
Smart Delivery Systems Responsive to Cathepsin B Activity for Cancer Treatment
 , ,
, ,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Cathepsin B Synthesis and Physiological Function
3. CtsB Activity and Targetability in Cancer
4. CtsB-Responsive Delivery Systems
4.1. CtsB-Sensitive Conjugates Based on GFLG and Other Peptides
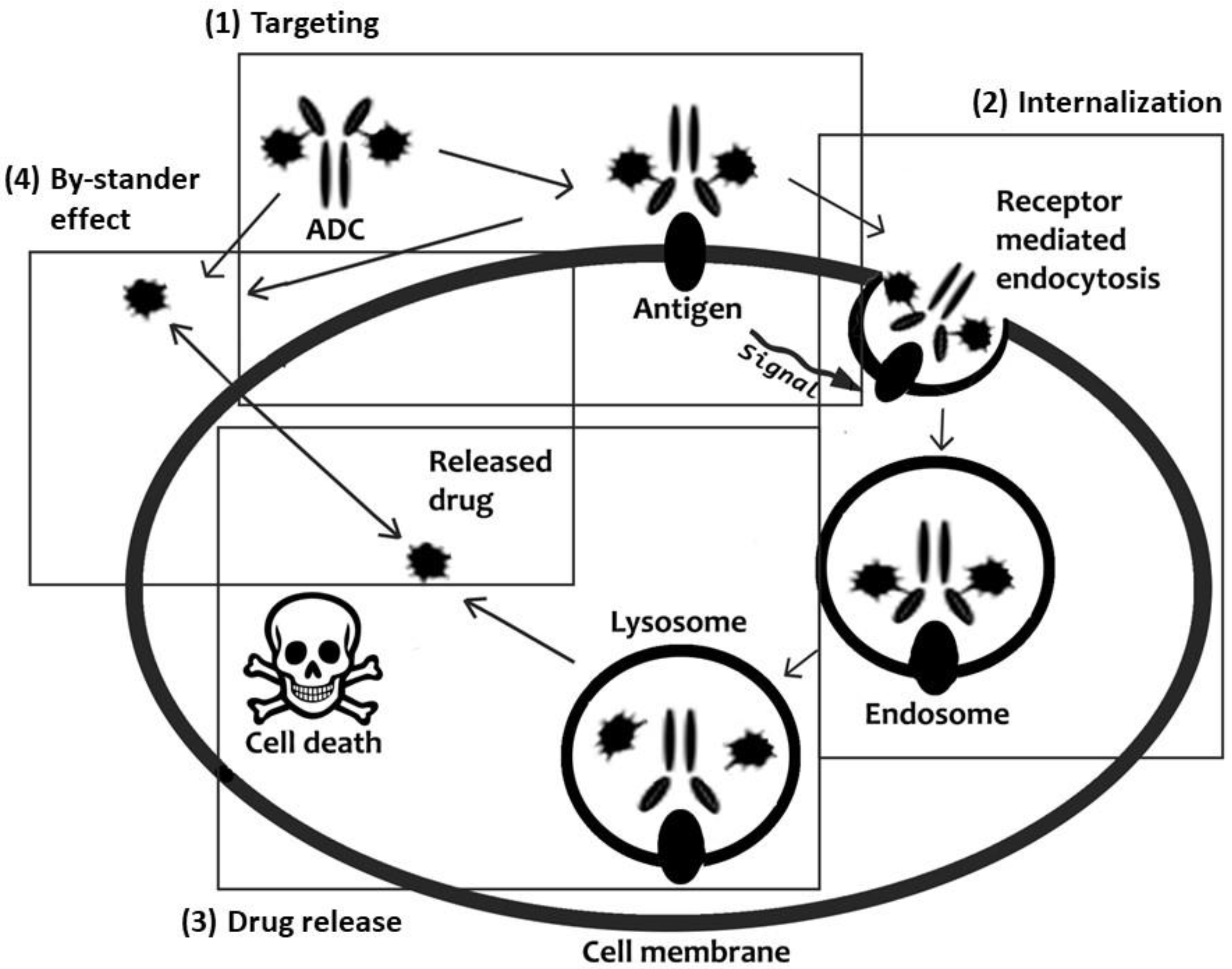
4.2. Antibody-Drug Conjugates
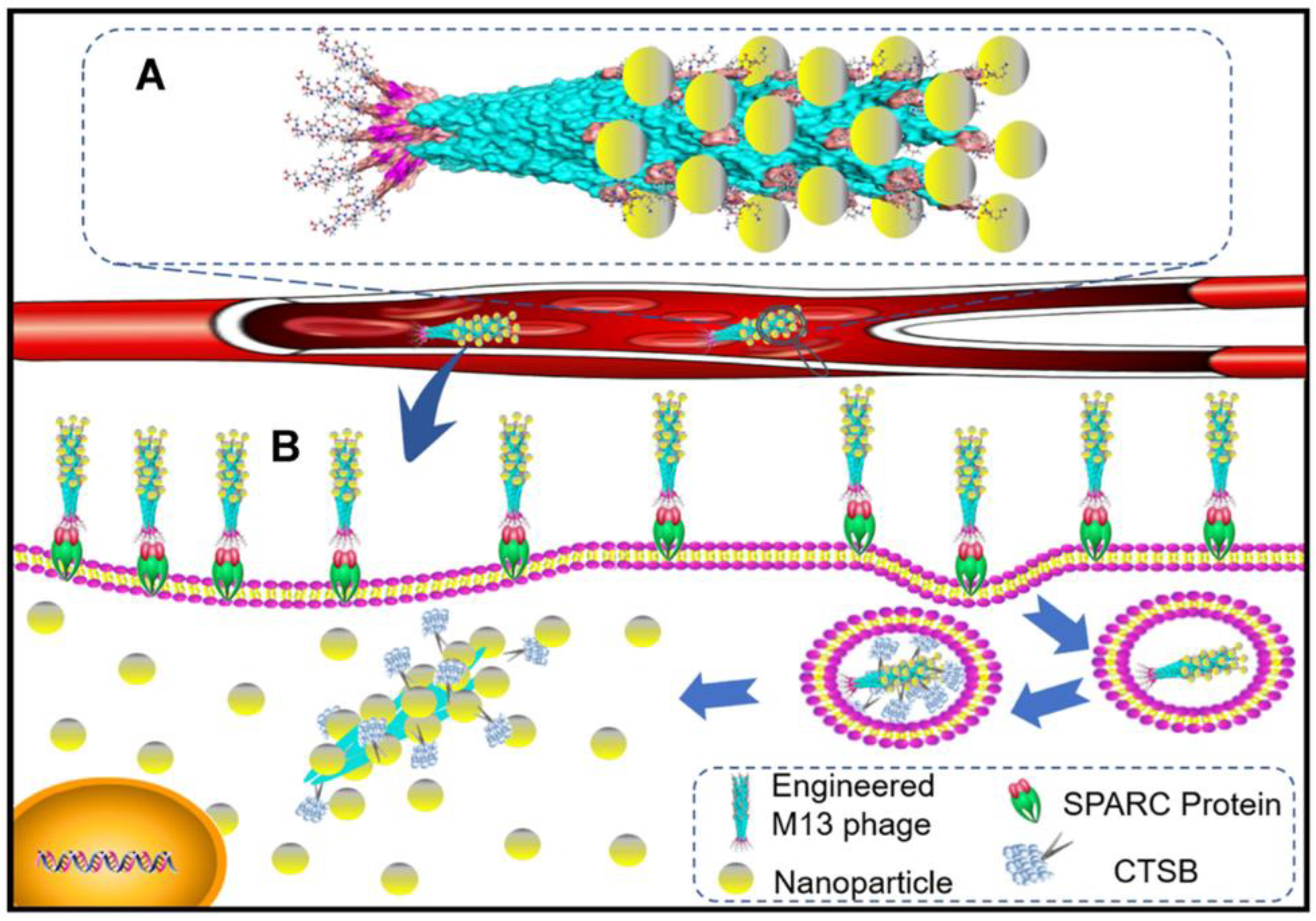
4.3. CtsB-Sensitive Nanoparticles
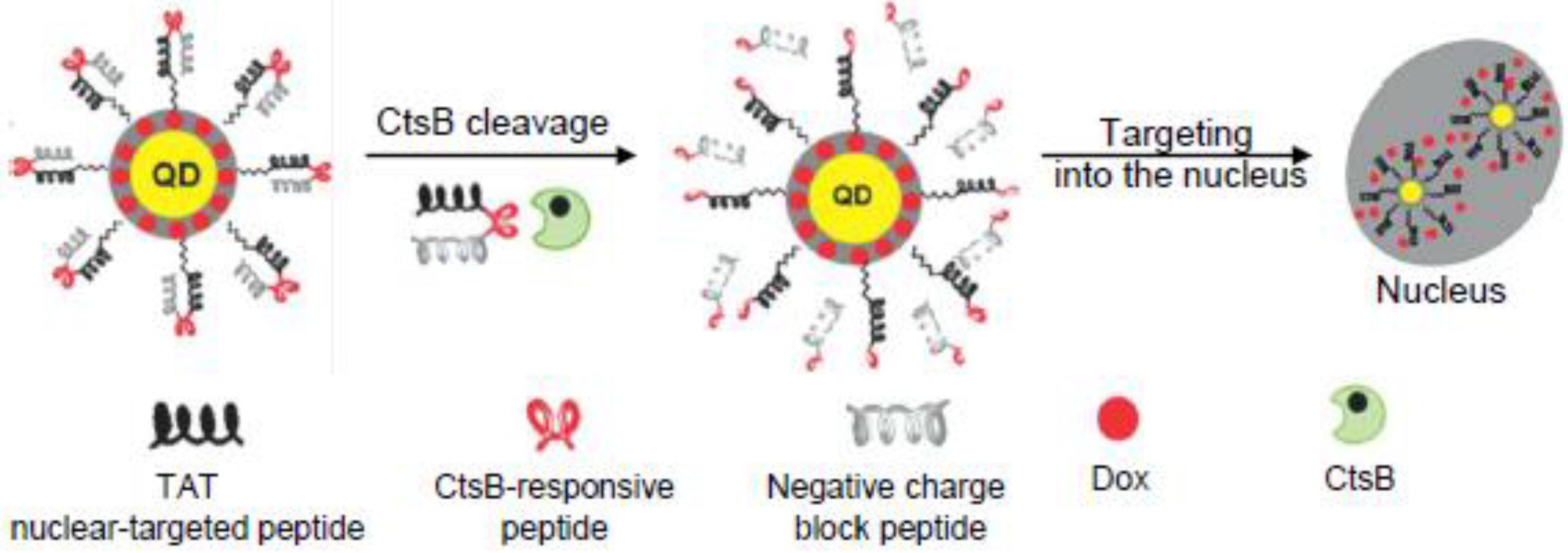
5. CtsB-Cleavable Surface Modifications
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parodi, A.; Kostyushev, D.; Brezgin, S.; Kostyusheva, A.; Borodina, T.; Akasov, R.; Frolova, A.; Chulanov, V.; Zamyatnin, A.A., Jr. Biomimetic approaches for targeting tumor inflammation. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2022. [Google Scholar] [CrossRef]
- Soltani, M.; Savvateeva, L.V.; Ganjalikhani-Hakemi, M.; Zamyatnin, A.A. Clinical Combinatorial Treatments Based on Cancer Vaccines: Combination with Checkpoint Inhibitors and Beyond. Curr. Drug Targets 2022, 23, 1072–1084. [Google Scholar] [PubMed]
- Soond, S.M.; Zamyatnin, A.A., Jr. Targeting G protein-coupled receptors in cancer therapy. Adv. Cancer Res. 2020, 145, 49–97. [Google Scholar] [PubMed]
- Vidak, E.; Javoršek, U.; Vizovišek, M.; Turk, B. Cysteine cathepsins and their extracellular roles: Shaping the microenvironment. Cells 2019, 8, 264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Withana, N.P.; Blum, G.; Sameni, M.; Slaney, C.; Anbalagan, A.; Olive, M.B.; Bidwell, B.N.; Edgington, L.; Wang, L.; Moin, K. Cathepsin B Inhibition Limits Bone Metastasis in Breast Cancer Cathepsin B Inhibition Reduces Bone Metastasis. Cancer Res. 2012, 72, 1199–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, E.; Mann, M.; Honda, K.; Vidimos, A.; Schluchter, M.D.; Straight, B.; Bogyo, M.; Popkin, D.; Basilion, J.P. Rapid visualization of nonmelanoma skin cancer. J. Am. Acad. Dermatol. 2017, 76, 209–216.e209. [Google Scholar] [CrossRef] [Green Version]
- Rudzińska, M.; Parodi, A.; Soond, S.M.; Vinarov, A.Z.; Korolev, D.O.; Morozov, A.O.; Daglioglu, C.; Tutar, Y.; Zamyatnin, A.A., Jr. The role of cysteine cathepsins in cancer progression and drug resistance. Int. J. Mol. Sci. 2019, 20, 3602. [Google Scholar] [CrossRef]
- Harbeck, N.; Alt, U.; Berger, U.; Krüger, A.; Thomssen, C.; Jänicke, F.; Höfler, H.; Kates, R.E.; Schmitt, M. Prognostic impact of proteolytic factors (urokinase-type plasminogen activator, plasminogen activator inhibitor 1, and cathepsins B, D, and L) in primary breast cancer reflects effects of adjuvant systemic therapy. Clin. Cancer Res. 2001, 7, 2757–2764. [Google Scholar]
- Kayser, K.; Richter, N.; Hufnagl, P.; Kayser, G.; Kos, J.; Werle, B. Expression, proliferation activity and clinical significance of cathepsin B and cathepsin L in operated lung cancer. Anticancer Res. 2003, 23, 2767–2772. [Google Scholar]
- Ma, K.; Chen, X.; Liu, W.; Chen, S.; Yang, C.; Yang, J. CTSB is a negative prognostic biomarker and therapeutic target associated with immune cells infiltration and immunosuppression in gliomas. Sci. Rep. 2022, 12, 4295. [Google Scholar] [CrossRef]
- Olson, O.C.; Joyce, J.A. Cysteine cathepsin proteases: Regulators of cancer progression and therapeutic response. Nat. Rev. Cancer 2015, 15, 712–729. [Google Scholar] [CrossRef]
- Sevenich, L.; Schurigt, U.; Sachse, K.; Gajda, M.; Werner, F.; Müller, S.; Vasiljeva, O.; Schwinde, A.; Klemm, N.; Deussing, J. Synergistic antitumor effects of combined cathepsin B and cathepsin Z deficiencies on breast cancer progression and metastasis in mice. Proc. Natl. Acad. Sci. USA 2010, 107, 2497–2502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Mei, T.; Han, S.; Han, T.; Sun, Y.; Zhang, H.; An, F. Cathepsin B-responsive nanodrug delivery systems for precise diagnosis and targeted therapy of malignant tumors. Chin. Chem. Lett. 2020, 31, 3027–3040. [Google Scholar] [CrossRef]
- Zamyatnin, A.A., Jr.; Gregory, L.C.; Townsend, P.A.; Soond, S.M. Beyond Basic Research: The Contribution of Cathepsin B to Cancer Development, Diagnosis and Therapy. Expert Opin. Ther. Targets 2022, 26, 963–977. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ding, Y.; Shi, C.; Yuan, F.; Sheng, X.; Liu, Y.; Xie, Y.; Lu, H.; Duan, C.; Hu, J. Identification of Cathepsin B as a Therapeutic Target for Ferroptosis of Macrophage after Spinal Cord Injury. Aging Dis. 2023. [Google Scholar]
- Tena Pérez, V.; Apaza Ticona, L.; Cabanillas, A.H.; Maderuelo Corral, S.; Rosero Valencia, D.F.; Martel Quintana, A.; Ortega Domenech, M.; Rumbero Sánchez, Á. Isolation of Nocuolin A and Synthesis of New Oxadiazine Derivatives. Design, Synthesis, Molecular Docking, Apoptotic Evaluation, and Cathepsin B Inhibition. Mar. Drugs 2023, 21, 284. [Google Scholar] [CrossRef]
