
Challenges and Opportunities in the Oral Delivery of Recombinant Biologics
 , , , and
, , , and
Abstract
:1. Introduction
2. Challenges Associated with Oral Delivery of Drugs
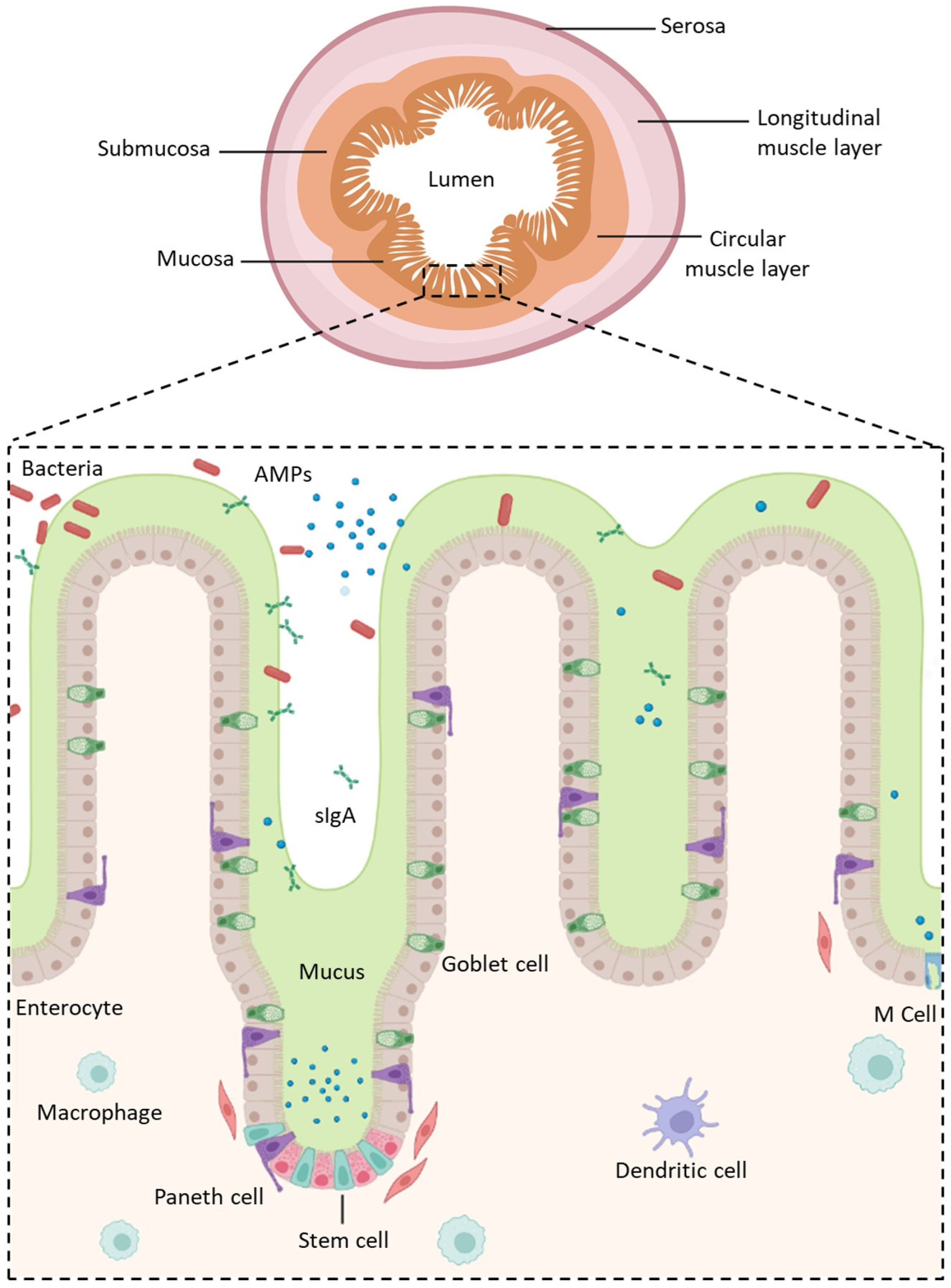
2.1. Gastro-Intestinal Barriers
2.1.1. Mucus and Glycocalyx
2.1.2. The Intestinal Epithelium
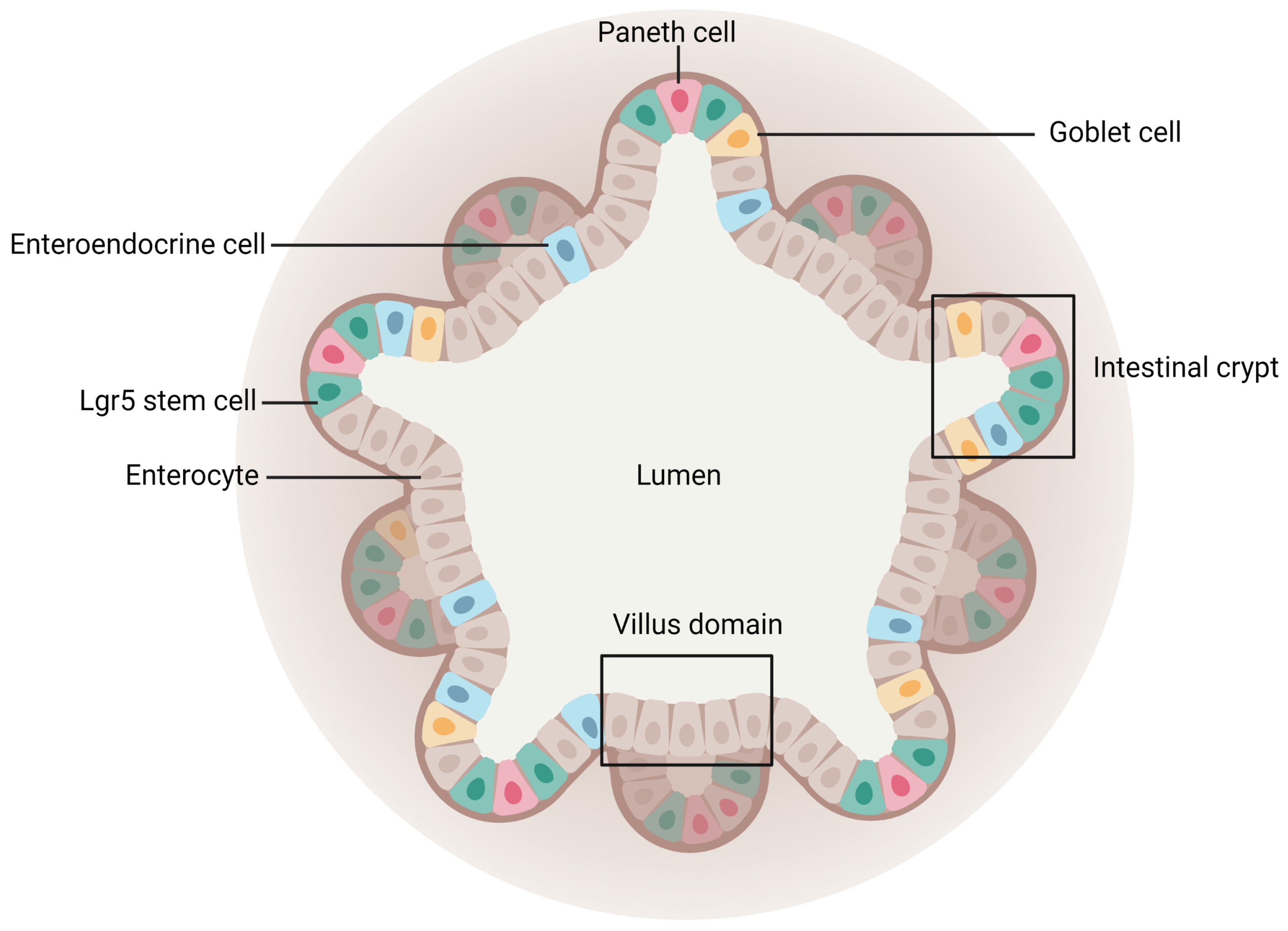
- Enterocytes, which are the predominant cell type found in the intestinal epithelium (90%). They have a microvilli network that increases their surface area for transport and forms a brush border on their apical surface. Many digestive enzymes, receptors, and transporters needed for the uptake and transport of molecules can be found on their microvilli [44];
- Goblet cells, which represent 10% of all IECs and are responsible for mucus production;
- Enteroendocrine cells, which are able to secrete and release intestinal hormones or peptides into the circulation upon stimulation [45];
- Long-lived Paneth cells, which produce and release antimicrobial peptides in the intestinal crypts of the monolayer [46];
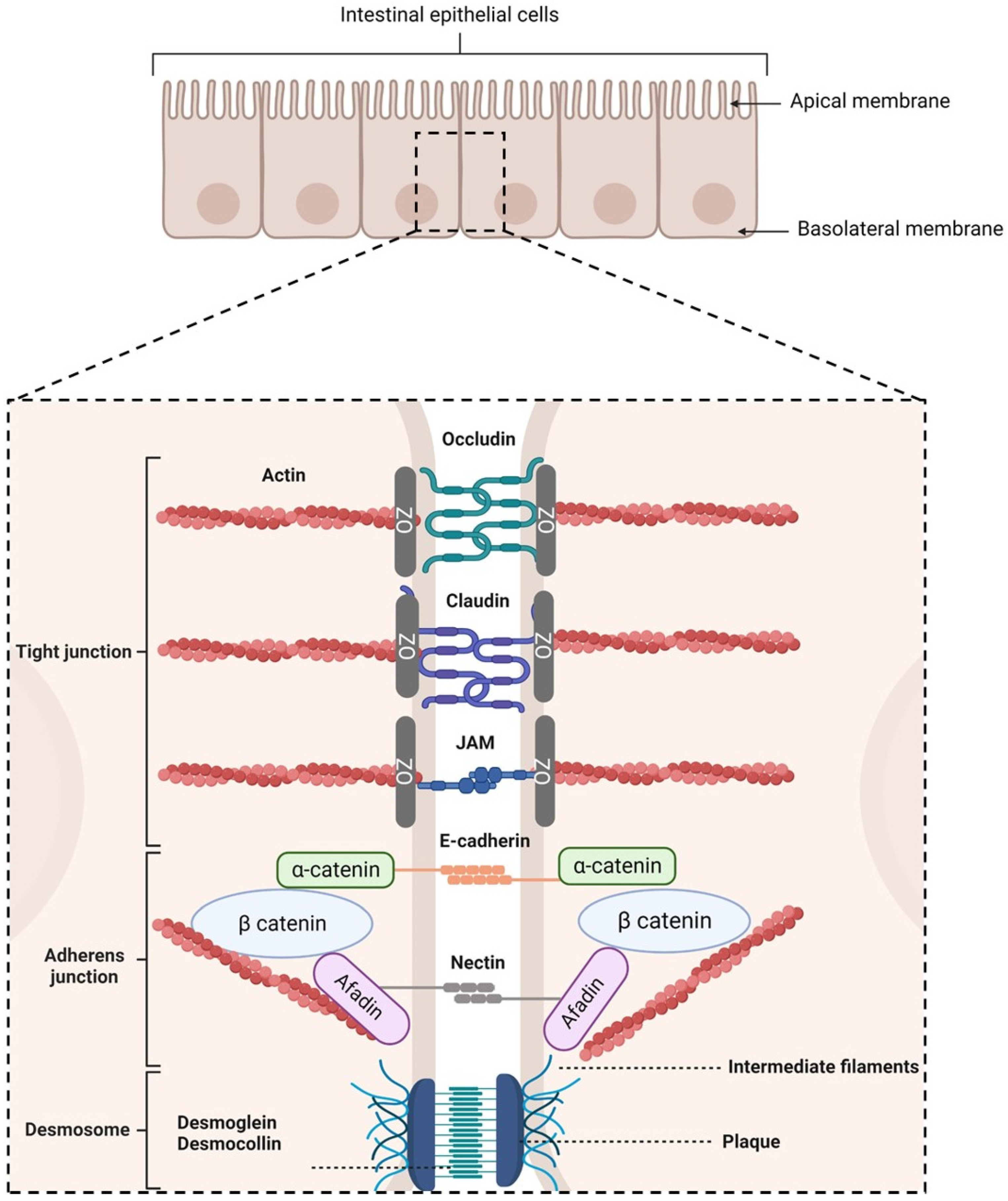
- Tight junctions (TJs), which are located at the most apical region of polarized IECs. They make paracellular transport mainly dependent on the size of a molecule due to their multiple protein–protein interactions between adjacent cells [53,54,55]. TJs are composed of transmembrane proteins (occludins, claudins, tricellulin, and junctional adhesion molecules) and plaque proteins (e.g., zonula occludens-1, -2, and -3), which act as bridges to connect integral membrane proteins to the actin cytoskeleton and to other signaling proteins. They are also composed of other regulatory proteins [56].
- Adherens junctions (AJs), which are located beneath TJs. They are involved in cell–cell adhesion stability, intracellular signaling, and interact with the actin cytoskeleton. E-cadherins are the major component of AJs. They interact with the E-cadherins of adjacent cells and with the actin cytoskeleton [57,58]. Nectin–afadin complexes are also important as they form homophilic and heterophilic strands with adjacent cells [59,60].
- Desmosomes, which are located at the most basolateral region of IECs. They are found in tissues that require mechanical forces. Indeed, they promote strong adhesive bonds between adjacent cells by connecting them to the intermediate filaments of the cytoskeleton. Its main constituents are cadherins, armadillo proteins, and plakins [61,62,63,64].
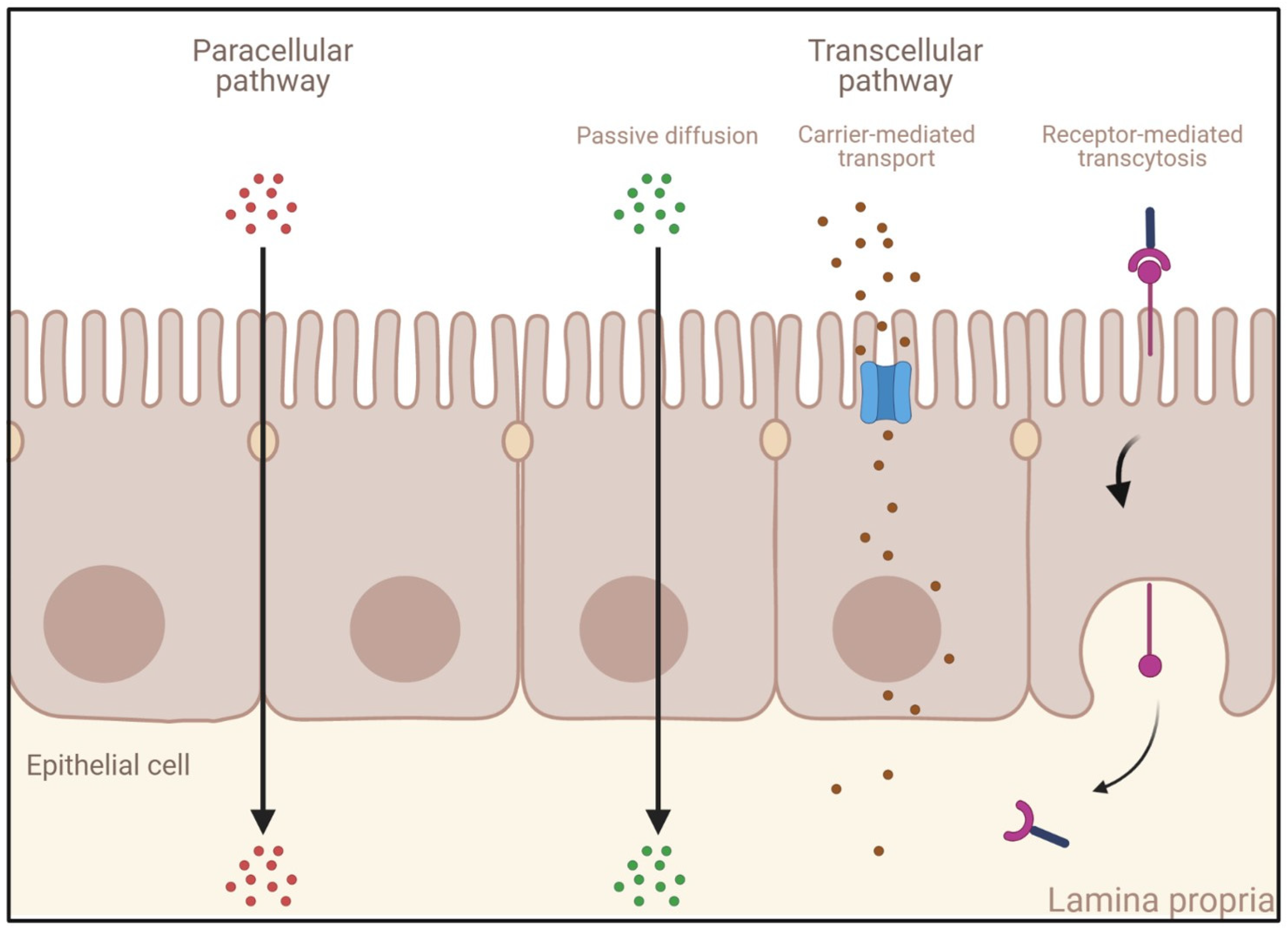
- Passive diffusion: This process, which does not require energy expenditure, is a non-selective, non-saturable, and non-carrier-mediated transport through the phospholipid membranes of IECs driven by the concentration gradient of a compound [65]. Passive diffusion only allows small lipophilic molecules to diffuse at significant rates (e.g., gases such as O2 and CO2, hydrophobic compounds, small polar but uncharged molecules).
- Carrier-mediated transport: Large uncharged polar molecules (e.g., glucose) and charged molecules of any size (e.g., small ions such as H+, Na+, K+, and Cl−) are not able to cross the membrane by passive diffusion. Hence, they require specific transporters and channel proteins situated at the cell surface to be transported across the epithelium. This transport can be achieved either by a reversible facilitated diffusion, which, similarly to passive diffusion, induces a movement of solutes across membranes from the side of high concentration to the side of low concentration without energy (e.g., carbohydrates, amino acids, nucleosides, ions), or by an active and saturable passage against the concentration gradient requiring chemical energy (e.g., ATP hydrolysis) [66]. Other molecules are transported against the concentration gradient by using the electrochemical potential difference created by pumping ions out of the cell (e.g., the Na+-K+ pump).
- Endocytosis: This mechanism induces the internalization of extracellular molecules via several processes involving the formation of intracellular vesicles and not limited by the size of the cargo [67,68] such as pinocytosis, phagocytosis, or receptor-mediated endocytosis (RME) [69]. Pinocytosis is a fluid-phase endocytosis pathway that is non-specific and non-saturable (cellular incorporation of molecules in the extracellular fluid via micropinocytosis or macropinocytosis) [70], whereas phagocytosis is triggered by the binding to phagocytic receptors of particles larger than 0.5 μm in diameter (e.g., microorganisms, foreign substances, apoptotic cells). They are engulfed to form large intracellular vesicles called phagosomes that will fuse to lysosomes to create phagolysosomes and induce the degradation of the particles [71]. RME is an uptake mechanism occurring in different cell types and triggered by the binding of ligands on their specific membrane-bound receptors [69,72]. When followed by transcytosis, it enables their transport and is thus of high interest for the specific delivery of large molecules into the bloodstream.
