
Exploring the Molecular Players behind the Potentiation of Chemotherapy Effects by Durvalumab in Lung Adenocarcinoma Cell Lines

Abstract
:1. Introduction
2. Materials and Methods
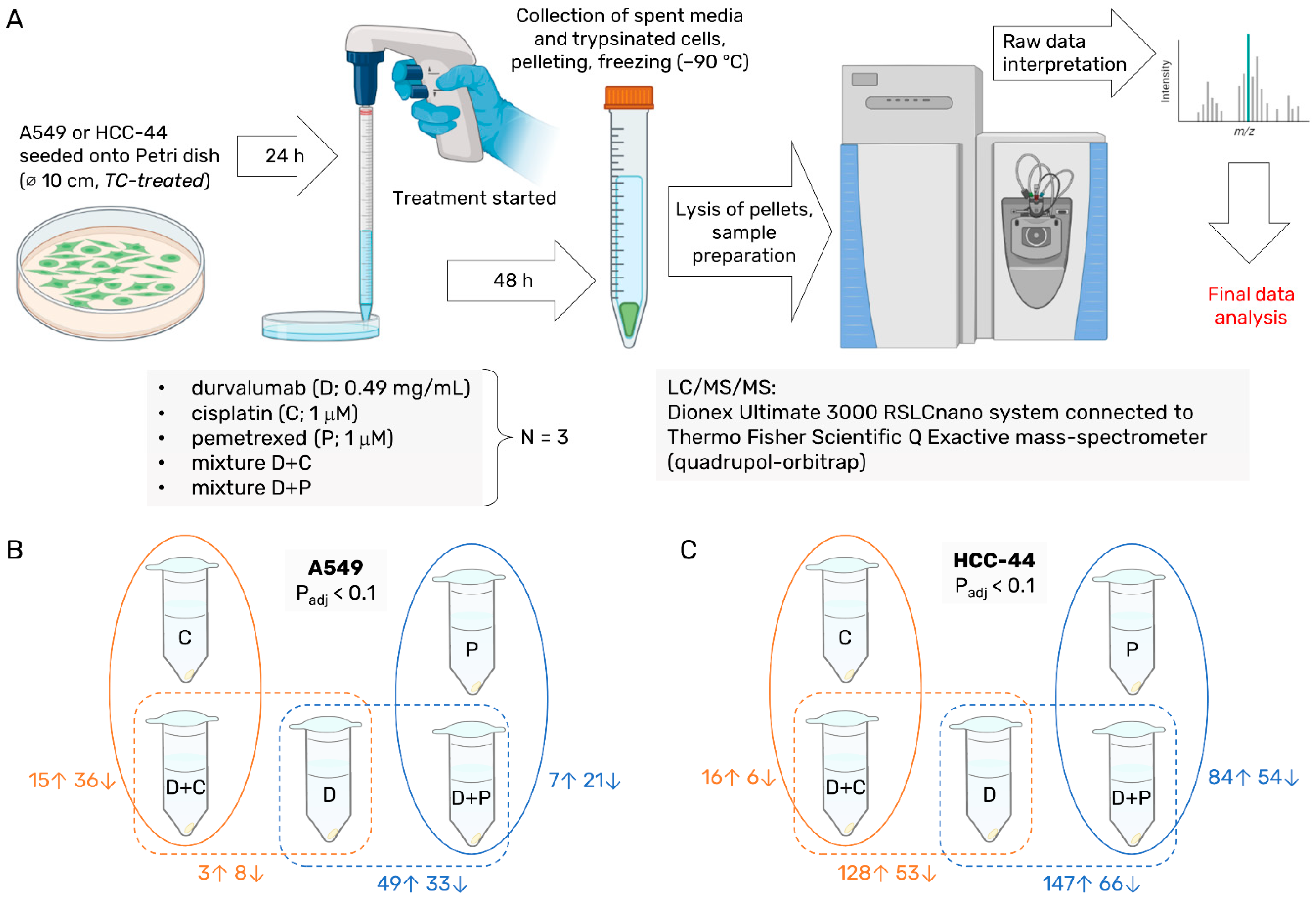
2.1. Chemicals, Cell Lines, and Equipment
2.2. Proteomics Sample Preparation
2.3. Label-Free Proteomics
2.4. Proteomics Data Analysis
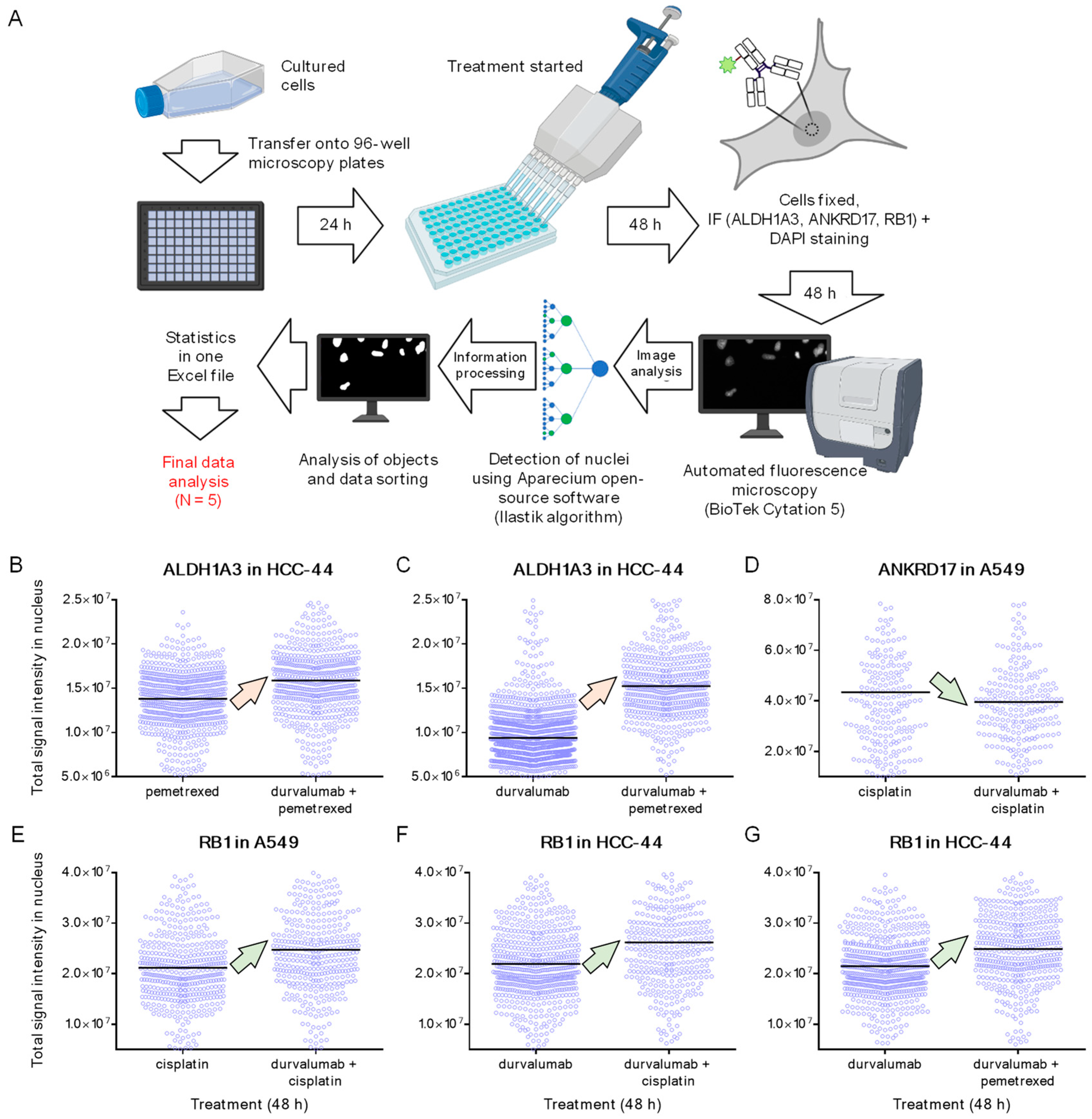
2.5. IF and Microscopy
2.6. IF Data Analysis
2.7. Other Software
3. Results
