
From the Discovery of Targets to Delivery Systems: How to Decipher and Improve the Metallodrugs’ Actions at a Molecular Level

 , , ,
, , ,
Abstract
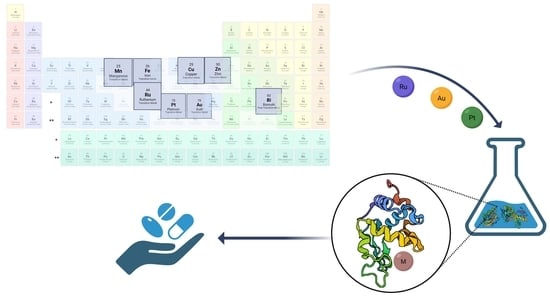
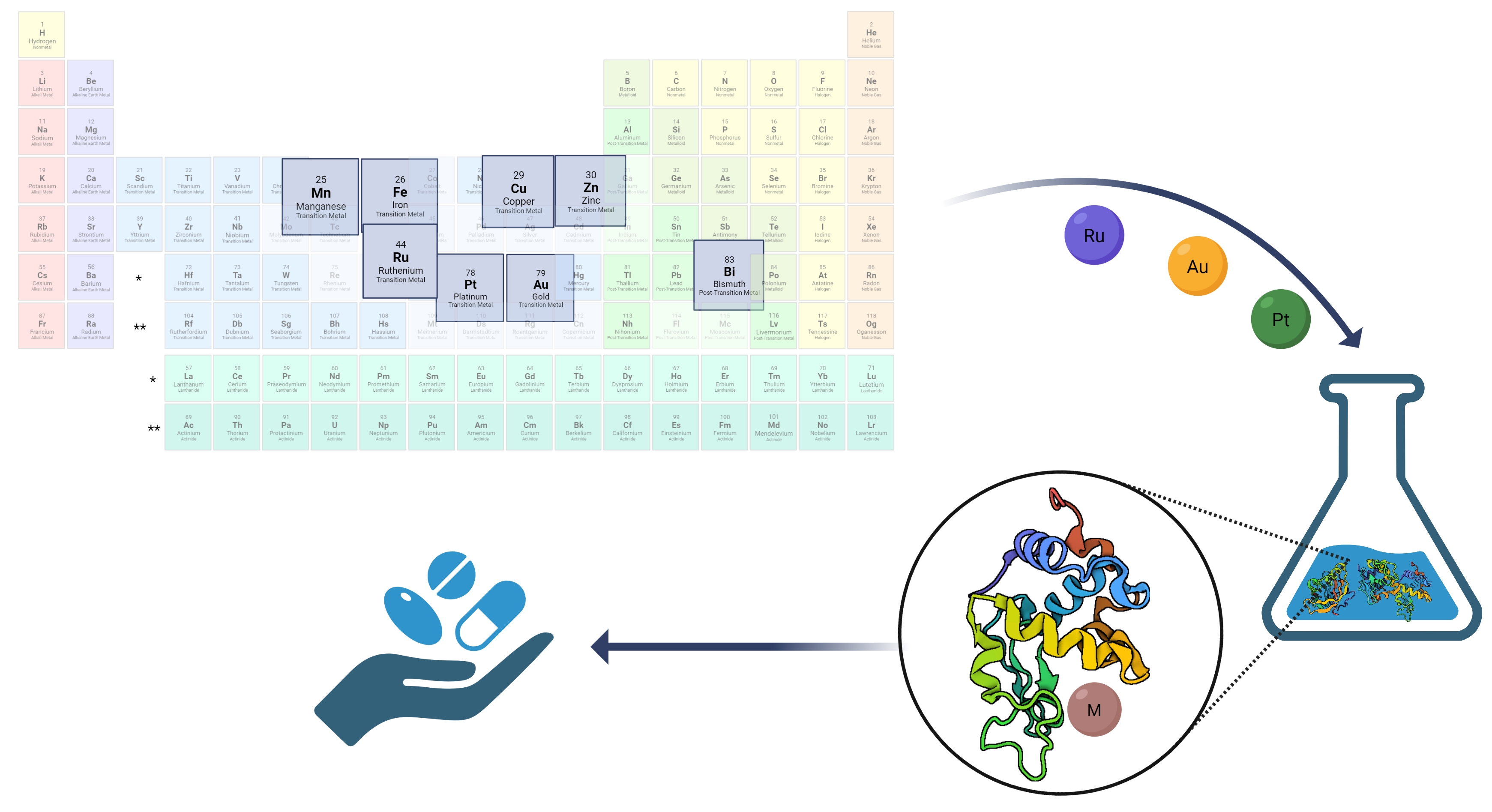
:
1. Introduction
2. Protein Target Identification through Proteomic Approaches
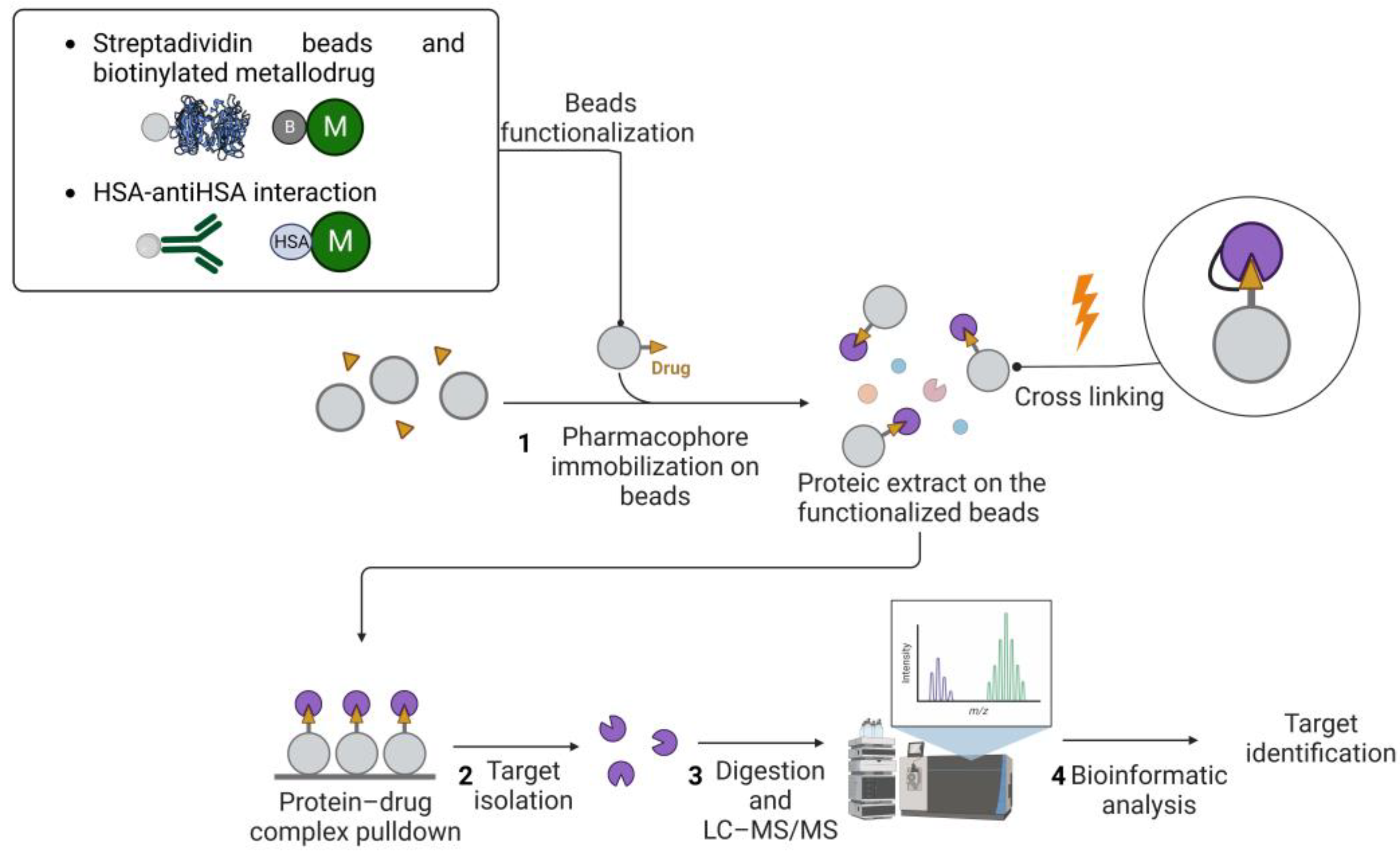
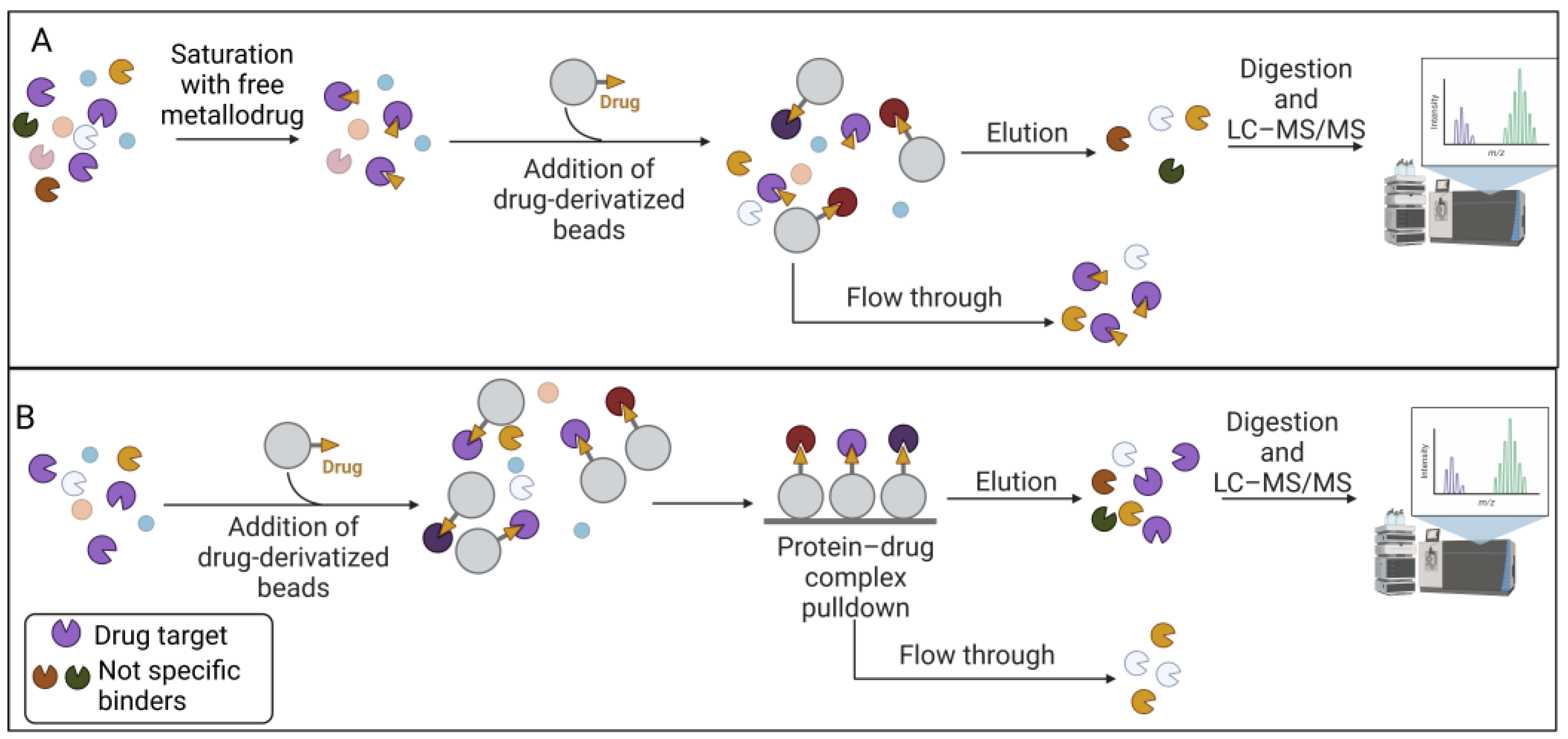
2.1. Affinity-Based Strategies
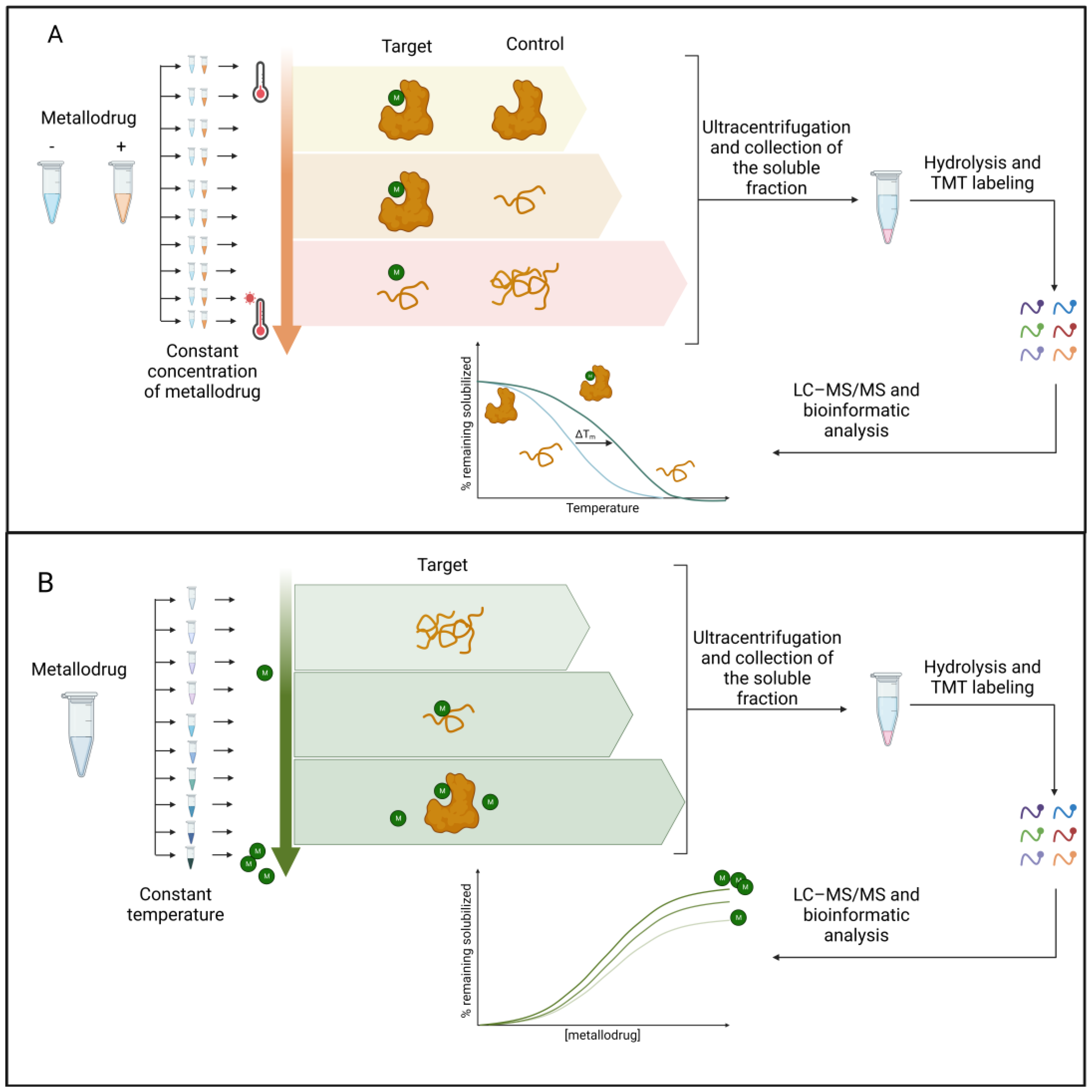
2.2. Label-Free Approaches
3. Methods to Enhance Metallodrugs Efficiency Administration
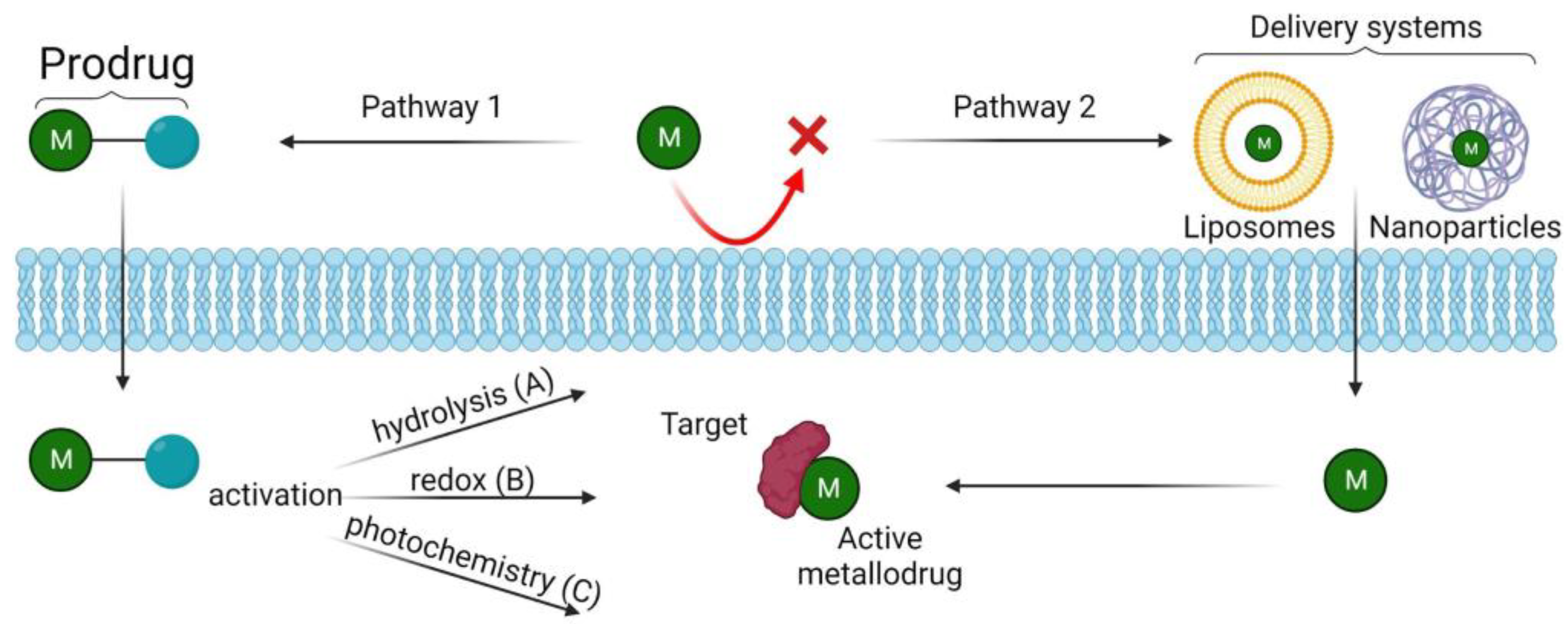
3.1. Pro-Metallodrugs
- (1)
- Activation via hydrolysis
- (2)
- Redox activation
- (3)
- Photoactivation (Light-activatable metallodrugs)
3.2. Delivery Systems
4. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kambe, T.; Tsuji, T.; Hashimoto, A.; Itsumura, N. The Physiological, Biochemical, and Molecular Roles of Zinc Transporters in Zinc Homeostasis and Metabolism. Physiol. Rev. 2015, 95, 749–784. [Google Scholar] [CrossRef] [Green Version]
- Bagheri-Mohammadi, S.; Farjami, M.; Suha, A.J.; Zarch, S.M.A.; Najafi, S.; Esmaeili, A. The Mitochondrial Calcium Signaling, Regulation, and Cellular Functions: A Novel Target for Therapeutic Medicine in Neurological Disorders. J. Cell. Biochem. 2023, 124, 635–655. [Google Scholar] [CrossRef]
- An, Y.; Li, S.; Huang, X.; Chen, X.; Shan, H.; Zhang, M. The Role of Copper Homeostasis in Brain Disease. Int. J. Mol. Sci. 2022, 23, 13850. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, L.M.; Libedinsky, A.; Elorza, A.A. Role of Copper on Mitochondrial Function and Metabolism. Front. Mol. Biosci. 2021, 8, 711227. [Google Scholar] [CrossRef] [PubMed]
- Opazo, C.M.; Greenough, M.A.; Bush, A.I. Copper: From Neurotransmission to Neuroproteostasis. Front. Aging Neurosci. 2014, 6, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Yin, Y.-L.; Liu, X.-Z.; Shen, P.; Zheng, Y.-G.; Lan, X.-R.; Lu, C.-B.; Wang, J.-Z. Current Understanding of Metal Ions in the Pathogenesis of Alzheimer’s Disease. Transl. Neurodegener. 2020, 9, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haywood, S. Brain–Barrier Regulation, Metal (Cu, Fe) Dyshomeostasis, and Neurodegenerative Disorders in Man and Animals. Inorganics 2019, 7, 108. [Google Scholar] [CrossRef] [Green Version]
- Franz, K.J.; Metzler-Nolte, N. Introduction: Metals in Medicine. Chem. Rev. 2019, 119, 727–729. [Google Scholar] [CrossRef] [Green Version]
- Galib; Mashru, M.; Patgiri, B.; Barve, M.; Jagtap, C.; Prajapati, P. Therapeutic Potentials of Metals in Ancient India: A Review through Charaka Samhita. J. Ayurveda Integr. Med. 2011, 2, 55. [Google Scholar] [CrossRef] [Green Version]
- Begley, T.P. Wiley Encyclopedia of Chemical Biology; John Wiley & Sons: Hoboken, NJ, USA, 2007; ISBN 978-0-470-04867-2. [Google Scholar]
- Sodhi, R.K. Metal Complexes in Medicine: An Overview and Update from Drug Design Perspective. Cancer Ther. Oncol. Int. J. 2019, 14, 555883. [Google Scholar] [CrossRef]
- Rosenberg, B.; Van Camp, L.; Krigas, T. Inhibition of Cell Division in Escherichia Coli by Electrolysis Products from a Platinum Electrode. Nature 1965, 205, 698–699. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, G.B.; Pentimalli, R.; Doldi, S.; Hall, M.D. Michele Peyrone (1813–1883), Discoverer of Cisplatin. Platin. Met. Rev. 2010, 54, 250–256. [Google Scholar] [CrossRef]
- Sigel, A.; Sigel, H.; Freisinger, E.; Sigel, R.K.O. (Eds.) Metallo-Drugs: Development and Action of Anticancer Agents; (Metal Ions in Life Sciences); De Gruyter: Berlin, Germany; Boston, MA, USA, 2018; ISBN 978-3-11-046984-4. [Google Scholar]