2.1.3. The Biochemical Barrier
2.2. Challenges of the Oral Delivery of Biologics
3. Models to Study Permeability of Biologics across the Intestinal Barrier
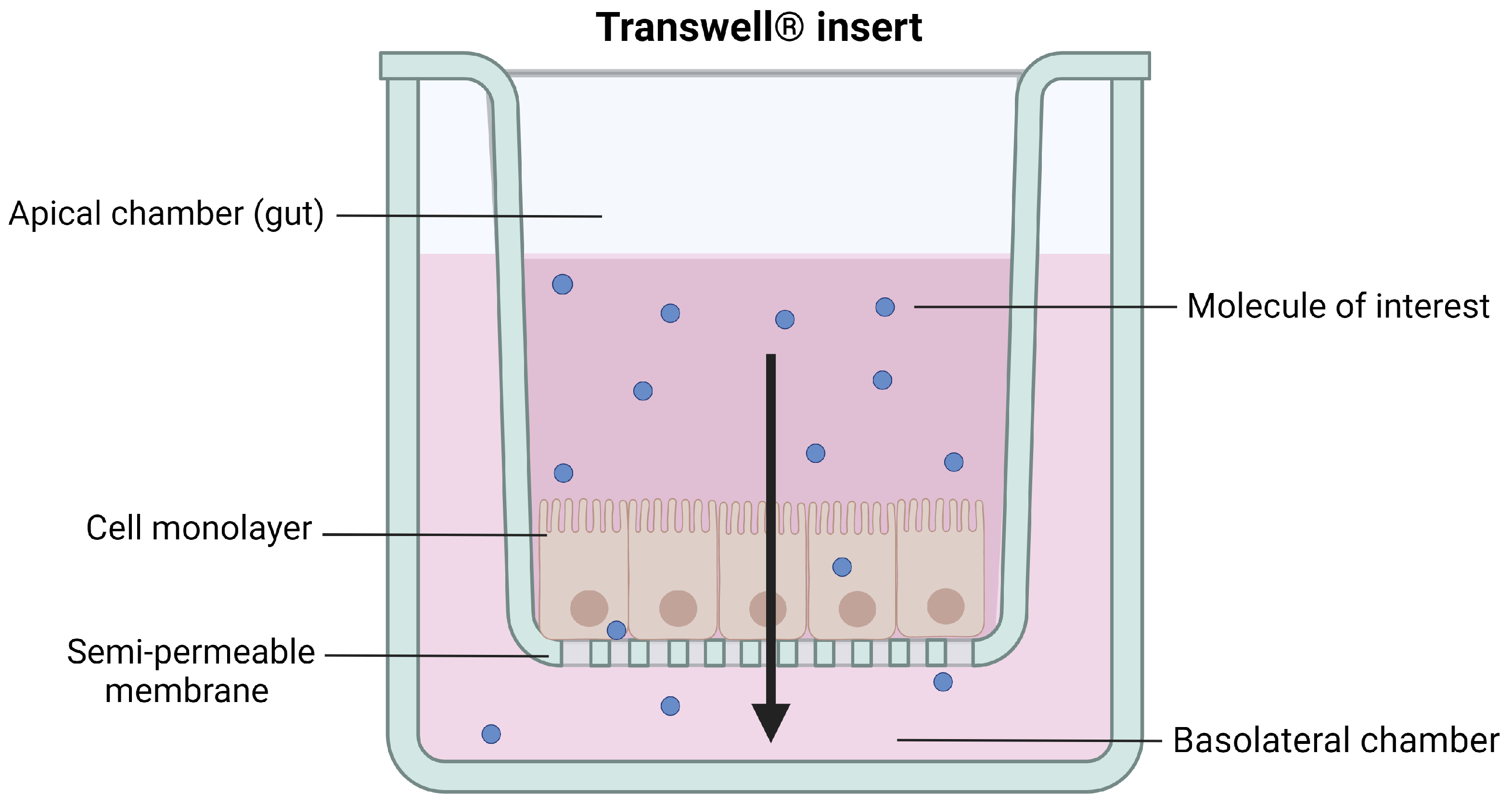
3.1. In Vitro Models
3.1.1. Caco-2 Cells
3.1.2. MDCK Cells
3.1.3. Caco-2/HT29 Cells
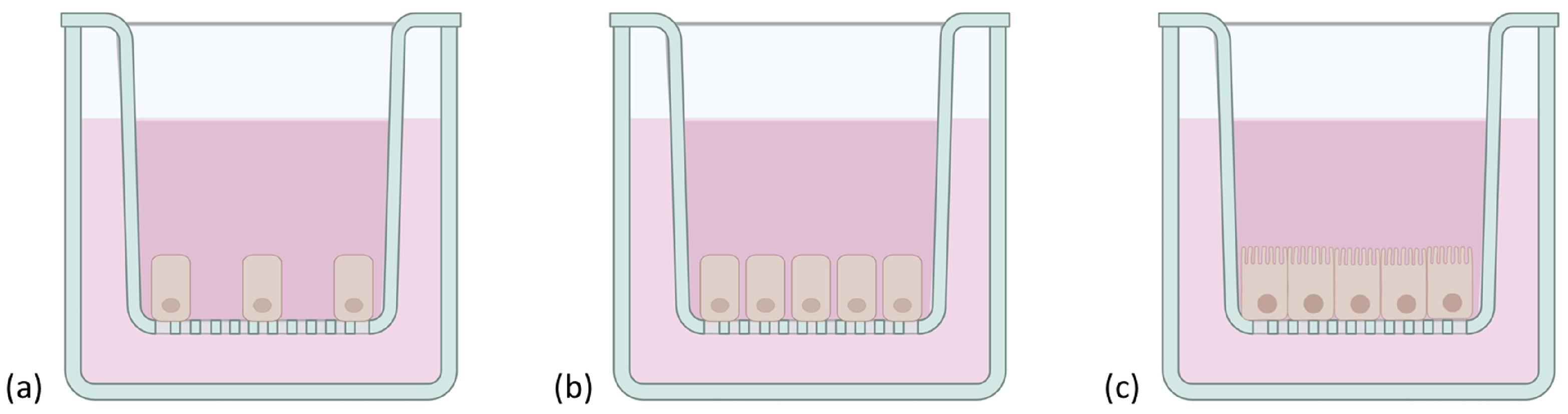
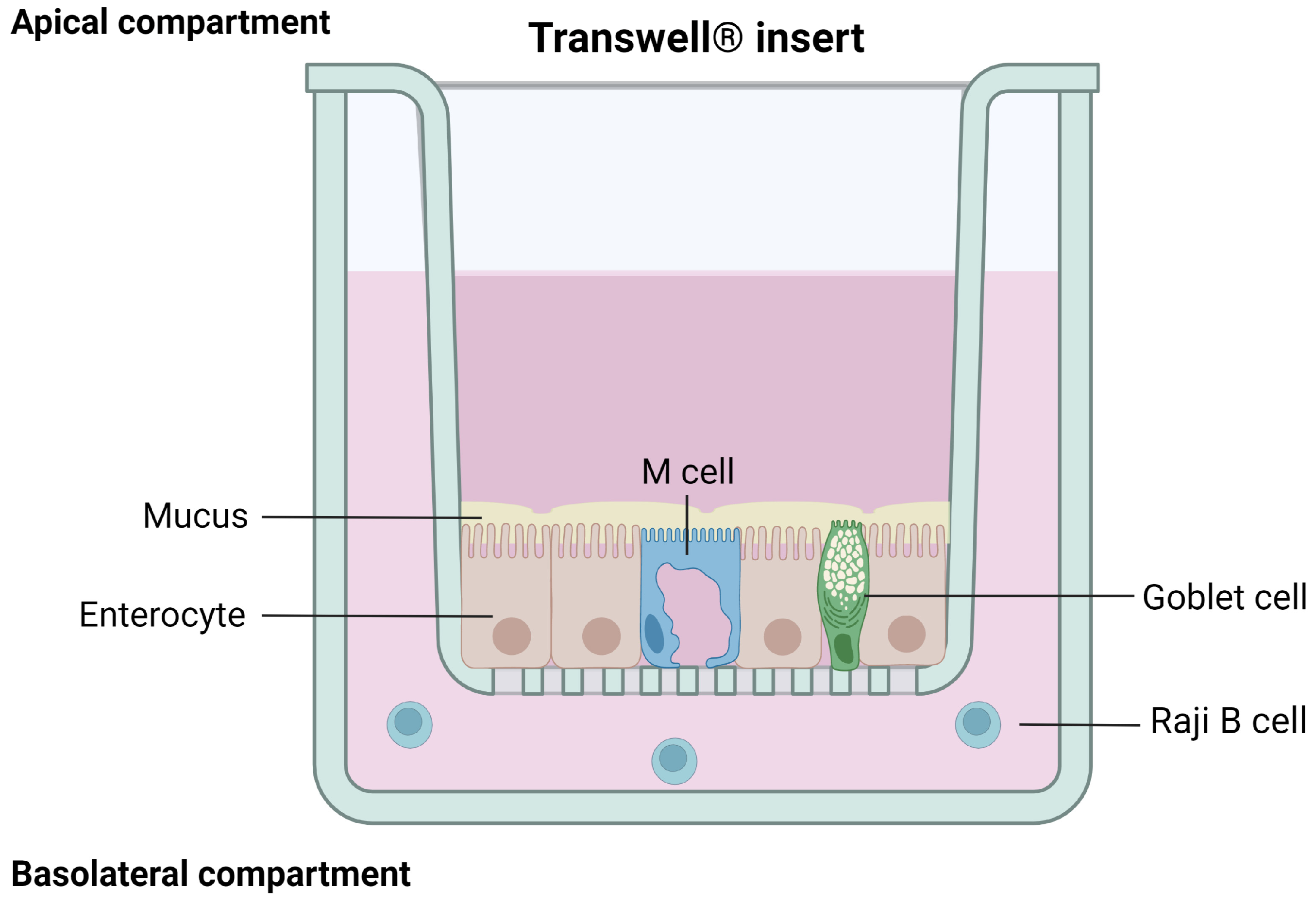
3.1.4. Caco-2/HT29/Raji B Cells
3.1.5. 3D Culture Models
EpiIntestinal
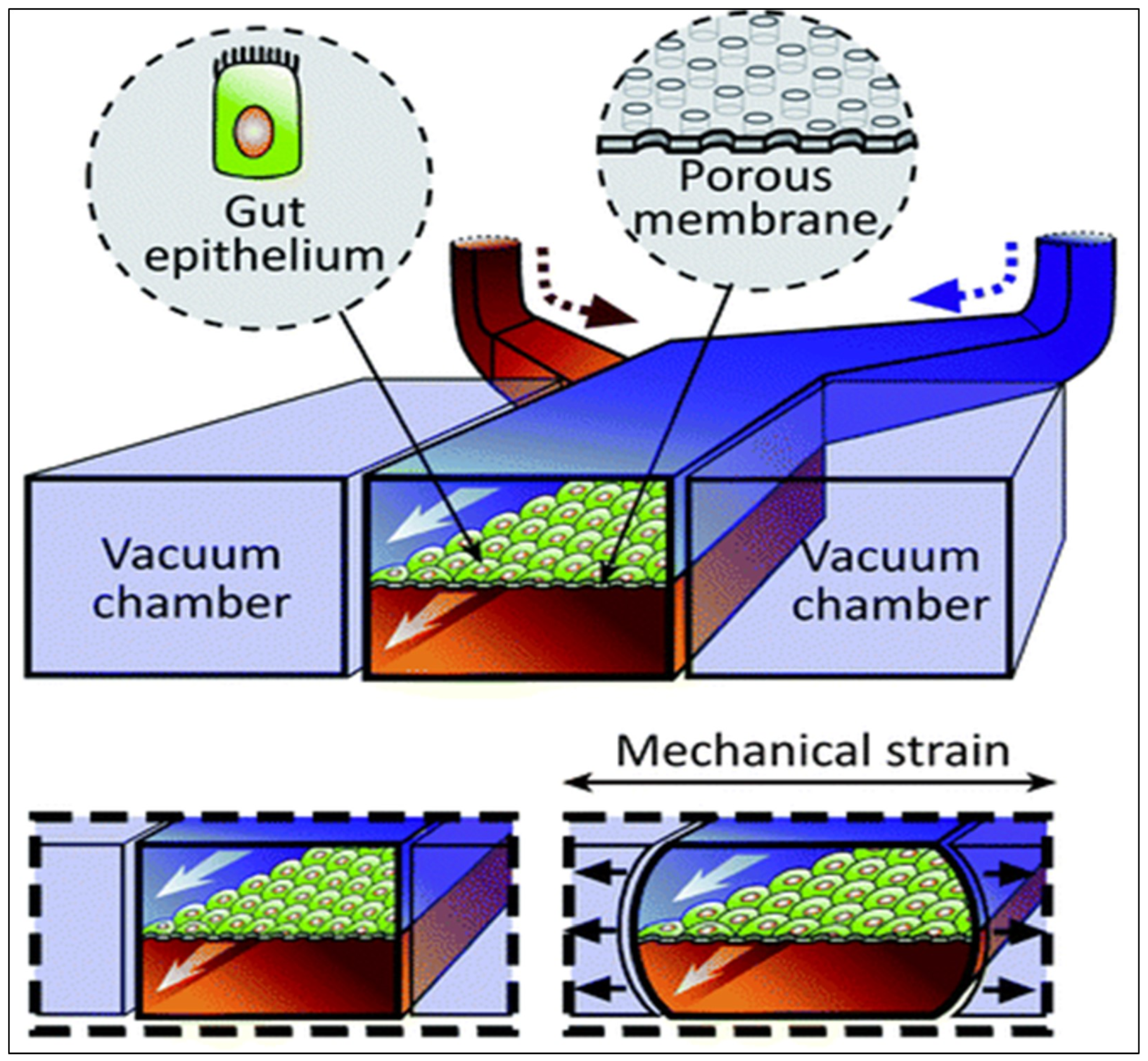
Gut-on-a-Chip

Intestinal Organoids

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Advantages | Limitations | References |
|---|---|---|---|
| Caco-2 cells |
|
| [85,101,102,104] |
| MDCK cells |
|
| [84,111,117,166] |
| Caco-2/HT29 cells |
|
| [11,118,121] |
| Caco-2/HT29/RajiB cells |
|
| [126,127] |
| EpiIntestinal |
|
| [131,132,134,135,166] |
| Gut-on-a-chip |
|
| [138,140,141] |
| Intestinal organoids |
|
| [155,156,158] |
3.2. Ex Vivo Models
3.2.1. Everted Intestinal Sacs
3.2.2. Diffusion Chambers
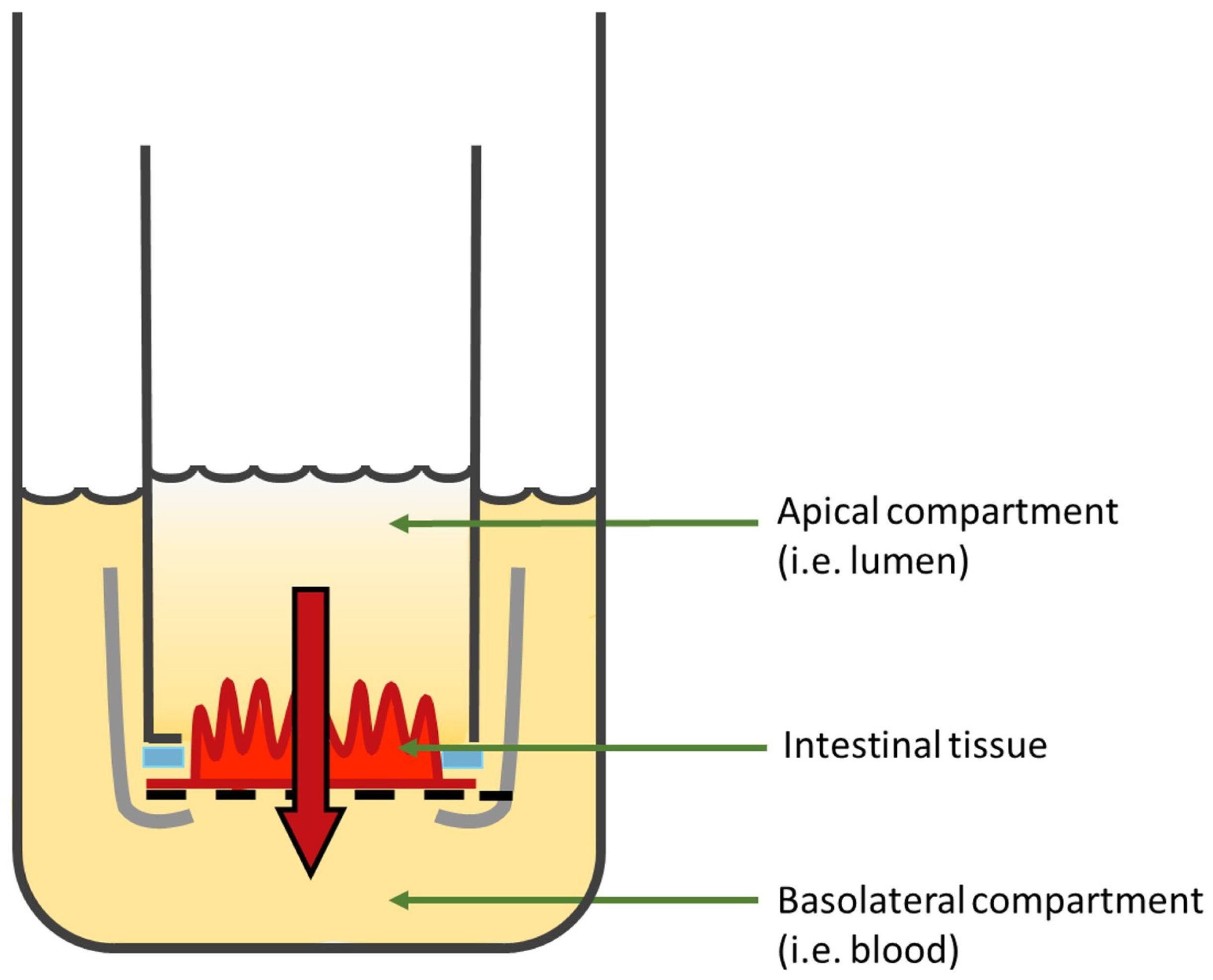
Ussing Chambers
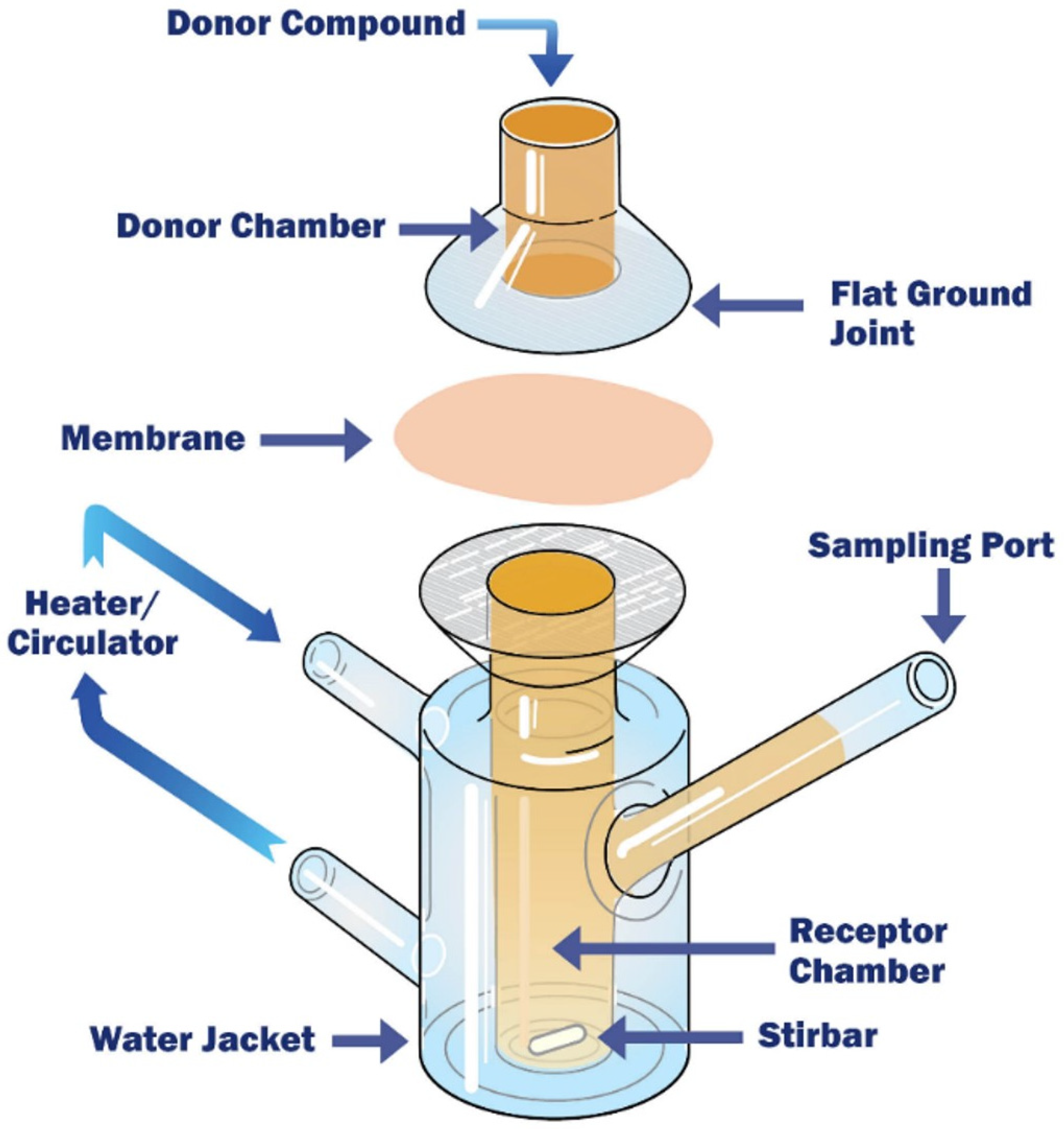
Franz Cells
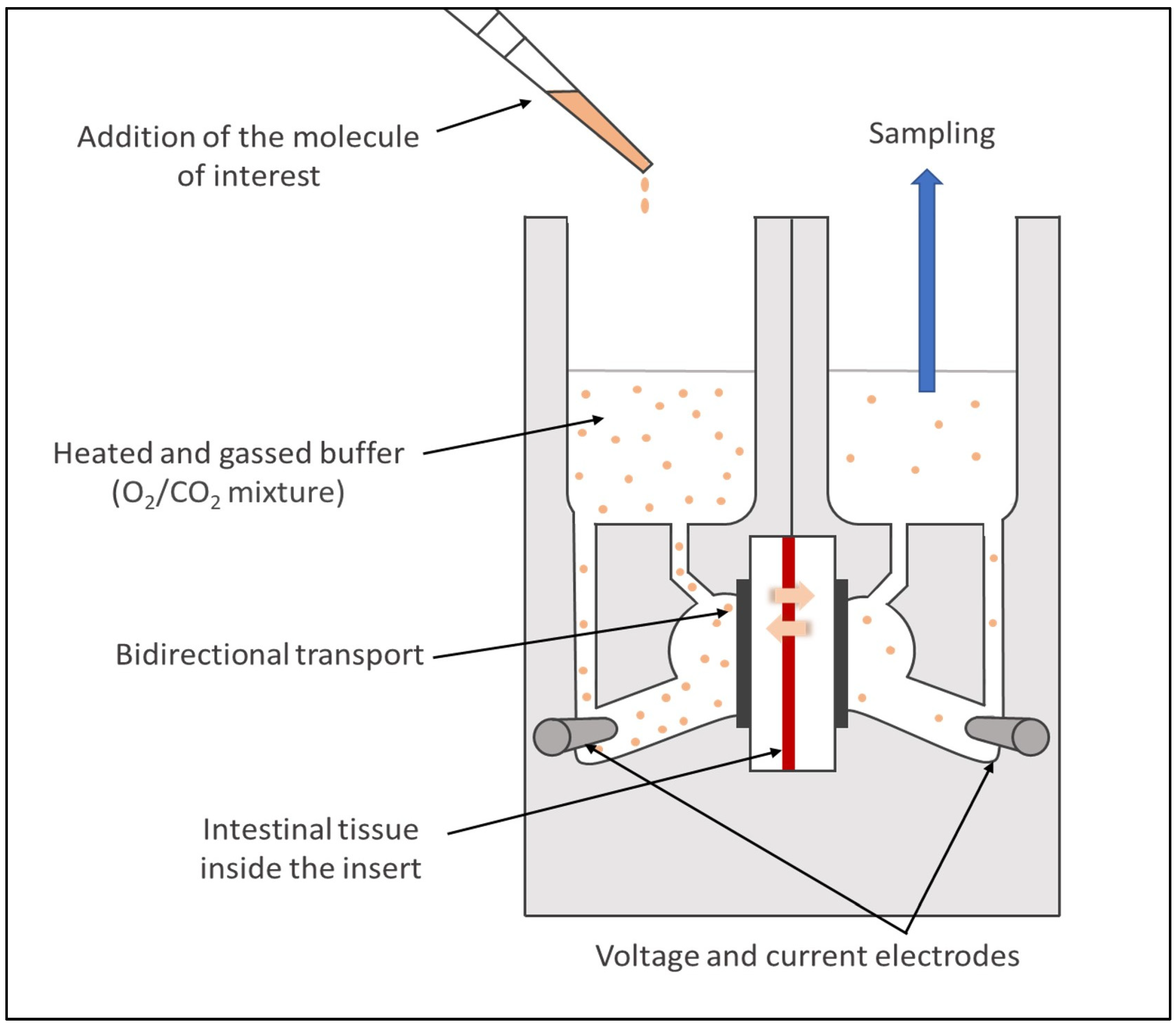
3.2.3. InTESTine™
| Model | Advantages | Limitations | References |
|---|---|---|---|
| Everted intestinal sacs |
|
| [14,168,169,171] |
| Ussing chambers |
|
| [14,180,196] |
| Franz diffusion cells |
|
| [188,189,191,197] |
| IntesTINETM |
|
| [193,195] |
4. Strategies to Improve the Oral Delivery of Biologics
4.1. Strategies to Increase Stability
4.1.1. Enteric Coating
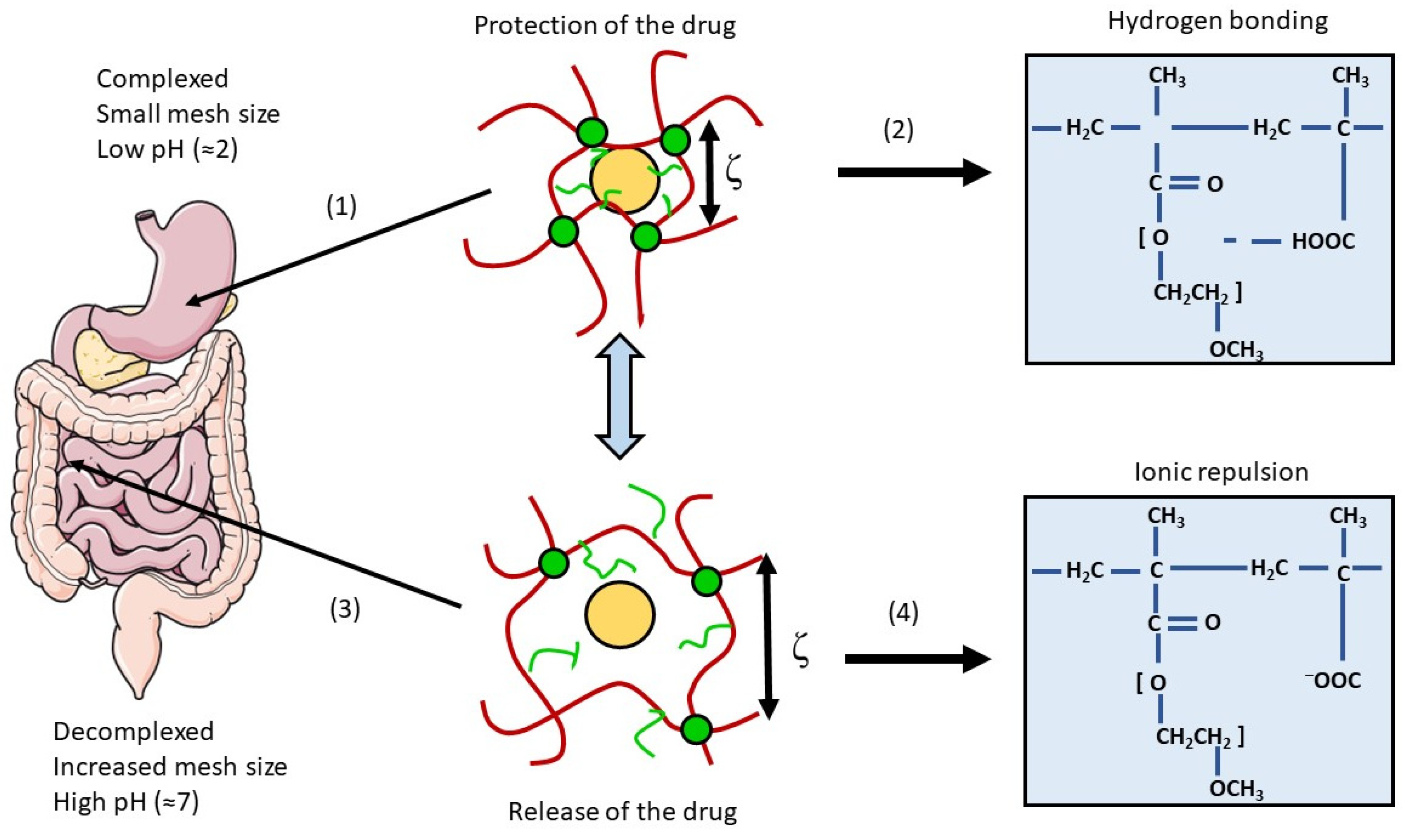
4.1.2. pH Modulators
4.1.3. Enzyme Inhibitors
4.2. Strategies to Penetrate the Mucus Layer
4.3. Strategies to Increase Contact Time with the Epithelium and Induce a Site-Specific Release
4.3.1. Thiolated Polymers
4.3.2. Intestinal Patches
- a pH-sensitive layer generally made of materials with pH-dependent solubility (e.g., Eudragit® polymers L or S [235]) to bypass the acidic environment of the stomach and avoid drug delivery before reaching the localized site in the small intestine;
- a mucoadhesive/drug reservoir layer with the molecule of interest (dissolved, suspended, or incorporated as microspheres into the layer); the mucoadhesive components (e.g., chitosan, thiomers); and other excipients (e.g., PEs, enzyme inhibitors). It induces the adhesion to the intestinal mucosa and enables a longer retention time at the specific site of drug release [236];
4.3.3. Intestinal Hydrogels
4.4. Strategies to Cross the Intestinal Epithelium
4.4.1. Polymeric Particles
4.4.2. Targeting a Specific Cell-Surface Receptor
Vitamin B12/IF/Cubilin Receptor
Transferrin/Transferrin Receptor
Immunoglobulin G/Nenonatal Fc Receptor
4.4.3. Permeation Enhancers
Chelating Agents
Surfactants
Cell-Penetrating Peptides
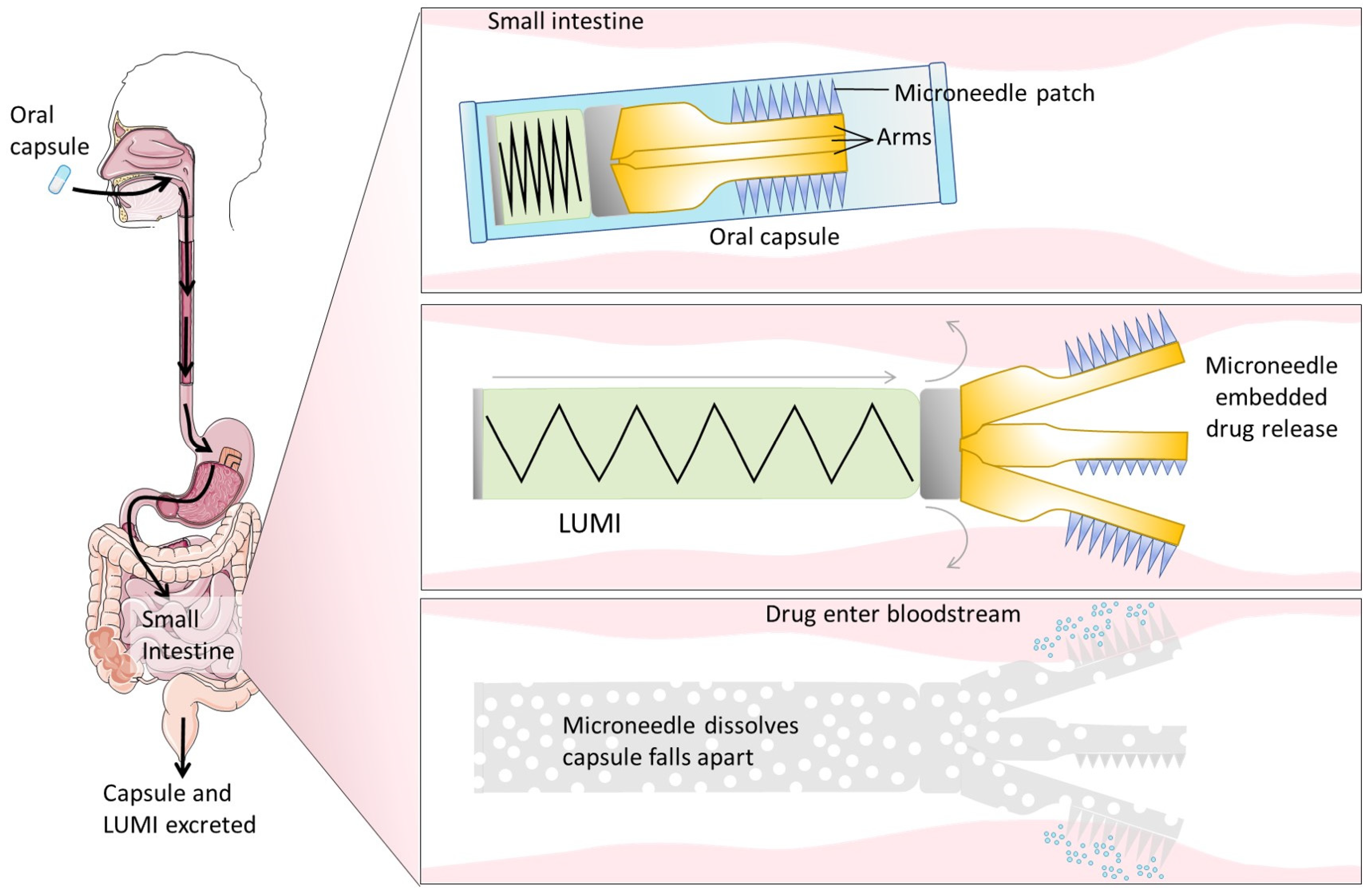
4.4.4. Devices for Physical Delivery
Microneedles
Ultrasounds
5. Current Landscape of Clinical Trials and Marketed Orally Delivered Biologics
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liang, W.; Pan, H.W.; Vllasaliu, D.; Lam, J.K.W. Pulmonary Delivery of Biological Drugs. Pharmaceutics 2020, 12, 1025. [Google Scholar] [CrossRef] [PubMed]
- de la Torre, B.G.; Albericio, F. The Pharmaceutical Industry in 2021. An Analysis of FDA Drug Approvals from the Perspective of Molecules. Molecules 2022, 27, 1075. [Google Scholar] [CrossRef] [PubMed]
- Kinch, M.S. An overview of FDA-approved biologics medicines. Drug Discov. Today 2015, 20, 393–398. [Google Scholar] [CrossRef]
- Thomaidou, E.; Ramot, Y. Injection site reactions with the use of biological agents. Dermatol. Ther. 2019, 32, e12817. [Google Scholar] [CrossRef] [PubMed]
- Richardson, T.; Kerr, D. Skin-related complications of insulin therapy: Epidemiology and emerging management strategies. Am. J. Clin. Dermatol. 2003, 4, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Eek, D.; Krohe, M.; Mazar, I.; Horsfield, A.; Pompilus, F.; Friebe, R.; Shields, A.L. Patient-reported preferences for oral versus intravenous administration for the treatment of cancer: A review of the literature. Patient Prefer. Adherence 2016, 10, 1609–1621. [Google Scholar]
- Groschwitz, K.R.; Hogan, S.P. Intestinal barrier function: Molecular regulation and disease pathogenesis. J. Allergy Clin. Immunol. 2009, 124, 3–20. [Google Scholar] [CrossRef]
- Schoultz, I.; Keita, Å.V. The Intestinal Barrier and Current Techniques for the Assessment of Gut Permeability. Cells 2020, 9, 1909. [Google Scholar] [CrossRef]