- Rudzińska, M.; Parodi, A.; Maslova, V.D.; Efremov, Y.M.; Gorokhovets, N.V.; Makarov, V.A.; Popkov, V.A.; Golovin, A.V.; Zernii, E.Y.; Zamyatnin, A.A., Jr. Cysteine cathepsins inhibition affects their expression and human renal cancer cell phenotype. Cancers 2020, 12, 1310. [Google Scholar] [CrossRef]
- Park, S.-H.; Lee, J.-H.; Yang, S.-B.; Lee, D.-N.; Kang, T.-B.; Park, J. Development of a Peptide-Based Nano-Sized Cathepsin B Inhibitor for Anticancer Therapy. Pharmaceutics 2023, 15, 1131. [Google Scholar] [CrossRef]
- Xie, Z.; Zhao, M.; Yan, C.; Kong, W.; Lan, F.; Narengaowa; Zhao, S.; Yang, Q.; Bai, Z.; Qing, H. Cathepsin B in programmed cell death machinery: Mechanisms of execution and regulatory pathways. Cell Death Dis. 2023, 14, 255. [Google Scholar] [CrossRef]
- Garland, M.; Yim, J.J.; Bogyo, M. A bright future for precision medicine: Advances in fluorescent chemical probe design and their clinical application. Cell Chem. Biol. 2016, 23, 122–136. [Google Scholar] [CrossRef] [Green Version]
- Linke, M.; Herzog, V.; Brix, K. Trafficking of lysosomal cathepsin B—Green fluorescent protein to the surface of thyroid epithelial cells involves the endosomal/lysosomal compartment. J. Cell Sci. 2002, 115, 4877–4889. [Google Scholar] [CrossRef] [Green Version]
- Mort, J.S.; Buttle, D.J. Cathepsin b. Int. J. Biochem. Cell Biol. 1997, 29, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Hook, G.; Reinheckel, T.; Ni, J.; Wu, Z.; Kindy, M.; Peters, C.; Hook, V. Cathepsin B Gene Knockout Improves Behavioral Deficits and Reduces Pathology in Models of Neurologic Disorders. Pharmacol. Rev. 2022, 74, 600–629. [Google Scholar] [CrossRef] [PubMed]
- Brömme, D. Papain-like cysteine proteases. Curr. Protoc. Protein Sci. 2000, 21, 21.22.14–21.22.21. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Ye, Z.; Li, F.; Mahato, R.I. HPMA polymer-based site-specific delivery of oligonucleotides to hepatic stellate cells. Bioconjugate Chem. 2009, 20, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
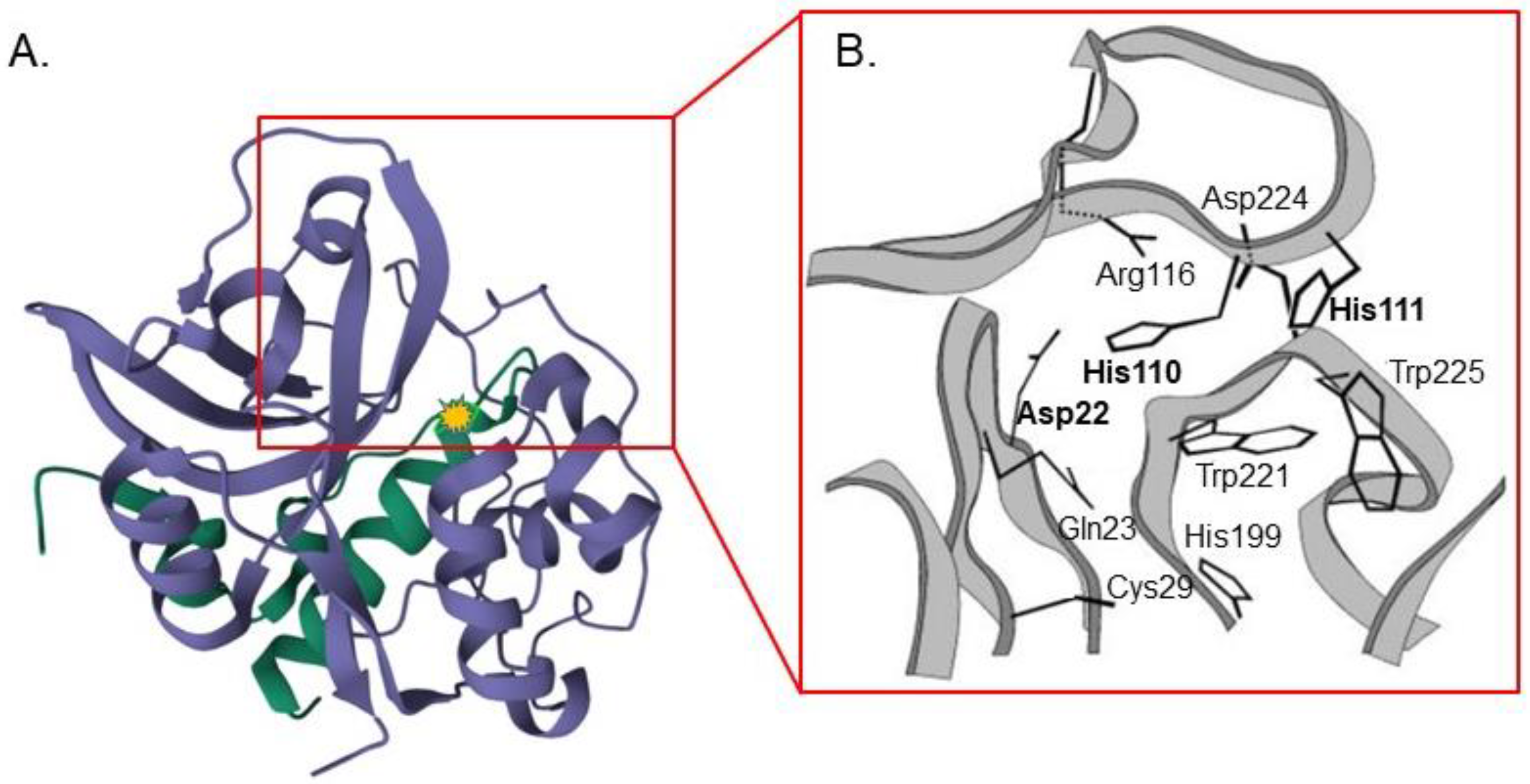
- Krupa, J.C.; Hasnain, S.; Nägler, D.K.; Menard, R.; Mort, J.S. S′ 2 substrate specificity and the role of His110 and His111 in the exopeptidase activity of human cathepsin B. Biochem. J. 2002, 361, 613–619. [Google Scholar] [CrossRef]
- Takahashi, T.; Dehdarani, A.H.; Yonezawa, S.; Tang, J. Porcine spleen cathepsin B is an exopeptidase. J. Biol. Chem. 1986, 261, 9375–9381. [Google Scholar] [CrossRef]
- Almeida, P.C.; Nantes, I.L.; Chagas, J.R.; Rizzi, C.C.; Faljoni-Alario, A.; Carmona, E.; Juliano, L.; Nader, H.B.; Tersariol, I.L. Cathepsin B activity regulation: Heparin-like glycosaminoglycans protect human cathepsin B from alkaline pH-induced inactivation. J. Biol. Chem. 2001, 276, 944–951. [Google Scholar] [CrossRef] [Green Version]
- Turk, D.; Podobnik, M.; Popovic, T.; Katunuma, N.; Bode, W.; Huber, R.; Turk, V. Crystal structure of cathepsin B inhibited with CA030 at 2.0-. ANG. resolution: A basis for the design of specific epoxysuccinyl inhibitors. Biochemistry 1995, 34, 4791–4797. [Google Scholar] [CrossRef]
- Bohley, P.; Seglen, P. Proteases and proteolysis in the lysosome. Experientia 1992, 48, 151–157. [Google Scholar] [CrossRef]
- Deussing, J.; Roth, W.; Saftig, P.; Peters, C.; Ploegh, H.L.; Villadangos, J.A. Cathepsins B and D are dispensable for major histocompatibility complex class II-mediated antigen presentation. Proc. Natl. Acad. Sci. USA 1998, 95, 4516–4521. [Google Scholar] [CrossRef] [Green Version]
- Ni, J.; Lan, F.; Xu, Y.; Nakanishi, H.; Li, X. Extralysosomal cathepsin B in central nervous system: Mechanisms and therapeutic implications. Brain Pathol. 2022, 32, e13071. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Niu, H.; Hu, Z.; Zhu, M.; Wang, L.; Han, L.; Qian, L.; Tian, K.; Yuan, H.; Lou, H. Targeting the lysosome by an aminomethylated Riccardin D triggers DNA damage through cathepsin B-mediated degradation of BRCA1. J. Cell. Mol. Med. 2019, 23, 1798–1812. [Google Scholar] [CrossRef] [Green Version]
- Brix, K.; Linke, M.; Tepel, C.; Herzog, V. Cysteine proteinases mediate extracellular prohormone processing in the thyroid. Biol. Chem. 2001, 382, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Friedrichs, B.; Tepel, C.; Reinheckel, T.; Deussing, J.; von Figura, K.; Herzog, V.; Peters, C.; Saftig, P.; Brix, K. Thyroid functions of mouse cathepsins B, K, and L. J. Clin. Investig. 2003, 111, 1733–1745. [Google Scholar] [CrossRef] [PubMed]
- Tran, A.P.; Sundar, S.; Yu, M.; Lang, B.T.; Silver, J. Modulation of receptor protein tyrosine phosphatase sigma increases chondroitin sulfate proteoglycan degradation through cathepsin B secretion to enhance axon outgrowth. J. Neurosci. 2018, 38, 5399–5414. [Google Scholar] [CrossRef]