- Khoury, A.; Deo, K.M.; Aldrich-Wright, J.R. Recent Advances in Platinum-Based Chemotherapeutics That Exhibit Inhibitory and Targeted Mechanisms of Action. J. Inorg. Biochem. 2020, 207, 111070. [Google Scholar] [CrossRef] [PubMed]
- Zhong, T.; Yu, J.; Pan, Y.; Zhang, N.; Qi, Y.; Huang, Y. Recent Advances of Platinum-Based Anticancer Complexes in Combinational Multimodal Therapy. Adv Healthc. Mater. 2023, 2300253. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, X.; Guo, Z. Functionalization of Platinum Complexes for Biomedical Applications. Acc. Chem. Res. 2015, 48, 2622–2631. [Google Scholar] [CrossRef] [PubMed]
- Barabas, K.; Milner, R.; Lurie, D.; Adin, C. Cisplatin: A Review of Toxicities and Therapeutic Applications. Vet. Comp. Oncol. 2008, 6, 1–18. [Google Scholar] [CrossRef]
- Rosenberg, B.; Vancamp, L.; Trosko, J.E.; Mansour, V.H. Platinum Compounds: A New Class of Potent Antitumour Agents. Nature 1969, 222, 385–386. [Google Scholar] [CrossRef]
- Williams, C.J.; Whitehouse, J.M. Cis-Platinum: A New Anticancer Agent. BMJ 1979, 1, 1689–1691. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Alcántar, G.; Picchetti, P.; Casini, A. Gold Complexes in Anticancer Therapy: From New Design Principles to Particle-Based Delivery Systems. Angew. Chem. Int. Ed. 2023, 62, e202218000. [Google Scholar] [CrossRef]
- Coffetti, G.; Moraschi, M.; Facchetti, G.; Rimoldi, I. The Challenging Treatment of Cisplatin-Resistant Tumors: State of the Art and Future Perspectives. Molecules 2023, 28, 3407. [Google Scholar] [CrossRef]
- Florio, D.; La Manna, S.; Annunziata, A.; Iacobucci, I.; Monaco, V.; Di Natale, C.; Mollo, V.; Ruffo, F.; Monti, M.; Marasco, D. Ruthenium Complexes Bearing Glucosyl Ligands Are Able to Inhibit the Amyloid Aggregation of Short Histidine-Peptides. Dalton Trans. 2023, 52, 8549–8557. [Google Scholar] [CrossRef]
- Paul, N.P.; Galván, A.E.; Yoshinaga-Sakurai, K.; Rosen, B.P.; Yoshinaga, M. Arsenic in Medicine: Past, Present and Future. Biometals 2023, 36, 283–301. [Google Scholar] [CrossRef]
- Florio, D.; Iacobucci, I.; Ferraro, G.; Mansour, A.M.; Morelli, G.; Monti, M.; Merlino, A.; Marasco, D. Role of the Metal Center in the Modulation of the Aggregation Process of Amyloid Model Systems by Square Planar Complexes Bearing 2-(2’-Pyridyl)Benzimidazole Ligands. Pharmaceuticals 2019, 12, 154. [Google Scholar] [CrossRef] [Green Version]
- Jaouen, G.; Metzler-Nolte, N.; Alberto, R. (Eds.) Medicinal Organometallic Chemistry; (Topics in Organometallic Chemistry); Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 2010; ISBN 978-3-642-13184-4. [Google Scholar]
- Zhou, Y.; Li, H.; Sun, H. Metalloproteomics for Biomedical Research: Methodology and Applications. Annu. Rev. Biochem. 2022, 91, 449–473. [Google Scholar] [CrossRef]
- Kostova, I. Ruthenium Complexes as Anticancer Agents. Curr. Med. Chem. 2006, 13, 1085–1107. [Google Scholar] [CrossRef]
- Huang, H.; Cao, K.; Kong, Y.; Yuan, S.; Liu, H.; Wang, Y.; Liu, Y. A Dual Functional Ruthenium Arene Complex Induces Differentiation and Apoptosis of Acute Promyelocytic Leukemia Cells. Chem. Sci. 2019, 10, 9721–9728. [Google Scholar] [CrossRef]
- Ahrweiler-Sawaryn, M.-C.; Biswas, A.; Frias, C.; Frias, J.; Wilke, N.L.; Wilke, N.; Berkessel, A.; Prokop, A. Novel Gold(I) Complexes Induce Apoptosis in Leukemia Cells via the ROS-Induced Mitochondrial Pathway with an Upregulation of Harakiri and Overcome Multi Drug Resistances in Leukemia and Lymphoma Cells and Sensitize Drug Resistant Tumor Cells to Apoptosis in Vitro. Biomed. Pharmacother. 2023, 161, 114507. [Google Scholar] [CrossRef]
