
Surface Functionalised Parenteral Nanoemulsions for Active and Homotypic Targeting to Melanoma
 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Experimental
2.1. Chemicals
2.2. Cells
2.3. Methods
2.3.1. Linkers Synthesis and Characterisation
2.3.2. IL Functionalisation with TRF
2.3.3. IL Wrapping with CMF
2.3.4. Characterisation of Nanoemulsions
Particle Size, Shape, Zeta Potential
Protein Analysis
2.3.5. HPLC
2.3.6. Cellular Uptake of Fluorescently Labelled Nanoemulsions
2.4. Statistical Analysis
3. Results
3.1. Physico-Chemical Characterisation of Nanoemulsions
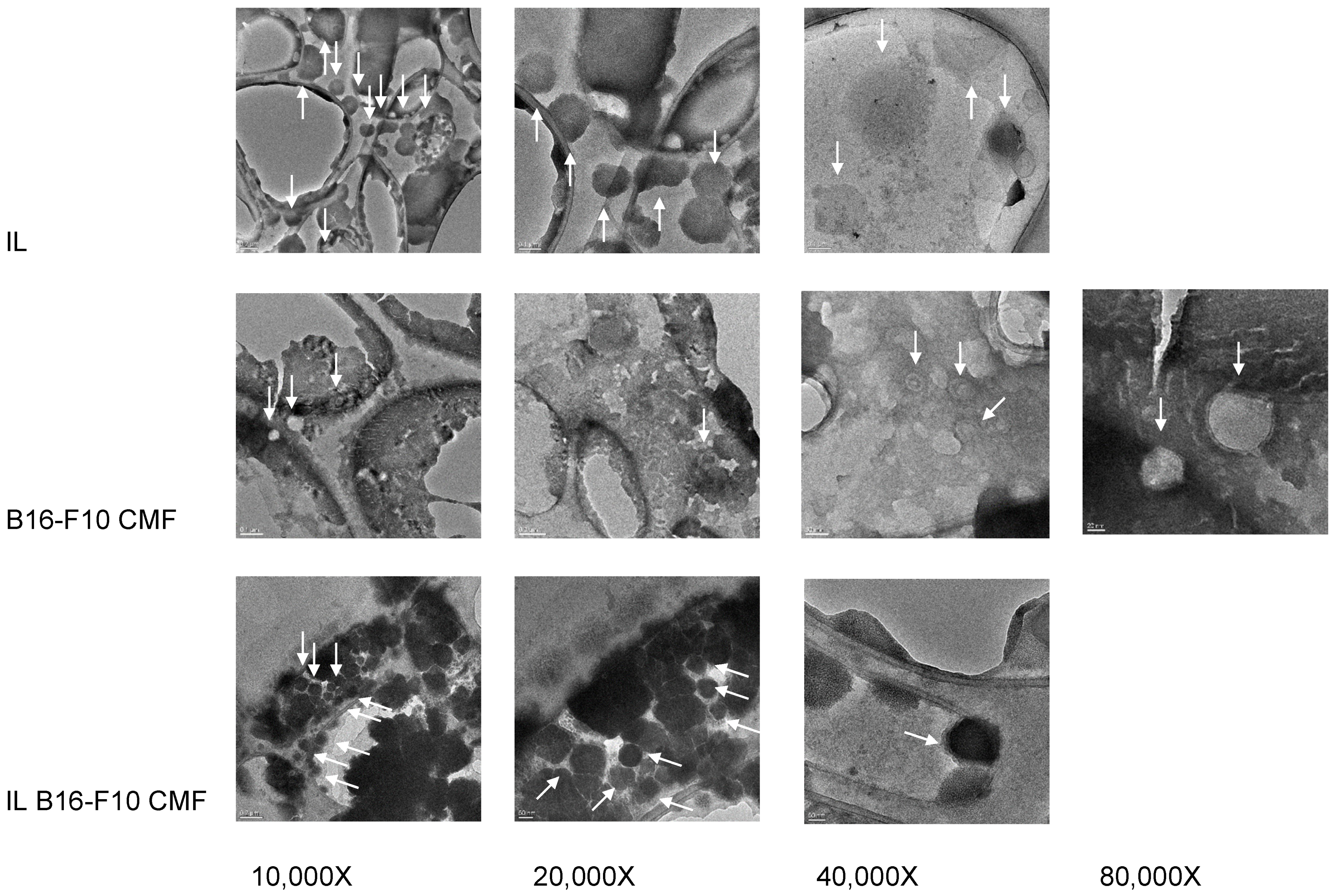
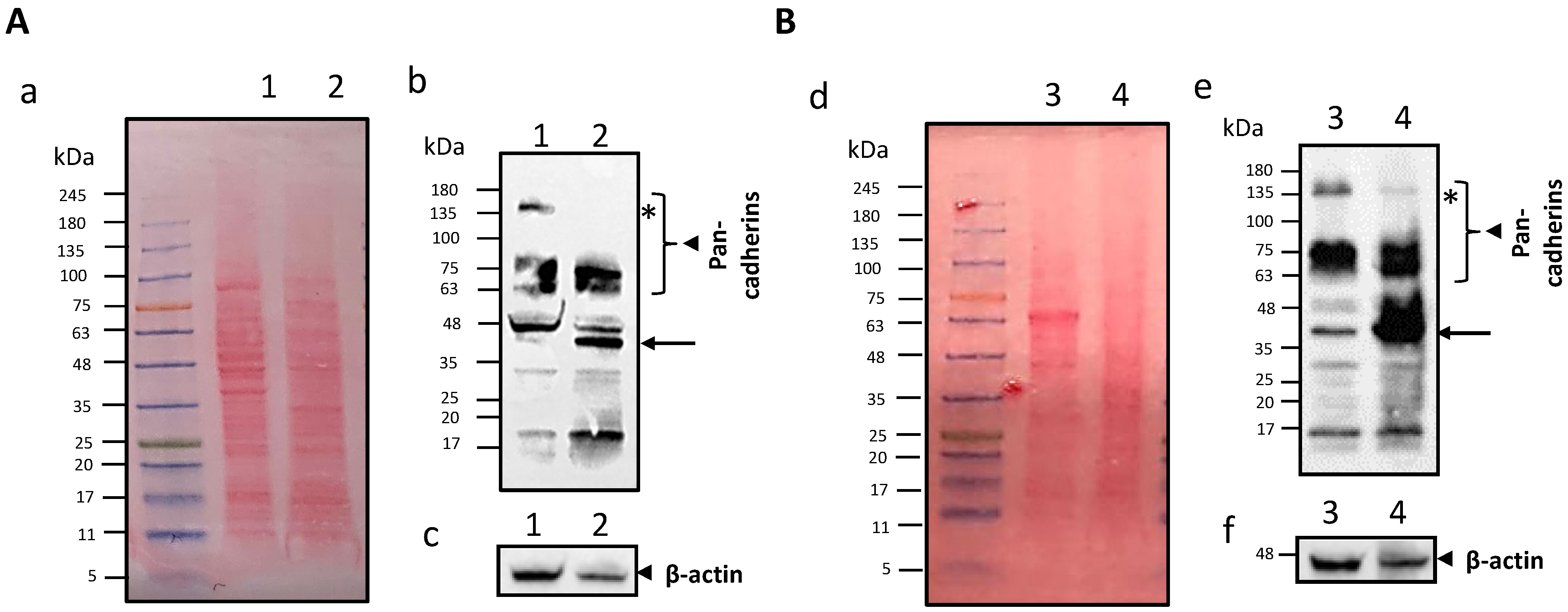
Surface Coating Characterisation of CMF-Wrapped IL
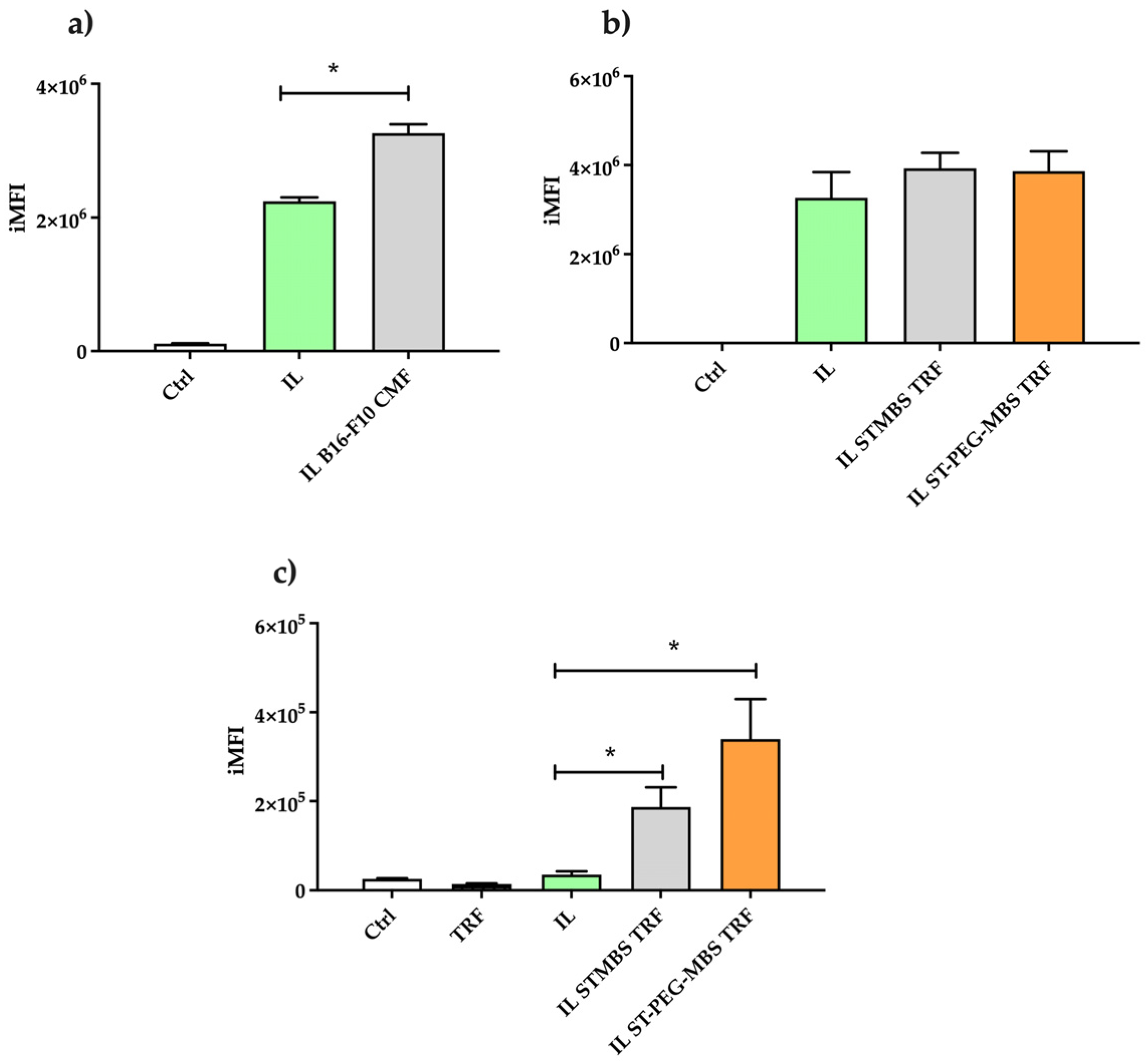
3.2. Cytofluorimetric Uptake Studies of Lipid Nanoemulsion in B16-F10 Cells
4. Discussion
4.1. Physico-Chemical Standpoint
4.2. Biological Standpoint
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Luke, J.J.; Schwartz, G.K. Chemotherapy in the management of advanced cutaneous malignant melanoma. Clin. Dermatol. 2013, 31, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.; George, J.; Aucoin, D.; Bower, J.; Burrell, S.; Gilbert, R.; Bower, N. The Pathogenesis and Clinical Management of Cutaneous Melanoma: An Evidence-Based Review. J. Med. Imaging Radiat. Sci. 2019, 50, 460–469.e1. [Google Scholar] [CrossRef]
- Battaglia, L.; Scomparin, A.; Dianzani, C.; Milla, P.; Muntoni, E.; Arpicco, S.; Cavalli, R. Nanotechnology Addressing Cutaneous Melanoma: The Italian Landscape. Pharmaceutics. 2021, 13, 1617. [Google Scholar] [CrossRef] [PubMed]
- Gmeiner, W.H.; Ghosh, S. Nanotechnology for cancer treatment. Nanotechnol. Rev. 2014, 3, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Mishra, H.; Mishra, P.K.; Ekielski, A.; Jaggi, M.; Iqbal, Z.; Talegaonkar, S. Melanoma treatment: From conventional to nanotechnology. J. Cancer Res. Clin. Oncol. 2018, 144, 2283–2302. [Google Scholar] [CrossRef] [PubMed]
- Boggio, E.; Gigliotti, C.L.; Stoppa, I.; Pantham, D.; Sacchetti, S.; Rolla, R.; Grattarola, M.; Monge, C.; Pizzimenti, S.; Dianzani, U.; et al. Exploiting Nanomedicine for Cancer Polychemotherapy: Recent Advances and Clinical Applications. Pharmaceutics 2023, 15, 937. [Google Scholar] [CrossRef]
- Li, M.; Yin, S.; Lin, M.; Chen, X.; Pan, Y.; Peng, Y.; Sun, J.; Kumar, A.; Liu, J. Current status and prospects of metal-organic frameworks for bone therapy and bone repair. J. Mater. Chem. B 2022, 10, 5105–5128. [Google Scholar] [CrossRef]