- Ménard, S.; Cerf-Bensussan, N.; Heyman, M. Multiple facets of intestinal permeability and epithelial handling of dietary antigens. Mucosal. Immunol. 2010, 3, 247–259. [Google Scholar] [CrossRef]
- Ricceri, L.; Vitale, A. The law through the eye of a needle. EMBO Rep. 2011, 12, 637–640. [Google Scholar] [CrossRef]
- Xu, Y.; Shrestha, N.; Préat, V.; Beloqui, A. An overview of in vitro, ex vivo and in vivo models for studying the transport of drugs across intestinal barriers. Adv. Drug Deliv. Rev. 2021, 175, 113795. [Google Scholar] [CrossRef] [PubMed]
- Polli, J.E. In Vitro Studies are Sometimes Better than Conventional Human Pharmacokinetic In Vivo Studies in Assessing Bioequivalence of Immediate-Release Solid Oral Dosage Forms. AAPS J. 2008, 10, 289–299. [Google Scholar] [CrossRef]
- Sjöberg, Å.; Lutz, M.; Tannergren, C.; Wingolf, C.; Borde, A.; Ungell, A.L. Comprehensive study on regional human intestinal permeability and prediction of fraction absorbed of drugs using the Ussing chamber technique. Eur. J. Pharm. Sci. 2013, 48, 166–180. [Google Scholar] [CrossRef]
- Nunes, R.; Silva, C.; Chaves, L. 4.2—Tissue-based in vitro and ex vivo models for intestinal permeability studies. In Concepts and Models for Drug Permeability Studies; Sarmento, B., Ed.; Woodhead Publishing: Sawston, UK, 2016; pp. 203–236. [Google Scholar]
- Russell-Jones, G.J.; Westwood, S.W.; Habberfield, A.D. Vitamin B12 mediated oral delivery systems for granulocyte-colony stimulating factor and erythropoietin. Bioconjug. Chem. 1995, 6, 459–465. [Google Scholar] [CrossRef]
- Amet, N.; Wang, W.; Shen, W.C. Human growth hormone-transferrin fusion protein for oral delivery in hypophysectomized rats. J. Control. Release 2010, 141, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Ann, D.K.; Shen, W.C. Recombinant granulocyte colony-stimulating factor-transferrin fusion protein as an oral myelopoietic agent. Proc. Natl. Acad. Sci. USA 2005, 102, 7292–7296. [Google Scholar] [CrossRef] [PubMed]
- Bernkop-Schnürch, A.; Kast, C.E.; Guggi, D. Permeation enhancing polymers in oral delivery of hydrophilic macromolecules: Thiomer/GSH systems. J. Control. Release 2003, 93, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Kast, C.E.; Guggi, D.; Langoth, N.; Bernkop-Schnürch, A. Development and in vivo evaluation of an oral delivery system for low molecular weight heparin based on thiolated polycarbophil. Pharm. Res. 2003, 20, 931–936. [Google Scholar] [CrossRef]
- Gupta, V.; Hwang, B.H.; Doshi, N.; Banerjee, A.; Anselmo, A.C.; Mitragotri, S. Delivery of Exenatide and Insulin Using Mucoadhesive Intestinal Devices. Ann. Biomed. Eng. 2016, 44, 1993–2007. [Google Scholar] [CrossRef]
- Banerjee, A.; Wong, J.; Gogoi, R.; Brown, T.; Mitragotri, S. Intestinal micropatches for oral insulin delivery. J. Drug Target. 2017, 25, 608–615. [Google Scholar] [CrossRef]
- Liu, C.; Kou, Y.; Zhang, X.; Cheng, H.; Chen, X.; Mao, S. Strategies and industrial perspectives to improve oral absorption of biological macromolecules. Expert Opin. Drug Deliv. 2018, 15, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Fattah, S.; Ismaiel, M.; Murphy, B.; Rulikowska, A.; Frias, J.M.; Winter, D.C.; Brayden, D.J. Salcaprozate sodium (SNAC) enhances permeability of octreotide across isolated rat and human intestinal epithelial mucosae in Ussing chambers. Eur. J. Pharm. Sci. 2020, 154, 105509. [Google Scholar] [CrossRef] [PubMed]
- Kamei, N.; Shigei, C.; Hasegawa, R.; Takeda-Morishita, M. Exploration of the Key Factors for Optimizing the In Vivo Oral Delivery of Insulin by Using a Noncovalent Strategy with Cell-Penetrating Peptides. Biol. Pharm. Bull. 2018, 41, 239–246. [Google Scholar] [CrossRef]
- Abramson, A.; Caffarel-Salvador, E.; Soares, V.; Minahan, D.; Tian, R.Y.; Lu, X.; Dellal, D.; Gao, Y.; Kim, S.; Wainer, J.; et al. A luminal unfolding microneedle injector for oral delivery of macromolecules. Nat. Med. 2019, 25, 1512–1518. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Nazzal, S.; Reddy, I.K.; Khan, M.A. Transport studies of insulin across rat jejunum in the presence of chicken and duck ovomucoids. J. Pharm. Pharmacol. 2001, 53, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- nir.Technology. Oramed Pharmaceuticals. Available online: https://www.oramed.com/technology/ (accessed on 14 March 2023).
