
Physiologically Based Pharmacokinetic Model Development and Verification for Bioequivalence Testing of Bempedoic Acid Oral Suspension and Reference Tablet Formulation
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Clinical Studies
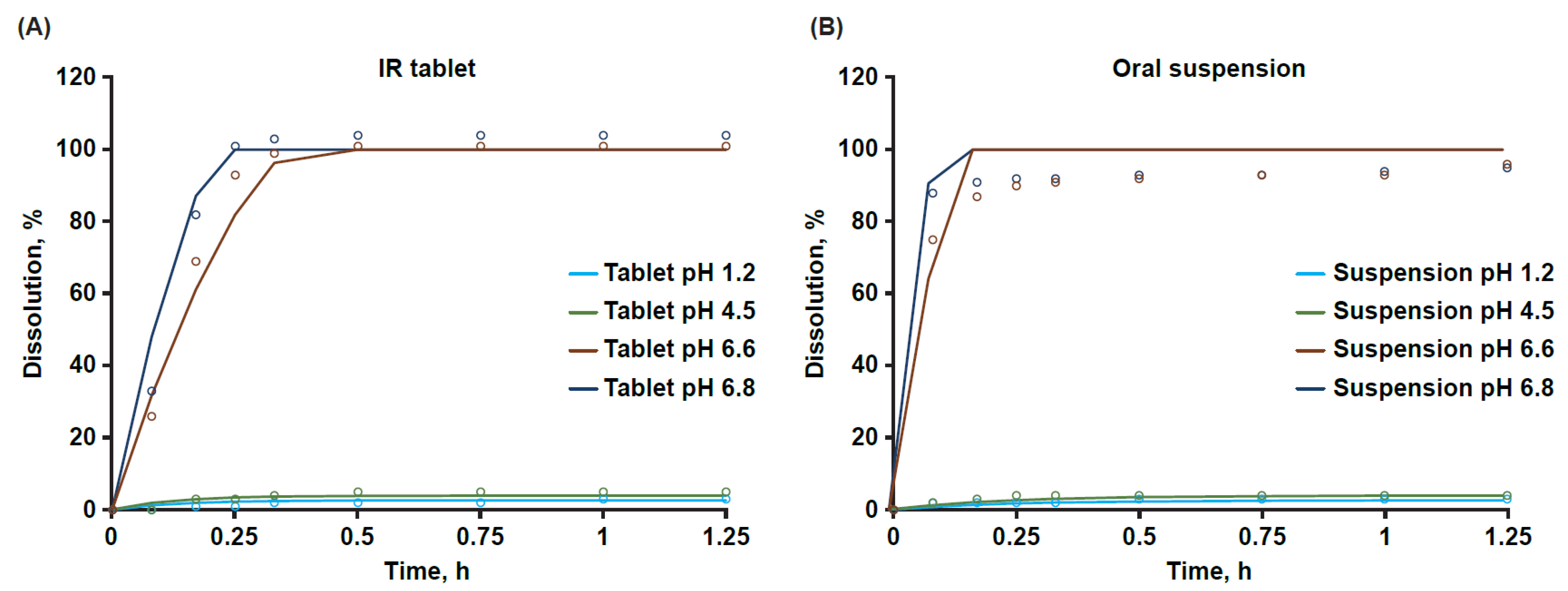
2.3. In Vitro Solubility and Dissolution
2.4. Modeling Strategy
2.5. Model Development and Verification
2.6. Model Application
3. Results
3.1. Aqueous Solubility and Dissolution
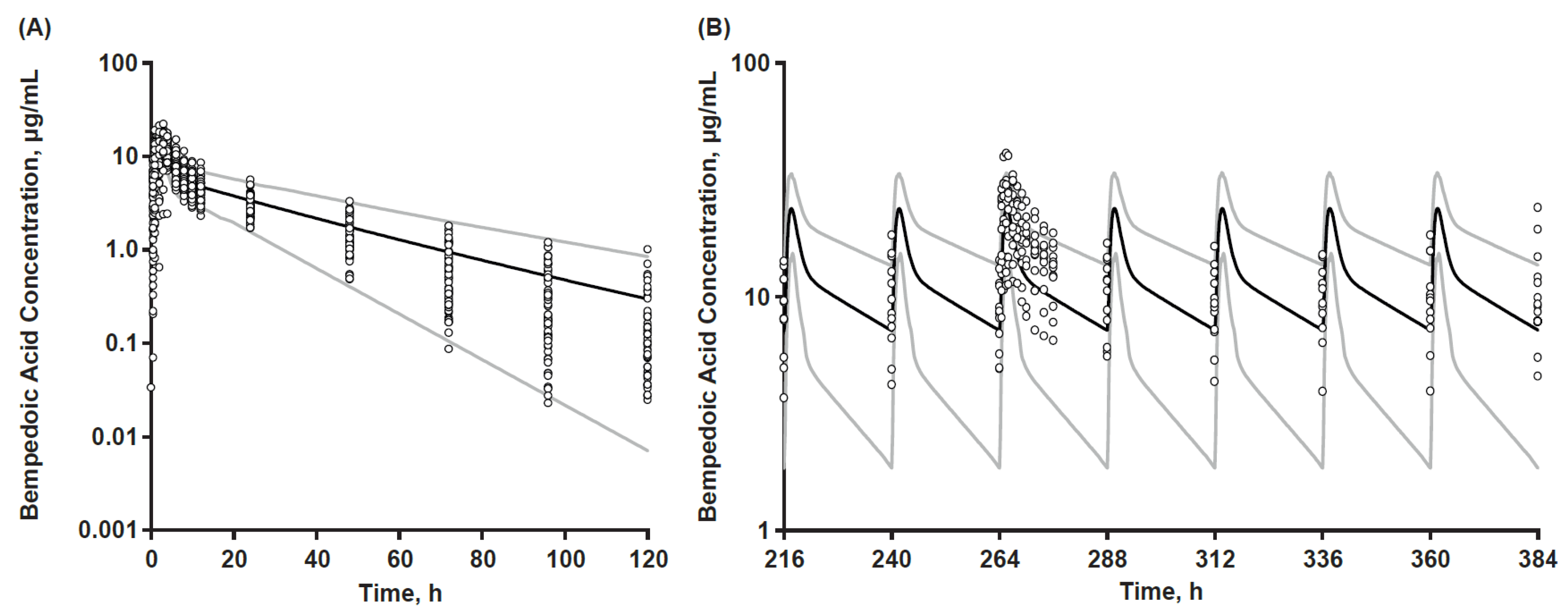
3.2. PBPK Base Model
3.3. Final Model
3.4. Determination of Virtual Population Sample Size
3.5. Final PBPK Model Sensitivity Analyses
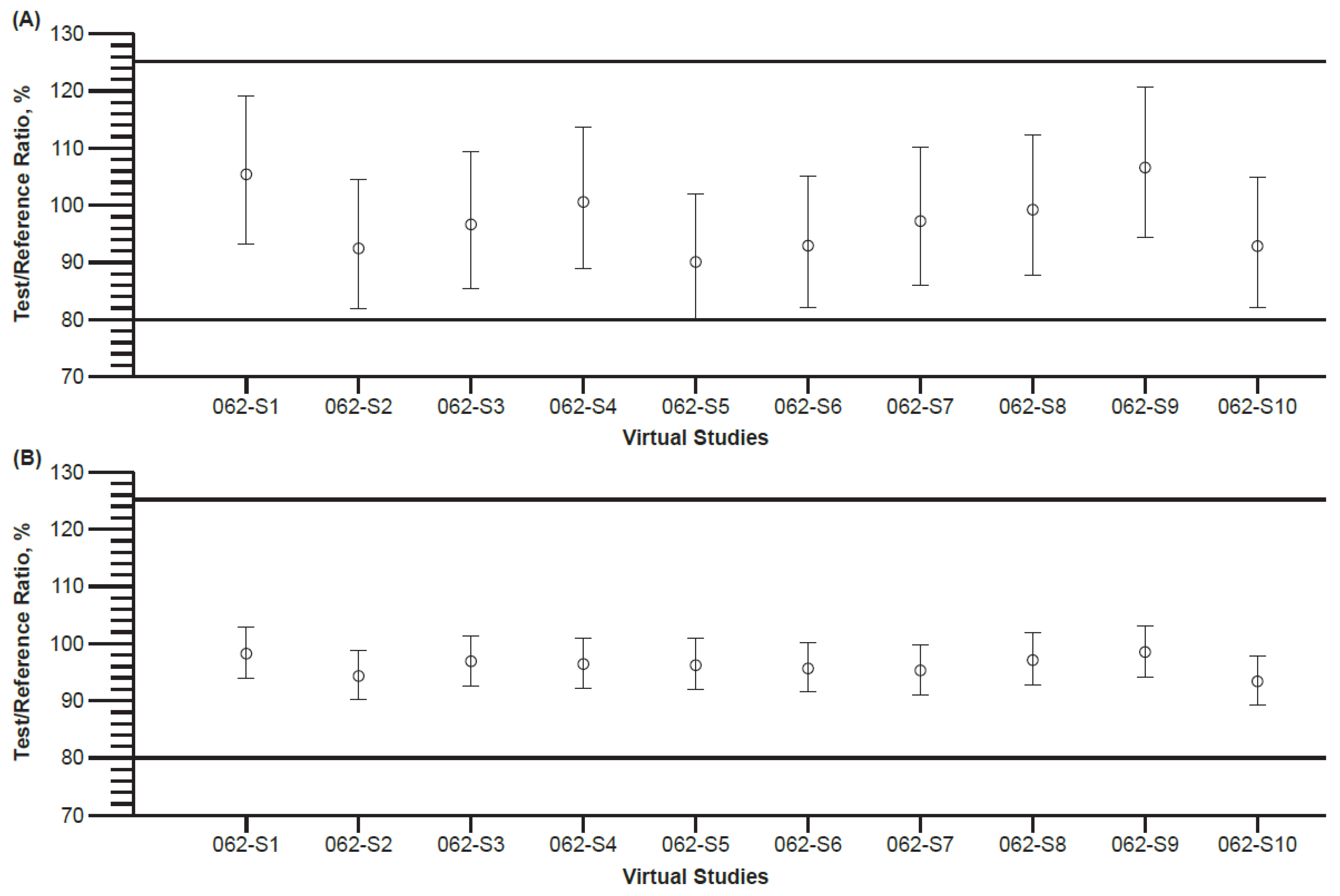
3.6. Final Model Application in Virtual Bioequivalence Assessment