- Ferraro, M.G.; Piccolo, M.; Misso, G.; Santamaria, R.; Irace, C. Bioactivity and Development of Small Non-Platinum Metal-Based Chemotherapeutics. Pharmaceutics 2022, 14, 954. [Google Scholar] [CrossRef]
- Peña, Q.; Wang, A.; Zaremba, O.; Shi, Y.; Scheeren, H.W.; Metselaar, J.M.; Kiessling, F.; Pallares, R.M.; Wuttke, S.; Lammers, T. Metallodrugs in Cancer Nanomedicine. Chem. Soc. Rev. 2022, 51, 2544–2582. [Google Scholar] [CrossRef]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The Side Effects of Platinum-Based Chemotherapy Drugs: A Review for Chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef]
- Casini, A.; Vessieres, A.; Meier-Menches, S.M. (Eds.) Metal-Based Anticancer Agents; (Metallobiology Series); Royal Society of Chemistry: London, UK, 2019; ISBN 978-1-78801-406-9. [Google Scholar]
- Mjos, K.D.; Orvig, C. Metallodrugs in Medicinal Inorganic Chemistry. Chem. Rev. 2014, 114, 4540–4563. [Google Scholar] [CrossRef] [PubMed]
- Barhamand, B.A. Difficulties Encountered in Implementing Guidelines for Handling Antineoplastics in the Physician’s Office. Cancer Nurs. 1986, 9, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Zanca, C.; Cozzolino, F.; Quintavalle, C.; Di Costanzo, S.; Ricci-Vitiani, L.; Santoriello, M.; Monti, M.; Pucci, P.; Condorelli, G. PED Interacts with Rac1 and Regulates Cell Migration/Invasion Processes in Human Non-Small Cell Lung Cancer Cells. J. Cell. Physiol. 2010, 225, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Fusco, S.; Aulitto, M.; Iacobucci, I.; Crocamo, G.; Pucci, P.; Bartolucci, S.; Monti, M.; Contursi, P. The Interaction between the F55 Virus-Encoded Transcription Regulator and the RadA Host Recombinase Reveals a Common Strategy in Archaea and Bacteria to Sense the UV-Induced Damage to the Host DNA. Biochim. Biophys. Acta (BBA)—Gene Regul. Mech. 2020, 1863, 194493. [Google Scholar] [CrossRef]
- Iacobucci, I.; Monaco, V.; Cozzolino, F.; Monti, M. From Classical to New Generation Approaches: An Excursus of -Omics Methods for Investigation of Protein-Protein Interaction Networks. J. Proteom. 2021, 230, 103990. [Google Scholar] [CrossRef]
- Cozzolino, F.; Iacobucci, I.; Monaco, V.; Monti, M. Protein–DNA/RNA Interactions: An Overview of Investigation Methods in the -Omics Era. J. Proteome Res. 2021, 20, 3018–3030. [Google Scholar] [CrossRef]
- Kumara, B.N.; Kalimuthu, P.; Prasad, K.S. Synthesis, Properties and Potential Applications of Photoluminescent Carbon Nanoparticles: A Review. Anal. Chim. Acta 2023, 1268, 341430. [Google Scholar] [CrossRef]
- Xia, Y.; Fu, S.; Ma, Q.; Liu, Y.; Zhang, N. Application of Nano-Delivery Systems in Lymph Nodes for Tumor Immunotherapy. Nano-Micro Lett. 2023, 15, 145. [Google Scholar] [CrossRef]
- Kargozar, S.; Moghanian, A.; Rashvand, A.; Miri, A.K.; Hamzehlou, S.; Baino, F.; Mozafari, M.; Wang, A.Z. Nanostructured Bioactive Glasses: A Bird’s Eye View on Cancer Therapy. WIREs Nanomed. Nanobiotechnol. 2023, e1905. [Google Scholar] [CrossRef]
- Fedorov, I.I.; Lineva, V.I.; Tarasova, I.A.; Gorshkov, M.V. Mass Spectrometry-Based Chemical Proteomics for Drug Target Discoveries. Biochemistry 2022, 87, 983–994. [Google Scholar] [CrossRef]
- Skos, L.; Borutzki, Y.; Gerner, C.; Meier-Menches, S.M. Methods to Identify Protein Targets of Metal-Based Drugs. Curr. Opin. Chem. Biol. 2023, 73, 102257. [Google Scholar] [CrossRef]