- Brayden, D.J.; Maher, S. Transient Permeation Enhancer® (TPE®) technology for oral delivery of octreotide: A technological evaluation. Expert Opin. Drug Deliv. 2021, 18, 1501–1512. [Google Scholar] [CrossRef]
- Semenya, A.M.; Wilson, S.A. Oral Semaglutide (Rybelsus) for the Treatment of Type 2 Diabetes Mellitus. Am. Fam. Physician 2020, 102, 627–628. [Google Scholar]
- Homayun, B.; Lin, X.; Choi, H.J. Challenges and Recent Progress in Oral Drug Delivery Systems for Biopharmaceuticals. Pharmaceutics 2019, 11, 129. [Google Scholar] [CrossRef]
- Vancamelbeke, M.; Vermeire, S. The intestinal barrier: A fundamental role in health and disease. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 821–834. [Google Scholar] [CrossRef]
- Moran, E.T. Nutrients central to maintaining intestinal absorptive efficiency and barrier integrity with fowl. Poult. Sci. 2017, 96, 1348–1363. [Google Scholar] [CrossRef]
- Dharmani, P.; Srivastava, V.; Kissoon-Singh, V.; Chadee, K. Role of intestinal mucins in innate host defense mechanisms against pathogens. J. Innate Immun. 2009, 1, 123–135. [Google Scholar] [CrossRef]
- Ensign, L.M.; Cone, R.; Hanes, J. Oral Drug Delivery with Polymeric Nanoparticles: The Gastrointestinal Mucus Barriers. Adv. Drug Deliv. Rev. 2012, 64, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Aranguez, A.; Argüeso, P. Structure and Biological Roles of Mucin-type O-glycans at the Ocular Surface. Ocul. Surf. 2010, 8, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Vllasaliu, D.; Thanou, M.; Stolnik, S.; Fowler, R. Recent advances in oral delivery of biologics: Nanomedicine and physical modes of delivery. Expert Opin. Drug Deliv. 2018, 15, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Bernkop-Schnürch, A.; Fragner, R. Investigations into the Diffusion Behaviour of Polypeptides in Native Intestinal Mucus with Regard to their Peroral Administration. Pharm. Pharmacol. Commun. 1996, 2, 361–363. [Google Scholar]
- Scott, P.G.; Leaver, A.G. The degradation of human dentine collagen by trypsin. Connect. Tissue Res. 1974, 2, 299–307. [Google Scholar] [CrossRef]
- Hansson, G.C. Mucins and the Microbiome. Annu. Rev. Biochem. 2020, 89, 769–793. [Google Scholar] [CrossRef]
- Bergstrom, K.S.B.; Xia, L. Mucin-type O-glycans and their roles in intestinal homeostasis. Glycobiology 2013, 23, 1026–1037. [Google Scholar] [CrossRef]
- Pelaseyed, T.; Bergström, J.H.; Gustafsson, J.K.; Ermund, A.; Birchenough, G.M.H.; Schütte, A.; van der Post, S.; Svensson, F.; Rodríguez-Piñeiro, A.M.; Nyström, E.E.L.; et al. The mucus and mucins of the goblet cells and enterocytes provide the first defense line of the gastrointestinal tract and interact with the immune system. Immunol. Rev. 2014, 260, 8–20. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Y.G. Intestinal epithelial plasticity and regeneration via cell dedifferentiation. Cell Regen. 2020, 9, 14. [Google Scholar] [CrossRef]
- Kong, S.; Zhang, Y.H.; Zhang, W. Regulation of Intestinal Epithelial Cells Properties and Functions by Amino Acids. Biomed. Res. Int. 2018, 2018, 2819154. [Google Scholar] [CrossRef] [PubMed]
- Snoeck, V.; Goddeeris, B.; Cox, E. The role of enterocytes in the intestinal barrier function and antigen uptake. Microbes Infect. 2005, 7, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Luyer, M.D.; Greve, J.W.M.; Hadfoune, M.; Jacobs, J.A.; Dejong, C.H.; Buurman, W.A. Nutritional stimulation of cholecystokinin receptors inhibits inflammation via the vagus nerve. J. Exp. Med. 2005, 202, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.C.; Bevins, C.L. Paneth cells: Maestros of the small intestinal crypts. Annu. Rev. Physiol. 2013, 75, 289–311. [Google Scholar] [CrossRef] [PubMed]
- Kanaya, T.; Sakakibara, S.; Jinnohara, T.; Hachisuka, M.; Tachibana, N.; Hidano, S.; Kobayashi, T.; Kimura, S.; Iwanaga, T.; Nakagawa, T.; et al. Development of intestinal M cells and follicle-associated epithelium is regulated by TRAF6-mediated NF-κB signaling. J. Exp. Med. 2018, 215, 501–519. [Google Scholar] [CrossRef] [PubMed]
- Allaire, J.M.; Crowley, S.M.; Law, H.T.; Chang, S.Y.; Ko, H.J.; Vallance, B.A. The Intestinal Epithelium: Central Coordinator of Mucosal Immunity. Trends Immunol. 2018, 39, 677–696. [Google Scholar] [CrossRef]
- Knipp, G.T.; Ho, N.F.; Barsuhn, C.L.; Borchardt, R.T. Paracellular diffusion in Caco-2 cell monolayers: Effect of perturbation on the transport of hydrophilic compounds that vary in charge and size. J. Pharm. Sci. 1997, 86, 1105–1110. [Google Scholar] [CrossRef]
- Ferraris, R.P.; Diamond, J. Regulation of intestinal sugar transport. Physiol. Rev. 1997, 77, 257–302. [Google Scholar] [CrossRef]
- Kunzelmann, K.; Mall, M. Electrolyte transport in the mammalian colon: Mechanisms and implications for disease. Physiol. Rev. 2002, 82, 245–289. [Google Scholar] [CrossRef]
- Blikslager, A.T.; Moeser, A.J.; Gookin, J.L.; Jones, S.L.; Odle, J. Restoration of barrier function in injured intestinal mucosa. Physiol. Rev. 2007, 87, 545–564. [Google Scholar] [CrossRef]
- Sawada, N.; Murata, M.; Kikuchi, K.; Osanai, M.; Tobioka, H.; Kojima, T.; Chiba, H. Tight junctions and human diseases. Med. Electron. Microsc. 2003, 36, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, C.; Schwietzer, Y.A.; Otani, T.; Furuse, M.; Ebnet, K. Physiological functions of junctional adhesion molecules (JAMs) in tight junctions. Biochim. Biophys. Acta (BBA)—Biomembr. 2020, 1862, 183299. [Google Scholar] [CrossRef] [PubMed]
- Yap, A.S.; Mullin, J.M.; Stevenson, B.R. Molecular Analyses of Tight Junction Physiology: Insights and Paradoxes. J. Membr. Biol. 1998, 163, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.M.; Anderson, J.M. Architecture of tight junctions and principles of molecular composition. Semin. Cell Dev. Biol. 2014, 36, 157–165. [Google Scholar] [CrossRef]
- Mehta, S.; Nijhuis, A.; Kumagai, T.; Lindsay, J.; Silver, A. Defects in the adherens junction complex (E-cadherin/β-catenin) in inflammatory bowel disease. Cell Tissue Res. 2015, 360, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Pokutta, S.; Herrenknecht, K.; Kemler, R.; Engel, J. Conformational changes of the recombinant extracellular domain of E-cadherin upon calcium binding. Eur. J. Biochem. 1994, 223, 1019–1026. [Google Scholar] [CrossRef]
- Niessen, C.M. Tight Junctions/Adherens Junctions: Basic Structure and Function. J. Investig. Dermatol. 2007, 127, 2525–2532. [Google Scholar] [CrossRef]
- Gehren, A.S.; Rocha, M.R.; de Souza, W.F.; Morgado-Díaz, J.A. Alterations of the apical junctional complex and actin cytoskeleton and their role in colorectal cancer progression. Tissue Barriers 2015, 3, e1017688. [Google Scholar] [CrossRef]
- Chitaev, N.A.; Troyanovsky, S.M. Direct Ca2+-dependent Heterophilic Interaction between Desmosomal Cadherins, Desmoglein and Desmocollin, Contributes to Cell–Cell Adhesion. J. Cell Biol. 1997, 138, 193–201. [Google Scholar] [CrossRef]
- Kang, H.; Weiss, T.M.; Bang, I.; Weis, W.I.; Choi, H.J. Structure of the Intermediate Filament-Binding Region of Desmoplakin. PLoS ONE 2016, 11, e0147641. [Google Scholar] [CrossRef]
- Chitaev, N.A.; Leube, R.E.; Troyanovsky, R.B.; Eshkind, L.G.; Franke, W.W.; Troyanovsky, S.M. The binding of plakoglobin to desmosomal cadherins: Patterns of binding sites and topogenic potential. J. Cell Biol. 1996, 133, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Gallicano, G.I.; Kouklis, P.; Bauer, C.; Yin, M.; Vasioukhin, V.; Degenstein, L.; Fuchs, E. Desmoplakin is required early in development for assembly of desmosomes and cytoskeletal linkage. J. Cell Biol. 1998, 143, 2009–2022. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Artursson, P.; Avdeef, A.; Benet, L.Z.; Houston, J.B.; Kansy, M.; Kerns, E.H.; Lennernäs, H.; Smith, D.A.; Sugano, K. The Critical Role of Passive Permeability in Designing Successful Drugs. Chem. Med. Chem. 2020, 15, 1862–1874. [Google Scholar] [CrossRef]
- Tsuji, A.; Tamai, I. Carrier-mediated intestinal transport of drugs. Pharm. Res. 1996, 13, 963–977. [Google Scholar] [CrossRef]
- Pusztai, A. Transport of proteins through the membranes of the adult gastro-intestinal tract—A potential for drug delivery? Adv. Drug Deliv. Rev. 1989, 3, 215–228. [Google Scholar] [CrossRef]
- Hornby, P.J.; Cooper, P.R.; Kliwinski, C.; Ragwan, E.; Mabus, J.R.; Harman, B.; Thompson, S.; Kauffman, A.L.; Yan, Z.; Tam, S.H.; et al. Human and non-human primate intestinal FcRn expression and immunoglobulin G transcytosis. Pharm. Res. 2014, 31, 908–922. [Google Scholar] [CrossRef]
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef]
- Kruth, H.S. Receptor-independent fluid-phase pinocytosis mechanisms for induction of foam cell formation with native LDL particles. Curr. Opin. Lipidol. 2011, 22, 386–393. [Google Scholar] [CrossRef]
- Morikawa, M.; Tsujibe, S.; Kiyoshima-Shibata, J.; Watanabe, Y.; Kato-Nagaoka, N.; Shida, K.; Matsumoto, S. Microbiota of the Small Intestine Is Selectively Engulfed by Phagocytes of the Lamina Propria and Peyer’s Patches. PLoS ONE 2016, 11, e0163607. [Google Scholar] [CrossRef]
- Hamman, J.H.; Demana, P.H.; Olivier, E.I. Targeting Receptors, Transporters and Site of Absorption to Improve Oral Drug Delivery. Drug Target Insights 2007, 2, 71–81. [Google Scholar] [CrossRef]
- Picot, D.; Layec, S.; Seynhaeve, E.; Dussaulx, L.; Trivin, F.; Carsin-Mahe, M. Chyme Reinfusion in Intestinal Failure Related to Temporary Double Enterostomies and Enteroatmospheric Fistulas. Nutrients 2020, 12, 1376. [Google Scholar] [CrossRef]
- Smart, A.L.; Gaisford, S.; Basit, A.W. Oral peptide and protein delivery: Intestinal obstacles and commercial prospects. Expert Opin. Drug Deliv. 2014, 11, 1323–1335. [Google Scholar] [CrossRef] [PubMed]
- Philip, A.K.; Philip, B. Colon Targeted Drug Delivery Systems: A Review on Primary and Novel Approaches. Oman Med. J. 2010, 25, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Hua, S. Advances in Oral Drug Delivery for Regional Targeting in the Gastrointestinal Tract—Influence of Physiological, Pathophysiological and Pharmaceutical Factors. Front. Pharmacol. 2020, 11, 524. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.D.; Whitehead, K.A.; Mitragotri, S. Materials for oral delivery of proteins and peptides. Nat. Rev. Mater. 2020, 5, 127–148. [Google Scholar] [CrossRef]
- Yun, Y.; Cho, Y.W.; Park, K. Nanoparticles for oral delivery: Targeted nanoparticles with peptidic ligands for oral protein delivery. Adv. Drug Deliv. Rev. 2013, 65, 822–832. [Google Scholar] [CrossRef]
- Capaldo, C.T.; Powell, D.N.; Kalman, D. Layered defense: How mucus and tight junctions seal the intestinal barrier. J. Mol. Med. 2017, 95, 927–934. [Google Scholar] [CrossRef]
- Cooper, G.M. Transport of Small Molecules. In The Cell: A Molecular Approach, 2nd ed.; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Zhang, X.; Han, Y.; Huang, W.; Jin, M.; Gao, Z. The influence of the gut microbiota on the bioavailability of oral drugs. Acta Pharm. Sin. B 2021, 11, 1789–1812. [Google Scholar] [CrossRef]
- Balimane, P.V.; Chong, S.; Morrison, R.A. Current methodologies used for evaluation of intestinal permeability and absorption. J. Pharmacol. Toxicol. Methods 2000, 44, 301–312. [Google Scholar] [CrossRef]
- Costa, J.; Ahluwalia, A. Advances and Current Challenges in Intestinal in vitro Model Engineering: A Digest. Front. Bioeng. Biotechnol. 2019, 7, 144. [Google Scholar] [CrossRef]
- Antunes, F.; Andrade, F.; Ferreira, D.; Nielsen, H.M.; Sarmento, B. Models to predict intestinal absorption of therapeutic peptides and proteins. Curr. Drug Metab. 2013, 14, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Angelis, I.D.; Turco, L. Caco-2 cells as a model for intestinal absorption. Curr. Protoc. Toxicol. 2011, 47, 20.6.1–20.6.15. [Google Scholar] [CrossRef] [PubMed]
- Van Beers, E.H.; Al, R.H.; Rings, E.H.; Einerhand, A.W.; Dekker, J.; Büller, H.A. Lactase and sucrase-isomaltase gene expression during Caco-2 cell differentiation. Biochem. J. 1995, 308 Pt 3, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Burton, P.S.; Conradi, R.A.; Hilgers, A.R.; Ho, N.F.H. Evidence for a Polarized Efflux System for Peptides in the Apical Membrane of Caco-2 Cells. Biochem. Biophys. Res. Commun. 1993, 190, 760–766. [Google Scholar] [CrossRef]
- Artursson, P.; Palm, K.; Luthman, K. Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv. Drug Deliv. Rev. 2012, 64, 280–289. [Google Scholar] [CrossRef]
- Hunter, J.; Jepson, M.A.; Tsuruo, T.; Simmons, N.L.; Hirst, B.H. Functional expression of P-glycoprotein in apical membranes of human intestinal Caco-2 cells. Kinetics of vinblastine secretion and interaction with modulators. J. Biol. Chem. 1993, 268, 14991–14997. [Google Scholar] [CrossRef]
- Srinivasan, B.; Kolli, A.R.; Esch, M.B.; Abaci, H.E.; Shuler, M.L.; Hickman, J.J. TEER measurement techniques for in vitro barrier model systems. J. Lab. Autom. 2015, 20, 107–126. [Google Scholar] [CrossRef]
- Gamboa, J.M.; Leong, K.W. In vitro and in vivo models for the study of oral delivery of nanoparticles. Adv. Drug Deliv. Rev. 2013, 65, 800–810. [Google Scholar] [CrossRef]
- Pires, C.L.; Praça, C.; Martins, P.A.T.; Batista de Carvalho, A.L.M.; Ferreira, L.; Marques, M.P.M.; Moreno, M.J. Re-Use of Caco-2 Monolayers in Permeability Assays—Validation Regarding Cell Monolayer Integrity. Pharmaceutics 2021, 13, 1563. [Google Scholar] [CrossRef]
- Konishi, Y.; Hagiwara, K.; Shimizu, M. Transepithelial transport of fluorescein in Caco-2 cell monolayers and use of such transport in in vitro evaluation of phenolic acid availability. Biosci. Biotechnol. Biochem. 2002, 66, 2449–2457. [Google Scholar] [CrossRef]
- Skolnik, S.; Lin, X.; Wang, J.; Chen, X.H.; He, T.; Zhang, B. Towards prediction of in vivo intestinal absorption using a 96-well Caco-2 assay. J. Pharm. Sci. 2010, 99, 3246–3265. [Google Scholar] [CrossRef] [PubMed]
- Yee, S. In vitro permeability across Caco-2 cells (colonic) can predict in vivo (small intestinal) absorption in man—Fact or myth. Pharm. Res. 1997, 14, 763–766. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.; Cheng, W.P.; Gadad, P.; Skene, K.; Smith, M.; Smith, G.; Smith, G.; McKinnon, A.; Knott, R. Uptake and transport of novel amphiphilic polyelectrolyte-insulin nanocomplexes by Caco-2 cells—Towards oral insulin. Pharm. Res. 2011, 28, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, H.; Peppas, N.A. Novel complexation hydrogels for oral peptide delivery: In vitro evaluation of their cytocompatibility and insulin-transport enhancing effects using Caco-2 cell monolayers. J. Biomed. Mater. Res. A 2003, 67, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Juul, C.B.; Fedosov, S.N.; Nexo, E.; Heegaard, C.W. Kinetic analysis of transcellular passage of the cobalamin–transcobalamin complex in Caco-2 monolayers. Mol. Biol. Cell 2019, 30, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.J.; Norouziyan, F.; Shen, W.C. Accumulation of Transferrin in Caco-2 Cells: A Possible Mechanism of Intestinal Transferrin Absorption. J. Control. Release 2007, 122, 393–398. [Google Scholar] [CrossRef]
- Pearce, S.C.; Coia, H.G.; Karl, J.P.; Pantoja-Feliciano, I.G.; Zachos, N.C.; Racicot, K. Intestinal in vitro and ex vivo Models to Study Host-Microbiome Interactions and Acute Stressors. Front. Physiol. 2018, 9, 1584. [Google Scholar] [CrossRef]