3.1. Proteomics in Lysates of HCC-44 and A549 following the 48 h Treatment
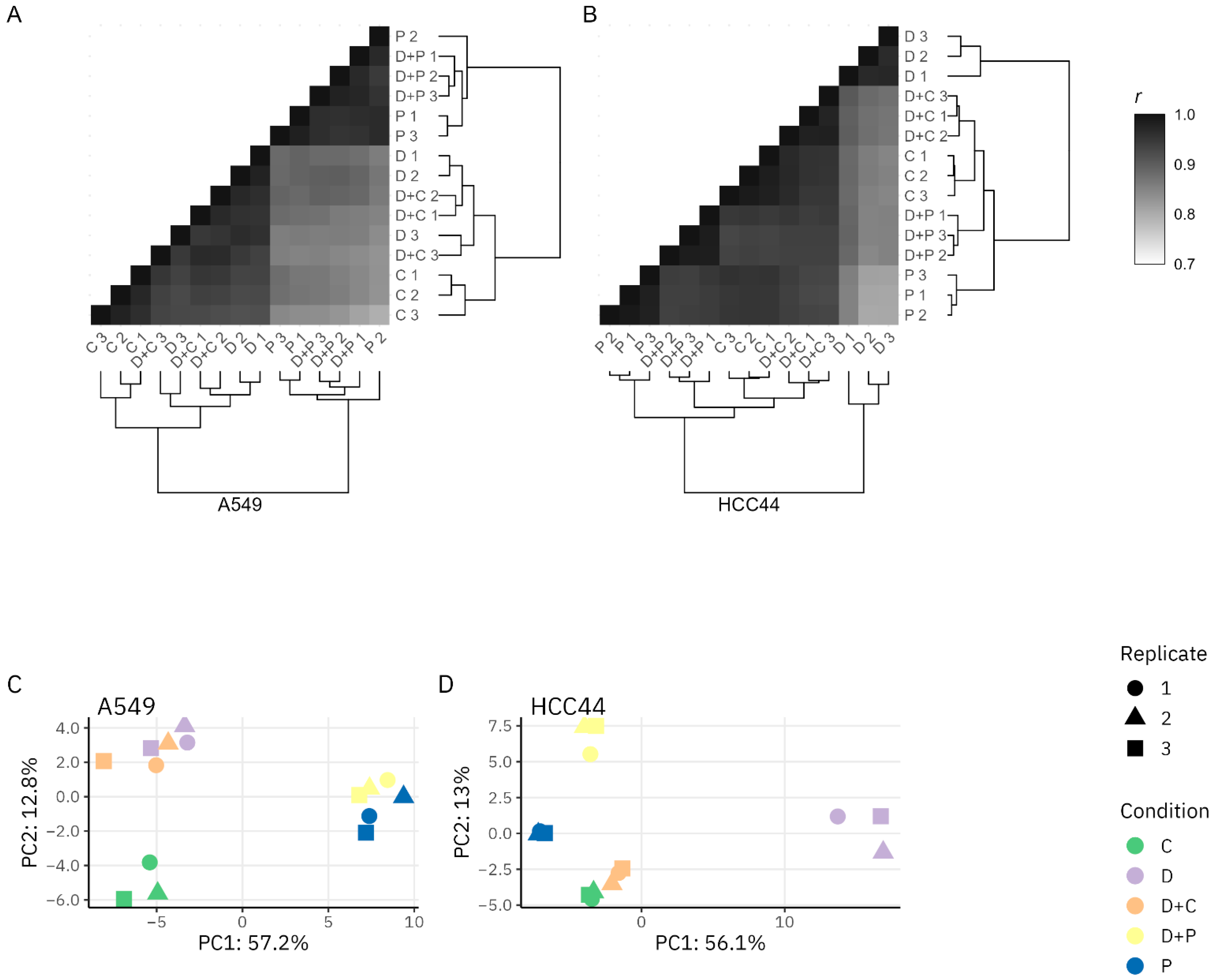
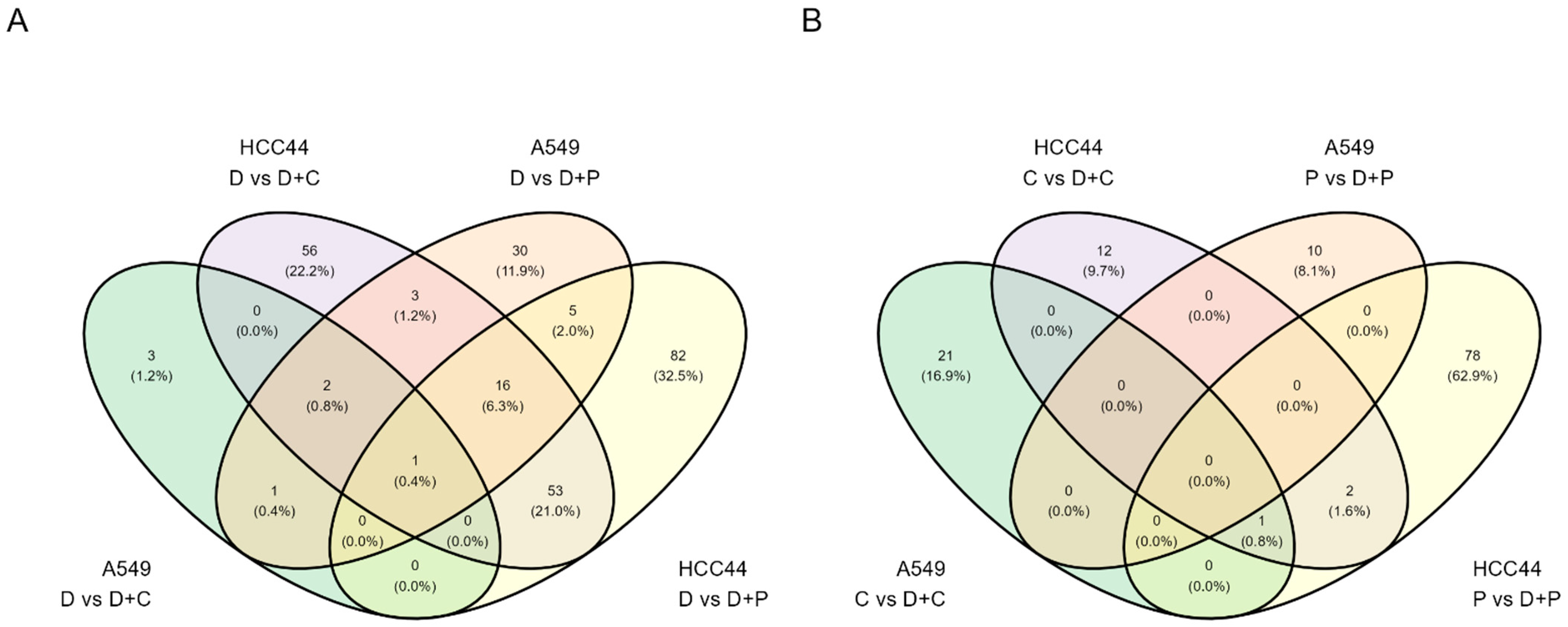
3.1.1. Proteomics Quality Assessment
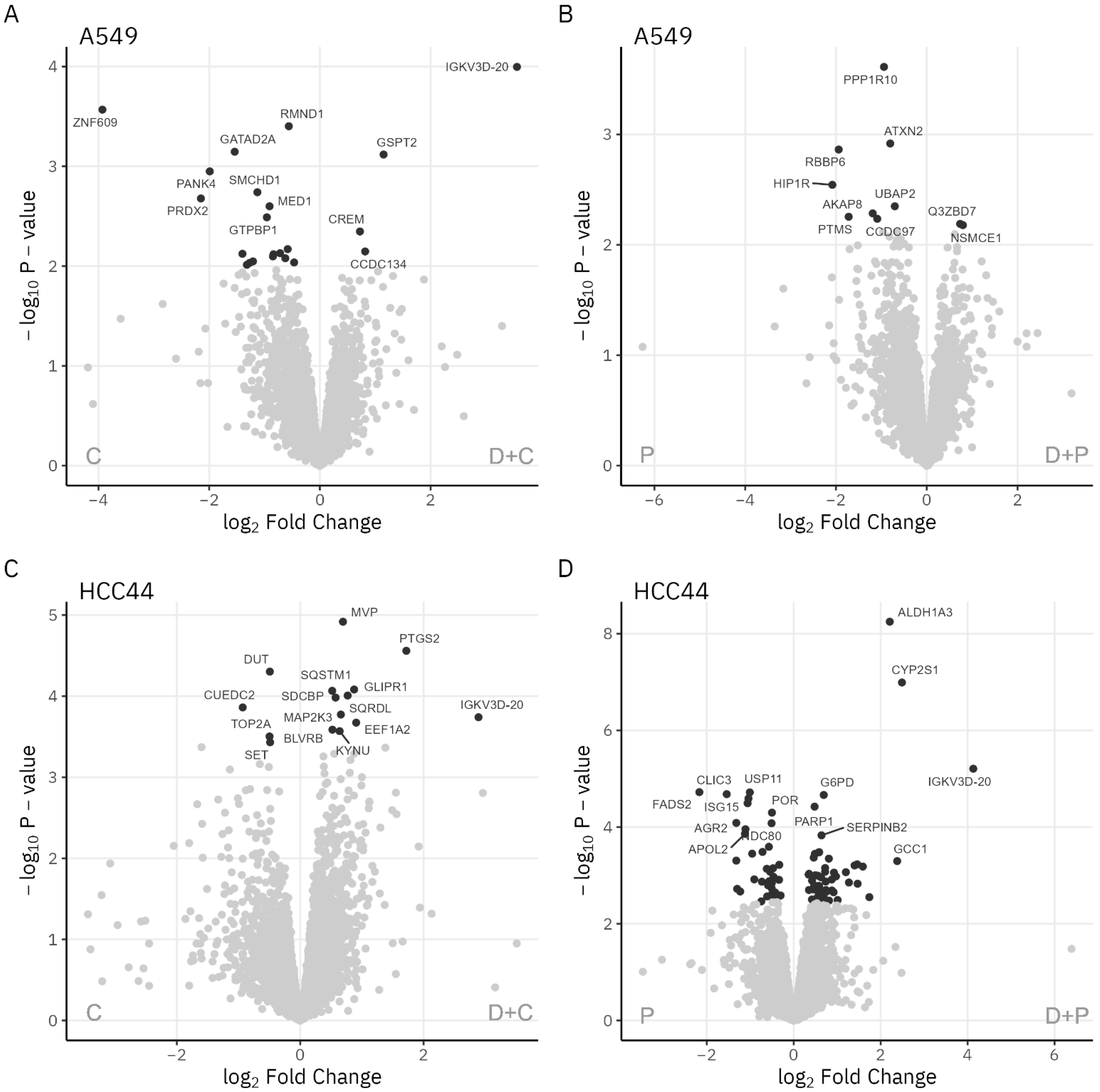
3.1.2. Analysis of Cellular Networks
3.2. IF in Fixed HCC-44 and A549 Cells following 48 h Treatment
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, B.; Zang, R.; Zhang, M.; Song, P.; Liu, L.; Bie, F.; Peng, Y.; Bai, G.; Gao, S. Worldwide Burden and Epidemiological Trends of Tracheal, Bronchus, and Lung Cancer: A Population-Based Study. eBioMedicine 2022, 78, 103951. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, L.E.; Kerr, K.M.; Menis, J.; Mok, T.S.; Nestle, U.; Passaro, A.; Peters, S.; Planchard, D.; Smit, E.F.; Solomon, B.J.; et al. Oncogene-Addicted Metastatic Non-Small-Cell Lung Cancer: ESMO Clinical Practice Guideline for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2023, 34, 339–357. [Google Scholar] [CrossRef] [PubMed]
- Postmus, P.E.; Kerr, K.M.; Oudkerk, M.; Senan, S.; Waller, D.A.; Vansteenkiste, J.; Escriu, C.; Peters, S. ESMO Guidelines Committee Early and Locally Advanced Non-Small-Cell Lung Cancer (NSCLC): ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2017, 28, iv1–iv21. [Google Scholar] [CrossRef] [PubMed]
- Non-Oncogene-Addicted Metastatic Non-Small-Cell Lung Cancer|ESMO. Available online: https://www.esmo.org/guidelines/guidelines-by-topic/lung-and-chest-tumours/non-oncogene-addicted-metastatic-non-small-cell-lung-cancer (accessed on 6 March 2023).