3.7. Sensitivity Analysis (Worst Case) of Virtual Bioequivalence Predictions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Banach, M.; Duell, P.B.; Gotto, A.M., Jr.; Laufs, U.; Leiter, L.A.; Mancini, G.B.J.; Ray, K.K.; Flaim, J.; Ye, Z.; Catapano, A.L. Association of bempedoic acid administration with atherogenic lipid levels in phase 3 randomized clinical trials of patients with hypercholesterolemia. JAMA Cardiol. 2020, 5, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- Pinkosky, S.L.; Newton, R.S.; Day, E.A.; Ford, R.J.; Lhotak, S.; Austin, R.C.; Birch, C.M.; Smith, B.K.; Filippov, S.; Groot, P.H.E.; et al. Liver-specific ATP-citrate lyase inhibition by bempedoic acid decreases LDL-C and attenuates atherosclerosis. Nat. Commun. 2016, 7, 13457. [Google Scholar] [CrossRef] [PubMed]
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Amore, B.M.; MacDougall, D.E.; Hanselman, J.C.; Emery, M.G. Phase 1, Single- and Multiple-Ascending-Dose, Food-Effect, and East Asian Subject Studies to Assess the Pharmacokinetics, Safety, and Tolerability of Bempedoic Acid, a Selective Inhibitor of ATP Citrate Lyase. Clin. Pharmacol. Drug Dev. 2023; submitted. [Google Scholar]
- Amore, B.M.; Cramer, C.; MacDougall, D.E.; Emery, M.G. The disposition and metabolism of bempedoic acid, a potent inhibitor of ATP citrate lyase, in healthy human subjects. Drug Metab. Dispos. 2023, 51, 599–609. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration. The Use of Physiologically Based Pharmacokinetic Analyses—Biopharmaceutics Applications for Oral Drug Product Development, Manufacturing Changes, and Controls. Available online: https://www.fda.gov/media/142500/download (accessed on 10 January 2023).