- Walter, E.; Kissel, T. Heterogeneity in the human intestinal cell line Caco-2 leads to differences in transepithelial transport. Eur. J. Pharm. Sci. 1995, 3, 215–230. [Google Scholar] [CrossRef]
- Lee, J.B.; Zgair, A.; Taha, D.A.; Zang, X.; Kagan, L.; Kim, T.H.; Kim, M.G.; Yun, H.-Y.; Fischer, P.M.; Gershkovich, P. Quantitative analysis of lab-to-lab variability in Caco-2 permeability assays. Eur. J. Pharm. Biopharm. 2017, 114, 38–42. [Google Scholar] [CrossRef]
- Behrens, I.; Stenberg, P.; Artursson, P.; Kissel, T. Transport of lipophilic drug molecules in a new mucus-secreting cell culture model based on HT29-MTX cells. Pharm. Res. 2001, 18, 1138–1145. [Google Scholar] [CrossRef]
- Sun, H.; Chow, E.C.; Liu, S.; Du, Y.; Pang, K.S. The Caco-2 cell monolayer: Usefulness and limitations. Expert Opin. Drug Metab. Toxicol. 2008, 4, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Xu, C.; Chen, P.; Hu, J.; Hu, R.; Huang, M.; Bi, H. Development, validation, and application of a novel 7-day Caco-2 cell culture system. J. Pharmacol. Toxicol. Methods 2014, 70, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Lentz, K.A.; Hayashi, J.; Lucisano, L.J.; Polli, J.E. Development of a more rapid, reduced serum culture system for Caco-2 monolayers and application to the biopharmaceutics classification system. Int. J. Pharm. 2000, 200, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Sevin, E.; Dehouck, L.; Fabulas-da Costa, A.; Cecchelli, R.; Dehouck, M.P.; Lundquist, S.; Culot, M. Accelerated Caco-2 cell permeability model for drug discovery. J. Pharmacol. Toxicol. Methods 2013, 68, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Turco, L.; Catone, T.; Caloni, F.; Consiglio, E.D.; Testai, E.; Stammati, A. Caco-2/TC7 cell line characterization for intestinal absorption: How reliable is this in vitro model for the prediction of the oral dose fraction absorbed in human? Toxicol. Vitr. 2011, 25, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Caro, I.; Boulenc, X.; Rousset, M.; Meunier, V.; Bourrié, M.; Julian, B.; Joyeux, H.; Roques, C.; Berger, Y.; Zweibaum, A.; et al. Characterisation of a newly isolated Caco-2 clone (TC-7), as a model of transport processes and biotransformation of drugs. Int. J. Pharm. 1995, 116, 147–158. [Google Scholar] [CrossRef]
- Woodcook, S.; Williamson, I.; Hassan, I.; Mackay, M. Isolation and characterisation of clones from the Caco-2 cell line displaying increased taurocholic acid transport. J. Cell Sci. 1991, 98 Pt 3, 323–332. [Google Scholar] [CrossRef]
- Irvine, J.D.; Takahashi, L.; Lockhart, K.; Cheong, J.; Tolan, J.W.; Selick, H.E.; Grove, J. MDCK (Madin-Darby canine kidney) cells: A tool for membrane permeability screening. J. Pharm. Sci. 1999, 88, 28–33. [Google Scholar] [CrossRef]
- Brändli, A.W.; Parton, R.G.; Simons, K. Transcytosis in MDCK cells: Identification of glycoproteins transported bidirectionally between both plasma membrane domains. J. Cell Biol. 1990, 111, 2909–2921. [Google Scholar] [CrossRef]
- Shah, D.; Guo, Y.; Ocando, J.; Shao, J. FITC labeling of human insulin and transport of FITC-insulin conjugates through MDCK cell monolayer. J. Pharm. Anal. 2019, 9, 400–405. [Google Scholar] [CrossRef]
- Wan, J.; Taub, M.E.; Shah, D.; Shen, W.C. Brefeldin A enhances receptor-mediated transcytosis of transferrin in filter-grown Madin-Darby canine kidney cells. J. Biol. Chem. 1992, 267, 13446–13450. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, T.S.; Detmer, S.A.; Martin, W.L.; Bjorkman, P.J. IgG transcytosis and recycling by FcRn expressed in MDCK cells reveals ligand-induced redistribution. EMBO J. 2002, 21, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Sockolosky, J.T.; Tiffany, M.R.; Szoka, F.C. Engineering neonatal Fc receptor-mediated recycling and transcytosis in recombinant proteins by short terminal peptide extensions. Proc. Natl. Acad. Sci. USA 2012, 109, 16095–16100. [Google Scholar] [CrossRef]
- Husted, R.F.; Welsh, M.J.; Stokes, J.B. Variability of functional characteristics of MDCK cells. Am. J. Physiol. 1986, 250 Pt 1, C214–C221. [Google Scholar] [CrossRef] [PubMed]
- Lesuffleur, T.; Barbat, A.; Dussaulx, E.; Zweibaum, A. Growth adaptation to methotrexate of HT-29 human colon carcinoma cells is associated with their ability to differentiate into columnar absorptive and mucus-secreting cells. Cancer Res. 1990, 50, 6334–6343. [Google Scholar] [PubMed]
- Béduneau, A.; Tempesta, C.; Fimbel, S.; Pellequer, Y.; Jannin, V.; Demarne, F.; Lamprecht, A. A tunable Caco-2/HT29-MTX co-culture model mimicking variable permeabilities of the human intestine obtained by an original seeding procedure. Eur. J. Pharm. Biopharm. 2014, 87, 290–298. [Google Scholar] [CrossRef]
- Shofner, J.P.; Phillips, M.A.; Peppas, N.A. Cellular Evaluation of Synthesized Insulin/Transferrin Bioconjugates for Oral Insulin Delivery Using Intelligent Complexation Hydrogels. Macromol. Biosci. 2010, 10, 299–306. [Google Scholar] [CrossRef]
- Chen, X.M.; Elisia, I.; Kitts, D.D. Defining conditions for the co-culture of Caco-2 and HT29-MTX cells using Taguchi design. J. Pharmacol. Toxicol. Methods 2010, 61, 334–342. [Google Scholar] [CrossRef]
- Gebert, A.; Rothkötter, H.J.; Pabst, R. M cells in Peyer’s patches of the intestine. Int. Rev. Cytol. 1996, 167, 91–159. [Google Scholar]
- Kernéis, S.; Bogdanova, A.; Kraehenbuhl, J.P.; Pringault, E. Conversion by Peyer’s patch lymphocytes of human enterocytes into M cells that transport bacteria. Science 1997, 277, 949–952. [Google Scholar] [CrossRef]
- Gullberg, E.; Leonard, M.; Karlsson, J.; Hopkins, A.M.; Brayden, D.; Baird, A.W.; Artursson, P. Expression of specific markers and particle transport in a new human intestinal M-cell model. Biochem. Biophys. Res. Commun. 2000, 279, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Lo, D.; Tynan, W.; Dickerson, J.; Scharf, M.; Cooper, J.; Byrne, D.; Brayden, D.; Higgins, L.; Evans, C.; Daniel, J. Cell culture modeling of specialized tissue: Identification of genes expressed specifically by follicle-associated epithelium of Peyer’s patch by expression profiling of Caco-2/Raji co-cultures. Int. Immunol. 2004, 16, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Araújo, F.; Sarmento, B. Towards the characterization of an in vitro triple co-culture intestine cell model for permeability studies. Int. J. Pharm. 2013, 458, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Lozoya-Agullo, I.; Araujo, F.; Gonzalez-Alvarez, I.; Merino-Sanjuan, M.; Gonzalez-Alvarez, M.; Bermejo, M.; Sarmento, B. Usefulness of Caco-2/HT29-MTX and Caco-2/HT29-MTX/Raji B coculture models to predict intestinal and colonic permeability compared to Caco-2 monoculture. Mol. Pharm. 2017, 14, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Antunes, F.; Andrade, F.; Araújo, F.; Ferreira, D.; Sarmento, B. Establishment of a triple co-culture in vitro cell models to study intestinal absorption of peptide drugs. Eur. J. Pharm. Biopharm. 2013, 83, 427–435. [Google Scholar] [CrossRef]
- Lopes, M.; Shrestha, N.; Correia, A.; Shahbazi, M.A.; Sarmento, B.; Hirvonen, J.; Veiga, F.; Seiça, R.; Ribeiro, A.; Santos, H.A. Dual chitosan/albumin-coated alginate/dextran sulfate nanoparticles for enhanced oral delivery of insulin. J. Control. Release 2016, 232, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Pampaloni, F.; Reynaud, E.G.; Stelzer, E.H.K. The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol. 2007, 8, 839–845. [Google Scholar] [CrossRef]
- Bruun Larsen, J.; Taebnia, N.; Dolatshahi-Pirouz, A.; Zebitz Eriksen, A.; Hjørringgaard, C.; Kristensen, K.; Larsen, N.W.; Larsen, N.B.; Marie, R.; Mündler, A.-K.; et al. Imaging therapeutic peptide transport across intestinal barriers. RSC Chem. Biol. 2021, 2, 1115–1143. [Google Scholar] [CrossRef]
- EpiIntestinal 3D In Vitro Microtissues. MatTek Life Sciences, 2015. Available online: https://www.mattek.com/products/epiintestinal/ (accessed on 17 February 2023).
- Markus, J.; Landry, T.; Stevens, Z.; Scott, H.; Llanos, P.; Debatis, M.; Armento, A.; Klausner, M.; Ayehunie, S. Human small intestinal organotypic culture model for drug permeation, inflammation, and toxicity assays. Vitr. Cell Dev. Biol. Anim. 2021, 57, 160–173. [Google Scholar] [CrossRef]
- Cui, Y.; Claus, S.; Schnell, D.; Runge, F.; MacLean, C. In-Depth Characterization of EpiIntestinal Microtissue as a Model for Intestinal Drug Absorption and Metabolism in Human. Pharmaceutics 2020, 12, 405. [Google Scholar] [CrossRef]
- Ayehunie, S.; Landry, T.; Stevens, Z.; Armento, A.; Hayden, P.; Klausner, M. Human Primary Cell-Based Organotypic Microtissues for Modeling Small Intestinal Drug Absorption. Pharm. Res. 2018, 35, 72. [Google Scholar] [CrossRef] [PubMed]
- Taverner, A.; MacKay, J.; Laurent, F.; Hunter, T.; Liu, K.; Mangat, K.; Song, L.; Seto, E.; Postlethwaite, S.; Alam, A.; et al. Cholix protein domain I functions as a carrier element for efficient apical to basal epithelial transcytosis. Tissue Barriers 2020, 8, 1710429. [Google Scholar] [CrossRef] [PubMed]
- Fay, N.C.; Muthusamy, B.P.; Nyugen, L.P.; Desai, R.C.; Taverner, A.; MacKay, J.; Seung, M.; Hunter, T.; Liu, K.; Chandalia, A.; et al. A Novel Fusion of IL-10 Engineered to Traffic across Intestinal Epithelium to Treat Colitis. J. Immunol. 2020, 205, 3191–3204. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Ingber, D.E. Gut-on-a-Chip microenvironment induces human intestinal cells to undergo villus differentiation. Integr. Biol. 2013, 5, 1130–1140. [Google Scholar] [CrossRef] [PubMed]
- Bhise, N.S.; Ribas, J.; Manoharan, V.; Zhang, Y.S.; Polini, A.; Massa, S.; Dokmeci, M.R.; Khademhosseini, A. Organ-on-a-Chip Platforms for Studying Drug Delivery Systems. J. Control. Release 2014, 190, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Sakharov, D.; Maltseva, D.; Knyazev, E.; Nikulin, S.; Poloznikov, A.; Shilin, S.; Baranova, A.; Tsypina, I.; Tonevitsky, A. Towards embedding Caco-2 model of gut interface in a microfluidic device to enable multi-organ models for systems biology. BMC Syst. Biol. 2019, 13 (Suppl. S1), 19. [Google Scholar] [CrossRef]
- Ashammakhi, N.; Nasiri, R.; Roberto De Barros, N.; Tebon, P.; Thakor, J.; Goudie, M.; Shamloo, A.; Martin, M.G.; Khademhosseini, A. Gut-on-a-chip: Current Progress and Future Opportunities. Biomaterials 2020, 255, 120196. [Google Scholar] [CrossRef]
- Henry, O.Y.F.; Villenave, R.; Cronce, M.J.; Leineweber, W.D.; Benz, M.A.; Ingber, D.E. Organs-on-chips with integrated electrodes for trans-epithelial electrical resistance (TEER) measurements of human epithelial barrier function. Lab. Chip. 2017, 17, 2264–2271. [Google Scholar] [CrossRef]
- Jalili-Firoozinezhad, S.; Gazzaniga, F.S.; Calamari, E.L.; Camacho, D.M.; Fadel, C.W.; Bein, A.; Swenor, B.; Nestor, B.; Cronce, M.J.; Tovaglieri, A.; et al. A complex human gut microbiome cultured in an anaerobic intestine-on-a-chip. Nat. Biomed. Eng. 2019, 3, 520–531. [Google Scholar] [CrossRef]
- Kim, H.J.; Huh, D.; Hamilton, G.; Ingber, D.E. Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab. Chip. 2012, 12, 2165–2174. [Google Scholar] [CrossRef]
- Prot, J.M.; Maciel, L.; Bricks, T.; Merlier, F.; Cotton, J.; Paullier, P.; Bois, F.Y.; Leclerc, E. First pass intestinal and liver metabolism of paracetamol in a microfluidic platform coupled with a mathematical modeling as a means of evaluating ADME processes in humans. Biotechnol. Bioeng. 2014, 111, 2027–2040. [Google Scholar] [CrossRef] [PubMed]
- Skardal, A.; Shupe, T.; Atala, A. Organoid-on-a-chip and body-on-a-chip systems for drug screening and disease modeling. Drug Discov. Today 2016, 21, 1399–1411. [Google Scholar] [CrossRef] [PubMed]
- Marrero, D.; Pujol-Vila, F.; Vera, D.; Gabriel, G.; Illa, X.; Elizalde-Torrent, A.; Alvarez, M.; Villa, R. Gut-on-a-chip: Mimicking and monitoring the human intestine. Biosens. Bioelectron. 2021, 181, 113156. [Google Scholar] [CrossRef] [PubMed]
- Kasendra, M.; Tovaglieri, A.; Sontheimer-Phelps, A.; Jalili-Firoozinezhad, S.; Bein, A.; Chalkiadaki, A.; Scholl, W.; Zhang, C.; Rickner, H.; Richmond, C.A.; et al. Development of a primary human Small Intestine-on-a-Chip using biopsy-derived organoids. Sci. Rep. 2018, 8, 2871. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.; Kim, H.J. Intestinal barrier dysfunction orchestrates the onset of inflammatory host–microbiome cross-talk in a human gut inflammation-on-a-chip. Proc. Natl. Acad. Sci. USA 2018, 115, E10539–E10547. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.; Kim, H.J. Pathomimetic modeling of human intestinal diseases and underlying host-gut microbiome interactions in a gut-on-a-chip. Methods Cell Biol. 2018, 146, 135–148. [Google Scholar]
- van der Flier, L.G.; Clevers, H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu. Rev. Physiol. 2009, 71, 241–260. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; Van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Fatehullah, A.; Tan, S.H.; Barker, N. Organoids as an in vitro model of human development and disease. Nat. Cell. Biol. 2016, 18, 246–254. [Google Scholar] [CrossRef]
- Fair, K.L.; Colquhoun, J.; Hannan, N.R.F. Intestinal organoids for modelling intestinal development and disease. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170217. [Google Scholar] [CrossRef]
- Sato, T.; Clevers, H. Growing self-organizing mini-guts from a single intestinal stem cell: Mechanism and applications. Science 2013, 340, 1190–1194. [Google Scholar] [CrossRef] [PubMed]
- Gómez, D.P.; Boudreau, F. Organoids and Their Use in Modeling Gut Epithelial Cell Lineage Differentiation and Barrier Properties During Intestinal Diseases. Front. Cell Dev. Biol. 2021, 9, 732137. [Google Scholar] [CrossRef] [PubMed]
- Lukovac, S.; Roeselers, G. Intestinal Crypt Organoids as Experimental Models. In The Impact of Food Bioactives on Health: In Vitro and Ex Vivo Models; Verhoeckx, K., Cotter, P., López-Expósito, I., Kleiveland, C., Lea, T., Mackie, A., Requena, T., Swiatecka, D., Wichers, H., Eds.; Springer: Cham, Switzerland, 2015. [Google Scholar]
- Lee, S.B.; Han, S.H.; Park, S. Long-Term Culture of Intestinal Organoids. Methods Mol. Biol. 2018, 1817, 123–135. [Google Scholar] [PubMed]
- Hughes, C.S.; Postovit, L.M.; Lajoie, G.A. Matrigel: A complex protein mixture required for optimal growth of cell culture. Proteomics 2010, 10, 1886–1890. [Google Scholar] [CrossRef]
- Karve, S.S.; Pradhan, S.; Ward, D.V.; Weiss, A.A. Intestinal organoids model human responses to infection by commensal and Shiga toxin producing Escherichia coli. PLoS ONE 2017, 12, e0178966. [Google Scholar] [CrossRef] [PubMed]
- Williamson, I.A.; Arnold, J.W.; Samsa, L.A.; Gaynor, L.; DiSalvo, M.; Cocchiaro, J.L.; Carroll, I.; Azcarate-Peril, M.A.; Rawls, J.F.; Allbritton, N.L.; et al. A High-Throughput Organoid Microinjection Platform to Study Gastrointestinal Microbiota and Luminal Physiology. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 301–319. [Google Scholar] [CrossRef]
- Roodsant, T.; Navis, M.; Aknouch, I.; Renes, I.B.; van Elburg, R.M.; Pajkrt, D.; Wolthers, K.C.; Schultsz, C.; Van Der Ark, K.C.H.; Sridhar, A.; et al. A Human 2D Primary Organoid-Derived Epithelial Monolayer Model to Study Host-Pathogen Interaction in the Small Intestine. Front. Cell Infect. Microbiol. 2020, 10, 272. [Google Scholar] [CrossRef]