- Droga-Mazovec, G.; Bojic, L.; Petelin, A.; Ivanova, S.; Repnik, U.; Salvesen, G.S.; Stoka, V.; Turk, V.; Turk, B. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J. Biol. Chem. 2008, 283, 19140–19150. [Google Scholar] [CrossRef] [Green Version]
- Ni, J.; Wu, Z.; Stoka, V.; Meng, J.; Hayashi, Y.; Peters, C.; Qing, H.; Turk, V.; Nakanishi, H. Increased expression and altered subcellular distribution of cathepsin B in microglia induce cognitive impairment through oxidative stress and inflammatory response in mice. Aging Cell 2019, 18, e12856. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhong, C.; Shi, L.; Guo, Y.; Fan, Z. Granulysin induces cathepsin B release from lysosomes of target tumor cells to attack mitochondria through processing of bid leading to Necroptosis. J. Immunol. 2009, 182, 6993–7000. [Google Scholar] [CrossRef] [Green Version]
- Meng, J.; Liu, Y.; Xie, Z.; Qing, H.; Lei, P.; Ni, J. Nucleus distribution of cathepsin B in senescent microglia promotes brain aging through degradation of sirtuins. Neurobiol. Aging 2020, 96, 255–266. [Google Scholar] [CrossRef]
- Vasiljeva, O.; Korovin, M.; Gajda, M.; Brodoefel, H.; Bojic, L.; Krüger, A.; Schurigt, U.; Sevenich, L.; Turk, B.; Peters, C. Reduced tumour cell proliferation and delayed development of high-grade mammary carcinomas in cathepsin B-deficient mice. Oncogene 2008, 27, 4191–4199. [Google Scholar] [CrossRef] [Green Version]
- Mijanović, O.; Branković, A.; Panin, A.N.; Savchuk, S.; Timashev, P.; Ulasov, I.; Lesniak, M.S. Cathepsin B: A sellsword of cancer progression. Cancer Lett. 2019, 449, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Kuester, D.; Lippert, H.; Roessner, A.; Krueger, S. The cathepsin family and their role in colorectal cancer. Pathol. Res. Pract. 2008, 204, 491–500. [Google Scholar] [CrossRef]
- Kolwijck, E.; Massuger, L.F.; Thomas, C.M.; Span, P.N.; Krasovec, M.; Kos, J.; Sweep, F.C. Cathepsins B, L and cystatin C in cyst fluid of ovarian tumors. J. Cancer Res. Clin. Oncol. 2010, 136, 771–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebert, M.P.; Krüger, S.; Fogeron, M.L.; Lamer, S.; Chen, J.; Pross, M.; Schulz, H.U.; Lage, H.; Heim, S.; Roessner, A. Overexpression of cathepsin B in gastric cancer identified by proteome analysis. Proteomics 2005, 5, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Rudzinska-Radecka, M.; Frolova, A.S.; Balakireva, A.V.; Gorokhovets, N.V.; Pokrovsky, V.S.; Sokolova, D.V.; Korolev, D.O.; Potoldykova, N.V.; Vinarov, A.Z.; Parodi, A. In Silico, In Vitro, and Clinical Investigations of Cathepsin B and Stefin A mRNA Expression and a Correlation Analysis in Kidney Cancer. Cells 2022, 11, 1455. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, M.M.; Day, M.L. Soluble E-cadherin: More than a symptom of disease. Front. Biosci. 2012, 17, 1948. [Google Scholar] [CrossRef] [Green Version]
- Mitrović, A.; Fonović, U.P.; Kos, J. Cysteine cathepsins B and X promote epithelial-mesenchymal transition of tumor cells. Eur. J. Cell Biol. 2017, 96, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Nettesheim, A.; Shim, M.S.; Dixon, A.; Raychaudhuri, U.; Gong, H.; Liton, P.B. Cathepsin B localizes in the caveolae and participates in the proteolytic cascade in trabecular meshwork cells. Potential new drug target for the treatment of glaucoma. J. Clin. Med. 2020, 10, 78. [Google Scholar] [CrossRef]
- Ruan, J.; Zheng, H.; Rong, X.; Rong, X.; Zhang, J.; Fang, W.; Zhao, P.; Luo, R. Over-expression of cathepsin B in hepatocellular carcinomas predicts poor prognosis of HCC patients. Mol. Cancer 2016, 15, 17. [Google Scholar] [CrossRef] [Green Version]
- Nakao, S.; Zandi, S.; Sun, D.; Hafezi-Moghadam, A. Cathepsin B-mediated CD18 shedding regulates leukocyte recruitment from angiogenic vessels. FASEB J. 2018, 32, 143. [Google Scholar] [CrossRef] [Green Version]
- Gondi, C.S.; Kandhukuri, N.; Kondraganti, S.; Gujrati, M.; Olivero, W.C.; Dinh, D.H.; Rao, J.S. RNA interference–mediated simultaneous down-regulation of urokinase-type plasminogen activator receptor and cathepsin B induces caspase-8–mediated apoptosis in SNB19 human glioma cells. Mol. Cancer Ther. 2006, 5, 3197–3208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gondi, C.S.; Lakka, S.S.; Yanamandra, N.; Olivero, W.C.; Dinh, D.H.; Gujrati, M.; Tung, C.; Weissleder, R.; Rao, J.S. Adenovirus-mediated expression of antisense urokinase plasminogen activator receptor and antisense cathepsin B inhibits tumor growth, invasion, and angiogenesis in gliomas. Cancer Res. 2004, 64, 4069–4077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malla, R.R.; Gopinath, S.; Gondi, C.S.; Alapati, K.; Dinh, D.H.; Gujrati, M.; Rao, J.S. Cathepsin B and uPAR knockdown inhibits tumor-induced angiogenesis by modulating VEGF expression in glioma. Cancer Gene Ther. 2011, 18, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Tummalapalli, P.; Spomar, D.; Gondi, C.S.; Olivero, W.C.; Gujrati, M.; Dinh, D.H.; Rao, J.S. RNAi-mediated abrogation of cathepsin B and MMP-9 gene expression in a malignant meningioma cell line leads to decreased tumor growth, invasion and angiogenesis. Int. J. Oncol. 2007, 31, 1039–1050. [Google Scholar] [PubMed] [Green Version]