- Engel, B.J.; Preusch, K.; Brown, C.; Cramer, C.T.; Shoup, R. Measurement of bempedoic acid and its keto metabolite in human plasma and urine using solid phase extraction and electrospray LC-MS/MS. J. Chromatogr. B Analyt. Technol. Biomed Life Sci. 2020, 1154, 122291. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Flanagan, D.R. General solution for diffusion-controlled dissolution of spherical particles. 1. Theory. J. Pharm. Sci. 1999, 88, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Sawada, Y.; Hanano, M.; Sugiyama, Y.; Harashima, H.; Iga, T. Prediction of the volumes of distribution of basic drugs in humans based on data from animals. J. Pharm. Biopharm. 1984, 12, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, T.; Rowland, M. Physiologically based pharmacokinetic modelling 2: Predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 2006, 95, 1238–1257. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, T.; Rowland, M. Mechanistic approaches to volume of distribution predictions: Understanding the processes. Pharm. Res. 2007, 24, 918–933. [Google Scholar] [CrossRef] [PubMed]
- Tubic-Grozdanis, M.; Bolger, M.B.; Langguth, P. Application of gastrointestinal simulation for extensions for biowaivers of highly permeable compounds. AAPS J. 2008, 10, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Tsume, Y.; Langguth, P.; Garcia-Arieta, A.; Amidon, G.L. In silico prediction of drug dissolution and absorption with variation in intestinal pH for BCS class II weak acid drugs: Ibuprofen and ketoprofen. Biopharm. Drug Dispos. 2012, 33, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wen, H.; Fan, J.; Vince, B.; Li, T.; Gao, W.; Kinjo, M.; Brown, J.; Sun, W.; Jiang, W.; et al. Integrating In Vitro, Modeling, and In Vivo Approaches to Investigate Warfarin Bioequivalence. CPT Pharm. Syst. Pharmacol. 2017, 6, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.M. Clinical pharmacokinetics of flurbiprofen and its enantiomers. Clin. Pharm. 1995, 28, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Faassen, F.; Vromans, H. Biowaivers for oral immediate-release products: Implications of linear pharmacokinetics. Clin. Pharm. 2004, 43, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Yazdanian, M.; Briggs, K.; Jankovsky, C.; Hawi, A. The “high solubility” definition of the current FDA Guidance on Biopharmaceutical Classification System may be too strict for acidic drugs. Pharm. Res. 2004, 21, 293–299. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Clinical Study a | Description | Bempedoic Acid Dose Regimen and PK Data Used to Support Model Development and Verification | Sample Timepoints of Bempedoic Acid Plasma Concentration Determinations |
|---|---|---|---|
| 001 | Bempedoic acid absorption, distribution, metabolism, and excretion in healthy subjects (n = 6) | Single 240 mg dose as an oral solution formulation. Single-dose plasma PK results were used | Day 1 (pre-dose and 2, 4, 6, 12, 24, 48, 96, 144, and 168 h) |