- Macedo, M.H.; Araújo, F.; Martínez, E.; Barrias, C.; Sarmento, B. iPSC-Derived Enterocyte-like Cells for Drug Absorption and Metabolism Studies. Trends Mol. Med. 2018, 24, 696–708. [Google Scholar] [CrossRef]
- Yoshida, S.; Miwa, H.; Kawachi, T.; Kume, S.; Takahashi, K. Generation of intestinal organoids derived from human pluripotent stem cells for drug testing. Sci. Rep. 2020, 10, 5989. [Google Scholar] [CrossRef]
- Creff, J.; Malaquin, L.; Besson, A. In vitro models of intestinal epithelium: Toward bioengineered systems. J. Tissue Eng. 2021, 12, 2041731420985202. [Google Scholar] [CrossRef]
- Fedi, A.; Vitale, C.; Ponschin, G.; Ayehunie, S.; Fato, M.; Scaglione, S. In vitro models replicating the human intestinal epithelium for absorption and metabolism studies: A systematic review. J. Control. Release 2021, 335, 247–268. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.H.; Wiseman, G. The use of sacs of everted small intestine for the study of the transference of substances from the mucosal to the serosal surface. J. Physiol. 1954, 123, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.A.; Al-Jenoobi, F.I.; Al-Mohizea, A.M. Everted gut sac model as a tool in pharmaceutical research: Limitations and applications. J. Pharm. Pharmacol. 2012, 64, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Liu, Y.; Zhao, B.; Tang, M.; Dong, H.; Zhang, L.; Lv, B.; Wei, L. Ex vivo and in situ approaches used to study intestinal absorption. J. Pharmacol. Toxicol. Methods 2013, 68, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Hillgren, K.M.; Kato, A.; Borchardt, R.T. In vitro systems for studying intestinal drug absorption. Med. Res. Rev. 1995, 15, 83–109. [Google Scholar] [CrossRef]
- Mateer, S.W.; Cardona, J.; Marks, E.; Goggin, B.J.; Hua, S.; Keely, S. Ex Vivo Intestinal Sacs to Assess Mucosal Permeability in Models of Gastrointestinal Disease. J. Vis. Exp. 2016, 108, 53250. [Google Scholar]
- Crane, R.K.; Wilson, T.H. In vitro method for the study of the rate of intestinal absorption of sugars. J. Appl. Physiol. 1958, 12, 145–146. [Google Scholar] [CrossRef]
- Kaplan, S.A.; Cotler, S. Use of Cannulated Everted Intestinal Sac for Serial Sampling as a Drug Absorbability (Permeability) Screen. J. Pharm. Sci. 1972, 61, 1361–1365. [Google Scholar] [CrossRef]
- Wolfe, D.L.; Forland, S.C.; Benet, L.Z. Drug Transfer across Intact Rat Intestinal Mucosa Following Surgical Removal of Serosa and Muscularis Externa. J. Pharm. Sci. 1973, 62, 200–205. [Google Scholar] [CrossRef]
- van de Kerkhof, E.G.; de Graaf, I.A.M.; Groothuis, G.M.M. In vitro methods to study intestinal drug metabolism. Curr. Drug Metab. 2007, 8, 658–675. [Google Scholar] [CrossRef]
- Ruan, L.P.; Chen, S.; Yu, B.Y.; Zhu, D.N.; Cordell, G.A.; Qiu, S.X. Prediction of human absorption of natural compounds by the non-everted rat intestinal sac model. Eur. J. Med. Chem. 2006, 41, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Giaginis, C.; Tsantili-Kakoulidou, A. Alternative Measures of Lipophilicity: From Octanol–Water Partitioning to IAM Retention. J. Pharm. Sci. 2008, 97, 2984–3004. [Google Scholar] [CrossRef] [PubMed]
- Ussing, H.H.; Zerahn, K. Active transport of sodium as the source of electric current in the short-circuited isolated frog skin. Acta Physiol. Scand. 1951, 23, 110–127. [Google Scholar] [CrossRef] [PubMed]
- Westerhout, J.; Wortelboer, H.; Verhoeckx, K. Ussing Chamber. In The Impact of Food Bioactives on Health: In Vitro and Ex Vivo Models; Verhoeckx, K., Cotter, P., López-Expósito, I., Kleiveland, C., Lea, T., Mackie, A., Requena, T., Swiatecka, D., Wichers, H., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 263–273. [Google Scholar]
- Grass, G.M.; Sweetana, S.A. In vitro measurement of gastrointestinal tissue permeability using a new diffusion cell. Pharm. Res. 1988, 5, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Benlounes, N.; Chedid, R.; Thuillier, F.; Desjeux, J.F.; Rousselet, F.; Heyman, M. Intestinal transport and processing of immunoglobulin G in the neonatal and adult rat. Biol. Neonate 1995, 67, 254–263. [Google Scholar] [CrossRef]
- Michiba, K.; Maeda, K.; Kurimori, K.; Enomoto, T.; Shimomura, O.; Takeuchi, T.; Nishiyama, H.; Oda, T.; Kusuhara, H. Characterization of the Human Intestinal Drug Transport with Ussing Chamber System Incorporating Freshly Isolated Human Jejunum. Drug Metab. Dispos. 2021, 49, 84–93. [Google Scholar] [CrossRef]
- Forner, K.; Roos, C.; Dahlgren, D.; Kesisoglou, F.; Konerding, M.A.; Mazur, J.; Lennernäs, H.; Langguth, P. Optimization of the Ussing chamber setup with excised rat intestinal segments for dissolution/permeation experiments of poorly soluble drugs. Drug Dev. Ind. Pharm. 2017, 43, 338–346. [Google Scholar] [CrossRef]
- Dumontier, A.; Brachet, P.; Huneau, J.F.; Tomé, D. Transport of putrescine in the isolated rabbit intestine. Pflüg. Arch. Eur. J. Physiol. 1992, 420, 329–335. [Google Scholar] [CrossRef]
- Arnold, Y.E.; Thorens, J.; Bernard, S.; Kalia, Y.N. Drug Transport across Porcine Intestine Using an Ussing Chamber System: Regional Differences and the Effect of P-Glycoprotein and CYP3A4 Activity on Drug Absorption. Pharmaceutics 2019, 11, 139. [Google Scholar] [CrossRef]
- Neupane, R.; Boddu, S.H.S.; Renukuntla, J.; Babu, R.J.; Tiwari, A.K. Alternatives to Biological Skin in Permeation Studies: Current Trends and Possibilities. Pharmaceutics 2020, 12, 152. [Google Scholar] [CrossRef]
- Ameri, M.; Lewis, H.; Lehman, P. Effect of Skin Model on In Vitro Performance of an Adhesive Dermally Applied Microarray Coated with Zolmitriptan. J. Pharm. 2018, 2018, 7459124. [Google Scholar] [CrossRef] [PubMed]
- Dezani, A.B.; Pereira, T.M.; Caffaro, A.M.; Reis, J.M.; Serra, C.H.d.R. Determination of lamivudine and zidovudine permeability using a different ex vivo method in Franz cells. J. Pharmacol. Toxicol. Methods 2013, 67, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhang, C.; Zhang, X.; Zhen, Z.; Wang, P.; Li, J.; Yi, D.; Jin, Y.; Yang, D. Permeation measurement of gestodene for some biodegradable materials using Franz diffusion cells. Saudi Pharm. J. 2015, 23, 413–420. [Google Scholar] [CrossRef]
- Franz Cell—The Original. PermeGear. Available online: https://permegear.com/franz-cells/ (accessed on 18 February 2023).
- Sánchez, A.B.; Calpena, A.C.; Mallandrich, M.; Clares, B. Validation of an Ex Vivo Permeation Method for the Intestinal Permeability of Different BCS Drugs and Its Correlation with Caco-2 In Vitro Experiments. Pharmaceutics 2019, 11, 638. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; Bouic, P.J.D. Permeation of Four Oral Drugs through Human Intestinal Mucosa. AAPS Pharm. Sci. Tech. 2009, 10, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Westerhout, J.; van de Steeg, E.; Grossouw, D.; Zeijdner, E.E.; Krul, C.A.M.; Verwei, M.; Wortelboer, H.M. A new approach to predict human intestinal absorption using porcine intestinal tissue and biorelevant matrices. Eur. J. Pharm. Sci. 2014, 63, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Ex_Vivo_InTESTine_Liver. Available online: http://www.tno-pharma.com/Intestine_en.html (accessed on 25 December 2022).
- Stevens, L.J.; van Lipzig, M.M.H.; Erpelinck, S.L.A.; Pronk, A.; van Gorp, J.; Wortelboer, H.M.; van de Steeg, E. A higher throughput and physiologically relevant two-compartmental human ex vivo intestinal tissue system for studying gastrointestinal processes. Eur. J. Pharm. Sci. 2019, 137, 104989. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.; Smart, K.; Somerville, M.S.; Lauder, S.N.; Appanna, G.; Horwood, J.; Raj, L.S.; Srivastava, B.; Durai, D.; Scurr, M.J.; et al. The Ussing chamber system for measuring intestinal permeability in health and disease. BMC Gastroenterol. 2019, 19, 98. [Google Scholar] [CrossRef]
- Ng, S.F.; Rouse, J.J.; Sanderson, F.D.; Meidan, V.; Eccleston, G.M. Validation of a Static Franz Diffusion Cell System for In Vitro Permeation Studies. AAPS Pharm. Sci. Tech. 2010, 11, 1432–1441. [Google Scholar] [CrossRef]
- Maderuelo, C.; Lanao, J.M.; Zarzuelo, A. Enteric coating of oral solid dosage forms as a tool to improve drug bioavailability. Eur. J. Pharm. Sci. 2019, 138, 105019. [Google Scholar] [CrossRef]
- Arbit, E.; Kidron, M. Oral Insulin Delivery in a Physiologic Context: Review. J. Diabetes Sci. Technol. 2017, 11, 825–832. [Google Scholar] [CrossRef]
- Martinsen, T.C.; Fossmark, R.; Waldum, H.L. The Phylogeny and Biological Function of Gastric Juice—Microbiological Consequences of Removing Gastric Acid. Int. J. Mol. Sci. 2019, 20, 6031. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Perry, B.A.; Labruno, S.; Lee, H.S.; Stern, W.; Falzone, L.M.; Sinko, P.J. Impact of regional intestinal pH modulation on absorption of peptide drugs: Oral absorption studies of salmon calcitonin in beagle dogs. Pharm. Res. 1999, 16, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Binkley, N.; Bolognese, M.; Sidorowicz-Bialynicka, A.; Vally, T.; Trout, R.; Miller, C.; Buben, C.E.; Gilligan, J.P.; Krause, D.S. A phase 3 trial of the efficacy and safety of oral recombinant calcitonin: The Oral Calcitonin in Postmenopausal Osteoporosis (ORACAL) trial. J. Bone Miner. Res. 2012, 27, 1821–1829. [Google Scholar] [CrossRef] [PubMed]
- Dvořáčková, K.; Doležel, P.; Mašková, E.; Muselík, J.; Kejdušová, M.; Vetchý, D. The Effect of Acid pH Modifiers on the Release Characteristics of Weakly Basic Drug from Hydrophlilic–Lipophilic Matrices. AAPS Pharm. Sci. Tech. 2013, 14, 1341–1348. [Google Scholar] [CrossRef]
- Bolourchian, N.; Dadashzadeh, S. pH-independent release of propranolol hydrochloride from HPMC-based matrices using organic acids. DARU J. Pharm. Sci. 2008, 16, 136–142. [Google Scholar]
- Choonara, B.F.; Choonara, Y.E.; Kumar, P.; Bijukumar, D.; du Toit, L.C.; Pillay, V. A review of advanced oral drug delivery technologies facilitating the protection and absorption of protein and peptide molecules. Biotechnol. Adv. 2014, 32, 1269–1282. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Tipton, K.F. Assessment of Enzyme Inhibition: A Review with Examples from the Development of Monoamine Oxidase and Cholinesterase Inhibitory Drugs. Molecules 2017, 22, 1192. [Google Scholar] [CrossRef]
- Belorgey, D.; Dirrig, S.; Amouric, M.; Figarella, C.; Bieth, J.G. Inhibition of human pancreatic proteinases by mucus proteinase inhibitor, eglin c and aprotinin. Biochem. J. 1996, 313 Pt 2, 555–560. [Google Scholar] [CrossRef]
- Vanchugova, L.V.; Valueva, T.A.; Romashkin, V.I.; Rozenfel’d, M.A.; Valuev, L.I. Interaction of ovomucoid from duck egg white with serine proteinases. Biokhimiia 1988, 53, 1455–1461. [Google Scholar]
- Fujii, S.; Yokoyama, T.; Ikegaya, K.; Sato, F.; Yokoo, N. Promoting effect of the new chymotrypsin inhibitor FK-448 on the intestinal absorption of insulin in rats and dogs. J. Pharm. Pharmacol. 1985, 37, 545–549. [Google Scholar] [CrossRef]
- Flavin, D.F. The effects of soybean trypsin inhibitors on the pancreas of animals and man: A review. Vet. Hum. Toxicol. 1982, 24, 25–28. [Google Scholar] [PubMed]
- Araújo, F.; Fonte, P.; Santos, H.A.; Sarmento, B. Oral Delivery of Glucagon-Like Peptide-1 and Analogs: Alternatives for Diabetes Control? J. Diabetes Sci. Technol. 2012, 6, 1486–1497. [Google Scholar] [CrossRef] [PubMed]
- Bernkop-Schnürch, A. The use of inhibitory agents to overcome the enzymatic barrier to perorally administered therapeutic peptides and proteins. J. Control. Release 1998, 52, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Reseland, J.E.; Holm, H.; Jacobsen, M.B.; Jenssen, T.G.; Hanssen, L.E. Proteinase inhibitors induce selective stimulation of human trypsin and chymotrypsin secretion. J. Nutr. 1996, 126, 634–642. [Google Scholar] [CrossRef]
- Otsuki, M.; Ohki, A.; Okabayashi, Y.; Suehiro, I.; Baba, S. Effect of synthetic protease inhibitor camostate on pancreatic exocrine function in rats. Pancreas 1987, 2, 164–169. [Google Scholar] [CrossRef]
- Melmed, R.N.; El-Aaser, A.A.; Holt, S.J. Hypertrophy and hyperplasia of the neonatal rat exocrine pancreas induced by orally administered soybean trypsin inhibitor. Biochim. Biophys. Acta 1976, 421, 280–288. [Google Scholar] [CrossRef]
- Del Curto, M.D.; Maroni, A.; Palugan, L.; Zema, L.; Gazzaniga, A.; Sangalli, M.E. Oral delivery system for two-pulse colonic release of protein drugs and protease inhibitor/absorption enhancer compounds. J. Pharm. Sci. 2011, 100, 3251–3259. [Google Scholar] [CrossRef]
- Wilkinson, M.; Sugumar, K.; Milan, S.J.; Hart, A.; Crockett, A.; Crossingham, I. Mucolytics for bronchiectasis. Cochrane Database Syst. Rev. 2014, 2014, CD001289. [Google Scholar] [CrossRef]
- Olmsted, S.S.; Padgett, J.L.; Yudin, A.I.; Whaley, K.J.; Moench, T.R.; Cone, R.A. Diffusion of macromolecules and virus-like particles in human cervical mucus. Biophys. J. 2001, 81, 1930–1937. [Google Scholar] [CrossRef]
- Maisel, K.; Ensign, L.; Reddy, M.; Cone, R.; Hanes, J. Effect of surface chemistry on nanoparticle interaction with gastrointestinal mucus and distribution in the gastrointestinal tract following oral and rectal administration in the mouse. J. Control. Release 2015, 197, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Popov, A.; Enlow, E.; Bourassa, J.; Chen, H. Mucus-penetrating nanoparticles made with “mucoadhesive” poly(vinyl alcohol). Nanomedicine 2016, 12, 1863–1871. [Google Scholar] [CrossRef] [PubMed]
- Huckaby, J.T.; Lai, S.K. PEGylation for enhancing nanoparticle diffusion in mucus. Adv. Drug Deliv. Rev. 2018, 124, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Wei, M.; Li, W.; Guo, M.; Guo, C.; Ma, M.; Wang, Y.; Yang, Z.; Li, M.; Fu, Q.; et al. Impacts of particle shapes on the oral delivery of drug nanocrystals: Mucus permeation, transepithelial transport and bioavailability. J. Control. Release 2019, 307, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Wang, J.; Yang, Y.; Zhu, C.; Su, Q.; Guo, S.; Sun, J.; Gan, Y.; Shi, X.; Gao, H. Rotation-Facilitated Rapid Transport of Nanorods in Mucosal Tissues. Nano Lett. 2016, 16, 7176–7182. [Google Scholar] [CrossRef]
- Shan, W.; Zhu, X.; Liu, M.; Li, L.; Zhong, J.; Sun, W.; Zhang, Z.; Huang, Y. Overcoming the diffusion barrier of mucus and absorption barrier of epithelium by self-assembled nanoparticles for oral delivery of insulin. ACS Nano 2015, 9, 2345–2356. [Google Scholar] [CrossRef]
- Ch’ng, H.S.; Park, H.; Kelly, P.; Robinson, J.R. Bioadhesive polymers as platforms for oral controlled drug delivery II: Synthesis and evaluation of some swelling, water-insoluble bioadhesive polymers. J. Pharm. Sci. 1985, 74, 399–405. [Google Scholar] [CrossRef]
- Bernkop-Schnürch, A. Thiomers: A new generation of mucoadhesive polymers. Adv. Drug Deliv. Rev. 2005, 57, 1569–1582. [Google Scholar] [CrossRef]
- Bernkop-Schnürch, A.; Hoffer, M.H.; Kafedjiiski, K. Thiomers for oral delivery of hydrophilic macromolecular drugs. Expert Opin. Drug Deliv. 2004, 1, 87–98. [Google Scholar] [CrossRef]
- Federer, C.; Kurpiers, M.; Bernkop-Schnürch, A. Thiolated Chitosans: A Multi-talented Class of Polymers for Various Applications. Biomacromolecules 2021, 22, 24–56. [Google Scholar] [CrossRef]
- Vetter, A.; Martien, R.; Bernkop-Schnürch, A. Thiolated polycarbophil as an adjuvant for permeation enhancement in nasal delivery of antisense oligonucleotides. J. Pharm. Sci. 2010, 99, 1427–1439. [Google Scholar] [CrossRef] [PubMed]