- Liu, W.; Yan, Q.; Xia, C.; Wang, X.; Kumar, A.; Wang, Y.; Liu, Y.; Pan, Y.; Liu, J. Recent advances in cell membrane coated metal-organic frameworks (MOFs) for tumor therapy. J. Mater. Chem. B 2021, 9, 4459–4474. [Google Scholar] [CrossRef]
- Qiu, Y.; Tan, G.; Fang, Y.; Liu, S.; Zhou, Y.; Kumar, A.; Trivedi, M.; Liu, D.; Liu, J. Biomedical applications of metal-organic framework (MOF)-based nano-enzymes. New J. Chem. 2021, 45, 20987–21000. [Google Scholar] [CrossRef]
- Battaglia, L.S.; Dorati, R.; Maestrelli, F.; Conti, B.; Gabriele, M.; Di Cesare Mannelli, L.; Selmin, F.; Cosco, D. Repurposing of parenterally administered active substances used to treat pain both systemically and locally. Drug Discov. Today 2022, 27, 103321. [Google Scholar] [CrossRef]
- Dianzani, C.; Monge, C.; Miglio, G.; Serpe, L.; Martina, K.; Cangemi, L.; Ferraris, C.; Mioletti, S.; Osella, S.; Gigliotti, C.L.; et al. Nanoemulsions as Delivery Systems for Poly-Chemotherapy Aiming at Melanoma Treatment. Cancers 2020, 12, 1198. [Google Scholar] [CrossRef]
- Monge, C.; Stoppa, I.; Ferraris, C.; Bozza, A.; Battaglia, L.; Cangemi, L.; Miglio, G.; Pizzimenti, S.; Clemente, N.; Gigliotti, C.L.; et al. Parenteral Nanoemulsions Loaded with Combined Immuno- and Chemo-Therapy for Melanoma Treatment. Nanomaterials 2022, 12, 4233. [Google Scholar] [CrossRef] [PubMed]
- Sarfati, G.; Dvir, T.; Elkabets, M.; Apte, R.N.; Cohen, S. Targeting of polymeric nanoparticles to lung metastases by surface-attachment of YIGSR peptide from laminin. Biomaterials 2011, 32, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Zhai, T.; Zhong, W.; Gao, Y.; Zhou, H.; Zhou, Z.; Liu, X.; Yang, S.; Yang, H. Tumor Microenvironment-Activated Nanoparticles Loadd with an Iron-Carbonyl Complex for Chemodynamic Immunotherapy of Lung Metastasis of Melanoma In Vivo. ACS Appl. Mater. Interfaces 2021, 13, 39100–39111. [Google Scholar] [CrossRef]
- Harris, J.C.; Scully, M.A.; Day, E.S. Cancer cell membrane-coated nanoparticles for cancer management. Cancers 2019, 11, 1836. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Qiu, Y.; Zhang, L.; Gao, H.; He, Q. Targeted delivery of transferrin and TAT co-modified liposomes encapsulating both paclitaxel and doxorubicin for melanoma. Drug Deliv. 2016, 23, 1171–1183. [Google Scholar] [CrossRef]
- Sakpakdeejaroen, I.; Somani, S.; Laskar, P.; Mullin, M.; Dufès, C. Regression of melanoma following intravenous injection of plumbagin entrapped in transferrin-conjugated, lipid–polymer hybrid nanoparticles. Int. J. Nanomed. 2021, 16, 2615–2631. [Google Scholar] [CrossRef]