- Steel, T.R.; Hartinger, C.G. Metalloproteomics for Molecular Target Identification of Protein-Binding Anticancer Metallodrugs. Metallomics 2020, 12, 1627–1636. [Google Scholar] [CrossRef]
- Ziegler, S.; Pries, V.; Hedberg, C.; Waldmann, H. Target Identification for Small Bioactive Molecules: Finding the Needle in the Haystack. Angew. Chem. Int. Ed. 2013, 52, 2744–2792. [Google Scholar] [CrossRef]
- Iacobucci, I.; Monaco, V.; Canè, L.; Bibbò, F.; Cioffi, V.; Cozzolino, F.; Guarino, A.; Zollo, M.; Monti, M. Spike S1 Domain Interactome in Non-Pulmonary Systems: A Role beyond the Receptor Recognition. Front. Mol. Biosci. 2022, 9, 975570. [Google Scholar] [CrossRef]
- Federico, A.; Sepe, R.; Cozzolino, F.; Piccolo, C.; Iannone, C.; Iacobucci, I.; Pucci, P.; Monti, M.; Fusco, A. The Complex CBX7-PRMT1 Has a Critical Role in Regulating E-Cadherin Gene Expression and Cell Migration. Biochim. Biophys. Acta (BBA)—Gene Regul. Mech. 2019, 1862, 509–521. [Google Scholar] [CrossRef]
- Cozzolino, F.; Vezzoli, E.; Cheroni, C.; Besusso, D.; Conforti, P.; Valenza, M.; Iacobucci, I.; Monaco, V.; Birolini, G.; Bombaci, M.; et al. ADAM10 Hyperactivation Acts on Piccolo to Deplete Synaptic Vesicle Stores in Huntington’s Disease. Hum. Mol. Genet. 2021, 30, 1175–1187. [Google Scholar] [CrossRef]
- Babak, M.V.; Meier, S.M.; Huber, K.V.M.; Reynisson, J.; Legin, A.A.; Jakupec, M.A.; Roller, A.; Stukalov, A.; Gridling, M.; Bennett, K.L.; et al. Target Profiling of an Antimetastatic RAPTA Agent by Chemical Proteomics: Relevance to the Mode of Action. Chem. Sci. 2015, 6, 2449–2456. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, M.; Gao, F.; Wei, W.; Qian, Y.; Liu, H.-K.; Zhao, J. Imaging of a Clickable Anticancer Iridium Catalyst. J. Inorg. Biochem. 2018, 180, 179–185. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, J.; Zhao, X.; Wei, W.; Zhao, J. Imaging and Proteomic Study of a Clickable Iridium Complex. Metallomics 2019, 11, 1344–1352. [Google Scholar] [CrossRef]
- Neuditschko, B.; King, A.P.; Huang, Z.; Janker, L.; Bileck, A.; Borutzki, Y.; Marker, S.C.; Gerner, C.; Wilson, J.J.; Meier-Menches, S.M. An Anticancer Rhenium Tricarbonyl Targets Fe−S Cluster Biogenesis in Ovarian Cancer Cells. Angew. Chem. Int. Ed. 2022, 61, e202209136. [Google Scholar] [CrossRef]
- Neuditschko, B.; Legin, A.A.; Baier, D.; Schintlmeister, A.; Reipert, S.; Wagner, M.; Keppler, B.K.; Berger, W.; Meier-Menches, S.M.; Gerner, C. Interaction with Ribosomal Proteins Accompanies Stress Induction of the Anticancer Metallodrug BOLD-100/KP1339 in the Endoplasmic Reticulum. Angew. Chem. Int. Ed. 2021, 60, 5063–5068. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, Z. Development and Applications of the Copper-Catalyzed Azide-Alkyne Cycloaddition (CuAAC) as a Bioorthogonal Reaction. Molecules 2016, 21, 1393. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, H.; Sun, H. Metalloproteomics for Unveiling the Mechanism of Action of Metallodrugs. Inorg. Chem. 2019, 58, 13673–13685. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Liu, Y.; Lai, Y.-T.; Tong, K.-C.; Fung, Y.-M.; Lok, C.-N.; Che, C.-M. Anticancer Gold(III) Porphyrins Target Mitochondrial Chaperone Hsp60. Angew. Chem. Int. Ed. 2016, 55, 1387–1391. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Tian, Y.; Wang, M.; Wang, M.; Sun, G.; Sun, X. Advanced Activity-Based Protein Profiling Application Strategies for Drug Development. Front. Pharmacol. 2018, 9, 353. [Google Scholar] [CrossRef] [PubMed]
- Lu, K. Chemoproteomics: Towards Global Drug Target Profiling. ChemBioChem 2020, 21, 3189–3191. [Google Scholar] [CrossRef]