- Forde, P.M.; Spicer, J.; Lu, S.; Provencio, M.; Mitsudomi, T.; Awad, M.M.; Felip, E.; Broderick, S.R.; Brahmer, J.R.; Swanson, S.J.; et al. Neoadjuvant Nivolumab plus Chemotherapy in Resectable Lung Cancer. N. Engl. J. Med. 2022, 386, 1973–1985. [Google Scholar] [CrossRef]
- Johnson, M.; Cho, B.C.; Luft, A.; Alatorre-Alexander, J.; Geater, S.L.; Laktionov, K.; Vasiliev, A.; Trukhin, D.; Kim, S.; Ursol, G.; et al. PL02.01 Durvalumab ± Tremelimumab + Chemotherapy as First-Line Treatment for MNSCLC: Results from the Phase 3 POSEIDON Study. J. Thorac. Oncol. 2021, 16, S844. [Google Scholar] [CrossRef]
- Sehgal, K. Hyperprogression in Patients with Cancer Receiving Immune Checkpoint Inhibitors. JAMA Netw. Open 2021, 4, e211839. [Google Scholar] [CrossRef]
- Saar, M.; Lavogina, D.; Lust, H.; Tamm, H.; Jaal, J. Immune Checkpoint Inhibitors Modulate the Cytotoxic Effect of Chemotherapy in Lung Adenocarcinoma Cells. Oncol. Lett. 2023, 25, 152. [Google Scholar] [CrossRef]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for Micro-Purification, Enrichment, Pre-Fractionation and Storage of Peptides for Proteomics Using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant Computational Platform for Mass Spectrometry-Based Shotgun Proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- European Bioinformatics Institute (EMBL-EBI); Swiss Institute of Bioinformatics (SIB); Protein Information Resource (PIR). UniProt. Available online: https://www.uniprot.org/ (accessed on 31 January 2023).
- Zhang, X.; Smits, A.H.; van Tilburg, G.B.; Ovaa, H.; Huber, W.; Vermeulen, M. Proteome-Wide Identification of Ubiquitin Interactions Using UbIA-MS. Nat. Protoc. 2018, 13, 530–550. [Google Scholar] [CrossRef]
- Oba, S.; Sato, M.; Takemasa, I.; Monden, M.; Matsubara, K.; Ishii, S. A Bayesian Missing Value Estimation Method for Gene Expression Profile Data. Bioinformatics 2003, 19, 2088–2096. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- SIB; CPR; EMBL. STRING: Functional Protein Association Networks. Available online: https://string-db.org/ (accessed on 27 April 2023).
- Lavogina, D.; Laasfeld, T.; Vardja, M.; Lust, H.; Jaal, J. Viability Fingerprint of Glioblastoma Cell Lines: Roles of Mitotic, Proliferative, and Epigenetic Targets. Sci. Rep. 2021, 11, 20338. [Google Scholar] [CrossRef] [PubMed]
- Veiksina, S.; Tahk, M.-J.; Laasfeld, T.; Link, R.; Kopanchuk, S.; Rinken, A. Fluorescence Anisotropy-Based Assay for Characterization of Ligand Binding Dynamics to GPCRs: The Case of Cy3B-Labeled Ligands Binding to MC4 Receptors in Budded Baculoviruses. In G Protein-Coupled Receptor Screening Assays: Methods and Protocols; Martins, S.A.M., Prazeres, D.M.F., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2021; pp. 119–136. ISBN 978-1-07-161221-7. [Google Scholar]
- University of Tartu. Aparecium—The GPCR Workgroup. Available online: https://gpcr.ut.ee/aparecium.html (accessed on 31 January 2023).
- BioRender. Available online: https://biorender.com/ (accessed on 31 January 2023).