- Sakpakdeejaroen, I.; Muanrit, P.; Panthong, S.; Ruangnoo, S. Alpha-Mangostin-Loaded Transferrin-Conjugated Lipid-Polymer Hybrid Nanoparticles: Development and Characterization for Tumor-Targeted Delivery. Sci. World J. 2022, 2022. [Google Scholar] [CrossRef]
- Tsavaler, L.; Stein, B.S.; Sussman, H.H. Demonstration of the specific binding of bovine transferrin to the human transferrin receptor in k562 cells: Evidence for interspecies transferrin internalization. J. Cell. Physiol. 1986, 128, 1–8. [Google Scholar] [CrossRef]
- Goldstein, D.; Nassar, T.; Lambert, G.; Kadouche, J.; Benita, S. The design and evaluation of a novel targeted drug delivery system using cationic emulsion-antibody conjugates. J. Control. Release 2005, 108, 418–432. [Google Scholar] [CrossRef]
- Ben-Akiva, E.; Meyer, R.A.; Yu, H.; Smith, J.T.; Pardoll, D.M.; Green, J.J. Biomimetic anisotropic polymeric nanoparticles coated with red blood cell membranes for enhanced circulation and toxin removal. Sci. Adv. 2020, 6, eaay9035. [Google Scholar] [CrossRef]
- Cao, H.; Dan, Z.; He, X.; Zhang, Z.; Yu, H.; Yin, Q.; Li, Y. Liposomes Coated with Isolated Macrophage Membrane Can Target Lung Metastasis of Breast Cancer. ACS Nano 2016, 10, 7738–7748. [Google Scholar] [CrossRef]
- Copp, J.A.; Fang, R.H.; Luk, B.T.; Hu, C.M.J.; Gao, W.; Zhang, K.; Zhang, L. Clearance of pathological antibodies using biomimetic nanoparticles. Proc. Natl. Acad. Sci. USA 2014, 111, 13481–13486. [Google Scholar] [CrossRef]
- Fan, Z.; Zhou, H.; Li, P.Y.; Speer, J.E.; Cheng, H. Structural elucidation of cell membrane-derived nanoparticles using molecular probes. J. Mater. Chem. B 2014, 2, 8231–8238. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.M.J.; Zhang, L.; Aryal, S.; Cheung, C.; Fang, R.H.; Zhang, L. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc. Natl. Acad. Sci. USA 2011, 108, 10980–10985. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Jin, K.; Luo, M.; Wang, X.; Zhu, X.; Liu, X.; Jiang, T.; Zhang, Q.; Wang, S.; Pang, Z. Size dependency of circulation and biodistribution of biomimetic nanoparticles: Red blood cell membrane-coated nanoparticles. Cells 2019, 8, 881. [Google Scholar] [CrossRef] [PubMed]
