TNF-α is exhaustible and dictated by tumor susceptibility. Oncotarget 2017, 8, 36492–36508. [Google Scholar] [CrossRef] [Green Version]
- Bueno, S.M.; González, P.A.; Carreño, L.J.; Tobar, J.A.; Mora, G.C.; Pereda, C.J.; Salazar-Onfray, F.; Kalergis, A.M. The capacity of Salmonella to survive inside dendritic cells and prevent antigen presentation to T cells is host specific. Immunology 2008, 124, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Wang, P.; Song, X.; Zhang, H.; Ma, S.; Wang, J.; Li, W.; Lv, R.; Liu, X.; Ma, S.; et al. Salmonella Typhimurium reprograms macrophage metabolism via T3SS effector SopE2 to promote intracellular replication and virulence. Nat. Commun. 2021, 12, 879. [Google Scholar] [CrossRef]
- Coombes, B.K.; Brown, N.F.; Valdez, Y.; Brumell, J.H.; Finlay, B.B. Expression and secretion of Salmonella pathogenicity island-2 virulence genes in response to acidification exhibit differential requirements of a functional type III secretion apparatus and SsaL. J. Biol. Chem. 2004, 279, 49804–49815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtz, J.R.; Goggins, J.A.; McLachlan, J.B. Salmonella infection: Interplay between the bacteria and host immune system. Immunol. Lett. 2017, 190, 42–50. [Google Scholar] [CrossRef]
- Yano, S.; Miwa, S.; Mii, S.; Hiroshima, Y.; Uehara, F.; Yamamoto, M.; Kishimoto, H.; Tazawa, H.; Bouvet, M.; Fujiwara, T.; et al. Invading cancer cells are predominantly in G0/G1 resulting in chemoresistance demonstrated by real-time FUCCI imaging. Cell Cycle 2014, 13, 953–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsao, Y.-T.; Kuo, C.-Y.; Cheng, S.-P.; Lee, C.-H. Downregulations of AKT/mTOR Signaling Pathway for Salmonella-Mediated Suppression of Matrix Metalloproteinases-9 Expression in Mouse Tumor Models. Int. J. Mol. Sci. 2018, 19, 1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakoob, R.; Pradeep, B.V. Bifidobacterium sp. as Probiotic Agent—Roles and Applications. J. Pure Appl. Microbiol. 2019, 13, 1407–1417. [Google Scholar] [CrossRef] [Green Version]
- Bifidobacterium (ID 13541)—Genome—NCBI. Available online: https://www.ncbi.nlm.nih.gov/genome/13541 (accessed on 24 February 2023).
- Fanning, S.; Hall, L.J.; Cronin, M.; Zomer, A.; MacSharry, J.; Goulding, D.; O’Connell, M.; Shanahan, F.; Nally, K.; Dougan, G.; et al. Bifidobacterial surface-exopolysaccharide facilitates commensal-host interaction through immune modulation and pathogen protection. Proc. Natl. Acad. Sci. USA 2012, 109, 2108–2113. [Google Scholar] [CrossRef]
- Ling, X.; Linglong, P.; Weixia, D.; Hong, W. Protective Effects of Bifidobacterium on Intestinal Barrier Function in LPS-Induced Enterocyte Barrier Injury of Caco-2 Monolayers and in a Rat NEC Model. PLoS ONE 2016, 11, e0161635. [Google Scholar] [CrossRef]
- Devi, T.B.; Devadas, K.; George, M.; Gandhimathi, A.; Chouhan, D.; Retnakumar, R.J.; Alexander, S.M.; Varghese, J.; Dharmaseelan, S.; Chandrika, S.K.; et al. Low Bifidobacterium Abundance in the Lower Gut Microbiota Is Associated With Helicobacter pylori-Related Gastric Ulcer and Gastric Cancer. Front. Microbiol. 2021, 12, 631140. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Shi, H.; Zhang, Y.; Fan, Y.; Wang, L.; Xiang, L.; Liu, Y.; Zhao, L.; Fu, S. Bacteria-driven hypoxia targeting delivery of chemotherapeutic drug proving outcome of breast cancer. J. Nanobiotechnol. 2022, 20, 178. [Google Scholar] [CrossRef]
- Lu, D.; Wang, L.; Wang, L.; An, L.; Huo, M.; Xu, H.; Shi, J. Probiotic Engineering and Targeted Sonoimmuno-Therapy Augmented by STING Agonist. Adv. Sci. 2022, 9, 2201711. [Google Scholar] [CrossRef] [PubMed]