- Plowright, A.T. Target Discovery and Validation: Methods and Strategies for Drug Discovery; (Methods and Principles in Medicinal Chemistry); Wiley-VCH: Weinheim, Germany, 2020; ISBN 978-3-527-34529-8. [Google Scholar]
- Savitski, M.M.; Reinhard, F.B.M.; Franken, H.; Werner, T.; Savitski, M.F.; Eberhard, D.; Molina, D.M.; Jafari, R.; Dovega, R.B.; Klaeger, S.; et al. Tracking Cancer Drugs in Living Cells by Thermal Profiling of the Proteome. Science 2014, 346, 1255784. [Google Scholar] [CrossRef] [Green Version]
- Saei, A.A.; Gullberg, H.; Sabatier, P.; Beusch, C.M.; Johansson, K.; Lundgren, B.; Arvidsson, P.I.; Arnér, E.S.J.; Zubarev, R.A. Comprehensive Chemical Proteomics for Target Deconvolution of the Redox Active Drug Auranofin. Redox Biol. 2020, 32, 101491. [Google Scholar] [CrossRef]
- Mateus, A.; Kurzawa, N.; Becher, I.; Sridharan, S.; Helm, D.; Stein, F.; Typas, A.; Savitski, M.M. Thermal Proteome Profiling for Interrogating Protein Interactions. Mol. Syst. Biol. 2020, 16, e9232. [Google Scholar] [CrossRef]
- Hu, D.; Yang, C.; Lok, C.; Xing, F.; Lee, P.; Fung, Y.M.E.; Jiang, H.; Che, C. An Antitumor Bis(N-Heterocyclic Carbene)Platinum(II) Complex That Engages Asparagine Synthetase as an Anticancer Target. Angew. Chem. Int. Ed. 2019, 58, 10914–10918. [Google Scholar] [CrossRef]
- Chernobrovkin, A.; Marin-Vicente, C.; Visa, N.; Zubarev, R.A. Functional Identification of Target by Expression Proteomics (FITExP) Reveals Protein Targets and Highlights Mechanisms of Action of Small Molecule Drugs. Sci. Rep. 2015, 5, 11176. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.F.S.; Chernobrovkin, A.; Rutishauser, D.; Allardyce, C.S.; Hacker, D.; Johnsson, K.; Zubarev, R.A.; Dyson, P.J. Expression Proteomics Study to Determine Metallodrug Targets and Optimal Drug Combinations. Sci. Rep. 2017, 7, 1590. [Google Scholar] [CrossRef] [Green Version]
- Strickland, E.C.; Geer, M.A.; Tran, D.T.; Adhikari, J.; West, G.M.; DeArmond, P.D.; Xu, Y.; Fitzgerald, M.C. Thermodynamic Analysis of Protein-Ligand Binding Interactions in Complex Biological Mixtures Using the Stability of Proteins from Rates of Oxidation. Nat. Protoc. 2013, 8, 148–161. [Google Scholar] [CrossRef] [Green Version]
- Park, C.; Marqusee, S. Pulse Proteolysis: A Simple Method for Quantitative Determination of Protein Stability and Ligand Binding. Nat. Methods 2005, 2, 207–212. [Google Scholar] [CrossRef]
- Lomenick, B.; Jung, G.; Wohlschlegel, J.A.; Huang, J. Target Identification Using Drug Affinity Responsive Target Stability (DARTS). Curr. Protoc. Chem. Biol. 2011, 3, 163–180. [Google Scholar] [CrossRef] [Green Version]
- Feng, F.; Zhang, W.; Chai, Y.; Guo, D.; Chen, X. Label-Free Target Protein Characterization for Small Molecule Drugs: Recent Advances in Methods and Applications. J. Pharm. Biomed. Anal. 2023, 223, 115107. [Google Scholar] [CrossRef]
- Jia, S.; Wang, R.; Wu, K.; Jiang, H.; Du, Z. Elucidation of the Mechanism of Action for Metal Based Anticancer Drugs by Mass Spectrometry-Based Quantitative Proteomics. Molecules 2019, 24, 581. [Google Scholar] [CrossRef] [Green Version]
- Roberts, E.A.; Sarkar, B. Metalloproteomics: Focus on Metabolic Issues Relating to Metals. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 425–430. [Google Scholar] [CrossRef]
- Anthony, E.J.; Bolitho, E.M.; Bridgewater, H.E.; Carter, O.W.L.; Donnelly, J.M.; Imberti, C.; Lant, E.C.; Lermyte, F.; Needham, R.J.; Palau, M.; et al. Metallodrugs Are Unique: Opportunities and Challenges of Discovery and Development. Chem. Sci. 2020, 11, 12888–12917. [Google Scholar] [CrossRef]