- Yaman, S.; Chintapula, U.; Rodriguez, E.; Ramachandramoorthy, H.; Nguyen, K.T. Cell-mediated and cell membrane-coated nanoparticles for drug delivery and cancer therapy. Cancer Drug Resist. 2020, 3, 879–911. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, M.H.; Steinberg, S.M.; Alexander, M.P.; Fisher, J.L.; Ernstoff, M.S.; Turk, M.J.; Mullins, D.W.; Brinckerhoff, C.E. Multiple murine BRaf(V600E) melanoma cell lines with sensitivity to PLX4032. Pigment Cell Melanoma Res. 2014, 27, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, E.; Martina, K.; Marini, E.; Giorgis, M.; Lazzarato, L.; Salaroglio, I.C.; Riganti, C.; Lanotte, M.; Battaglia, L. Methotrexate-Loaded Solid Lipid Nanoparticles: Protein Functionalization to Improve Brain Biodistribution. Pharmaceutics 2019, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Visser, C.C.; Voorwinden, L.H.; Harders, L.R.; Eloualid, M.; van Bloois, L.; Crommelin, D.J.; Danhof, M.; de Boer, A.G. Coupling of metal containing homing devices to liposomes via a maleimide linker: Use of TCEP to stabilize thiol-groups without scavenging metals. J. Drug Target. 2004, 12, 569–573. [Google Scholar] [CrossRef]
- Fang, R.H.; Hu, C.M.; Luk, B.T.; Gao, W.; Copp, J.A.; Tai, Y.; O’Connor, D.E.; Zhang, L. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014, 14, 2181–2188. [Google Scholar] [CrossRef]
- Finke, J.H.; Richter, C.; Gothsch, T.; Kwade, A.; Büttgenbach, S.; Müller-Goymann, C.C. Coumarin 6 as a fluorescent model drug: How to identify properties of lipid colloidal drug delivery systems via fluorescence spectroscopy? Eur. J. Lipid Sci. Technol. 2014, 116, 1234–1246. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, B.; Kebebe, D.; Guo, L.; Guo, H.; Li, N.; Pi, J.; Qi, D.; Guo, P.; Liu, Z. Preparation, optimization and cellular uptake study of tanshinone I nanoemulsion modified with lactoferrin for brain drug delivery. Pharm. Dev. Technol. 2019, 24, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhang, L.; Guo, Y.; Bai, L.; Luo, Y.; Wang, B.; Kuang, M.; Liu, X.; Sun, M.; Wang, C.; et al. Nanoemulsion Co-Loaded with XIAP siRNA and Gambogic Acid for Inhalation Therapy of Lung Cancer. Int. J. Mol. Sci. 2022, 23, 14294. [Google Scholar] [CrossRef] [PubMed]
- Argenziano, M.; Foglietta, F.; Canaparo, R.; Spagnolo, R.; Della Pepa, C.; Caldera, F.; Trotta, F.; Serpe, L.; Cavalli, R. Biological Effect Evaluation of Glutathione-Responsive Cyclodextrin-Based Nanosponges: 2D and 3D Studies. Molecules 2020, 25, 2775. [Google Scholar] [CrossRef] [PubMed]
- Kushiro, K.; Núñez, N.P. Ethanol inhibits B16-BL6 melanoma metastasis and cell phenotypes associated with metastasis. In Vivo 2012, 26, 47–58. [Google Scholar]