- Nalla, A.K.; Gorantla, B.; Gondi, C.S.; Lakka, S.S.; Rao, J.S. Targeting MMP-9, uPAR, and cathepsin B inhibits invasion, migration and activates apoptosis in prostate cancer cells. Cancer Gene Ther. 2010, 17, 599–613. [Google Scholar] [CrossRef] [Green Version]
- Bengsch, F.; Buck, A.; Günther, S.; Seiz, J.; Tacke, M.; Pfeifer, D.; Von Elverfeldt, D.; Sevenich, L.; Hillebrand, L.; Kern, U. Cell type-dependent pathogenic functions of overexpressed human cathepsin B in murine breast cancer progression. Oncogene 2014, 33, 4474–4484. [Google Scholar] [CrossRef] [Green Version]
- Wei, B.; Gunzner-Toste, J.; Yao, H.; Wang, T.; Wang, J.; Xu, Z.; Chen, J.; Wai, J.; Nonomiya, J.; Tsai, S.P. Discovery of peptidomimetic antibody–drug conjugate linkers with enhanced protease specificity. J. Med. Chem. 2018, 61, 989–1000. [Google Scholar] [CrossRef]
- Patel, D.K.; Menon, D.V.; Patel, D.H.; Dave, G. Linkers: A synergistic way for the synthesis of chimeric proteins. Protein Expr. Purif. 2022, 191, 106012. [Google Scholar] [CrossRef]
- Schmitz, J.; Gilberg, E.; Löser, R.; Bajorath, J.; Bartz, U.; Gütschow, M. Cathepsin B: Active site mapping with peptidic substrates and inhibitors. Bioorganic Med. Chem. 2019, 27, 1–15. [Google Scholar] [CrossRef]
- Loganzo, F.; Sung, M.; Gerber, H.-P. Mechanisms of resistance to antibody–drug conjugates. Mol. Cancer Ther. 2016, 15, 2825–2834. [Google Scholar] [CrossRef] [Green Version]
- Yin, Q.; Shen, J.; Zhang, Z.; Yu, H.; Li, Y. Reversal of multidrug resistance by stimuli-responsive drug delivery systems for therapy of tumor. Adv. Drug Deliv. Rev. 2013, 65, 1699–1715. [Google Scholar] [CrossRef] [PubMed]
- Shim, M.K.; Moon, Y.; Yang, S.; Kim, J.; Cho, H.; Lim, S.; Yoon, H.Y.; Seong, J.-K.; Kim, K. Cancer-specific drug-drug nanoparticles of pro-apoptotic and cathepsin B-cleavable peptide-conjugated doxorubicin for drug-resistant cancer therapy. Biomaterials 2020, 261, 120347. [Google Scholar] [CrossRef] [PubMed]
- Rejmanová, P.; Kopeček, J.; Pohl, J.; Baudyš, M.; Kostka, V. Polymers containing enzymatically degradable bonds, 8. Degradation of oligopeptide sequences in N-(2-hydroxypropyl) methacrylamide copolymers by bovine spleen cathepsin B. Die Makromol. Chem. 1983, 184, 2009–2020. [Google Scholar] [CrossRef]
- Omelyanenko, V.; Gentry, C.; Kopečková, P.; Kopeček, J. HPMA copolymer–anticancer drug–OV-TL16 antibody conjugates. II. Processing in epithelial ovarian carcinoma cells in vitro. Int. J. Cancer 1998, 75, 600–608. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, P.; Cheetham, A.G.; Moon, J.H.; Moxley, J.W., Jr.; Lin, Y.-a.; Cui, H. Controlled release of free doxorubicin from peptide–drug conjugates by drug loading. J. Control. Release 2014, 191, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Dubikovskaya, E.A.; Thorne, S.H.; Pillow, T.H.; Contag, C.H.; Wender, P.A. Overcoming multidrug resistance of small-molecule therapeutics through conjugation with releasable octaarginine transporters. Proc. Natl. Acad. Sci. USA 2008, 105, 12128–12133. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Lock, L.L.; Cheetham, A.G.; Cui, H. Enhanced cellular entry and efficacy of tat conjugates by rational design of the auxiliary segment. Mol. Pharm. 2014, 11, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Lammers, T.; Peschke, P.; Kühnlein, R.; Subr, V.; Ulbrich, K.; Debus, J.; Huber, P.; Hennink, W.; Storm, G. Effect of radiotherapy and hyperthermia on the tumor accumulation of HPMA copolymer-based drug delivery systems. J. Control. Release 2007, 117, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Minko, T.; Kopečková, P.; Kopeček, J. Comparison of the anticancer effect of free and HPMA copolymer-bound adriamycin in human ovarian carcinoma cells. Pharm. Res. 1999, 16, 986–996. [Google Scholar] [CrossRef]
- Drobnik, J.; Kopeček, J.; Labský, J.; Rejmanová, P.; Exner, J.; Saudek, V.; Kálal, J. Enzymatic cleavage of side chains of synthetic water-soluble polymers. Die Makromol. Chem. Macromol. Chem. Phys. 1976, 177, 2833–2848. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, R.; Pan, H.; Li, Y.; Fang, Y.; Zhang, L.; Kopeček, J.i. Backbone degradable N-(2-hydroxypropyl) methacrylamide copolymer conjugates with gemcitabine and paclitaxel: Impact of molecular weight on activity toward human ovarian carcinoma xenografts. Mol. Pharm. 2017, 14, 1384–1394. [Google Scholar] [CrossRef]