- Laiño, J.; Villena, J.; Kanmani, P.; Kitazawa, H. Immunoregulatory Effects Triggered by Lactic Acid Bacteria Exopolysaccharides: New Insights into Molecular Interactions with Host Cells. Microorganisms 2016, 4, 27. [Google Scholar] [CrossRef] [Green Version]
- Altmann, F.; Kosma, P.; O’Callaghan, A.; Leahy, S.; Bottacini, F.; Molloy, E.; Plattner, S.; Schiavi, E.; Gleinser, M.; Groeger, D.; et al. Genome Analysis and Characterisation of the Exopolysaccharide Produced by Bifidobacterium longum subsp. longum 35624TM. PLoS ONE 2016, 11, e0162983. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Zheng, W.; Yang, K.; Harris, K.G.; Ni, K.; Xue, L.; Lin, W.; Chang, E.B.; Weichselbaum, R.R.; Fu, Y.-X. Intratumoral accumulation of gut microbiota facilitates CD47-based immunotherapy via STING signaling. J. Exp. Med. 2020, 217, e20192282. [Google Scholar] [CrossRef]
- Shioya, K.; Matsumura, T.; Seki, Y.; Shimizu, H.; Nakamura, T.; Taniguchi, S. Potentiated antitumor effects of APS001F/5-FC combined with anti-PD-1 antibody in a CT26 syngeneic mouse model. Biosci. Biotechnol. Biochem. 2021, 85, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Justesen, U.S.; Holt, H.M.; Thiesson, H.C.; Blom, J.; Nielsen, X.C.; Dargis, R.; Kemp, M.; Christensen, J.J. Report of the First Human Case of Caulobacter sp. Infection. J. Clin. Microbiol. 2007, 45, 1366–1369. [Google Scholar] [CrossRef] [Green Version]
- Caulobacter (ID 13664)—Genome—NCBI. Available online: https://www.ncbi.nlm.nih.gov/genome/13664 (accessed on 25 February 2023).
- Bhatnagar, P.K.; Awasthi, A.; Nomellini, J.F.; Smit, J.; Suresh, M.R. Anti-tumor effects of the bacterium caulobacter crescentus in murine tumor models. Cancer Biol. Ther. 2006, 5, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Duval, M.; Lewis, C.J.; Nomellini, J.F.; Horwitz, M.S.; Smit, J.; Cavacini, L.A.; Pea, F.; Cojutti, P.; Petrosillo, N.; Furlanut, M.; et al. Enhanced Neutralization of HIV by Antibodies Displayed on the S-Layer of Caulobacter crescentus. Antimicrob. Agents Chemother. 2011, 55, 5547–5552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smit, J.; Kaltashov, I.A.; Cotter, R.J.; Vinogradov, E.; Perry, M.B.; Haider, H.; Qureshi, N. Structure of a novel lipid A obtained from the lipopolysaccharide of Caulobacter crescentus. Innate Immun. 2008, 14, 25–36. [Google Scholar] [CrossRef]
- Aksel, E.G.; Akyüz, B. Effect of LPS and LTA stimulation on the expression of TLR-pathway genes in PBMCs of Akkaraman lambs in vivo. Trop. Anim. Health Prod. 2021, 53, 65. [Google Scholar] [CrossRef] [PubMed]
- Moore, G.M.; Gitai, Z. Both clinical and environmental Caulobacter species are virulent in the Galleria mellonella infection model. PLoS ONE 2020, 15, e0230006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Bayona, L.; Guo, M.S.; Laub, M.T. Contact-dependent killing by Caulobacter crescentus via cell surface-associated, glycine zipper proteins. eLife 2017, 6, e24869. [Google Scholar] [CrossRef] [Green Version]
- Adeleye, M.; Bhatnagar, P.; Suresh, M.; Smit, J. Caulobacter in cancer immunotherapy. Clin. Cancer Res. 2014, 13, A60. [Google Scholar]
- Loo, E.W.-L. Activation of Natural Killer T Cells and Dendritic Cells with Caulobacter Crescentus: Implications for Developing Tumour Immunity; University of Alberta: Edmonton, AB, Canada, 2013. [Google Scholar] [CrossRef]
- Goldstein, E.J.C.; Tyrrell, K.L.; Citron, D.M. Lactobacillus Species: Taxonomic Complexity and Controversial Susceptibilities. Clin. Infect. Dis. 2015, 60, S98–S107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lactobacillus (ID 13564)—Genome—NCBI. Available online: https://www.ncbi.nlm.nih.gov/genome/13564 (accessed on 25 February 2023).