- Ruan, J.S.; Liu, Y.P.; Zhang, L.; Yan, L.G.; Fan, F.T.; Shen, C.S.; Wang, A.Y.; Zheng, S.Z.; Wang, S.M.; Lu, Y. Luteolin reduces the invasive potential of malignant melanoma cells by targeting β3 integrin and the epithelial-mesenchymal transition. Acta Pharmacol. Sin. 2012, 33, 1325–1331. [Google Scholar] [CrossRef] [PubMed]
- Lobos-González, L.; Aguilar, L.; Diaz, J.; Diaz, N.; Urra, H.; Torres, V.A.; Silva, V.; Fitzpatrick, C.; Lladser, A.; Hoek, K.S.; et al. E-cadherin determines Caveolin-1 tumor suppression or metastasis enhancing function in melanoma cells. Pigment Cell Melanoma Res. 2013, 26, 555–570. [Google Scholar] [CrossRef]
- Shields, B.D.; Koss, B.; Taylor, E.M.; Storey, A.J.; West, K.L.; Byrum, S.D.; Mackintosh, S.G.; Edmondson, R.; Mahmoud, F.; Shalin, S.C.; et al. Loss of E-Cadherin Inhibits CD103 Antitumor Activity and Reduces Checkpoint Blockade Responsiveness in Melanoma. Cancer Res. 2019, 79, 1113–1123. [Google Scholar] [CrossRef]
- Díaz-Valdivia, N.; Simón, S.; Díaz, J.; Martinez-Meza, S.; Contreras, P.; Burgos-Ravanal, R.; Pérez, V.I.; Frei, B.; Leyton, L.; Quest, A.F. Mitochondrial Dysfunction and the Glycolytic Switch Induced by Caveolin-1 Phosphorylation Promote Cancer Cell Migration, Invasion, and Metastasis. Cancers 2022, 14, 2862. [Google Scholar] [CrossRef]
- Geiger, B.; Volberg, T.; Ginsberg, D.; Bitzur, S.; Sabanay, I.; Hynes, R.O. Broad spectrum pan-cadherin antibodies, reactive with the C-terminal 24 amino acid residues of N-cadherin. J. Cell. Sci. 1990, 97, 607–614. [Google Scholar] [CrossRef]
- Liwosz, A.; Lei, T.; Kukuruzinska, M.A. N-glycosylation affects the molecular organization and stability of E-cadherin junctions. J. Biol. Chem. 2006, 281, 23138–23149. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.L.; Lapyckyj, L.; Rosso, M.; Besso, M.J.; Mencucci, M.V.; Briggiler, C.I.; Giustina, S.; Furlong, L.I.; Vazquez-Levin, M.H. Identification of a Novel Human E-Cadherin Splice Variant and Assessment of Its Effects Upon EMT-Related Events. J. Cell. Physiol. 2017, 232, 1368–1386. [Google Scholar] [CrossRef] [PubMed]
- David, J.M.; Rajasekaran, A.K. Dishonorable discharge: The oncogenic roles of cleaved E-cadherin fragments. Cancer Res. 2012, 72, 2917–2923. [Google Scholar] [CrossRef] [PubMed]
- Bauer, R.; Hein, R.; Bosserhoff, A.K. A secreted form of P-cadherin is expressed in malignant melanoma. Exp. Cell. Res. 2005, 305, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Shore, E.M.; Nelson, W.J. Biosynthesis of the cell adhesion molecule uvomorulin (E-cadherin) in Madin-Darby canine kidney epithelial cells. J. Biol. Chem. 1991, 266, 19672–19680. [Google Scholar] [CrossRef]