- Vasey, P.A.; Kaye, S.B.; Morrison, R.; Twelves, C.; Wilson, P.; Duncan, R.; Thomson, A.H.; Murray, L.S.; Hilditch, T.E.; Murray, T. Phase I clinical and pharmacokinetic study of PK1 [N-(2-hydroxypropyl) methacrylamide copolymer doxorubicin]: First member of a new class of chemotherapeutic agents—Drug-polymer conjugates. Clin. Cancer Res. 1999, 5, 83–94. [Google Scholar]
- Santos, S.d.S.; Gonzaga, R.V.; Silva, J.V.; Savino, D.F.; Prieto, D.; Shikay, J.M.; Silva, R.S.; Paulo, L.H.d.A.; Ferreira, E.I.; Giarolla, J. Peptide dendrimers: Drug/gene delivery and other approaches. Can. J. Chem. 2017, 95, 907–916. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Zhuang, X.; Tang, Z.; Tian, H.; Chen, X. Stimuli-sensitive synthetic polypeptide-based materials for drug and gene delivery. Adv. Healthc. Mater. 2012, 1, 48–78. [Google Scholar] [CrossRef]
- Lee, C.C.; Gillies, E.R.; Fox, M.E.; Guillaudeu, S.J.; Fréchet, J.M.; Dy, E.E.; Szoka, F.C. A single dose of doxorubicin-functionalized bow-tie dendrimer cures mice bearing C-26 colon carcinomas. Proc. Natl. Acad. Sci. USA 2006, 103, 16649–16654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Gao, H.; Zhao, Z.; Rostami, I.; Wang, C.; Zhu, L.; Yang, Y. Improved tumor targeting and penetration by a dual-functional poly (amidoamine) dendrimer for the therapy of triple-negative breast cancer. J. Mater. Chem. B 2019, 7, 3724–3736. [Google Scholar] [CrossRef]
- Wu, Z.-Y.; Shen, J.-M.; Lang, H.; Yue, T.; Sun, C. pH/Enzyme dual sensitive and nucleus-targeting dendrimer nanoparticles to enhance the antitumour activity of doxorubicin. Pharm. Dev. Technol. 2022, 27, 357–371. [Google Scholar] [CrossRef]
- Jain, M.; Bouilloux, J.; Borrego, I.; Cook, S.; van den Bergh, H.; Lange, N.; Wagnieres, G.; Giraud, M.-N. Cathepsin B-Cleavable Polymeric Photosensitizer Prodrug for Selective Photodynamic Therapy: In Vitro Studies. Pharmaceuticals 2022, 15, 564. [Google Scholar] [CrossRef]
- Wilcox, D.; Mason, R.W. Inhibition of cysteine proteinases in lysosomes and whole cells. Biochem. J. 1992, 285, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Sun, Z.; Wang, K.; Li, N.; Chen, H.; Tan, X.; Li, L.; He, Z.; Sun, J. Multifunctional tumor-targeting cathepsin B-sensitive gemcitabine prodrug covalently targets albumin in situ and improves cancer therapy. Bioconjugate Chem. 2018, 29, 1852–1858. [Google Scholar] [CrossRef]
- Ford, C.; Newman, C.; Johnson, J.; Woodhouse, C.; Reeder, T.; Rowland, G.; Simmonds, R. Localisation and toxicity study of a vindesine-anti-CEA conjugate in patients with advanced cancer. Br. J. Cancer 1983, 47, 35–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponziani, S.; Di Vittorio, G.; Pitari, G.; Cimini, A.M.; Ardini, M.; Gentile, R.; Iacobelli, S.; Sala, G.; Capone, E.; Flavell, D.J. Antibody-drug conjugates: The new frontier of chemotherapy. Int. J. Mol. Sci. 2020, 21, 5510. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Smith, S.W.; Ghone, S.; Tomczuk, B. Current ADC linker chemistry. Pharm. Res. 2015, 32, 3526–3540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, J.T.; Harris, P.W.; Brimble, M.A.; Kavianinia, I. An insight into FDA approved antibody-drug conjugates for cancer therapy. Molecules 2021, 26, 5847. [Google Scholar] [CrossRef] [PubMed]
- Bryden, F.; Martin, C.; Letast, S.; Lles, E.; Viéitez-Villemin, I.; Rousseau, A.; Colas, C.; Brachet-Botineau, M.; Allard-Vannier, E.; Larbouret, C. Impact of cathepsin B-sensitive triggers and hydrophilic linkers on in vitro efficacy of novel site-specific antibody–drug conjugates. Org. Biomol. Chem. 2018, 16, 1882–1889. [Google Scholar] [CrossRef]
- Dubowchik, G.M.; Mosure, K.; Knipe, J.O.; Firestone, R.A. Cathepsin B-sensitive dipeptide prodrugs. 2. Models of anticancer drugs paclitaxel (Taxol®), mitomycin C and doxorubicin. Bioorganic Med. Chem. Lett. 1998, 8, 3347–3352. [Google Scholar] [CrossRef]
- Fu, Y.; Urban, D.J.; Nani, R.R.; Zhang, Y.F.; Li, N.; Fu, H.; Shah, H.; Gorka, A.P.; Guha, R.; Chen, L. Glypican-3-specific antibody drug conjugates targeting hepatocellular carcinoma. Hepatology 2019, 70, 563–576. [Google Scholar] [CrossRef]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody–drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.-B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J. Trastuzumab deruxtecan in previously treated HER2-positive breast cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef]
- Shitara, K.; Bang, Y.-J.; Iwasa, S.; Sugimoto, N.; Ryu, M.-H.; Sakai, D.; Chung, H.-C.; Kawakami, H.; Yabusaki, H.; Lee, J. Trastuzumab deruxtecan in previously treated HER2-positive gastric cancer. N. Engl. J. Med. 2020, 382, 2419–2430. [Google Scholar] [CrossRef]
- Narang, P. ENHERTU® Approved in the US as the First HER2 Directed Therapy for Patients with Previously Treated HER2 Mutant Metastatic Non-Small Cell Lung Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-fam-trastuzumab-deruxtecan-nxki-her2-mutant-non-small-cell-lung (accessed on 15 June 2023).
