
Combination of Chemotherapy and Mild Hyperthermia Using Targeted Nanoparticles: A Potential Treatment Modality for Breast Cancer
 ,
,  , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Fabrication of pSiNPs
2.3. Gold Nanocluster (AuNC) Synthesis
2.4. Loading and Modification of pSiNPs
2.4.1. Thermal Hydrosilylation of pSiNPs
2.4.2. Loading of Therapeutics
2.4.3. Antibody Conjugation
2.5. Loading and Release Measurements
2.6. Dynamic Light Scattering and ζ Potential Measurements
2.7. Transmission Electron Microscopy
2.8. Fourier Transform Infrared Spectroscopy
2.9. Determination of pSi Concentration
2.10. RF Setup
2.11. Cell Culture
2.12. Confocal Microscopy
2.13. Flow Cytometry
2.14. Cell Viability
2.15. ROS Generation
2.16. Statistical Analysis
3. Results and Discussion
3.1. Synthesis and Characterisation of pSiNPs and AuNCs
3.2. Loading and Release of Therapeutics
3.3. Design and Characterisation of the Radiofrequency (RF) Field Generator
3.4. Heating Properties of AuNCs under RF at 1 GHz
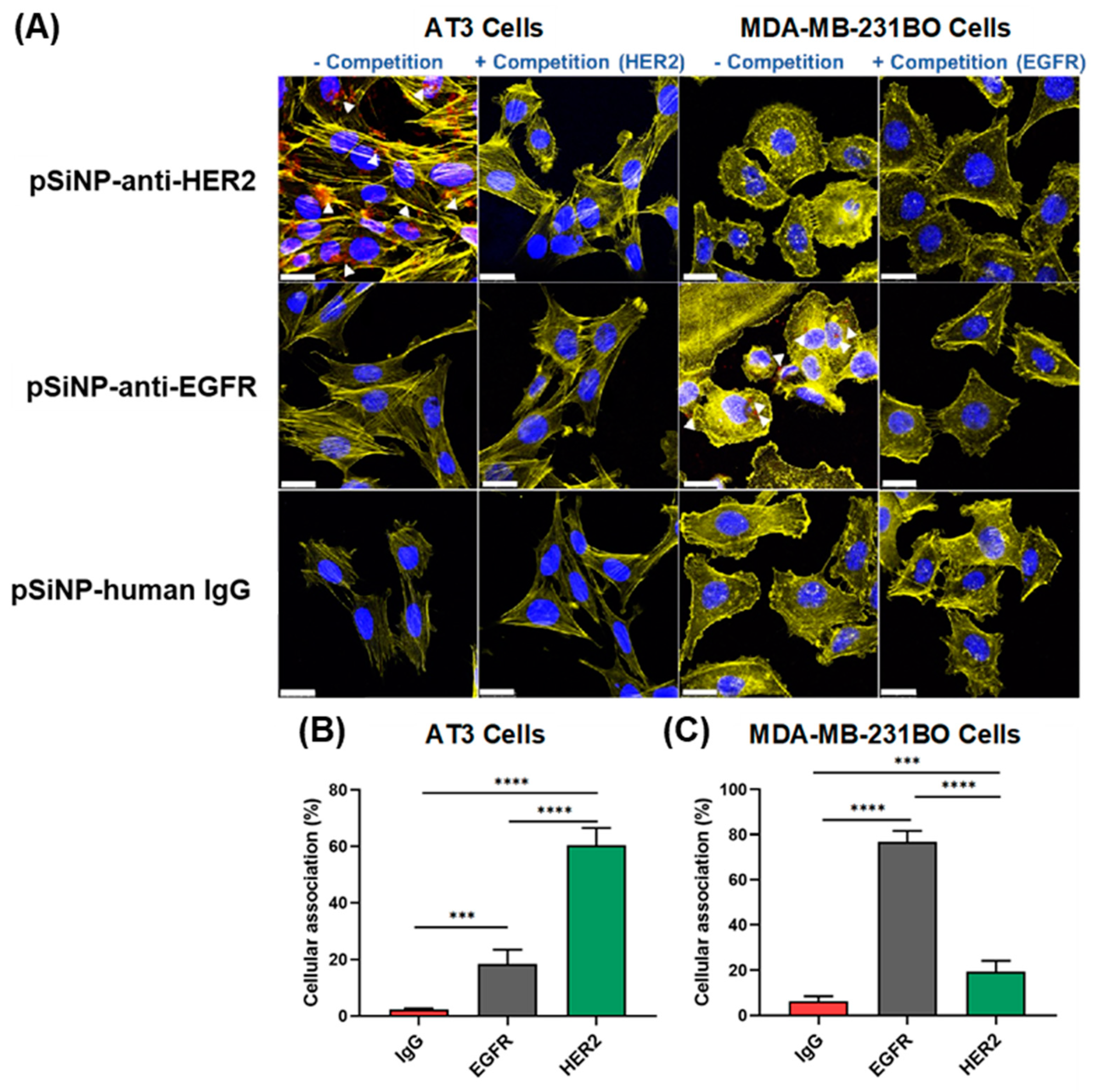
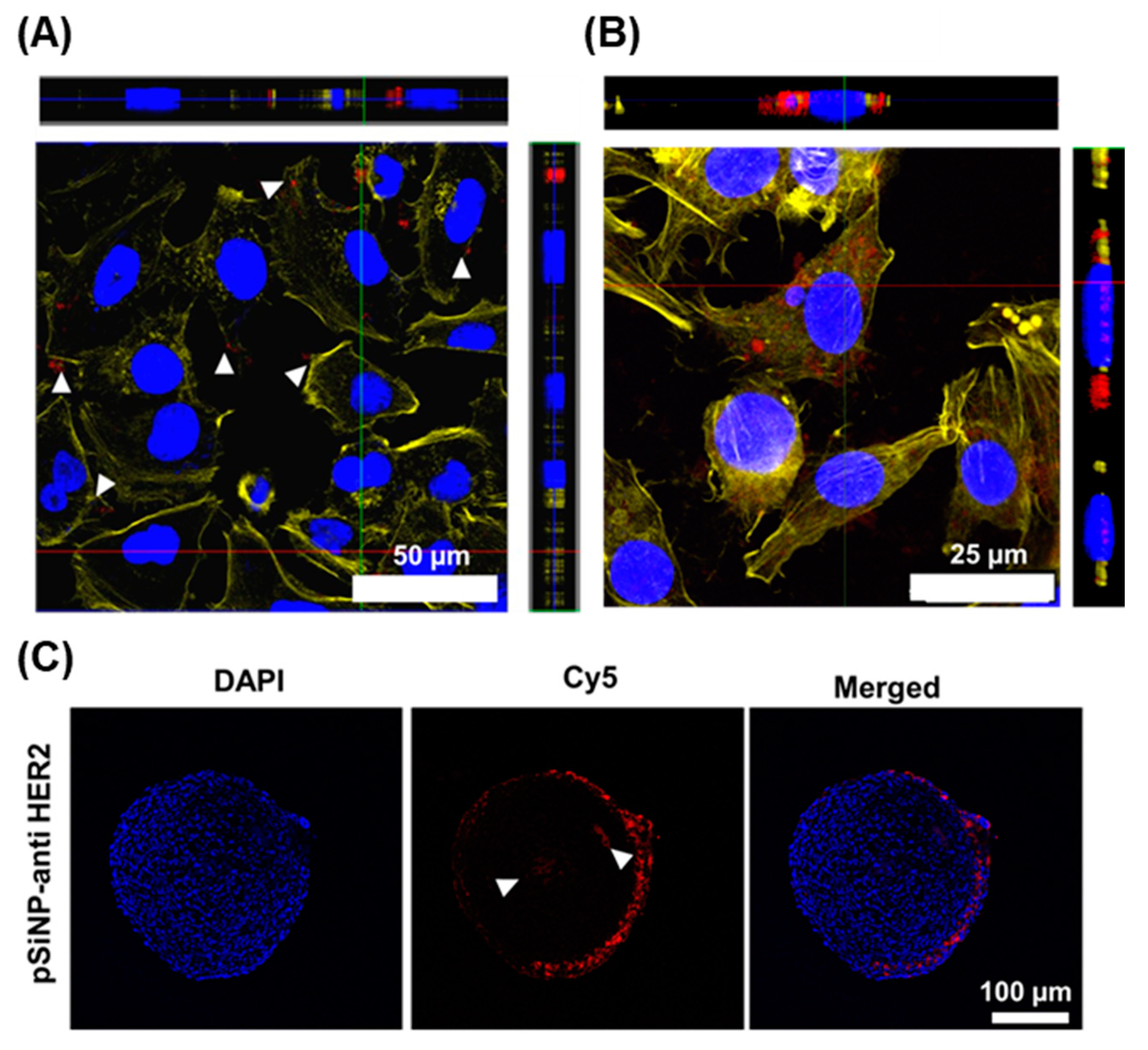
3.5. Cellular Association and Uptake of Antibody-Targeted pSiNPs
3.6. Cell Viability
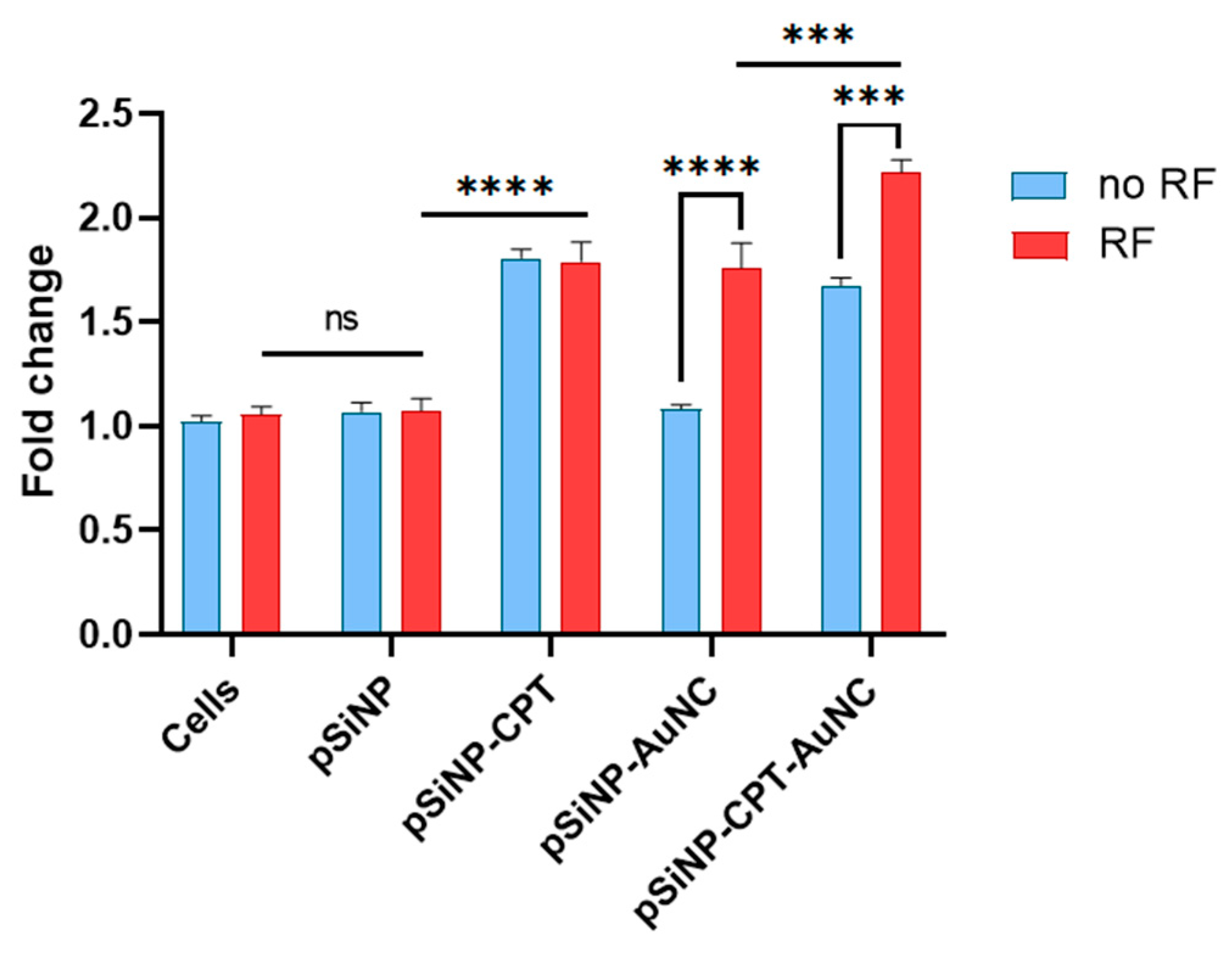
3.7. ROS Production in Cells
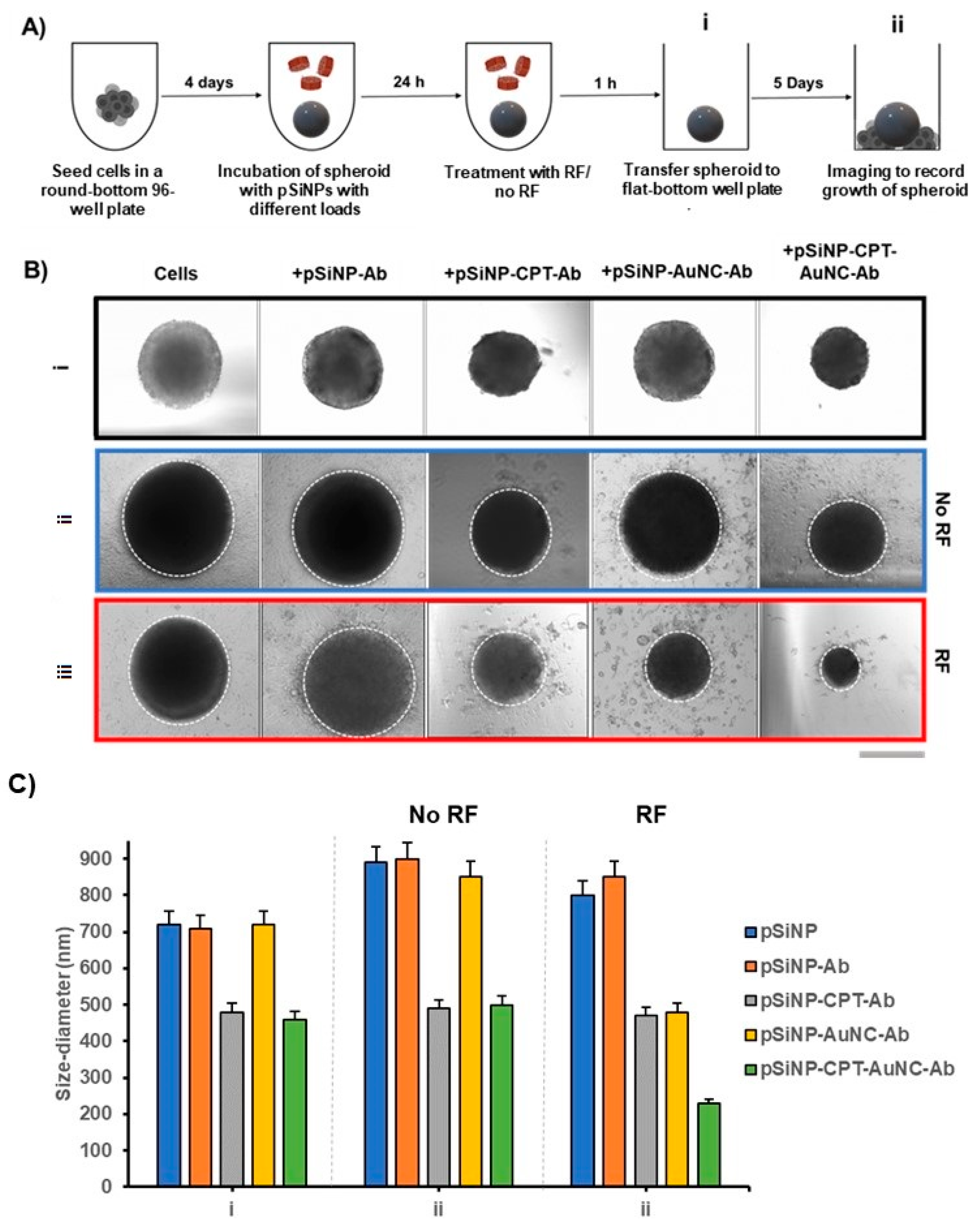
3.8. Cell Growth after Treatment in 3D Spheroids
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maughan, K.L.; Lutterbie, M.A.; Ham, P.S. Treatment of breast cancer. Am. Fam. Physician 2010, 81, 1339–1346. [Google Scholar]
- Wu, Q.; Yang, Z.; Nie, Y.; Shi, Y.; Fan, D. Multi-drug resistance in cancer chemotherapeutics: Mechanisms and lab approaches. Cancer Lett. 2014, 347, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Herrero, E.; Fernández-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef] [PubMed]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Bimbo, L.M.; Mäkilä, E.; Villanova, F.; Kaasalainen, M.; Herranz-Blanco, B.; Caramella, C.M.; Lehto, V.-P.; Salonen, J.; Herzig, K.-H.; et al. Co-delivery of a hydrophobic small molecule and a hydrophilic peptide by porous silicon nanoparticles. J. Control. Release 2013, 170, 268–278. [Google Scholar] [CrossRef]
- Maeda, H. The enhanced permeability and retention (EPR) effect in tumor vasculature: The key role of tumor-selective macromolecular drug targeting. Adv. Enzym. Regul. 2001, 41, 189–207. [Google Scholar] [CrossRef]