- Phillips, A.M.F.; Pombeiro, A.J.L. Transition Metal-Based Prodrugs for Anticancer Drug Delivery. Curr. Med. Chem. 2020, 26, 7476–7519. [Google Scholar] [CrossRef]
- Karnthaler-Benbakka, C.; Groza, D.; Kryeziu, K.; Pichler, V.; Roller, A.; Berger, W.; Heffeter, P.; Kowol, C.R. Tumor-Targeting of EGFR Inhibitors by Hypoxia-Mediated Activation. Angew. Chem. Int. Ed. 2014, 53, 12930–12935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hufziger, K.T.; Thowfeik, F.S.; Charboneau, D.J.; Nieto, I.; Dougherty, W.G.; Kassel, W.S.; Dudley, T.J.; Merino, E.J.; Papish, E.T.; Paul, J.J. Ruthenium Dihydroxybipyridine Complexes Are Tumor Activated Prodrugs Due to Low PH and Blue Light Induced Ligand Release. J. Inorg. Biochem. 2014, 130, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathak, R.K.; Marrache, S.; Choi, J.H.; Berding, T.B.; Dhar, S. The Prodrug Platin-A: Simultaneous Release of Cisplatin and Aspirin. Angew. Chem. Int. Ed. 2014, 53, 1963–1967. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Shi, H.; Wang, H.; Min, Y.; Wang, J.; Liu, Y. The Ligation of Aspirin to Cisplatin Demonstrates Significant Synergistic Effects on Tumor Cells. Chem. Commun. 2014, 50, 7427–7430. [Google Scholar] [CrossRef]
- Chen, S.-H.; Chang, J.-Y. New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment. Int. J. Mol. Sci. 2019, 20, 4136. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Wen, P.; Li, J.; Kataoka, K. Targeted Nanomedicine in Cisplatin-Based Cancer Therapeutics. J. Control. Release 2022, 345, 709–720. [Google Scholar] [CrossRef]
- Yao, Y.; Zhou, Y.; Liu, L.; Xu, Y.; Chen, Q.; Wang, Y.; Wu, S.; Deng, Y.; Zhang, J.; Shao, A. Nanoparticle-Based Drug Delivery in Cancer Therapy and Its Role in Overcoming Drug Resistance. Front. Mol. Biosci. 2020, 7, 193. [Google Scholar] [CrossRef]
- Osada, A. NC-6004, a Novel Cisplatin Nanoparticle, in Combination with Pembrolizumab for Head and Neck Cancer. Intergr. Clin. Med. 2019, 3. [Google Scholar] [CrossRef] [Green Version]
- Coverdale, J.; Laroiya-McCarron, T.; Romero-Canelón, I. Designing Ruthenium Anticancer Drugs: What Have We Learnt from the Key Drug Candidates? Inorganics 2019, 7, 31. [Google Scholar] [CrossRef] [Green Version]
- Ravera, M.; Gabano, E.; Zanellato, I.; Rangone, B.; Perin, E.; Ferrari, B.; Bottone, M.G.; Osella, D. Cis,Cis,Trans-[PtIVCl2(NH3)2(perillato)2], a Dual-Action Prodrug with Excellent Cytotoxic and Antimetastatic Activity. Dalton Trans. 2021, 50, 3161–3177. [Google Scholar] [CrossRef]
- Kenche, V.B.; Hung, L.W.; Perez, K.; Volitakes, I.; Ciccotosto, G.; Kwok, J.; Critch, N.; Sherratt, N.; Cortes, M.; Lal, V.; et al. Development of a Platinum Complex as an Anti-Amyloid Agent for the Therapy of Alzheimer’s Disease. Angew. Chem. Int. Ed. 2013, 52, 3374–3378. [Google Scholar] [CrossRef]
- Chen, Y.; Bai, L.; Zhang, P.; Zhao, H.; Zhou, Q. The Development of Ru(II)-Based Photoactivated Chemotherapy Agents. Molecules 2021, 26, 5679. [Google Scholar] [CrossRef]
- Kulkarni, G.S.; Lilge, L.; Nesbitt, M.; Dumoulin-White, R.J.; Mandel, A.; Jewett, M.A.S. A Phase 1b Clinical Study of Intravesical Photodynamic Therapy in Patients with Bacillus Calmette-Guérin–Unresponsive Non–Muscle-Invasive Bladder Cancer. Eur. Urol. Open Sci. 2022, 41, 105–111. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, Y.-S.; Liu, Y.-C.; Wang, N.; Zeng, X.-T.; Zhang, L.-L. Emerging Photodynamic/Sonodynamic Therapies for Urological Cancers: Progress and Challenges. J. Nanobiotechnol. 2022, 20, 437. [Google Scholar] [CrossRef]