- Powles, T.; Rosenberg, J.E.; Sonpavde, G.P.; Loriot, Y.; Durán, I.; Lee, J.-L.; Matsubara, N.; Vulsteke, C.; Castellano, D.; Wu, C. Enfortumab vedotin in previously treated advanced urothelial carcinoma. New Engl. J. Med. 2021, 384, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Tisotumab vedotin: First approval. Drugs 2021, 81, 2141–2147. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Liu, Y.; Zhou, X.; Shen, P.; Xue, R.; Zhang, M. Disitamab vedotin: A novel antibody-drug conjugates for cancer therapy. Drug Deliv. 2022, 29, 1335–1344. [Google Scholar] [CrossRef] [PubMed]
- Caculitan, N.G.; Chuh, J.D.C.; Ma, Y.; Zhang, D.; Kozak, K.R.; Liu, Y.; Pillow, T.H.; Sadowsky, J.; Cheung, T.K.; Phung, Q. Cathepsin B is dispensable for cellular processing of cathepsin B-cleavable antibody-drug conjugates. Cancer Res 2017, 77, 7027–7037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kung Sutherland, M.S.; Walter, R.B.; Jeffrey, S.C.; Burke, P.J.; Yu, C.; Kostner, H.; Stone, I.; Ryan, M.C.; Sussman, D.; Lyon, R.P. SGN-CD33A: A novel CD33-targeting antibody–drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood J. Am. Soc. Hematol. 2013, 122, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.K.; Forero-Torres, A.; Thompson, J.A.; Morris, J.C.; Chhabra, S.; Hoimes, C.J.; Vogelzang, N.J.; Boyd, T.; Bergerot, P.G.; Adashek, J.J. A phase 1 trial of SGN-CD70A in patients with CD70-positive, metastatic renal cell carcinoma. Cancer 2019, 125, 1124–1132. [Google Scholar] [CrossRef]
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody–drug conjugates: The last decade. Pharmaceuticals 2020, 13, 245. [Google Scholar] [CrossRef]
- Li, F.; Sutherland, M.K.; Yu, C.; Walter, R.B.; Westendorf, L.; Valliere-Douglass, J.; Pan, L.; Cronkite, A.; Sussman, D.; Klussman, K. Characterization of SGN-CD123A, A Potent CD123-Directed Antibody–Drug Conjugate for Acute Myeloid LeukemiaPreclinical Evaluation of SGN-CD123A. Mol. Cancer Ther. 2018, 17, 554–564. [Google Scholar] [CrossRef] [Green Version]
- Kahl, B.S.; Hamadani, M.; Radford, J.; Carlo-Stella, C.; Caimi, P.; Reid, E.; Feingold, J.M.; Ardeshna, K.M.; Solh, M.; Heffner, L.T. A Phase I Study of ADCT-402 (Loncastuximab Tesirine), a Novel Pyrrolobenzodiazepine-Based Antibody–Drug Conjugate, in Relapsed/Refractory B-Cell Non-Hodgkin Lymphoma Phase I Study of ADCT-402 in Relapsed/Refractory B-Cell NHL. Clin. Cancer Res. 2019, 25, 6986–6994. [Google Scholar] [CrossRef] [Green Version]
- Hamadani, M.; Collins, G.P.; Caimi, P.F.; Samaniego, F.; Spira, A.; Davies, A.; Radford, J.; Menne, T.; Karnad, A.; Zain, J.M. Camidanlumab tesirine in patients with relapsed or refractory lymphoma: A phase 1, open-label, multicentre, dose-escalation, dose-expansion study. Lancet Haematol. 2021, 8, e433–e445. [Google Scholar] [CrossRef]
- Dorywalska, M.; Dushin, R.; Moine, L.; Farias, S.E.; Zhou, D.; Navaratnam, T.; Lui, V.; Hasa-Moreno, A.; Casas, M.G.; Tran, T.-T. Molecular Basis of Valine-Citrulline-PABC Linker Instability in Site-Specific ADCs and Its Mitigation by Linker DesignMolecular Basis of VC-PABC Linker Instability. Mol. Cancer Ther. 2016, 15, 958–970. [Google Scholar] [CrossRef] [Green Version]
- Anami, Y.; Yamazaki, C.M.; Xiong, W.; Gui, X.; Zhang, N.; An, Z.; Tsuchikama, K. Glutamic acid–valine–citrulline linkers ensure stability and efficacy of antibody–drug conjugates in mice. Nat. Commun. 2018, 9, 2512. [Google Scholar] [CrossRef] [Green Version]
- ud Din, F.; Aman, W.; Ullah, I.; Qureshi, O.S.; Mustapha, O.; Shafique, S.; Zeb, A. Effective use of nanocarriers as drug delivery systems for the treatment of selected tumors. Int. J. Nanomed. 2017, 12, 7291. [Google Scholar] [CrossRef] [Green Version]
- Chamundeeswari, M.; Jeslin, J.; Verma, M.L. Nanocarriers for drug delivery applications. Environ. Chem. Lett. 2019, 17, 849–865. [Google Scholar] [CrossRef]
- Sun, T.; Zhang, Y.S.; Pang, B.; Hyun, D.C.; Yang, M.; Xia, Y. Engineered nanoparticles for drug delivery in cancer therapy. Nanomater. Neoplasms 2021, 53, 31–142. [Google Scholar]
- Ye, Z.; Zhang, Y.; Liu, Y.; Liu, Y.; Tu, J.; Shen, Y. EGFR targeted cetuximab-valine-citrulline (vc)-doxorubicin immunoconjugates-loaded bovine serum albumin (BSA) nanoparticles for colorectal tumor therapy. Int. J. Nanomed. 2021, 16, 2443. [Google Scholar] [CrossRef] [PubMed]
- Kolesova, E.P.; Egorova, V.S.; Syrocheva, A.O.; Frolova, A.S.; Kostyushev, D.; Kostyusheva, A.; Brezgin, S.; Trushina, D.B.; Fatkhutdinova, L.; Zyuzin, M. Proteolytic Resistance Determines Albumin Nanoparticle Drug Delivery Properties and Increases Cathepsin B, D, and G Expression. Int. J. Mol. Sci. 2023, 24, 10245. [Google Scholar] [CrossRef] [PubMed]
- Satsangi, A.; Roy, S.S.; Satsangi, R.K.; Tolcher, A.W.; Vadlamudi, R.K.; Goins, B.; Ong, J.L. Synthesis of a novel, sequentially active-targeted drug delivery nanoplatform for breast cancer therapy. Biomaterials 2015, 59, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Satsangi, A.; Roy, S.S.; Satsangi, R.K.; Vadlamudi, R.K.; Ong, J.L. Design of a paclitaxel prodrug conjugate for active targeting of an enzyme upregulated in breast cancer cells. Mol. Pharm. 2014, 11, 1906–1918. [Google Scholar] [CrossRef]
- Cha, H.; Yoon, J.H.; Yoon, S. Probing quantum plasmon coupling using gold nanoparticle dimers with tunable interparticle distances down to the subnanometer range. ACS Nano 2014, 8, 8554–8563. [Google Scholar] [CrossRef]
- Zhang, N.; Wu, H.; Liang, Y.; Ye, J.; Zhang, H.; Miao, Y.; Luo, Y.; Fan, H.; Yue, T. Design and Preparation of “corn-like” SPIONs@ DFK-SBP-M13 Assembly for Improvement of Effective Internalization. Int. J. Nanomed. 2021, 16, 7091. [Google Scholar] [CrossRef] [PubMed]
- Ehrsam, D.; Porta, F.; Hussner, J.; Seibert, I.; Meyer zu Schwabedissen, H.E. PDMS-PMOXA-Nanoparticles featuring a cathepsin B-triggered release mechanism. Materials 2019, 12, 2836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montaser, M.; Lalmanach, G.; Mach, L. CA-074, But not Its Methyl Ester CA-074Me, is a Selective Inhibitor of Cathepsin B within Living Cells; Walter de Gruyter: Berlin, Germany, 2002. [Google Scholar]