- Millotti, G.; Vetter, A.; Leithner, K.; Sarti, F.; Shahnaz Bano, G.; Augustijns, P.; Bernkop-Schnürch, A. Development of thiolated poly(acrylic acid) microparticles for the nasal administration of exenatide. Drug Dev. Ind. Pharm. 2014, 40, 1677–1682. [Google Scholar] [CrossRef] [PubMed]
- Bernkop-Schnürch, A.; Walker, G. Multifunctional matrices for oral peptide delivery. Crit. Rev. Ther. Drug Carrier Syst. 2001, 18, 459–501. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Ding, J.; He, C.; Cui, L.; Tang, C.; Yin, C. Drug permeability and mucoadhesion properties of thiolated trimethyl chitosan nanoparticles in oral insulin delivery. Biomaterials 2009, 30, 5691–5700. [Google Scholar] [CrossRef] [PubMed]
- Teutonico, D.; Ponchel, G. Patches for improving gastrointestinal absorption: An overview. Drug Discov. Today 2011, 16, 991–997. [Google Scholar] [CrossRef]
- Bagan, J.; Paderni, C.; Termine, N.; Campisi, G.; Lo Russo, L.; Compilato, D.; Di Fede, O. Mucoadhesive polymers for oral transmucosal drug delivery: A review. Curr. Pharm. Des. 2012, 18, 5497–5514. [Google Scholar] [CrossRef]
- Thakral, S.; Thakral, N.K.; Majumdar, D.K. Eudragit: A technology evaluation. Expert Opin. Drug Deliv. 2013, 10, 131–149. [Google Scholar] [CrossRef]
- Rossi, S.; Ferrari, F.; Bonferoni, M.C.; Caramella, C. Characterization of chitosan hydrochloride–mucin interaction by means of viscosimetric and turbidimetric measurements. Eur. J. Pharm. Sci. 2000, 10, 251–257. [Google Scholar] [CrossRef]
- Banerjee, A.; Mitragotri, S. Intestinal patch systems for oral drug delivery. Curr. Opin. Pharmacol. 2017, 36, 58–65. [Google Scholar] [CrossRef]
- Kirsch, K.; Hanke, U.; Weitschies, W. An overview of intestinal wafers for oral drug delivery. Eur. J. Pharm. Biopharm. 2017, 114, 135–144. [Google Scholar] [CrossRef]
- Toorisaka, E.; Watanabe, K.; Ono, H.; Hirata, M.; Kamiya, N.; Goto, M. Intestinal patches with an immobilized solid-in-oil formulation for oral protein delivery. Acta Biomater. 2012, 8, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Hwang, B.H.; Lee, J.; Anselmo, A.C.; Doshi, N.; Mitragotri, S. Mucoadhesive intestinal devices for oral delivery of salmon calcitonin. J. Control. Release 2013, 172, 753–762. [Google Scholar] [CrossRef]
- Ito, Y.; Tosh, B.; Togashi, Y.; Amagase, K.; Kishida, T.; Kishida, T.; Sugioka, N.; Shibata, N.; Takada, K. Absorption of interferon alpha from patches in rats. J. Drug Target. 2005, 13, 383–390. [Google Scholar] [CrossRef]
- Venkatesan, N.; Uchino, K.; Amagase, K.; Ito, Y.; Shibata, N.; Takada, K. Gastro-intestinal patch system for the delivery of erythropoietin. J. Control. Release 2006, 111, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Eiamtrakarn, S.; Itoh, Y.; Kishimoto, J.; Yoshikawa, Y.; Shibata, N.; Murakami, M.; Takada, K. Gastrointestinal mucoadhesive patch system (GI-MAPS) for oral administration of G-CSF, a model protein. Biomaterials 2002, 23, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Peppas, N.A.; Wood, K.M.; Blanchette, J.O. Hydrogels for oral delivery of therapeutic proteins. Expert Opin. Biol. Ther. 2004, 4, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Peppas, N.A.; Sahlin, J.J. Hydrogels as mucoadhesive and bioadhesive materials: A review. Biomaterials 1996, 17, 1553–1561. [Google Scholar] [CrossRef]
- Mastropietro, D.J.; Omidian, H.; Park, K. Drug delivery applications for superporous hydrogels. Expert Opin. Drug Deliv. 2012, 9, 71–89. [Google Scholar] [CrossRef]
- Li, J.; Mooney, D.J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 1–17. [Google Scholar] [CrossRef]
- Sharpe, L.A.; Daily, A.M.; Horava, S.D.; Peppas, N.A. Therapeutic applications of hydrogels in oral drug delivery. Expert Opin. Drug Deliv. 2014, 11, 901–915. [Google Scholar] [CrossRef]
- Kim, B.; La Flamme, K.; Peppas, N.A. Dynamic swelling behavior of pH-sensitive anionic hydrogels used for protein delivery. J. Appl. Polym. Sci. 2003, 89, 1606–1613. [Google Scholar] [CrossRef]
- Nakamura, K.; Murray, R.J.; Joseph, J.I.; Peppas, N.A.; Morishita, M.; Lowman, A.M. Oral insulin delivery using P(MAA-g-EG) hydrogels: Effects of network morphology on insulin delivery characteristics. J. Control. Release 2004, 95, 589–599. [Google Scholar] [CrossRef]
- Kamei, N.; Morishita, M.; Chiba, H.; Kavimandan, N.J.; Peppas, N.A.; Takayama, K. Complexation hydrogels for intestinal delivery of interferon beta and calcitonin. J. Control. Release 2009, 134, 98–102. [Google Scholar] [CrossRef]
- Yin, L.; Ding, J.; Zhang, J.; He, C.; Tang, C.; Yin, C. Polymer integrity related absorption mechanism of superporous hydrogel containing interpenetrating polymer networks for oral delivery of insulin. Biomaterials 2010, 31, 3347–3356. [Google Scholar] [CrossRef]
- Basan, H.; Gümüşderelioğlu, M.; Tevfik Orbey, M. Release characteristics of salmon calcitonin from dextran hydrogels for colon-specific delivery. Eur. J. Pharm. Biopharm. 2007, 65, 39–46. [Google Scholar] [CrossRef]
- Lai, S.K.; O’Hanlon, D.E.; Harrold, S.; Man, S.T.; Wang, Y.Y.; Cone, R.; Hanes, J. Rapid transport of large polymeric nanoparticles in fresh undiluted human mucus. Proc. Natl. Acad. Sci. USA 2007, 104, 1482–1487. [Google Scholar] [CrossRef]
- Yu, M.K.; Park, J.; Jon, S. Targeting strategies for multifunctional nanoparticles in cancer imaging and therapy. Theranostics 2012, 2, 3–44. [Google Scholar] [CrossRef]
- des Rieux, A.; Fievez, V.; Garinot, M.; Schneider, Y.J.; Préat, V. Nanoparticles as potential oral delivery systems of proteins and vaccines: A mechanistic approach. J. Control. Release 2006, 116, 1–27. [Google Scholar] [CrossRef]
- Desai, M.P.; Labhasetwar, V.; Amidon, G.L.; Levy, R.J. Gastrointestinal uptake of biodegradable microparticles: Effect of particle size. Pharm. Res. 1996, 13, 1838–1845. [Google Scholar] [CrossRef]
- Desai, M.P.; Labhasetwar, V.; Walter, E.; Levy, R.J.; Amidon, G.L. The mechanism of uptake of biodegradable microparticles in Caco-2 cells is size dependent. Pharm. Res. 1997, 14, 1568–1573. [Google Scholar] [CrossRef]
- Soppimath, K.S.; Aminabhavi, T.M.; Kulkarni, A.R.; Rudzinski, W.E. Biodegradable polymeric nanoparticles as drug delivery devices. J. Control. Release 2001, 70, 1–20. [Google Scholar] [CrossRef]
- Petrus, A.K.; Vortherms, A.R.; Fairchild, T.J.; Doyle, R.P. Vitamin B12 as a carrier for the oral delivery of insulin. Chem. Med. Chem. 2007, 2, 1717–1721. [Google Scholar] [CrossRef]
- Fyfe, J.C.; Madsen, M.; Højrup, P.; Christensen, E.I.; Tanner, S.M.; de la Chapelle, A.; He, Q.; Moestrup, S.K. The functional cobalamin (vitamin B12)–intrinsic factor receptor is a novel complex of cubilin and amnionless. Blood 2004, 103, 1573–1579. [Google Scholar] [CrossRef]
- Russell-Jones, G.J.; Westwood, S.W.; Farnworth, P.G.; Findlay, J.K.; Burger, H.G. Synthesis of LHRH antagonists suitable for oral administration via the vitamin B12 uptake system. Bioconjug. Chem. 1995, 6, 34–42. [Google Scholar] [CrossRef]
- Chalasani, K.B.; Russell-Jones, G.J.; Jain, A.K.; Diwan, P.V.; Jain, S.K. Effective oral delivery of insulin in animal models using vitamin B12-coated dextran nanoparticles. J. Control. Release 2007, 122, 141–150. [Google Scholar] [CrossRef]
- Pawar, V.K.; Meher, J.G.; Singh, Y.; Chaurasia, M.; Surendar Reddy, B.; Chourasia, M.K. Targeting of gastrointestinal tract for amended delivery of protein/peptide therapeutics: Strategies and industrial perspectives. J. Control. Release 2014, 196, 168–183. [Google Scholar] [CrossRef]
- Willson, J. Transferrin’ across the blood–brain barrier. Nat. Rev. Drug Discov. 2020, 19, 444. [Google Scholar] [CrossRef]
- Neves, A.R.; van der Putten, L.; Queiroz, J.F.; Pinheiro, M.; Reis, S. Transferrin-functionalized lipid nanoparticles for curcumin brain delivery. J. Biotechnol. 2021, 331, 108–117. [Google Scholar] [CrossRef]
- Pardridge, W.M. Blood-Brain Barrier and Delivery of Protein and Gene Therapeutics to Brain. Front. Aging Neurosci. 2019, 11, 373. [Google Scholar] [CrossRef]
- Bien-Ly, N.; Yu, Y.J.; Bumbaca, D.; Elstrott, J.; Boswell, C.A.; Zhang, Y.; Luk, W.; Lu, Y.; Dennis, M.S.; Weimer, R.M.; et al. Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants. J. Exp. Med. 2014, 211, 233–244. [Google Scholar] [CrossRef]
- Ma, Y.; Yeh, M.; Yeh, K.-Y.; Glass, J. Iron Transport in Intestinal Epithelial Cells Occurs by Transcytosis. A Fluorescent Metal-Sensor Study. Blood 2005, 106, 3583. [Google Scholar] [CrossRef]
- Du, W.; Fan, Y.; Zheng, N.; He, B.; Yuan, L.; Zhang, H.; Wang, X.; Wang, J.; Zhang, X.; Zhang, Q. Transferrin receptor specific nanocarriers conjugated with functional 7peptide for oral drug delivery. Biomaterials 2013, 34, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.Q.; Wang, J.; Shen, W.C. Hypoglycemic Effect of Insulin-Transferrin Conjugate in Streptozotocin-Induced Diabetic Rats. J. Pharmacol. Exp. Ther. 2000, 295, 594–600. [Google Scholar] [PubMed]
- Chen, Y.S.; Zaro, J.L.; Zhang, D.; Huang, N.; Simon, A.; Shen, W.C. Characterization and Oral Delivery of Proinsulin-Transferrin Fusion Protein Expressed Using ExpressTec. Int. J. Mol. Sci. 2018, 19, 378. [Google Scholar] [CrossRef]
- Yong, J.M.; Mantaj, J.; Cheng, Y.; Vllasaliu, D. Delivery of Nanoparticles across the Intestinal Epithelium via the Transferrin Transport Pathway. Pharmaceutics 2019, 11, 298. [Google Scholar] [CrossRef] [PubMed]
- Roopenian, D.C.; Sun, V.Z. Clinical ramifications of the MHC family Fc receptor FcRn. J. Clin. Immunol. 2010, 30, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Claypool, S.M.; Wagner, J.S.; Mizoguchi, E.; Mizoguchi, A.; Roopenian, D.C.; I Lencer, W.; Blumberg, R.S. Human Neonatal Fc Receptor Mediates Transport of IgG into Luminal Secretions for Delivery of Antigens to Mucosal Dendritic Cells. Immunity 2004, 20, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Hornby, P.J.; Cooper, P.R.; Kliwinski, C.; Ragwan, E.; Mabus, J.R.; Harman, B.; Dorai, H.; Giles-Komar, J. FcRn Expression and Antibody Transcytosis in Adult Human and Non-Human Primate Intestine. FASEB J. 2013, 27, 1093.3. [Google Scholar] [CrossRef]
- Claypool, S.M.; Dickinson, B.L.; Wagner, J.S.; Johansen, F.E.; Venu, N.; Borawski, J.A.; Lencer, W.I.; Blumberg, R.S. Bidirectional Transepithelial IgG Transport by a Strongly Polarized Basolateral Membrane Fcγ-Receptor. Mol. Biol. Cell 2004, 15, 1746–1759. [Google Scholar] [CrossRef]
- Rodewald, R. pH-dependent binding of immunoglobulins to intestinal cells of the neonatal rat. J. Cell Biol. 1976, 71, 666–669. [Google Scholar] [CrossRef]
- Vaughn, D.E.; Bjorkman, P.J. Structural basis of pH-dependent antibody binding by the neonatal Fc receptor. Structure 1998, 6, 63–73. [Google Scholar] [CrossRef]
- Ulloa-Aguirre, A.; Zariñán, T.; Jardón-Valadez, E.; Gutiérrez-Sagal, R.; Dias, J.A. Structure-Function Relationships of the Follicle-Stimulating Hormone Receptor. Front. Endocrinol. 2018, 9, 707. [Google Scholar] [CrossRef]
- Low, S.C.; Nunes, S.L.; Bitonti, A.J.; Dumont, J.A. Oral and pulmonary delivery of FSH–Fc fusion proteins via neonatal Fc receptor-mediated transcytosis. Hum. Reprod. 2005, 20, 1805–1813. [Google Scholar] [CrossRef]
- Saxena, A.; Wu, D. Advances in Therapeutic Fc Engineering—Modulation of IgG-Associated Effector Functions and Serum Half-life. Front. Immunol. 2016, 7, 580. [Google Scholar] [CrossRef]
- Pridgen, E.M.; Alexis, F.; Kuo, T.T.; Levy-Nissenbaum, E.; Karnik, R.; Blumberg, R.S.; Langer, R.; Farokhzad, O.C. Transepithelial Transport of Fc -Targeted Nanoparticles by the Neonatal Fc Receptor for Oral Delivery. Sci. Transl. Med. 2013, 5, 213ra167. [Google Scholar] [CrossRef]
- Maher, S.; Mrsny, R.J.; Brayden, D.J. Intestinal permeation enhancers for oral peptide delivery. Adv. Drug Deliv. Rev. 2016, 106 Pt B, 277–319. [Google Scholar] [CrossRef]
- Shen, L.; Zhao, H.Y.; Du, J.; Wang, F. Anti-tumor activities of four chelating agents against human neuroblastoma cells. In Vivo 2005, 19, 233–236. [Google Scholar]
- Su, F.Y.; Lin, K.J.; Sonaje, K.; Wey, S.P.; Yen, T.C.; Ho, Y.C.; Panda, N.; Chuang, E.Y.; Maiti, B.; Sung, H.W. Protease inhibition and absorption enhancement by functional nanoparticles for effective oral insulin delivery. Biomaterials 2012, 33, 2801–2811. [Google Scholar] [CrossRef]
- Zhu, Q.; Chen, Z.; Paul, P.K.; Lu, Y.; Wu, W.; Qi, J. Oral delivery of proteins and peptides: Challenges, status quo and future perspectives. Acta Pharm. Sin. B 2021, 11, 2416–2448. [Google Scholar] [CrossRef]
- Ilbäck, N.G.; Stålhandske, T.; Lindh, U. Effects of EDTA on trace elements and cardiovascular function in the anesthetised rabbit. Biol. Trace. Elem. Res. 2000, 76, 133–148. [Google Scholar] [CrossRef]
- Welling, S.H.; Hubálek, F.; Jacobsen, J.; Brayden, D.J.; Rahbek, U.L.; Buckley, S.T. The role of citric acid in oral peptide and protein formulations: Relationship between calcium chelation and proteolysis inhibition. Eur. J. Pharm. Biopharm. 2014, 86, 544–551. [Google Scholar] [CrossRef]
- Sadeghi, A.M.M.; Dorkoosh, F.A.; Avadi, M.R.; Weinhold, M.; Bayat, A.; Delie, F.; Gurny, R.; Larijani, B.; Rafieetehrani, M.; Junginger, H.E. Permeation enhancer effect of chitosan and chitosan derivatives: Comparison of formulations as soluble polymers and nanoparticulate systems on insulin absorption in Caco-2 cells. Eur. J. Pharm. Biopharm. 2008, 70, 270–278. [Google Scholar] [CrossRef]
- Thanou, M.; Verhoef, J.C.; Junginger, H.E. Oral drug absorption enhancement by chitosan and its derivatives. Adv. Drug Deliv. Rev. 2001, 52, 117–126. [Google Scholar] [CrossRef]
- Ghosh, S.; Ray, A.; Pramanik, N. Self-assembly of surfactants: An overview on general aspects of amphiphiles. Biophys. Chem. 2020, 265, 106429. [Google Scholar] [CrossRef]
- Twarog, C.; Fattah, S.; Heade, J.; Maher, S.; Fattal, E.; Brayden, D.J. Intestinal Permeation Enhancers for Oral Delivery of Macromolecules: A Comparison between Salcaprozate Sodium (SNAC) and Sodium Caprate (C10). Pharmaceutics 2019, 11, 78. [Google Scholar] [CrossRef]
- Weber, S.L.; Gkonos, P.J.; Skyler, J.S. Combined octreotide and insulin therapy in acromegaly. Endocr. Pract. 1997, 3, 19–21. [Google Scholar] [CrossRef]
- Rivera, T.M.; Leone-Bay, A.; Paton, D.R.; Leipold, H.R.; Baughman, R.A. Oral delivery of heparin in combination with sodium N-[8-(2-hydroxybenzoyl)amino]caprylate: Pharmacological considerations. Pharm. Res. 1997, 14, 1830–1834. [Google Scholar] [CrossRef]
- Malkov, D.; Angelo, R.; Wang, H.Z.; Flanders, E.; Tang, H.; Gomez-Orellana, I. Oral delivery of insulin with the eligen technology: Mechanistic studies. Curr. Drug Deliv. 2005, 2, 191–197. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Riis, B.J.; Mehta, N.; Stern, W.; Arbit, E.; Christiansen, C.; Henriksen, K. Lessons learned from the clinical development of oral peptides. Br. J. Clin. Pharmacol. 2015, 79, 720–732. [Google Scholar] [CrossRef]
- Nakada, Y.; Awata, N.; Nakamichi, C.; Sugimoto, I. The effect of additives on the oral mucosal absorption of human calcitonin in rats. J. Pharmacobiodyn. 1988, 11, 395–401. [Google Scholar] [CrossRef]
- Oh, C.K.; Ritschel, W.A. Absorption characteristics of insulin through the buccal mucosa. Methods Find. Exp. Clin. Pharmacol. 1990, 12, 275–279. [Google Scholar] [PubMed]