- Xiong, Y.; Liu, L.; Zhu, S.; Zhang, B.; Qin, Y.; Yao, R.; Zhou, H.; Gao, D.S. Precursor N-cadherin mediates glial cell line-derived neurotrophic factor-promoted human malignant glioma. Oncotarget 2017, 8, 24902–24914. [Google Scholar] [CrossRef] [PubMed]
- Masterson, J.; O’Dea, S. Posttranslational truncation of E-cadherin and significance for tumour progression. Cells Tissues Organs 2007, 185, 175–179. [Google Scholar] [CrossRef]
- Muntoni, E.; Marini, E.; Ferraris, C.; Garelli, S.; Capucchio, M.T.; Colombino, E.; Panciani, P.P.; Battaglia, L. Intranasal lipid nanocarriers: Uptake studies with fluorescently labeled formulations. Colloids Surf. B Biointerfaces 2022, 214, 112470. [Google Scholar] [CrossRef]
- Huebers, E.; Nelson, N.J.; Huebers, H.A.; Rasey, J.S. Removal of transferrin from fetal bovine serum. J. Lab. Clin. Med. 1987, 110, 719–725. [Google Scholar]
- Hippalgaonkar, K.; Majumdar, S.; Kansara, V. Injectable lipid emulsions-advancements, opportunities and challenges. AAPS PharmSciTech 2010, 11, 1526–1540. [Google Scholar] [CrossRef] [PubMed]
- Renault, K.; Fredy, J.W.; Renard, P.Y.; Sabot, C. Covalent Modification of Biomolecules through Maleimide-Based Labeling Strategies. Bioconjug. Chem. 2018, 29, 2497–2513. [Google Scholar] [CrossRef] [PubMed]
- Pichler, V.; Mayr, J.; Heffeter, P.; Dömötör, O.; Enyedy, É.A.; Hermann, G.; Groza, D.; Köllensperger, G.; Galanksi, M.; Berger, W.; et al. Maleimide-functionalised platinum(IV) complexes as a synthetic platform for targeted drug delivery. Chem. Commun. 2013, 49, 2249–2251. [Google Scholar] [CrossRef]
- Rana, S.; Yeh, Y.; Rotello, V.M. Engineering the nanoparticle–protein interface: Applications and possibilities. Curr. Opin. Chem. Biol. 2010, 14, 828–834. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.X.; Karnik, R.; Wang, A.Z.; Alexis, F.; Levy-Nissenbaum, E.; Hong, S.; Langer, R.S.; Farokhzad, O.C. Targeted nanoparticles for cancer therapy. Nano Today 2007, 2, 14–21. [Google Scholar] [CrossRef]
- Sutherland, R.; Delia, D.; Schneider, C.; Newman, R.; Kemshead, J.; Greaves, M. Ubiquitous cell-surface glycoprotein on tumor cells is proliferation-associated receptor for transferrin. Proc. Natl. Acad. Sci. USA 1981, 78, 4515–4519. [Google Scholar] [CrossRef]