- Ai, X.; Ho, C.J.H.; Aw, J.; Attia, A.B.E.; Mu, J.; Wang, Y.; Wang, X.; Wang, Y.; Liu, X.; Chen, H. In vivo covalent cross-linking of photon-converted rare-earth nanostructures for tumour localization and theranostics. Nat. Commun. 2016, 7, 10432. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, A.; Böttger, R.; Li, S.-D. Recent trends in bioresponsive linker technologies of Prodrug-Based Self-Assembling nanomaterials. Biomaterials 2021, 275, 120955. [Google Scholar] [CrossRef]
- Dong, X.; Brahma, R.K.; Fang, C.; Yao, S. Stimulus-responsive self-assembled prodrugs in cancer therapy. Chem. Sci. 2022, 13, 4239–4269. [Google Scholar] [CrossRef]
- German, H.W.; Uyaver, S.; Hansmann, U.H. Self-assembly of phenylalanine-based molecules. J. Phys. Chem. A 2014, 119, 1609–1615. [Google Scholar] [CrossRef]
- Ben-Nun, Y.; Fichman, G.; Adler-Abramovich, L.; Turk, B.; Gazit, E.; Blum, G. Cathepsin nanofiber substrates as potential agents for targeted drug delivery. J. Control. Release 2017, 257, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Shim, N.; Jeon, S.I.; Yang, S.; Park, J.Y.; Jo, M.; Kim, J.; Choi, J.; Yun, W.S.; Kim, J.; Lee, Y. Comparative study of cathepsin B-cleavable linkers for the optimal design of cathepsin B-specific doxorubicin prodrug nanoparticles for targeted cancer therapy. Biomaterials 2022, 289, 121806. [Google Scholar] [CrossRef]
- Huang, C.-H.; Chang, E.; Zheng, L.; Raj, J.G.J.; Wu, W.; Pisani, L.J.; Daldrup-Link, H.E. Tumor protease-activated theranostic nanoparticles for MRI-guided glioblastoma therapy. Theranostics 2023, 13, 1745. [Google Scholar] [CrossRef]
- Shi, Z.; Yang, Y.; Guo, Z.; Feng, S.; Wan, Y. A cathepsin B/GSH dual-responsive fluorinated peptide for effective siRNA delivery to cancer cells. Bioorganic Chem. 2023, 135, 106485. [Google Scholar] [CrossRef]
- He, X.; Alves, C.S.; Oliveira, N.; Rodrigues, J.; Zhu, J.; Bányai, I.; Tomás, H.; Shi, X. RGD peptide-modified multifunctional dendrimer platform for drug encapsulation and targeted inhibition of cancer cells. Colloids Surf. B Biointerfaces 2015, 125, 82–89. [Google Scholar] [CrossRef] [Green Version]
- Agostini, A.; Mondragón, L.; Coll, C.; Aznar, E.; Marcos, M.D.; Martínez-Máñez, R.; Sancenón, F.; Soto, J.; Pérez-Payá, E.; Amorós, P. Dual enzyme-triggered controlled release on capped nanometric silica mesoporous supports. ChemistryOpen 2012, 1, 17–20. [Google Scholar] [CrossRef]
- Singh, N.; Karambelkar, A.; Gu, L.; Lin, K.; Miller, J.S.; Chen, C.S.; Sailor, M.J.; Bhatia, S.N. Bioresponsive mesoporous silica nanoparticles for triggered drug release. J. Am. Chem. Soc. 2011, 133, 19582–19585. [Google Scholar] [CrossRef]
- Stephen, S.; Gorain, B.; Choudhury, H.; Chatterjee, B. Exploring the role of mesoporous silica nanoparticle in the development of novel drug delivery systems. Drug Deliv. Transl. Res. 2021, 12, 105–123. [Google Scholar] [CrossRef]
- Cheng, Y.-J.; Luo, G.-F.; Zhu, J.-Y.; Xu, X.-D.; Zeng, X.; Cheng, D.-B.; Li, Y.-M.; Wu, Y.; Zhang, X.-Z.; Zhuo, R.-X. Enzyme-induced and tumor-targeted drug delivery system based on multifunctional mesoporous silica nanoparticles. ACS Appl. Mater. Interfaces 2015, 7, 9078–9087. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, C.; Mondragón, L.; Coll, C.; Sancenón, F.; Marcos, M.D.; Martínez-Máñez, R.; Amorós, P.; Pérez-Payá, E.; Orzáez, M. Cathepsin-B induced controlled release from peptide-capped mesoporous silica nanoparticles. Chem. A Eur. J. 2014, 20, 15309–15314. [Google Scholar] [CrossRef]
- Li, J.; Liu, F.; Shao, Q.; Min, Y.; Costa, M.; Yeow, E.K.; Xing, B. Enzyme-responsive cell-penetrating peptide conjugated mesoporous silica quantum dot nanocarriers for controlled release of nucleus-targeted drug molecules and real-time intracellular fluorescence imaging of tumor cells. Adv. Healthc. Mater. 2014, 3, 1230–1239. [Google Scholar] [CrossRef] [PubMed]
- Zhi, X.; Jiang, Y.; Xie, L.; Li, Y.; Fang, C.-J. Gold Nanorods Functionalized with Cathepsin B Targeting Peptide and Doxorubicin for Combinatorial Therapy against Multidrug Resistance. ACS Appl. Bio Mater. 2019, 2, 5697–5706. [Google Scholar] [CrossRef]
- Gotov, O.; Battogtokh, G.; Ko, Y.T. Docetaxel-loaded hyaluronic acid–cathepsin b-cleavable-peptide–gold nanoparticles for the treatment of cancer. Mol. Pharm. 2018, 15, 4668–4676. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Song, C.; Zhang, J.; Zhao, X. Targeted delivery and apoptosis induction activity of peptide-transferrin targeted mesoporous silica encapsulated resveratrol in MCF-7 cells. J. Pharm. Pharmacol. 2023, 75, 49–56. [Google Scholar] [CrossRef]
- Lee, A. Loncastuximab tesirine: First approval. Drugs 2021, 81, 1229–1233. [Google Scholar] [CrossRef]
- Shultes, K.C. Loncastuximab Tesirine-lpyl (Zynlonta®). Oncol. Times 2022, 44, 14. [Google Scholar] [CrossRef]
- Younes, A.; Yasothan, U.; Kirkpatrick, P. Brentuximab vedotin. Nat. Rev. Drug Discov. 2012, 11, 19. [Google Scholar] [CrossRef]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Polatuzumab vedotin: First global approval. Drugs 2019, 79, 1467–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raedler, L.A. Padcev (Enfortumab Vedotin-ejfv) FDA Approved for the Treatment of Metastatic Urothelial Carcinoma. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-regular-approval-enfortumab-vedotin-ejfv-locally-advanced-or-metastatic-urothelial-cancer (accessed on 15 June 2023).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Linker | Chemical Structure | Molecular Weight | Net Charge | Isoelectrical Point |
|---|---|---|---|---|
| GFLG |  | 392.2 | 0 | 5.60 |
| GAGRRAAG |  | 714.4 | +2 | 12.49 |
| VC |  | 220.1 | 0 | 4.95 |
| DFK |  | 408.2 | 0 | 6.77 |
| VA |  | 188.1 | 0 | 5.60 |
| FFKF |  | 587.3 | +1 | 9.93 |
| PGFK |  | 447.2 | +1 | 10.59 |
| GIVRAK |  | 642.4 | +2 | 11.56 |
| Peptide Linker | Drug | Delivery System | Cancer | Outcomes | Ref. |
|---|---|---|---|---|---|
| GFLG | Doxorubicin | Conjugate | HepG2 cells | The conjugate structure had an opposite effect on DOX release and tumor accumulation. The synergistic effect of these properties exhibited the highest antitumor efficacy | [66] |
| GFLG | Paclitaxel and gemcitabine | Conjugate with HPMA dendrimers | A2780 human ovarian carcinoma cells | The combination of PTX, GEM and diblock structures yielded the highest inhibition efficacy of tumor growth | [72] |