- Chen, C.-Y.; Chang, Y.-L.; Shih, J.-Y.; Lin, J.-W.; Chen, K.-Y.; Yang, C.-H.; Yu, C.-J.; Yang, P.-C. Thymidylate Synthase and Dihydrofolate Reductase Expression in Non-Small Cell Lung Carcinoma: The Association with Treatment Efficacy of Pemetrexed. Lung Cancer 2011, 74, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Pan, C.C.; Yu, R.J.; Long, Y.; Cai, H.X.; Yin, D.X.; Hao, Q.L.; Luo, L.L. Association between TYMS Expression and Efficacy of Pemetrexed-Based Chemotherapy in Advanced Non-Small Cell Lung Cancer: A Meta-Analysis. PLoS ONE 2013, 8, e74284. [Google Scholar] [CrossRef]
- Ryan, S.-L.; Dave, K.A.; Beard, S.; Gyimesi, M.; McTaggart, M.; Sahin, K.B.; Molloy, C.; Gandhi, N.S.; Boittier, E.; O’Leary, C.G.; et al. Identification of Proteins Deregulated by Platinum-Based Chemotherapy as Novel Biomarkers and Therapeutic Targets in Non-Small Cell Lung Cancer. Front. Oncol. 2021, 11, 23. [Google Scholar] [CrossRef]
- Cavazzoni, A.; Digiacomo, G.; Alfieri, R.; La Monica, S.; Fumarola, C.; Galetti, M.; Bonelli, M.; Cretella, D.; Barili, V.; Zecca, A.; et al. Pemetrexed Enhances Membrane PD-L1 Expression and Potentiates T Cell-Mediated Cytotoxicity by Anti-PD-L1 Antibody Therapy in Non-Small-Cell Lung Cancer. Cancers 2020, 12, 666. [Google Scholar] [CrossRef]
- Bhateja, P.; Chiu, M.; Wildey, G.; Lipka, M.B.; Fu, P.; Yang, M.C.L.; Ardeshir-Larijani, F.; Sharma, N.; Dowlati, A. Retinoblastoma Mutation Predicts Poor Outcomes in Advanced Non Small Cell Lung Cancer. Cancer Med. 2019, 8, 1459–1466. [Google Scholar] [CrossRef]
- Arakawa, S.; Yoshida, T.; Shirasawa, M.; Takayanagi, D.; Yagishita, S.; Motoi, N.; Ohe, Y. RB1 Loss Induced Small Cell Lung Cancer Transformation as Acquired Resistance to Pembrolizumab in an Advanced NSCLC Patient. Lung Cancer 2021, 151, 101–103. [Google Scholar] [CrossRef]
- Niederst, M.J.; Sequist, L.V.; Poirier, J.T.; Mermel, C.H.; Lockerman, E.L.; Garcia, A.R.; Katayama, R.; Costa, C.; Ross, K.N.; Moran, T.; et al. RB Loss in Resistant EGFR Mutant Lung Adenocarcinomas That Transform to Small-Cell Lung Cancer. Nat. Commun. 2015, 6, 6377. [Google Scholar] [CrossRef]
- Rebollido-Rios, R.; Venton, G.; Sánchez-Redondo, S.; Iglesias i Felip, C.; Fournet, G.; González, E.; Romero Fernández, W.; Borroto Escuela, D.O.; Di Stefano, B.; Penarroche-Díaz, R.; et al. Dual Disruption of Aldehyde Dehydrogenases 1 and 3 Promotes Functional Changes in the Glutathione Redox System and Enhances Chemosensitivity in Nonsmall Cell Lung Cancer. Oncogene 2020, 39, 2756–2771. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Sullivan, J.P.; Girard, L.; Augustyn, A.; Yenerall, P.; Rodriguez, J.; Liu, H.; Behrens, C.; Shay, J.W.; Wistuba, I.I.; et al. Essential Role of Aldehyde Dehydrogenase 1A3 (ALDH1A3) for the Maintenance of Non-Small Cell Lung Cancer Stem Cells Is Associated with the STAT3 Pathway. Clin. Cancer Res. 2014, 20, 4154–4166. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.-J.; Cai, J.; Guo, Y.-F.; Bian, X.-W.; Yu, S.-C. ALDH1A3, a Metabolic Target for Cancer Diagnosis and Therapy. Int. J. Cancer 2016, 139, 965–975. [Google Scholar] [CrossRef] [PubMed]
- ELIXIR; SciLifeLab; Uppsala Universitet; Karolinska Institutet; KTH. The Human Protein Atlas. Available online: https://www.proteinatlas.org/ (accessed on 31 January 2023).
- de Almeida, B.O.; Machado-Neto, J.A. Emerging Functions for ANKHD1 in Cancer-Related Signaling Pathways and Cellular Processes. BMB Rep. 2020, 53, 413–418. [Google Scholar] [CrossRef]
- Wang, Z.; Pei, H.; Liang, H.; Zhang, Q.; Wei, L.; Shi, D.; Chen, Y.; Zhang, J. Construction and Analysis of a CircRNA-Mediated CeRNA Network in Lung Adenocarcinoma. OTT 2021, 14, 3659–3669. [Google Scholar] [CrossRef]
- Expression of ANKRD17 in Lung Cancer—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000132466-ANKRD17/pathology/lung+cancer (accessed on 31 January 2023).