- Daniels, T.R.; Delgado, T.; Helguera, G.; Penichet, M.L. The transferrin receptor part II: Targeted delivery of therapeutic agents into cancer cells. Clin. Immunol. 2006, 121, 159–176. [Google Scholar] [CrossRef]
- Yhee, J.Y.; Lee, S.J.; Lee, S.; Song, S.; Min, H.S.; Kang, S.W.; Son, S.; Jeong, S.Y.; Kwon, I.C.; Kim, S.H.; et al. Tumor-targeting transferrin nanoparticles for systemic polymerized siRNA delivery in tumor-bearing mice. Bioconjug. Chem. 2013, 24, 1850–1860. [Google Scholar] [CrossRef]
- De Pasquale, D.; Marino, A.; Tapeinos, C.; Pucci, C.; Rocchiccioli, S.; Michelucci, E.; Finamore, F.; McDonnell, L.; Scarpellini, A.; Lauciello, S.; et al. Homotypic targeting and drug delivery in glioblastoma cells through cell membrane-coated boron nitride nanotubes. Mater. Des. 2020, 192, 108742. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.H.; Gao, W.; Zhang, L. Targeting drugs to tumours using cell membrane-coated nanoparticles. Nat. Rev. Clin. Oncol. 2023, 20, 33–48. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Z.; Liu, C.; Hao, M.; Kong, C.; Zhao, X.; Gao, Y.; Zhang, Y.; Cui, W.; Zhang, C.; et al. B16 Membrane-Coated Vesicles for Combined Photodynamic Therapy and Immunotherapy Shift Immune Microenvironment of Melanoma. Int. J. Nanomed. 2022, 17, 855–868. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, C.; Kiel, C. Cell Adhesion Molecules in Normal Skin and Melanoma. Biomolecules 2021, 11, 1213. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, D.J.; Lawson, D.H.; Richards, J.M.; Conry, R.M.; Miller, D.M.; Treisman, J.; Gailani, F.; Riley, L.; Conlon, K.; Pockaj, B.; et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N. Engl. J. Med. 2011, 364, 2119–2127. [Google Scholar] [CrossRef]
- Bidkar, A.P.; Sanpui, P.; Ghosh, S.S. Transferrin-Conjugated Red Blood Cell Membrane-Coated Poly(lactic-co-glycolic acid) Nanoparticles for the Delivery of Doxorubicin and Methylene Blue. ACS Appl. Nano Mater. 2020, 3, 3807–3819. [Google Scholar] [CrossRef]
- Lehrich, B.M.; Liang, Y.; Fiandaca, M.S. Foetal bovine serum influence on in vitro extracellular vesicle analyses. J. Extracell. Vesicles 2021, 10, e12061. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.; Coombs, K.M. Serum-reduced media impacts on cell viability and protein expression in human lung epithelial cells. J. Cell. Physiol. 2019, 234, 7718–7724. [Google Scholar] [CrossRef] [PubMed]