- Farokhzad, O.C.; Langer, R. Impact of nanotechnology on drug delivery. ACS Nano 2009, 3, 16–20. [Google Scholar] [CrossRef]
- Zhang, D.-X.; Esser, L.; Vasani, R.B.; Thissen, H.; Voelcker, N.H. Porous silicon nanomaterials: Recent advances in surface engineering for controlled drug-delivery applications. Nanomedicine 2019, 14, 3213–3230. [Google Scholar] [CrossRef] [PubMed]
- Tieu, T.; Alba, M.; Elnathan, R.; Cifuentes-Rius, A.; Voelcker, N.H. Advances in Porous Silicon–Based Nanomaterials for Diagnostic and Therapeutic Applications. Adv. Ther. 2019, 2, 1800095. [Google Scholar] [CrossRef]
- Acharya, S.; Dilnawaz, F.; Sahoo, S.K. Targeted epidermal growth factor receptor nanoparticle bioconjugates for breast cancer therapy. Biomaterials 2009, 30, 5737–5750. [Google Scholar] [CrossRef] [PubMed]
- Shadidi, M.; Sioud, M. Selective targeting of cancer cells using synthetic peptides. Drug Resist. Updat. 2003, 6, 363–371. [Google Scholar] [CrossRef]
- Low, P.S.; Kularatne, S.A. Folate-targeted therapeutic and imaging agents for cancer. Curr. Opin. Chem. Biol. 2009, 13, 256–262. [Google Scholar] [CrossRef]
- Danhier, F.; Feron, O.; Préat, V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J. Control. Release 2010, 148, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.L.; Cobleigh, M.A.; Tripathy, D.; Gutheil, J.C.; Harris, L.N.; Fehrenbacher, L.; Slamon, D.J.; Murphy, M.; Novotny, W.F.; Burchmore, M.; et al. First-line Herceptin monotherapy in metastatic breast cancer. Oncology 2001, 61 (Suppl. S2), 37–42. [Google Scholar] [CrossRef]
- Landgraf, M.; Lahr, C.A.; Kaur, I.; Shafiee, A.; Sanchez-Herrero, A.; Janowicz, P.W.; Ravichandran, A.; Howard, C.B.; Cifuentes-Rius, A.; McGovern, J.A.; et al. Targeted camptothecin delivery via silicon nanoparticles reduces breast cancer metastasis. Biomaterials 2020, 240, 119791. [Google Scholar] [CrossRef] [PubMed]
- Cancer multidrug resistance. Nat. Biotechnol. 2000, 18, IT18–IT20. [CrossRef]
- Wu, H.; Jin, H.; Wang, C.; Zhang, Z.; Ruan, H.; Sun, L.; Yang, C.; Li, Y.; Qin, W.; Wang, C. Synergistic Cisplatin/Doxorubicin Combination Chemotherapy for Multidrug-Resistant Cancer via Polymeric Nanogels Targeting Delivery. ACS Appl. Mater. Interfaces 2017, 9, 9426–9436. [Google Scholar] [CrossRef] [PubMed]
- Yardley, D.A. Drug resistance and the role of combination chemotherapy in improving patient outcomes. Int. J. Breast Cancer 2013, 2013, 137414. [Google Scholar] [CrossRef] [PubMed]