- Karges, J. Clinical Development of Metal Complexes as Photosensitizers for Photodynamic Therapy of Cancer. Angew. Chem. Int. Ed. 2022, 61, e202112236. [Google Scholar] [CrossRef]
- Poursharifi, M.; Wlodarczyk, M.T.; Mieszawska, A.J. Nano-Based Systems and Biomacromolecules as Carriers for Metallodrugs in Anticancer Therapy. Inorganics 2018, 7, 2. [Google Scholar] [CrossRef] [Green Version]
- Boulikas, T. Clinical Overview on Lipoplatin TM: A Successful Liposomal Formulation of Cisplatin. Expert Opin. Investig. Drugs 2009, 18, 1197–1218. [Google Scholar] [CrossRef]
- Blunden, B.M.; Stenzel, M.H. Incorporating Ruthenium into Advanced Drug Delivery Carriers—An Innovative Generation of Chemotherapeutics. J. Chem. Technol. Biotechnol. 2015, 90, 1177–1195. [Google Scholar] [CrossRef]
- Fischer, B.; Heffeter, P.; Kryeziu, K.; Gille, L.; Meier, S.M.; Berger, W.; Kowol, C.R.; Keppler, B.K. Poly(Lactic Acid) Nanoparticles of the Lead Anticancer Ruthenium Compound KP1019 and Its Surfactant-Mediated Activation. Dalton Trans. 2014, 43, 1096–1104. [Google Scholar] [CrossRef] [Green Version]
- Alves, S.R.; Colquhoun, A.; Wu, X.Y.; De Oliveira Silva, D. Synthesis of Terpolymer-Lipid Encapsulated Diruthenium(II,III)-Anti-Inflammatory Metallodrug Nanoparticles to Enhance Activity against Glioblastoma Cancer Cells. J. Inorg. Biochem. 2020, 205, 110984. [Google Scholar] [CrossRef]
- Monti, D.M.; Ferraro, G.; Merlino, A. Ferritin-Based Anticancer Metallodrug Delivery: Crystallographic, Analytical and Cytotoxicity Studies. Nanomed. Nanotechnol. Biol. Med. 2019, 20, 101997. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name/Description | Mechanism of Action | Application (ID) | Clinical Trial Phase |
|---|---|---|---|
| Picoplatin | DNA damage, cell division arrest, and apoptosis | Treatment of: Solid tumor (NCT00710697); Bladder cancer Breast cancer Colorectal cancer (NCT00465725); Lymphoma Small intestine cancer Unspecified adult solid tumor, protocol specific (NCT00016172) | Phase I Phase II Phase I |
| Satraplatin | DNA-adduct alteration and translational DNA synthesis inhibition | Treatment of: Prostate cancer (NCT01289067, NCT00634647, NCT00069745); Lung cancer (NCT00370383, NCT00268970); Breast cancer (NCT00265655); Brain cancer (NCT01259479); Advanced cancers (NCT00473720) | Phase II/II/III Phase II/II Phase II Phase I, Phase I |
| Lipoplatin/(Liposomal form of cisplatin) | DNA damage, ERK pathway, and apoptosis | Treatment of: Pleural effusion, Malignant (NCT02702700) | Phase I |
| Triplatin tetranitrate/BBR3464/CT-3610 | DNA damage and apoptosis | Treatment of: Pancreatic cancer (NCT00024362) Lung cancer (NCT00014547) | Phase II Phase II |
| Aroplatin | N.A. | Treatment of: Pancreatic neoplasms (NCT00081549) | Phase I/II |
| Nedaplatin | DNA damage and apoptosis and oxidative stress induction | Treatment of: Nasopharyngeal carcinoma (NCT04834206) | Phase II |
| NKP1339/Sodium trans-tetrachloridobis(1H-indazole)ruthenate(III) | Disturbance of the cellular redox balance, G2/M cell cycle arrest, blockage of DNA synthesis, and induction of apoptosis via the mitochondrial pathway | Treatment of: Solid tumor (NCT01415297) | Phase I |
| TLD-1433/[Ru(4,4′-dmb)2(IP-3T)]Cl2 (4,4′-dmb = 4,4′-dimethyl-2,2′-bipyridine); IP = imidazo[4,5-f][1,10]phenanthroline]; 3T = α-terthienyl | Ideal photophysical properties to act as photodynamic therapy (PDT) | Treatment of: Non-muscle invasive bladder cancer (NMIBC) (NCT03053635) | Phase I |