- Zou, D.; Arora, M.; Ganugula, R.; Kumar, M.; Scott, E.M.; Shah, D.; Kumar, M.N.V.R. Nanoparticles that do not compete with endogenous ligands-Molecular characterization in vitro, acute safety in canine, and interspecies pharmacokinetics modeling to humans. J. Control. Release 2021, 332, 64–73. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Linker | Purification | Mean Size (nm) | PDI | Zeta Potential (mV) | Proteins in Functionalized IL | ||||
|---|---|---|---|---|---|---|---|---|---|
| μg Used in 1 mL | Recovered | ||||||||
| TRF | CMF | Lipid (μg/g) | Supernatant (μg/mL) | ||||||
| Blank IL | - | - | 290.0 ± 1.9 | 0.005 | −39.53 ± 2.07 | - | - | - | - |
| IL TRF | ST-MBS | UR | 263.5 ± 1.3 | 0.185 | −40.27 ± 3.77 | 200 | - | 40 ± 3.5 | 6.3 ± 0.4 |
| ST-PEG-MBS | UR | 262.8 ± 3.6 | 0.192 | −35.81 ± 1.81 | 200 | - | 16 ± 1.4 | 14.7 ± 1.2 | |
| ST-MBS | UR | 247.4 ± 3.0 | 0.022 | −24.11 ± 4.31 * | 1000 | - | 74 ± 8.7 | 70.7 ± 2.3 | |
| ST-PEG-MBS | UR | 252.3 ± 2.7 | 0.137 | −35.35 ± 1.80 | 1000 | - | 60 ± 7.1 | 15.5 ± 1.6 | |
| ST-MBS | SE | 251.1 ± 2.0 | 0.131 | −30.06 ± 4.55 | 1000 | - | 125 ± 11.8 | 26.7 ± 3.0 | |
| ST-PEG-MBS | SE | 256.1 ± 1.7 | 0.159 | −40.33 ± 3.85 | 1000 | - | 265 ± 24.5 | 48.5 ± 4.5 | |
| B16-F10 CMF | - | - | 642.5 ± 30.7 | 0.521 | −18.48 ± 3.17 ** | - | - | - | - |
| IL B16-F10 CMF | - | UR | 235.7 ± 2.8 | 0.072 | −32.81 ± 3.92 | - | 5.8 | 20.7 ± 1.7 | 2.5 ± 0.2 |
| D4M CMF | - | - | 748.3 ± 31.1 | 0.391 | −49.03 ± 4.40 * | - | - | - | - |
| IL D4M CMF | - | UR | 220.1 ± 3.2 | 0.180 | −42.83 ± 2.70 | - | 7.4 | 36.2 ± 16.9 | 2.8 ± 0.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Foglietta, F.; Bozza, A.; Ferraris, C.; Cangemi, L.; Bordano, V.; Serpe, L.; Martina, K.; Lazzarato, L.; Pizzimenti, S.; Grattarola, M.; et al. Surface Functionalised Parenteral Nanoemulsions for Active and Homotypic Targeting to Melanoma. Pharmaceutics 2023, 15, 1358. https://doi.org/10.3390/pharmaceutics15051358
Foglietta F, Bozza A, Ferraris C, Cangemi L, Bordano V, Serpe L, Martina K, Lazzarato L, Pizzimenti S, Grattarola M, et al. Surface Functionalised Parenteral Nanoemulsions for Active and Homotypic Targeting to Melanoma. Pharmaceutics. 2023; 15(5):1358. https://doi.org/10.3390/pharmaceutics15051358
Chicago/Turabian StyleFoglietta, Federica, Annalisa Bozza, Chiara Ferraris, Luigi Cangemi, Valentina Bordano, Loredana Serpe, Katia Martina, Loretta Lazzarato, Stefania Pizzimenti, Margherita Grattarola, and et al. 2023. "Surface Functionalised Parenteral Nanoemulsions for Active and Homotypic Targeting to Melanoma" Pharmaceutics 15, no. 5: 1358. https://doi.org/10.3390/pharmaceutics15051358