- Miles, D.; von Minckwitz, G.; Seidman, A.D. Combination versus sequential single-agent therapy in metastatic breast cancer. Oncologist 2002, 7 (Suppl. S6), 13–19. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Michor, F. Pharmacokinetics and Drug Interactions Determine Optimum Combination Strategies in Computational Models of Cancer Evolution. Cancer Res. 2017, 77, 3908–3921. [Google Scholar] [CrossRef]
- Hu, C.M.; Aryal, S.; Zhang, L. Nanoparticle-assisted combination therapies for effective cancer treatment. Ther. Deliv. 2010, 1, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.M.; Zhang, L. Nanoparticle-based combination therapy toward overcoming drug resistance in cancer. Biochem Pharm. 2012, 83, 1104–1111. [Google Scholar] [CrossRef] [PubMed]
- Klimanov, M.Y.; Syvak, L.A.; Orel, V.E.; Lavryk, G.V.; Tarasenko, T.Y.; Orel, V.B.; Rykhalskyi, A.Y.; Stegnii, V.V.; Nesterenko, A.O. Efficacy of Combined Regional Inductive Moderate Hyperthermia and Chemotherapy in Patients With Multiple Liver Metastases From Breast Cancer. Technol. Cancer Res. Treat. 2018, 17, 1533033818806003. [Google Scholar] [CrossRef]
- Phung, D.C.; Nguyen, H.T.; Phuong Tran, T.T.; Jin, S.G.; Yong, C.S.; Truong, D.H.; Tran, T.H.; Kim, J.O. Combined hyperthermia and chemotherapy as a synergistic anticancer treatment. J. Pharm. Investig. 2019, 49, 519–526. [Google Scholar] [CrossRef]
- Roti Roti, J.L. Cellular responses to hyperthermia (40–46 °C): Cell killing and molecular events. Int. J. Hyperth. 2008, 24, 3–15. [Google Scholar] [CrossRef]
- Januszewski, A.; Stebbing, J. Hyperthermia in cancer: Is it coming of age? Lancet Oncol. 2014, 15, 565–566. [Google Scholar] [CrossRef] [PubMed]
- Dewey, W.C.; Hopwood, L.E.; Sapareto, S.A.; Gerweck, L.E. Cellular responses to combinations of hyperthermia and radiation. Radiology 1977, 123, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Pellosi, D.S.; Macaroff, P.P.; Morais, P.C.; Tedesco, A.C. Magneto low-density nanoemulsion (MLDE): A potential vehicle for combined hyperthermia and photodynamic therapy to treat cancer selectively. Mater. Sci. Eng. C 2018, 92, 103–111. [Google Scholar] [CrossRef]
- Bienia, A.; Wiecheć-Cudak, O.; Murzyn, A.A.; Krzykawska-Serda, M. Photodynamic Therapy and Hyperthermia in Combination Treatment—Neglected Forces in the Fight against Cancer. Pharmaceutics 2021, 13, 1147. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Ye, M.; Wu, Y.; Wu, L.; Lan, K.; Wu, Z. Hyperthermia combined with immune checkpoint inhibitor therapy: Synergistic sensitization and clinical outcomes. Cancer Med. 2023, 12, 3201–3221. [Google Scholar] [CrossRef]