- Saar, M.; Narits, J.; Mägi, L.; Aaspõllu, H.; Vapper, A.; Kase, M.; Minajeva, A.; Vooder, T.; Tamm, H.; Buldakov, M.; et al. Expression of Immune Checkpoint PD-1 in Non-Small Cell Lung Cancer Is Associated with Tumor Cell DNA-Dependent Protein Kinase. Mol. Clin. Oncol. 2021, 15, 211. [Google Scholar] [CrossRef]
- de Campos-Nebel, M.; Larripa, I.; González-Cid, M. Topoisomerase II-Mediated DNA Damage Is Differently Repaired during the Cell Cycle by Non-Homologous End Joining and Homologous Recombination. PLoS ONE 2010, 5, e12541. [Google Scholar] [CrossRef]
- Maekawa, M.; Higashiyama, S. The Roles of SPOP in DNA Damage Response and DNA Replication. Int. J. Mol. Sci. 2020, 21, 7293. [Google Scholar] [CrossRef]
- Zámborszky, J.; Szikriszt, B.; Gervai, J.Z.; Pipek, O.; Póti, Á.; Krzystanek, M.; Ribli, D.; Szalai-Gindl, J.M.; Csabai, I.; Szallasi, Z.; et al. Loss of BRCA1 or BRCA2 Markedly Increases the Rate of Base Substitution Mutagenesis and Has Distinct Effects on Genomic Deletions. Oncogene 2017, 36, 746–755. [Google Scholar] [CrossRef]
- Redon, C.E.; Nakamura, A.J.; Zhang, Y.; Ji, J.; Bonner, W.M.; Kinders, R.; Parchment, R.; Doroshow, J.H.; Pommier, Y. Histone ΓH2AX and Poly(ADP Ribose) as Clinical Pharmacodynamic Biomarkers. Clin. Cancer Res. 2010, 16, 4532–4542. [Google Scholar] [CrossRef]
- Harel, M.; Lahav, C.; Jacob, E.; Dahan, N.; Sela, I.; Elon, Y.; Shoval, S.R.; Yahalom, G.; Kamer, I.; Zer, A.; et al. Longitudinal Plasma Proteomic Profiling of Patients with Non-Small Cell Lung Cancer Undergoing Immune Checkpoint Blockade. J. Immunother. Cancer 2022, 10, e004582. [Google Scholar] [CrossRef] [PubMed]
- Loriot, Y.; Marabelle, A.; Guégan, J.P.; Danlos, F.X.; Besse, B.; Chaput, N.; Massard, C.; Planchard, D.; Robert, C.; Even, C.; et al. Plasma Proteomics Identifies Leukemia Inhibitory Factor (LIF) as a Novel Predictive Biomarker of Immune-Checkpoint Blockade Resistance. Ann. Oncol. 2021, 32, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Kim, M.J.; Choi, Y.; Kim, N.H.; Kim, L.; Hong, S.P.D.; Cho, H.-G.; Yu, E.; Chae, Y.K. Role of Mass Spectrometry-Based Serum Proteomics Signatures in Predicting Clinical Outcomes and Toxicity in Patients with Cancer Treated with Immunotherapy. J. Immunother. Cancer 2022, 10, e003566. [Google Scholar] [CrossRef]
- Grigorieva, J.; Asmellash, S.; Net, L.; Tsypin, M.; Roder, H.; Roder, J. Mass Spectrometry-Based Multivariate Proteomic Tests for Prediction of Outcomes on Immune Checkpoint Blockade Therapy: The Modern Analytical Approach. Int. J. Mol. Sci. 2020, 21, 838. [Google Scholar] [CrossRef] [PubMed]
- Lim, Z.; Wu, X.; Hafez, M.; Albandar, H.; Zhu, L.; Yang, H.; Mackay, S.; Chen, J.; Zhou, J.; Ma, P. P2.04-26 Single Cell Proteomics Profiling of Live T-Cells in KRAS+ and MET-Amp NSCLC to Predict Immune Checkpoint Inhibitor Response. J. Thorac. Oncol. 2019, 14, S718. [Google Scholar] [CrossRef]
- Brocco, D.; Lanuti, P.; Pieragostino, D.; Cufaro, M.C.; Simeone, P.; Bologna, G.; Di Marino, P.; De Tursi, M.; Grassadonia, A.; Irtelli, L.; et al. Phenotypic and Proteomic Analysis Identifies Hallmarks of Blood Circulating Extracellular Vesicles in NSCLC Responders to Immune Checkpoint Inhibitors. Cancers 2021, 13, 585. [Google Scholar] [CrossRef]
- Novikova, S.E.; Soloveva, N.A.; Farafonova, T.E.; Tikhonova, O.V.; Liao, P.-C.; Zgoda, V.G. Proteomic Signature of Extracellular Vesicles for Lung Cancer Recognition. Molecules 2021, 26, 6145. [Google Scholar] [CrossRef]
- Liebler, D.C.; Holzer, T.R.; Haragan, A.; Morrison, R.D.; O’Neill Reising, L.; Ackermann, B.L.; Fill, J.A.; Schade, A.E.; Gruver, A.M. Analysis of Immune Checkpoint Drug Targets and Tumor Proteotypes in Non-Small Cell Lung Cancer. Sci. Rep. 2020, 10, 9805. [Google Scholar] [CrossRef]
- Fountzilas, E.; Kurzrock, R.; Vo, H.H.; Tsimberidou, A.-M. Wedding of Molecular Alterations and Immune Checkpoint Blockade: Genomics as a Matchmaker. J. Natl. Cancer Inst. 2021, 113, 1634–1647. [Google Scholar] [CrossRef]
- Végvári, Á.; Rodriguez, J.E.; Zubarev, R.A. Single-Cell Chemical Proteomics (SCCP) Interrogates the Timing and Heterogeneity of Cancer Cell Commitment to Death. Anal. Chem. 2022, 94, 9261–9269. [Google Scholar] [CrossRef]