- Kesarwani, K.; Gupta, R. Bioavailability enhancers of herbal origin: An overview. Asian Pac. J. Trop. Biomed. 2013, 3, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Coufalová, L.; Mrózek, L.; Rárová, L.; Plaček, L.; Opatřilová, R.; Dohnal, J.; Král’ová, K.; Paleta, O.; Král, V.; Drašar, P.; et al. New propanoyloxy derivatives of 5β-cholan-24-oic acid as drug absorption modifiers. Steroids 2013, 78, 435–453. [Google Scholar] [CrossRef] [PubMed]
- Fein, K.C.; Gleeson, J.P.; Newby, A.N.; Whitehead, K.A. Intestinal permeation enhancers enable oral delivery of macromolecules up to 70 kDa in size. Eur. J. Pharm. Biopharm. 2022, 170, 70–76. [Google Scholar] [CrossRef]
- Brayden, D.J.; Stuettgen, V. Sodium glycodeoxycholate and sodium deoxycholate as epithelial permeation enhancers: In vitro and ex vivo intestinal and buccal bioassays. Eur. J. Pharm. Sci. 2021, 159, 105737. [Google Scholar]
- Song, K.H.; Chung, S.J.; Shim, C.K. Enhanced intestinal absorption of salmon calcitonin (sCT) from proliposomes containing bile salts. J. Control. Release 2005, 106, 298–308. [Google Scholar] [CrossRef]
- Moghimipour, E.; Jalali, A.; Sajjadi Tabassi, S.A.; Lbenberg, R. The Enhancing Effect of Sodium Glycocholate and Sodium Salicylate on Rats Gastro-intestinal Permeability to Insulin. Iran. J. Pharm. Res. 2010, 3, 87–91. [Google Scholar]
- Niu, M.; Tan, Y.; Guan, P.; Hovgaard, L.; Lu, Y.; Qi, J.; Lian, R.; Li, X.; Wu, W. Enhanced oral absorption of insulin-loaded liposomes containing bile salts: A mechanistic study. Int. J. Pharm. 2014, 460, 119–130. [Google Scholar] [CrossRef]
- Hu, S.; Niu, M.; Hu, F.; Lu, Y.; Qi, J.; Yin, Z.; Wu, W. Integrity and stability of oral liposomes containing bile salts studied in simulated and ex vivo gastrointestinal media. Int. J. Pharm. 2013, 441, 693–700. [Google Scholar] [CrossRef]
- Christiaens, B.; Grooten, J.; Reusens, M.; Joliot, A.; Goethals, M.; Vandekerckhove, J.; Prochiantz, A.; Rosseneu, M. Membrane interaction and cellular internalization of penetratin peptides. Eur. J. Biochem. 2004, 271, 1187–1197. [Google Scholar] [CrossRef]
- Foged, C.; Nielsen, H.M. Cell-penetrating peptides for drug delivery across membrane barriers. Expert Opin. Drug Deliv. 2008, 5, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Säälik, P.; Elmquist, A.; Hansen, M.; Padari, K.; Saar, K.; Viht, K.; Langel, Ü.; Pooga, M. Protein cargo delivery properties of cell-penetrating peptides. A comparative study. Bioconjug. Chem. 2004, 15, 1246–1253. [Google Scholar] [CrossRef] [PubMed]
- Böhmová, E.; Machová, D.; Pechar, M.; Pola, R.; Venclíková, K.; Janoušková, O.; Etrych, T. Cell-penetrating peptides: A useful tool for the delivery of various cargoes into cells. Physiol. Res. 2018, 67 (Suppl. S2), S267–S279. [Google Scholar] [CrossRef] [PubMed]
- Zorko, M.; Langel, U. Cell-penetrating peptides: Mechanism and kinetics of cargo delivery. Adv. Drug Deliv. Rev. 2005, 57, 529–545. [Google Scholar] [CrossRef]
- Vivès, E.; Brodin, P.; Lebleu, B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef]
- Rothbard, J.B.; Garlington, S.; Lin, Q.; Kirschberg, T.; Kreider, E.; McGrane, P.L.; Wender, P.A.; Khavari, P.A. Conjugation of arginine oligomers to cyclosporin A facilitates topical delivery and inhibition of inflammation. Nat. Med. 2000, 6, 1253–1257. [Google Scholar] [CrossRef]
- Mo, R.H.; Zaro, J.L.; Shen, W.C. Comparison of cationic and amphipathic cell penetrating peptides for siRNA delivery and efficacy. Mol. Pharm. 2012, 9, 299–309. [Google Scholar] [CrossRef]
- Schwarze, S.R.; Ho, A.; Vocero-Akbani, A.; Dowdy, S.F. In vivo protein transduction: Delivery of a biologically active protein into the mouse. Science 1999, 285, 1569–1572. [Google Scholar] [CrossRef]
- Liang, J.F.; Yang, V.C. Insulin-cell penetrating peptide hybrids with improved intestinal absorption efficiency. Biochem. Biophys. Res. Commun. 2005, 335, 734–738. [Google Scholar] [CrossRef]
- Nielsen, E.J.B.; Yoshida, S.; Kamei, N.; Iwamae, R.; Khafagy, E.S.; Olsen, J.; Rahbek, U.L.; Pedersen, B.L.; Takayama, K.; Takeda-Morishita, M. In vivo proof of concept of oral insulin delivery based on a co-administration strategy with the cell-penetrating peptide penetratin. J. Control. Release 2014, 189, 19–24. [Google Scholar] [CrossRef]
- Kamei, N.; Nielsen, E.J.B.; Khafagy, E.S.; Takeda-Morishita, M. Noninvasive insulin delivery: The great potential of cell-penetrating peptides. Ther. Deliv. 2013, 4, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, J.; Xu, D. Cell-penetrating peptides as noninvasive transmembrane vectors for the development of novel multifunctional drug-delivery systems. J. Control. Release 2016, 229, 130–139. [Google Scholar] [CrossRef]
- Khafagy, E.S.; Morishita, M.; Kamei, N.; Eda, Y.; Ikeno, Y.; Takayama, K. Efficiency of cell-penetrating peptides on the nasal and intestinal absorption of therapeutic peptides and proteins. Int. J. Pharm. 2009, 381, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Waghule, T.; Singhvi, G.; Dubey, S.K.; Pandey, M.M.; Gupta, G.; Singh, M.; Dua, K. Microneedles: A smart approach and increasing potential for transdermal drug delivery system. Biomed. Pharmacother. 2019, 109, 1249–1258. [Google Scholar] [CrossRef]
- Kim, Y.C.; Park, J.H.; Prausnitz, M.R. Microneedles for drug and vaccine delivery. Adv. Drug Deliv. Rev. 2012, 64, 1547–1568. [Google Scholar] [CrossRef] [PubMed]
- Schoellhammer, C.M.; Langer, R.; Traverso, G. Of microneedles and ultrasound: Physical modes of gastrointestinal macromolecule delivery. Tissue Barriers 2016, 4, e1150235. [Google Scholar] [CrossRef] [PubMed]
- Traverso, G.; Schoellhammer, C.M.; Schroeder, A.; Maa, R.; Lauwers, G.Y.; Polat, B.E.; Anderson, D.G.; Blankschtein, D.; Langer, R. Microneedles for Drug Delivery via the Gastrointestinal Tract. J. Pharm. Sci. 2015, 104, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Wei, L.; Wu, F.; Wu, Z.; Chen, L.; Liu, Z.; Yuan, W. Dissolving and biodegradable microneedle technologies for transdermal sustained delivery of drug and vaccine. Drug Des. Dev. Ther. 2013, 7, 945–952. [Google Scholar]
- Prausnitz, M.R.; Gomaa, Y.; Li, W. Microneedle patch drug delivery in the gut. Nat. Med. 2019, 25, 1471–1472. [Google Scholar] [CrossRef]
- Dhalla, A.K.; Al-Shamsie, Z.; Beraki, S.; Dasari, A.; Fung, L.C.; Fusaro, L.; Garapaty, A.; Gutierrez, B.; Gratta, D.; Hashim, M.; et al. A robotic pill for oral delivery of biotherapeutics: Safety, tolerability, and performance in healthy subjects. Drug Deliv. Transl. Res. 2022, 12, 294–305. [Google Scholar] [CrossRef]
- Seah, B.C.Q.; Teo, B.M. Recent advances in ultrasound-based transdermal drug delivery. Int. J. Nanomed. 2018, 13, 7749–7763. [Google Scholar] [CrossRef] [PubMed]
- Polat, B.E.; Hart, D.; Langer, R.; Blankschtein, D. Ultrasound-Mediated Transdermal Drug Delivery: Mechanisms, Scope, and Emerging Trends. J. Control. Release 2011, 152, 330–348. [Google Scholar] [CrossRef] [PubMed]
- Apfel, R.E. Acoustic cavitation: A possible consequence of biomedical uses of ultrasound. Br. J. Cancer Suppl. 1982, 5, 140–146. [Google Scholar]
- Mitragotri, S.; Blankschtein, D.; Langer, R. Ultrasound-mediated transdermal protein delivery. Science 1995, 269, 850–853. [Google Scholar] [CrossRef] [PubMed]
- Schoellhammer, C.M.; Lauwers, G.Y.; Goettel, J.A.; Oberli, M.A.; Cleveland, C.; Park, J.Y.; Minahan, D.; Chen, Y.; Anderson, D.G.; Jaklenec, A.; et al. Ultrasound-Mediated Delivery of RNA to Colonic Mucosa of Live Mice. Gastroenterology 2017, 152, 1151–1160. [Google Scholar] [CrossRef]
- Stewart, F.; Cummins, G.; Turcanu, M.V.; Cox, B.F.; Prescott, A.; Clutton, E.; Newton, I.P.; Desmulliez, M.P.Y.; Thanou, M.; Mulvana, H.; et al. Ultrasound mediated delivery of quantum dots from a proof of concept capsule endoscope to the gastrointestinal wall. Sci. Rep. 2021, 11, 2584. [Google Scholar] [CrossRef]
- Pridgen, E.M.; Alexis, F.; Farokhzad, O.C. Polymeric nanoparticle drug delivery technologies for oral delivery applications. Expert Opin. Drug Deliv. 2015, 12, 1459–1473. [Google Scholar] [CrossRef]
- Garinot, M.; Fiévez, V.; Pourcelle, V.; Stoffelbach, F.; des Rieux, A.; Plapied, L.; Theate, I.; Freichels, H.; Jérôme, C.; Marchand-Brynaert, J.; et al. PEGylated PLGA-based nanoparticles targeting M cells for oral vaccination. J. Control. Release 2007, 120, 195–204. [Google Scholar] [CrossRef]
- Xu, X.; Vugmeyster, Y. Challenges and Opportunities in Absorption, Distribution, Metabolism, and Excretion Studies of Therapeutic Biologics. AAPS J. 2012, 14, 781–791. [Google Scholar] [CrossRef]
- Su, J.; Mazzeo, J.; Subbarao, N.; Jin, T. Pharmaceutical development of biologics: Fundamentals, challenges and recent advances. Ther. Deliv. 2011, 2, 865–871. [Google Scholar] [CrossRef]
- Shire, S.J. Formulation and manufacturability of biologics. Curr. Opin. Biotechnol. 2009, 20, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Truong-Le, V.; Lovalenti, P.M.; Abdul-Fattah, A.M. Stabilization challenges and formulation strategies associated with oral biologic drug delivery systems. Adv. Drug Deliv. Rev. 2015, 93, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Neoral: A Microemulsion Cyclosporine—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/9157923/ (accessed on 20 April 2023).
- Taesch, S.; Niese, D.; Mueller, E.A. Sandimmun neoral, a new oral formulation of cyclosporin A with improved pharmacokinetic characteristics: Safety and tolerability in renal transplant patients. Transpl. Proc. 1994, 26, 3147–3149. [Google Scholar]
- Kosman, M.E. Evaluation of a new antidiuretic agent, desmopressin acetate (DDAVP). JAMA 1978, 240, 1896–1897. [Google Scholar] [CrossRef]
- Khomane, K.S.; Meena, C.L.; Jain, R.; Bansal, A.K. Novel thyrotropin-releasing hormone analogs: A patent review. Expert Opin. Ther. Pat. 2011, 21, 1673–1691. [Google Scholar] [CrossRef]
- Prokai-Tatrai, K.; Prokai, L. Prodrugs of Thyrotropin-Releasing Hormone and Related Peptides as Central Nervous System Agents. Molecules 2009, 14, 633–654. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Sato, N.; Fujimura, Y.; Kihara, T.; Sugita, K.; Takahashi, K.; Koike, K.; Sugawara, T.; Tada, Y.; Nakai, H.; et al. Discovery of the Orally Effective Thyrotropin-Releasing Hormone Mimetic: 1-{N-[(4S,5S)-(5-Methyl-2-oxooxazolidine-4-yl)carbonyl]-3-(thiazol-4-yl)-l-alanyl}-(2R)-2-methylpyrrolidine Trihydrate (Rovatirelin Hydrate). ACS Omega 2018, 3, 13647–13666. [Google Scholar] [CrossRef] [PubMed]
- Oramed Announces Top-line Results from Phase 3 Trial of ORMD-0801 for the Treatment of Type 2 Diabetes—Oramed Pharmaceuticals. Available online: https://oramed.com/oramed-announces-top-line-results-from-phase-3-trial-of-ormd-0801-for-the-treatment-of-type-2-diabetes/ (accessed on 20 April 2023).












| Parameter to Improve | Strategy | Goals | Limitations | References |
|---|---|---|---|---|
| Stability | Enteric coating | To bypass the harsh gastric conditions | Not sufficient alone to protect from the biochemical barrier of the GI tract | [7,77,198] |
| pH modulators | To modulate the microenvironment’s pH in order to hinder the activation of proteolytic enzymes | Can influence the dissolution of enteric coating, shifts in pH could alter the cargo | [7,77,203,204] | |
| Enzyme inhibitors | To reduce the activity or inactivate proteolytic enzymes by the binding to their specific site | Possible side effects in long-term therapies (e.g., absorption of unwanted molecules, damages to the GI tract) | [205,211,212] | |
| Mucus penetration | Mucus penetrating systems | To cross the mucus layer in order to reach epithelial cells (mucolytics, NPs) | Not sufficient alone to increase intestinal permeability, side effects for mucolytics | [34,219,224] |
| Contact time/site-specific delivery | Thiomers | To increase contact time with the mucosa by binding to the mucus | Prone to oxidation, dependent on the mucus turn-over | [77,226,231] |
| Intestinal patches | To create a high concentration gradient, to increase the residence time at the site-specific drug release, and to confer protection from the harsh GI environment | Importance of the formulation (e.g., size, polymers) to avoid side effects | [233,234,237] | |
| Hydrogels | To promote a site-specific release and to confer protection from harsh GI environment | Swelling dependent on diffusion of water | [244,245,248] | |
| Permeability | NPs | Among others, to cross the intestinal epithelium by modulating optimal physicochemical properties and/or active targeting of a receptor | Depending on the polymers used: toxicity or instability of the drugs | [22,256,335,336] |
| Targeting of receptors | To trigger RMT across the intestinal epithelium | Optimal properties of the ligands to trigger RMT unknown (e.g., affinity, epitope targeted) | [7,262,270,271,280] | |
| PEs | To increase paracellular or transcellular transport across the intestinal barrier | Toxicity at high level concentrations, non-selective techniques | [284,285,308] | |
| Microneedles | To physically deliver the molecules via insertion into the intestinal mucosa | Potential use of metal, safety in long-term therapies not validated, amount of drug that can be loaded per microneedle | [324,325,328] | |
| Ultrasounds | To permeabilize tissues (reversibly) | Miniaturization of the device that needs further optimization | [329,333,334] |
| Biological Molecule | Company | Indication | Strategies |
|---|---|---|---|
| Insulin (Phase III: NCT04754334) | Oramed Pharmaceuticals (Jerusalem, Israel) | Type 2 Diabetes Mellitus | PE in insulin prodrug |
| Insulin (Phase II/III: NCT00814294 and NCT03096392) | Diasome Pharmaceuticals Inc. (Cleveland, OH, USA) | Type 2 Diabetes Mellitus | Liver-targeted liposomes |
| Insulin (Phase II/III: NCT03430856) | Biocon Ltd. (Bangalore, Karnataka, India) | Type 1 Diabetes Mellitus | PE (sodium caprate) included as permeability enhancer in insulin prodrug |
| Insulin (Phase II: NCT01973920) | Oshadi Drug Administration Ltd. (Rehovot, Israel) | Type 1 Diabetes Mellitus | NPs |
| Insulin (Phase II: NCT02470039) | Generex Biotechnology Corp. (Burlington, Canada) | Type 2 Diabetes Mellitus | Microemulsion systems (fatty acids) and enteric coating |
| Parathyroid hormone (PTH) (Phase II: NCT03516773) | Proxima Concepts Ltd./Diabetology (London, UK) | Hypoparathyroidism | PEs |
| Salmon calcitonin (Phase III: NCT00959764) | Tarsa Therapeutics (Philadelphia, PA, USA) | Osteoporosis, Postmenopausal | pH modulator |
| Leuprolide (Phase II: NCT05096065) | Enteris Biopharm (Boonton, NJ, USA) | Endometriosis | PE, pH modulator, and enzyme inhibitor |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masloh, S.; Culot, M.; Gosselet, F.; Chevrel, A.; Scapozza, L.; Zeisser Labouebe, M. Challenges and Opportunities in the Oral Delivery of Recombinant Biologics. Pharmaceutics 2023, 15, 1415. https://doi.org/10.3390/pharmaceutics15051415
Masloh S, Culot M, Gosselet F, Chevrel A, Scapozza L, Zeisser Labouebe M. Challenges and Opportunities in the Oral Delivery of Recombinant Biologics. Pharmaceutics. 2023; 15(5):1415. https://doi.org/10.3390/pharmaceutics15051415
Chicago/Turabian StyleMasloh, Solene, Maxime Culot, Fabien Gosselet, Anne Chevrel, Leonardo Scapozza, and Magali Zeisser Labouebe. 2023. "Challenges and Opportunities in the Oral Delivery of Recombinant Biologics" Pharmaceutics 15, no. 5: 1415. https://doi.org/10.3390/pharmaceutics15051415