- Huang, L.; Li, Y.; Du, Y.; Zhang, Y.; Wang, X.; Ding, Y.; Yang, X.; Meng, F.; Tu, J.; Luo, L.; et al. Mild photothermal therapy potentiates anti-PD-L1 treatment for immunologically cold tumors via an all-in-one and all-in-control strategy. Nat. Commun. 2019, 10, 4871. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, M.; Stauffer, P. Hyperthermia, radiation and chemotherapy: The role of heat in multidisciplinary cancer care. Semin. Oncol. 2014, 41, 714–729. [Google Scholar] [CrossRef] [PubMed]
- Hanson, G.W.; Monreal, R.C.; Apell, S.P. Electromagnetic absorption mechanisms in metal nanospheres: Bulk and surface effects in radiofrequency-terahertz heating of nanoparticles. J. Appl. Phys. 2011, 109, 124306. [Google Scholar] [CrossRef]
- Nasseri, B.; Kocum, I.C.; Seymen, C.M.; Rabiee, N. Penetration Depth in Nanoparticles Incorporated Radiofrequency Hyperthermia into the Tissue: Comprehensive Study with Histology and Pathology Observations. IET Nanobiotechnol. 2019, 13, 634–639. [Google Scholar] [CrossRef]
- Jin, R. Atomically precise metal nanoclusters: Stable sizes and optical properties. Nanoscale 2015, 7, 1549–1565. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes-Rius, A.; Deepagan, V.G.; Xie, J.; Voelcker, N.H. Bright Future of Gold Nanoclusters in Theranostics. ACS Appl. Mater. Interfaces 2021, 13, 49581–49588. [Google Scholar] [CrossRef]
- Zare, I.; Chevrier, D.M.; Cifuentes-Rius, A.; Moradi, N.; Xianyu, Y.; Ghosh, S.; Trapiella-Alfonso, L.; Tian, Y.; Shourangiz-Haghighi, A.; Mukherjee, S.; et al. Protein-protected metal nanoclusters as diagnostic and therapeutic platforms for biomedical applications. Mater. Today 2021. [Google Scholar] [CrossRef]
- Cifuentes-Rius, A.; Ivask, A.; Das, S.; Penya-Auladell, N.; Fabregas, L.; Fletcher, N.L.; Houston, Z.H.; Thurecht, K.J.; Voelcker, N.H. Gold Nanocluster-Mediated Cellular Death under Electromagnetic Radiation. ACS Appl. Mater. Interfaces 2017, 9, 41159–41167. [Google Scholar] [CrossRef]
- Cifuentes-Rius, A.; Ivask, A.; Sporleder, E.; Kaur, I.; Assan, Y.; Rao, S.; Warther, D.; Prestidge, C.A.; Durand, J.O.; Voelcker, N.H. Dual-Action Cancer Therapy with Targeted Porous Silicon Nanovectors. Small 2017, 13, 1701201. [Google Scholar] [CrossRef]