| 002 | Bioequivalence of 2 tablet formulations in healthy subjects (n = 58) | Single 180 mg dose as a tablet. Plasma PK results for the reference formulation (development tablet) of a 2-period crossover were used | Days 1 and 15; reference tablet PK only (pre-dose and 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 24, 48, 72, 96, and 120 h) |
| 003 | Drug–drug interaction of steady-state bempedoic acid with concomitant single-dose statin therapy in healthy subjects (n = 48) | Multiple dosing at 180 mg/day. Day 12 steady-state plasma PK results were used | Days 10 and 11 of bempedoic acid dosing (pre-dose) and Day 12 (pre-dose and 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, 12, 18, 24, 36, 48, 72, 96, and 120 h) |
| 004 | Crossover bioequivalence study with 2 tablet formulations in healthy subjects (n = 59) | Single 180 mg dose crossover of commercial (Formulation 2; test) and development (Formulation 1; reference) tablets. Plasma PK results from both groups were used | Days 1 and 15 (pre-dose and 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 24, 48, 72, 96, and 120 h) |
| Bempedoic Acid 180 mg Regimen | Clinical Study | Concomitant Treatment (No. Subjects) | Estimate | Cmax, μg/mL | AUCinf (Single Dose) or AUC24h (Repeat Dose) a, μg·h/mL | tmax b, h |
|---|---|---|---|---|---|---|
| Single Dose | 002 | NA (N = 58) | Predicted | 18.0 (2.60) | 273 (102) | 1.55 (1.15–3.50) |
| Observed | 13.5 (3.19) | 225 (70.3) | 3.00 (1.00–6.00) | |||
| Pred/Obs Ratio | 1.33 | 1.21 | 0.52 | |||
| Repeat Dose (Steady-State) | 003 | Atorva80 (N = 12) | Predicted | 24.5 (5.82) | 267 (111) | 1.60 (1.15–3.50) |
| Observed | 27.3 (6.98) | 348 (95.7) | 1.50 (1.00–4.03) | |||
| Pred/Obs Ratio | 0.90 | 0.77 | 1.07 | |||
| Simva40 (N = 12) | Predicted | 23.8 (5.46) | 260 (105) | 1.60 (1.15–3.50) | ||
| Observed | 24.7 (6.99) | 276 (64.5) | 2.00 (1.00–3.00) | |||
| Pred/Obs Ratio | 0.96 | 0.94 | 0.80 | |||
| Prava80 (N = 12) | Predicted | 25.1 (6.06) | 274 (116) | 1.55 (1.15–3.45) | ||
| Observed | 23.7 (5.59) | 289 (106) | 2.00 (1.00–3.00) | |||
| Pred/Obs Ratio | 1.06 | 0.95 | 0.78 | |||
| Rosuva40 (N = 12) | Predicted | 24.3 (5.76) | 265 (108) | 1.60 (1.15–3.50) | ||
| Observed | 21.9 (9.60) | 264 (129) | 1.75 (1.00–4.00) | |||
| Pred/Obs Ratio | 1.11 | 1.01 | 0.92 |
| Parameter | Estimate | Comment or Parameter Source |
|---|---|---|
| Molecular weight | 344.5 | Internal data on file |
| Ionization | Diprotic acid | Internal data on file |
| pKa1 | 4.88 | Internal data on file |
| pKa2 | 5.60 | Internal data on file |
| Intrinsic solubility, mg/mL | 0.0051 | Internal data on file |
| Log P | 4.328 | Internal data on file |
| DLM scalar (tablet/suspension) | 0.07/0.3 | Model-defined parameter |
| Caco-2 permeability, ×10−6 cm/s | 11.5 | Internal data on file |
| Permeability estimate, ×10−4 cm/s | 3 | Model-defined parameter |