- Jacouton, E.; Michel, M.-L.; Torres-Maravilla, E.; Chain, F.; Langella, P.; Bermúdez-Humarán, L.G. Elucidating the Immune-Related Mechanisms by Which Probiotic Strain Lactobacillus casei BL23 Displays Anti-tumoral Properties. Front. Microbiol. 2019, 9, 3281. [Google Scholar] [CrossRef] [PubMed]
- Khazaie, K.; Zadeh, M.; Khan, M.W.; Bere, P.; Gounari, F.; Dennis, K.; Blatner, N.R.; Owen, J.L.; Klaenhammer, T.R.; Mohamadzadeh, M. Abating colon cancer polyposis by Lactobacillus acidophilus deficient in lipoteichoic acid. Proc. Natl. Acad. Sci. USA 2012, 109, 10462–10467. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, H.; Liu, J.; Zhao, Y.; Gao, K.; Zhang, J. Adhesive ability means inhibition activities for lactobacillus against pathogens and S-layer protein plays an important role in adhesion. Anaerobe 2013, 22, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Oh, S.; Yun, H.S.; Oh, S.; Kim, S.H. Cell-bound exopolysaccharide from probiotic bacteria induces autophagic cell death of tumour cells: Antitumour activity of cb-EPS via autophagy. Lett. Appl. Microbiol. 2010, 51, 123–130. [Google Scholar] [CrossRef]
- Aindelis, G.; Tiptiri-Kourpeti, A.; Lampri, E.; Spyridopoulou, K.; Lamprianidou, E.; Kotsianidis, I.; Ypsilantis, P.; Pappa, A.; Chlichlia, K. Immune Responses Raised in an Experimental Colon Carcinoma Model Following Oral Administration of Lactobacillus casei. Cancers 2020, 12, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Liao, L.; Yin, H.; Nakamura, H.; Shin, T.; Maeda, H. Enhanced Bacterial Tumor Delivery by Modulating the EPR Effect and Therapeutic Potential of Lactobacillus casei. J. Pharm. Sci. 2014, 103, 3235–3243. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roe, J.M.; Seely, K.; Bussard, C.J.; Eischen Martin, E.; Mouw, E.G.; Bayles, K.W.; Hollingsworth, M.A.; Brooks, A.E.; Dailey, K.M. Hacking the Immune Response to Solid Tumors: Harnessing the Anti-Cancer Capacities of Oncolytic Bacteria. Pharmaceutics 2023, 15, 2004. https://doi.org/10.3390/pharmaceutics15072004
Roe JM, Seely K, Bussard CJ, Eischen Martin E, Mouw EG, Bayles KW, Hollingsworth MA, Brooks AE, Dailey KM. Hacking the Immune Response to Solid Tumors: Harnessing the Anti-Cancer Capacities of Oncolytic Bacteria. Pharmaceutics. 2023; 15(7):2004. https://doi.org/10.3390/pharmaceutics15072004
Chicago/Turabian StyleRoe, Jason M., Kevin Seely, Caleb J. Bussard, Emily Eischen Martin, Elizabeth G. Mouw, Kenneth W. Bayles, Michael A. Hollingsworth, Amanda E. Brooks, and Kaitlin M. Dailey. 2023. "Hacking the Immune Response to Solid Tumors: Harnessing the Anti-Cancer Capacities of Oncolytic Bacteria" Pharmaceutics 15, no. 7: 2004. https://doi.org/10.3390/pharmaceutics15072004