- Collins, C.B.; McCoy, R.S.; Ackerson, B.J.; Collins, G.J.; Ackerson, C.J. Radiofrequency heating pathways for gold nanoparticles. Nanoscale 2014, 6, 8459–8472. [Google Scholar] [CrossRef]
- Deepagan, V.G.; Leiske, M.N.; Fletcher, N.L.; Rudd, D.; Tieu, T.; Kirkwood, N.; Thurecht, K.J.; Kempe, K.; Voelcker, N.H.; Cifuentes-Rius, A. Engineering Fluorescent Gold Nanoclusters Using Xanthate-Functionalized Hydrophilic Polymers: Toward Enhanced Monodispersity and Stability. Nano Lett. 2021, 21, 476–484. [Google Scholar] [CrossRef]
- Chen, L.Y.; Wang, C.W.; Yuan, Z.; Chang, H.T. Fluorescent gold nanoclusters: Recent advances in sensing and imaging. Anal. Chem. 2015, 87, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Ma, H.; Liu, J.; Huo, S.; Kumar, A.; Wei, T.; Zhang, X.; Jin, S.; Gan, Y.; Wang, P.C.; et al. Size-dependent localization and penetration of ultrasmall gold nanoparticles in cancer cells, multicellular spheroids, and tumors in vivo. ACS Nano 2012, 6, 4483–4493. [Google Scholar] [CrossRef]
- Li, H.; Ménard, M.; Vardanyan, A.; Charnay, C.; Raehm, L.; Oliviero, E.; Seisenbaeva, G.A.; Pleixats, R.; Durand, J.-O. Synthesis of triethoxysilylated cyclen derivatives, grafting on magnetic mesoporous silica nanoparticles and application to metal ion adsorption. RSC Adv. 2021, 11, 10777–10784. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-A.J.; Yang, T.-Y.; Lee, C.-H.; Huang, S.H.; Sperling, R.A.; Zanella, M.; Li, J.K.; Shen, J.-L.; Wang, H.-H.; Yeh, H.-I.; et al. Synthesis, Characterization, and Bioconjugation of Fluorescent Gold Nanoclusters toward Biological Labeling Applications. ACS Nano 2009, 3, 395–401. [Google Scholar] [CrossRef] [PubMed]
- McCoy, R.S.; Choi, S.; Collins, G.; Ackerson, B.J.; Ackerson, C.J. Superatom Paramagnetism Enables Gold Nanocluster Heating in Applied Radiofrequency Fields. ACS Nano 2013, 7, 2610–2616. [Google Scholar] [CrossRef]
- Fukumura, H.; Sato, M.; Kezuka, K.; Sato, I.; Feng, X.; Okumura, S.; Fujita, T.; Yokoyama, U.; Eguchi, H.; Ishikawa, Y.; et al. Effect of ascorbic acid on reactive oxygen species production in chemotherapy and hyperthermia in prostate cancer cells. J. Physiol. Sci. 2012, 62, 251–257. [Google Scholar] [CrossRef]
- Tieu, T.; Dhawan, S.; Haridas, V.; Butler, L.M.; Thissen, H.; Cifuentes-Rius, A.; Voelcker, N.H. Maximizing RNA Loading for Gene Silencing Using Porous Silicon Nanoparticles. ACS Appl. Mater. Interfaces 2019, 11, 22993–23005. [Google Scholar] [CrossRef]