| GFLG | Doxorubicin | Conjugate with polymer | Lung carcinoma, colorectal cancer and anthracycline-resistant breast cancer | Antitumor activity in refractory cancers was demonstrated, and polymer-drug conjugation has been shown to reduce DOX dose-limiting toxicity | [73] |
| GFLG | Doxorubicin | Conjugate with polymer | H22 mice tumor | The conjugates were successfully internalized into the cell nuclei, resulting in an inhibition efficiency of ~90% for the tumor | [78] |
| GAGRRAAG | Pheophorbide | Conjugate | Bone marrow cells | The photodynamic effect was demonstrated to be greater than 60%, and the system could be used as a sensor for cathepsin activity | [79] |
| VC | Doxorubicin | Nanoparticle | RKO colon carcinoma cells | Conjugates can efficiently bind to and be internalized by EGFR-overexpressing cancer cells. This strategy could be used to reduce systemic toxicity | [108] |
| DFK | SPION | Nanoparticle | MDA-MB-231 breast cancer cells | The increased efficiency of NP internalization and spion release following exposure to CtsB were demonstrated | [113] |
| GSGFLGSC | PTX | Nanoparticle | OVCAR-3 adenocarcinoma cells and OVCAR-5 ovarian cancer cells | The time-dependent PTX release and a 25-fold reduction in IC50 compared to pure PTX were demonstrated | [114] |
| Ac-FKC(StBu)AC(SH)-CBT | Chlorin e6 | Nanoparticle | H-29 human colorectal adenicarcinoma cells | CtsB induced NPs self-assembly, resulting in an increased singlet oxygen generation and a significant enhancement of the photodynamic effect | [116] |
| FFKF | Doxorubicin | Self-assembled nanoparticle | Tumor lysates | A library of FFKF peptides with various N-terminal capping groups was studied, and their self-assembly and sensitivity to cathepsin B and L were analyzed. Cbz-FFKF-OH showed the highest potential and a release of 92% of DOX within 8 h | [120] |
| VA | Duocarmycin and pyrrolobenzodiazepine | ADC | Hepatocellular carcinoma | Using dipeptides, VC and VA, conjugating Duocarmycin SA and PBD dimers to antibodies targeting GPC3 on hepatocellular carcinoma cells advances in liver cancer therapy were achieved | [88] |
| VC | (Pyrrolo [2,1-c][1,4]benzodiazepine dimer) | ADC | BT474 carcinoma cells, KPL-4 breast cancer cells and BJAM lymphoma cells | The targeting agent used is of more importance for the effectiveness of ADC than the efficiency of linker cleavage | [96] |
| VC | Auristatin-based | ADC | Expi293 cells | Carboxylesterase 1C was identified as the enzyme responsible for the plasmatic hydrolysis of (VC-PABC)-based linkers | [103] |
| VC | Monomethyl auristatin E | Nanoparticle | U87 glioblastoma cells | The system provided efficient cellular uptake and high toxic effect on glioblastoma cells. The maximal anticancer effect was achieved using NPs and radiotherapy | [122] |
| GFLG | Anti-VEGF siRNA | Nanoparticle | HeLa cells | Efficient siRNA release and VEGF deregulation in HeLa cells were achieved | [123] |
| GFLG | Doxorubicin | Functionalized nanoparticle | HeLa cells | 80% of DOX release was observed in 24 h in the presence of CtsB | [128] |
| GIVRAKEAEGIVRAK | Safranin O or DOX | Functionalized nanoparticle | Hela cells | A 5-fold increase in the release of Safarin O was observed in the presence of lysosomal extract, leading to a CtsB-dependent cytotoxic effect | [129] |
| PGFK | Doxorubicin | Functionalized nanoparticle | A549 human non-small cell lung cancer cells, NIH-3T3 mouse fibroblast cells, A2780 human ovarian cancer cells | At acidic pH, CtsB led to a four-fold increase in DOX release and consequent higher toxicity | [130] |
| GFLG | Doxorubicin | Functionalized nanoparticle | MCF-7 human breast cancer cell | The nanoparticles represented a promising system to overcome MDR phenomena | [131] |
| GFLGC | Docetaxel | Functionalized nanoparticle | HeLa and MCF-7 breast cells | The systems showed higher circulation properties, efficacy and safety | [132] |
| DEGFLGED | Resveratrol | Functionalized nanoparticle | MCF-7 breast cells | Anticancer activity exceeded 80% | [133] |
| VA | SG3199 | ADC | B-cell non-Hodgkin Lymphoma | Zynlonta® is FDA-approved ADC for the treatment of large B-cell lymphoma (USA) | [134,135] |
| VC | Monomethyl auristatin E | ADC | Hodgkin lymphoma | Adcetris® was approved ADC for the treatment of Hodgkin lymphoma (USA) | [136,137] |
| VC | Monomethyl auristatin E | ADC | Large B-cell lymphoma | Polivy® was approved for the treatment of large B-cell lymphoma (USA) | [138] |
| VC | Monomethyl auristatin E | ADC | Metastatic urothelial cancer | Padcev® was approved for the treatment of metastatic urothelial cancer (USA) | [139] |
| VC | Monomethyl auristatin E | ADC | Metastatic cervical cancer | Tivdak® was approved for the treatment of metastatic cervical cancer (USA) | [94] |
| VC | Monomethyl auristatin E | ADC | HER-2 positive solid tumors | RC-48® was approved for the treatment of metastatic cervical cancer (China) | [95] |
| VA | SGD-1882 | ADC | Positive acute myeloid leukemia | Clinical trials were stopped because of severe adverse events and increased patient mortality | [97] |
| VA | SGD-1882 | ADC | Non-Hodgkin Lymphoma and Renal Cell Carcinoma | Clinical trials were stopped because of severe adverse events and increased patient mortality | [98] |
| VA | SG3199 | ADC | Large B-cell lymphoma | Phase 2 of clinical trials of ADCT-402 (Loncastuximab Tesirine), NCT05296070 NCT05249959 | [101] |
| VA | SG3199 | ADC | Hodgkin lymphoma | Phase 2 of clinical trials of Camidanlumab tesirine NCT04052997 | [102] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Egorova, V.S.; Kolesova, E.P.; Lopus, M.; Yan, N.; Parodi, A.; Zamyatnin, A.A., Jr. Smart Delivery Systems Responsive to Cathepsin B Activity for Cancer Treatment. Pharmaceutics 2023, 15, 1848. https://doi.org/10.3390/pharmaceutics15071848
Egorova VS, Kolesova EP, Lopus M, Yan N, Parodi A, Zamyatnin AA Jr. Smart Delivery Systems Responsive to Cathepsin B Activity for Cancer Treatment. Pharmaceutics. 2023; 15(7):1848. https://doi.org/10.3390/pharmaceutics15071848
Chicago/Turabian StyleEgorova, Vera S., Ekaterina P. Kolesova, Manu Lopus, Neng Yan, Alessandro Parodi, and Andrey A. Zamyatnin, Jr. 2023. "Smart Delivery Systems Responsive to Cathepsin B Activity for Cancer Treatment" Pharmaceutics 15, no. 7: 1848. https://doi.org/10.3390/pharmaceutics15071848