| Ferroquine | Negatively regulates Akt kinase and hypoxia-inducible factor-1α (HIF-1α) and redox mechanism | Treatment of: Plasmodium falciparum infection (NCT03660839) Malaria (NCT00563914, NCT05911828) | Phase II Phase I/II |
| GC-4419/Avasopasem manganese | Superoxide dismutase’s catalytic site as a radiation therapy intervention | Treatment of: Renal impairment (NCT05412472) Squamous cell carcinoma of the oral cavity (NCT01921426) | Phase II Phase II |
| Rostaporfin Purlytin/Ethyl etiopurpurin (SnET2) | PDT kills cancer cells catalytically in the presence of oxygen and PS produces reactive oxygen species (ROS) and causes cancerous tissues to apoptosis | Treatment of: HIV infections (NCT00002167) | Phase II/III |
| Aurothiomalate | Inhibit T cells and CD4+ T activation | Treatment of: Non-small-cell lung carcinoma (NSCLC) (NCT00575393) | Phase I |
| Auranofin/TetraO-acetyl-b-D-(glucopyranosyl)-thio-triethylphosphine | Thioredoxin reductase inhibition (TrxR), which is a crucial enzyme for maintaining redox homeostasis, managing ROS levels, and preventing DNA damage | Treatment of: Amoebic dysentery (NCT02089048) Chronic lymphocytic leukemia (CLL) (NCT01419691) Giardiasis (NCT02736968) Recurrent fallopian tube cancer, Recurrent ovarian epithelial cancer, Recurrent primary peritoneal cavity cancer (NCT01747798) Lung carcinoma (NCT01737502) HIV (NCT02961829) | Phase I Phase II Phase II Phase II Phase I/Phase II Phase II |
| Cu-ATSM | Due to its low molecular weight, high membrane permeability, and low redox potential, Cu (II)-ATSM easily penetrates cells | Treatment of: Amyotrophic lateral sclerosis (NCT02870634, NCT03136809) Parkinson’s disease (NCT03204929) Diagnosis of: Cervical cancer (NCT00794339) | Phase I, Phase II, Phase I Phase II |
| Tetrathiomolybdate | Binds copper ions | Treatment of: Wilson disease (NCT00004339) Primary biliary cirrhosis (NCT00805805) Idiophatic pulmunary fibrosis (NCT00189176) Prostate cancer (NCT00150995) Non-small lung cancer (NCT01837329) Esophageal carcinoma (NCT00176800) Psoriasis (NCT00113542) Breast cancer (NCT00195091) | Phase III Phase III Phase I/Phase II Phase II Phase I Phase II Phase II Phase II |
| Cu64-DOTA-alendronate | Detection of small calcium deposits | Diagnosis of: Breast carcinoma (calcification) (NCT03542695) | Early Phase I |
| Gallium Ga68-labeled GRPR Antagonist BAY86-7548 | Detection of regional nodal and distant metastases in patients with intermediate- or high-risk prostate cancer | Diagnosis of: Prostate adenocarcinoma (NCT03113617) | Phase II |
| Sn-117m-DTPA | Targeting of bones affected by metastases | Treatment of: Metastatic prostate adenocarcinoma (NCT04616547) | Phase II |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iacobucci, I.; La Manna, S.; Cipollone, I.; Monaco, V.; Canè, L.; Cozzolino, F. From the Discovery of Targets to Delivery Systems: How to Decipher and Improve the Metallodrugs’ Actions at a Molecular Level. Pharmaceutics 2023, 15, 1997. https://doi.org/10.3390/pharmaceutics15071997
Iacobucci I, La Manna S, Cipollone I, Monaco V, Canè L, Cozzolino F. From the Discovery of Targets to Delivery Systems: How to Decipher and Improve the Metallodrugs’ Actions at a Molecular Level. Pharmaceutics. 2023; 15(7):1997. https://doi.org/10.3390/pharmaceutics15071997
Chicago/Turabian StyleIacobucci, Ilaria, Sara La Manna, Irene Cipollone, Vittoria Monaco, Luisa Canè, and Flora Cozzolino. 2023. "From the Discovery of Targets to Delivery Systems: How to Decipher and Improve the Metallodrugs’ Actions at a Molecular Level" Pharmaceutics 15, no. 7: 1997. https://doi.org/10.3390/pharmaceutics15071997