- Aragón, F.H.; Coaquira, J.A.H.; Villegas-Lelovsky, L.; da Silva, S.W.; Cesar, D.F.; Nagamine, L.C.C.M.; Cohen, R.; Menéndez-Proupin, E.; Morais, P.C. Evolution of the doping regimes in the Al-doped SnO2 nanoparticles prepared by a polymer precursor method. J. Phys. Condens. Matter 2015, 27, 095301. [Google Scholar] [CrossRef]
- Alhmoud, H.; Brodoceanu, D.; Elnathan, R.; Kraus, T.; Voelcker, N. A MACEing Silicon: Towards single-step etching of defined porous nanostructures for biomedicine. Prog. Mater. Sci. 2019, 116, 100636. [Google Scholar] [CrossRef]
- Liu, T.; Xiao, Z. Dynamic Light Scattering of Rigid Rods—A Universal Relationship on the Apparent Diffusion Coefficient as Revealed by Numerical Studies and Its Use for Rod Length Determination. Macromol. Chem. Phys. 2012, 213, 1697–1705. [Google Scholar] [CrossRef]
- Secret, E.; Smith, K.; Dubljevic, V.; Moore, E.; Macardle, P.; Delalat, B.; Rogers, M.-L.; Johns, T.G.; Durand, J.-O.; Cunin, F.; et al. Antibody-Functionalized Porous Silicon Nanoparticles for Vectorization of Hydrophobic Drugs. Adv. Healthc. Mater. 2013, 2, 718–727. [Google Scholar] [CrossRef]
- Coffinier, Y.; Olivier, C.; Perzyna, A.; Grandidier, B.; Wallart, X.; Durand, J.O.; Melnyk, O.; Stiévenard, D. Semicarbazide-functionalized Si(111) surfaces for the site-specific immobilization of peptides. Langmuir 2005, 21, 1489–1496. [Google Scholar] [CrossRef]
- Salonen, J.; Laitinen, L.; Kaukonen, A.M.; Tuura, J.; Björkqvist, M.; Heikkilä, T.; Vähä-Heikkilä, K.; Hirvonen, J.; Lehto, V.P. Mesoporous silicon microparticles for oral drug delivery: Loading and release of five model drugs. J. Control. Release 2005, 108, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Venditto, V.J.; Simanek, E.E. Cancer therapies utilizing the camptothecins: A review of the in vivo literature. Mol. Pharm. 2010, 7, 307–349. [Google Scholar] [CrossRef]
- Kwon, I.K.; Lee, S.C.; Han, B.; Park, K. Analysis on the current status of targeted drug delivery to tumors. J. Control. Release 2012, 164, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Attaluri, A.; Mallipudi, R.; Cornejo, C.; Bordelon, D.; Armour, M.; Morua, K.; Deweese, T.L.; Ivkov, R. Method to reduce non-specific tissue heating of small animals in solenoid coils. Int. J. Hyperth. 2013, 29, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Pantano, P.; Harrison, C.D.; Poulose, J.; Urrabazo, D.; Norman, T.Q.; Braun, E.I.; Draper, R.K.; Overzet, L.J. Factors affecting the 13.56-MHz radio-frequency-mediated heating of gold nanoparticles. Appl. Spectrosc. Rev. 2017, 52, 821–836. [Google Scholar] [CrossRef]