| Tablet, particle radius, µm | 18.2 | Internal data on file (D50 measured: 36.4 µm) a |
| Suspension, particle radius, µm | 25 | Internal data on file (D50 measured: 50 µm) |
| Suspension, bempedoic acid, mg/mL | 20 | Internal data on file |
| Suspension, drug fraction dissolved | 0.01% | Model-defined parameter |
| Suspension, viscosity, cps | 118.8 | Internal data on file |
| Blood:Plasma ratio | 0.55 | Study 1002-001 |
| Plasma unbound fraction | 0.026 | Internal data on file |
| fa | 0.97 | Study 1002-001 |
| fg | 1.0 | 100% gut availability assumed |
| Kp scalar | 2 | Model-defined parameter |
| Vsac, L/kg | 0.1 | Model-defined parameter |
| CLin (L/h)/CLout, L/h | 3.16/1.32 | Model-defined parameters |
| Vss, L/kg | 0.14 | Model-defined parameter |
| CLoral, L/h | 0.81 | Study 1002-001 |
| CLrenal, L/h | 0.03 | Study 1002-001 |
| CLu,int,H, µL/min/mg protein | 8.17 | Model-defined parameter |
| Gastrointestinal Physiology Parameter | Cmax | tmax | AUCinf |
|---|---|---|---|
| Gastric TT | −0.32 | 0.40 | 0.07 |
| Stomach pH (fasted) | 0.00 | −0.02 | −0.01 |
| Small Intestine TT | −0.09 | 0.17 | −0.02 |
| Colon TT | −0.11 | 0.00 | −0.12 |
| Virtual Study Design | Parameter | Pilot Scale Suspension GM Estimate (%CV) | Commercial Tablet GM Estimate (%CV) | Test/Reference Ratio (90% CI) |
|---|---|---|---|---|
| Crossover | Cmax, µg/mL | 19.6 (11.5) | 19.6 (12.7) | 99.7% (96.1–103) |
| tmax, h | 1.57 (38.0) | 1.56 (38.3) | nc | |
| AUClast, µg·h/mL | 313 (33.2) | 313 (33.2) | 100% (90.6–110) | |
| AUCinf, µg·h/mL | 329 (39.2) | 329 (39.2) | 100% (89.1–112) | |
| Parallel | Cmax, µg/mL | 17.2 (13.0) | 17.8 (16.8) | 96.9% (92.6–101) |
| tmax, h | 1.85 (34.2) | 1.68 (36.3) | nc | |
| AUClast, µg·h/mL | 259 (38.8) | 264 (41.4) | 98.2% (87.3–111) | |
| AUCinf, µg·h/mL | 270 (42.6) | 274 (45.9) | 98.4% (86.4–112) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amore, B.M.; Patel, N.; Batheja, P.; Templeton, I.E.; Jones, H.M.; Louie, M.J.; Emery, M.G. Physiologically Based Pharmacokinetic Model Development and Verification for Bioequivalence Testing of Bempedoic Acid Oral Suspension and Reference Tablet Formulation. Pharmaceutics 2023, 15, 1476. https://doi.org/10.3390/pharmaceutics15051476
Amore BM, Patel N, Batheja P, Templeton IE, Jones HM, Louie MJ, Emery MG. Physiologically Based Pharmacokinetic Model Development and Verification for Bioequivalence Testing of Bempedoic Acid Oral Suspension and Reference Tablet Formulation. Pharmaceutics. 2023; 15(5):1476. https://doi.org/10.3390/pharmaceutics15051476
Chicago/Turabian StyleAmore, Benny M., Nikunjkumar Patel, Priya Batheja, Ian E. Templeton, Hannah M. Jones, Michael J. Louie, and Maurice G. Emery. 2023. "Physiologically Based Pharmacokinetic Model Development and Verification for Bioequivalence Testing of Bempedoic Acid Oral Suspension and Reference Tablet Formulation" Pharmaceutics 15, no. 5: 1476. https://doi.org/10.3390/pharmaceutics15051476