- Gaetani, M.; Sabatier, P.; Saei, A.A.; Beusch, C.M.; Yang, Z.; Lundström, S.L.; Zubarev, R.A. Proteome Integral Solubility Alteration: A High-Throughput Proteomics Assay for Target Deconvolution. J. Proteome Res. 2019, 18, 4027–4037. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE Database and Related Tools and Resources in 2019: Improving Support for Quantification Data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Compared Treatments a | Protein Hits and Clusters b,c |
|---|---|---|
| HCC-44 | D + C vs. C | DDX58, MAP1LC3B, SQSTM1 DUT, SET, TOP2A EEF1A2, PLEC MAP2K3, PTGS2, SERPINB2 |
| D + C vs. D | Cell cycle-related cluster (including AURKA, AURKB, BUB1B, CCNB1, KIF11, PLK1, TOP2A, TPX2, TYMS) Cluster-containing DDX58, MAP1LC3B, MAP2K3, PTGS2, RB1, SERPINB2, SQSTM1 Cytoskeleton-related cluster (including EHBP1) Histone cluster (including HIST1H4A, HIST1H2AJ, HIST2H3A) RNA-binding protein cluster (including RBM34) Transcriptional activity-related cluster (including SUPT5H) | |
| D + P vs. P | Cell adhesion and cell–cell communication-related cluster (including CD274, ITGA2, SERPINB2) DNA synthesis and damage response-related cluster (including BRCA2, DHFR, PARP1, PCNA, TOP2A) Growth factor-related cluster (including IGF1R, TGFB2) Metabolic enzyme cluster 1 (including ACAT2, DHCR7, FDPS, MVD) Metabolic enzyme cluster 3 (including ALDH1A3, CYP2S1, IDH1, TXNRD1) | |
| D + P vs. D | Cell cycle-related cluster (including AURKA, AURKB, BUB1B, CCNB1, KIF11, PLK1, TOP2A, TPX2) DNA synthesis-related cluster (including DHFR, TYMS) Histone and transcription regulation cluster (including HIST1H2BC, HIST1H2AJ, RB1) Cluster containing MAP1LC3B, MAP1B, SERPINB2, SQSTM1 Cell adhesion and cell–cell communication-related cluster 1 (including CTNNA1, ITGA2, ICAM1, EPHA2) Metabolic enzyme cluster 1 (including ALDH1A3, NAMPT, NNMT) Metabolic enzyme cluster 2 (including PGM2L1) | |
| A549 | D + C vs. C | AKAP1, DPY30 ATP5MF-PTCD1, CHCHD1 CBX4, GATAD2A, RB1 DTCN5, KIF3A FAM96B, GTPBP1, MLF2 INTS6, ZNF609 MAD1, MED11 RAB5A, RIN1, UVRAG |
| D + C vs. D | FDXR, RETSAT HIST1H1B, HIST1H1C, HIST2H3A, KIF4A, MCM2 | |
| D + P vs. P | TWISTNB, WDR55 | |
| D + P vs. D | Cell cycle-related cluster (including CCNB1, KIF11, TOP2A, TPX2) DNA synthesis-related cluster (including DHFR, SHMT2, TYMS) Histone cluster (including HIST1H1B, HIST1H4A) |
| Validated Protein | Cell Line and Treatment Conditions a | Trend in IF (Difference in Protein Abundance) b | Statistical Significance of Difference in Protein Abundance c | Relationship to the ICI Treatment Response d |
|---|---|---|---|---|
| ALDH1A3 | HCC-44, P vs. D + P | ↑ | p < 0.001 | Increase shows resistance |
| ALDH1A3 | HCC-44, D vs. D + P | ↑ | p < 0.001 | Increase shows resistance |
| ANKRD17 | A549, C vs. D + C | ↓ | ns | Decline shows sensitivity |
| RB1 | A549, C vs. D + C | ↑ | p < 0.001 | Increase shows sensitivity |
| RB1 | HCC-44, D vs. D + C | ↑ | p < 0.001 | Increase shows sensitivity |
| RB1 | HCC-44, D vs. D + P | ↑ | p < 0.001 | Increase shows sensitivity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saar, M.; Jaal, J.; Meltsov, A.; Laasfeld, T.; Lust, H.; Kasvandik, S.; Lavogina, D. Exploring the Molecular Players behind the Potentiation of Chemotherapy Effects by Durvalumab in Lung Adenocarcinoma Cell Lines. Pharmaceutics 2023, 15, 1485. https://doi.org/10.3390/pharmaceutics15051485
Saar M, Jaal J, Meltsov A, Laasfeld T, Lust H, Kasvandik S, Lavogina D. Exploring the Molecular Players behind the Potentiation of Chemotherapy Effects by Durvalumab in Lung Adenocarcinoma Cell Lines. Pharmaceutics. 2023; 15(5):1485. https://doi.org/10.3390/pharmaceutics15051485
Chicago/Turabian StyleSaar, Marika, Jana Jaal, Alvin Meltsov, Tõnis Laasfeld, Helen Lust, Sergo Kasvandik, and Darja Lavogina. 2023. "Exploring the Molecular Players behind the Potentiation of Chemotherapy Effects by Durvalumab in Lung Adenocarcinoma Cell Lines" Pharmaceutics 15, no. 5: 1485. https://doi.org/10.3390/pharmaceutics15051485