- Pearce, J.A.; Cook, J.R. Heating mechanisms in gold nanoparticles at radio frequencies. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. 2011, 2011, 5577–5580. [Google Scholar] [CrossRef] [PubMed]
- Corr, S.J.; Raoof, M.; Mackeyev, Y.; Phounsavath, S.; Cheney, M.A.; Cisneros, B.T.; Shur, M.; Gozin, M.; McNally, P.J.; Wilson, L.J.; et al. Citrate-capped gold nanoparticle electrophoretic heat production in response to a time-varying radiofrequency electric-field. J. Phys. Chem. C 2012, 116, 24380–24389. [Google Scholar] [CrossRef] [PubMed]
- Beyk, J.; Tavakoli, H. Selective radiofrequency ablation of tumor by magnetically targeting of multifunctional iron oxide–gold nanohybrid. J. Cancer Res. Clin. Oncol. 2019, 145, 2199–2209. [Google Scholar] [CrossRef]
- Haraz, O.M.; Almorqi, S.; Sebak, A.; Alshebeili, S.A. High-Gain Broadband Antennas for 60-GHz Short-Range Wireless Communications. In Wideband, Multiband, and Smart Reconfigurable Antennas for Modern Wireless Communications; IGI Global: Hershey, PA, USA, 2016. [Google Scholar]
- Takabatake, D.; Fujita, T.; Shien, T.; Kawasaki, K.; Taira, N.; Yoshitomi, S.; Takahashi, H.; Ishibe, Y.; Ogasawara, Y.; Doihara, H. Tumor inhibitory effect of gefitinib (ZD1839, Iressa) and taxane combination therapy in EGFR-overexpressing breast cancer cell lines (MCF7/ADR, MDA-MB-231). Int. J. Cancer 2007, 120, 181–188. [Google Scholar] [CrossRef]
- Nahta, R.; Esteva, F.J. HER2 therapy: Molecular mechanisms of trastuzumab resistance. Breast Cancer Res. 2006, 8, 215. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.F. Cetuximab: An epidermal growth factor receptor monoclonal antibody for the treatment of colorectal cancer. Clin. Ther. 2005, 27, 684–694. [Google Scholar] [CrossRef]
- Luo, M.; Lewik, G.; Ratcliffe, J.C.; Choi, C.H.J.; Mäkilä, E.; Tong, W.Y.; Voelcker, N.H. Systematic Evaluation of Transferrin-Modified Porous Silicon Nanoparticles for Targeted Delivery of Doxorubicin to Glioblastoma. ACS Appl. Mater. Interfaces 2019, 11, 33637–33649. [Google Scholar] [CrossRef]
- Habash, R.W.; Bansal, R.; Krewski, D.; Alhafid, H.T. Thermal therapy, part 2: Hyperthermia techniques. Crit. Rev. Biomed. Eng. 2006, 34, 491–542. [Google Scholar] [CrossRef]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Riffle, S.; Pandey, R.N.; Albert, M.; Hegde, R.S. Linking hypoxia, DNA damage and proliferation in multicellular tumor spheroids. BMC Cancer 2017, 17, 338. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, B.; Darley-Usmar, V.; Davies, K.J.; Dennery, P.A.; Forman, H.J.; Grisham, M.B.; Mann, G.E.; Moore, K.; Roberts, L.J., 2nd; Ischiropoulos, H. Measuring reactive oxygen and nitrogen species with fluorescent probes: Challenges and limitations. Free Radic. Biol. Med. 2012, 52, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Jayasooriya, R.G.P.; Dilshara, M.; Molagoda, N.; Park, C.; Park, S.; Lee, S.; Choi, Y.; Gi Young, K. Camptothecin induces G2/M phase arrest through the ATM-Chk2-Cdc25C axis as a result of autophagy-induced cytoprotection: Implications of reactive oxygen species. Oncotarget 2018, 9, 21744–21757. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample * | CPT (%w) | Au (%w) |
|---|---|---|
| pSiNP | 0 | 0 |
| pSiNP–CPT | 41.5 ± 0.5 | 0 |
| pSiNP–CPT–Ab | 39.8 ± 0.2 | 0 |
| pSiNP–AuNC | 0 | 9.1 ± 0.1 |
| pSiNP–AuNC–Ab | 0 | 8.8 ± 1 |
| pSiNP–CPT–AuNC | 33.9 ± 0.3 | 8.4 ± 0.2 |
| pSiNP–CPT–AuNC–Ab | 31.1 ± 0.1 | 8.5 ± 0.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaur, I.; Tieu, T.; Deepagan, V.G.; Ali, M.A.; Alsunaydih, F.; Rudd, D.; Moghaddam, M.A.; Bourgeois, L.; Adams, T.E.; Thurecht, K.J.; et al. Combination of Chemotherapy and Mild Hyperthermia Using Targeted Nanoparticles: A Potential Treatment Modality for Breast Cancer. Pharmaceutics 2023, 15, 1389. https://doi.org/10.3390/pharmaceutics15051389
Kaur I, Tieu T, Deepagan VG, Ali MA, Alsunaydih F, Rudd D, Moghaddam MA, Bourgeois L, Adams TE, Thurecht KJ, et al. Combination of Chemotherapy and Mild Hyperthermia Using Targeted Nanoparticles: A Potential Treatment Modality for Breast Cancer. Pharmaceutics. 2023; 15(5):1389. https://doi.org/10.3390/pharmaceutics15051389
Chicago/Turabian StyleKaur, Ishdeep, Terence Tieu, Veerasikku G. Deepagan, Muhammad A. Ali, Fahad Alsunaydih, David Rudd, Maliheh A. Moghaddam, Laure Bourgeois, Timothy E. Adams, Kristofer J. Thurecht, and et al. 2023. "Combination of Chemotherapy and Mild Hyperthermia Using Targeted Nanoparticles: A Potential Treatment Modality for Breast Cancer" Pharmaceutics 15, no. 5: 1389. https://doi.org/10.3390/pharmaceutics15051389