
Drug Delivery Strategies for the Treatment of Pancreatic Cancer

Abstract
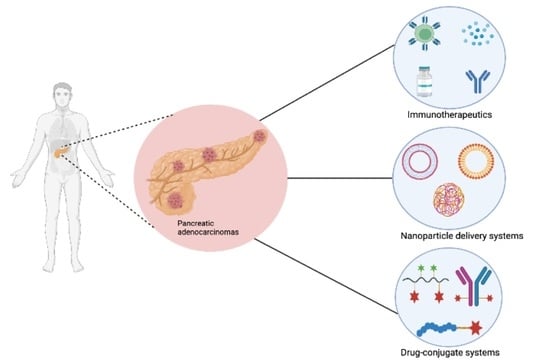
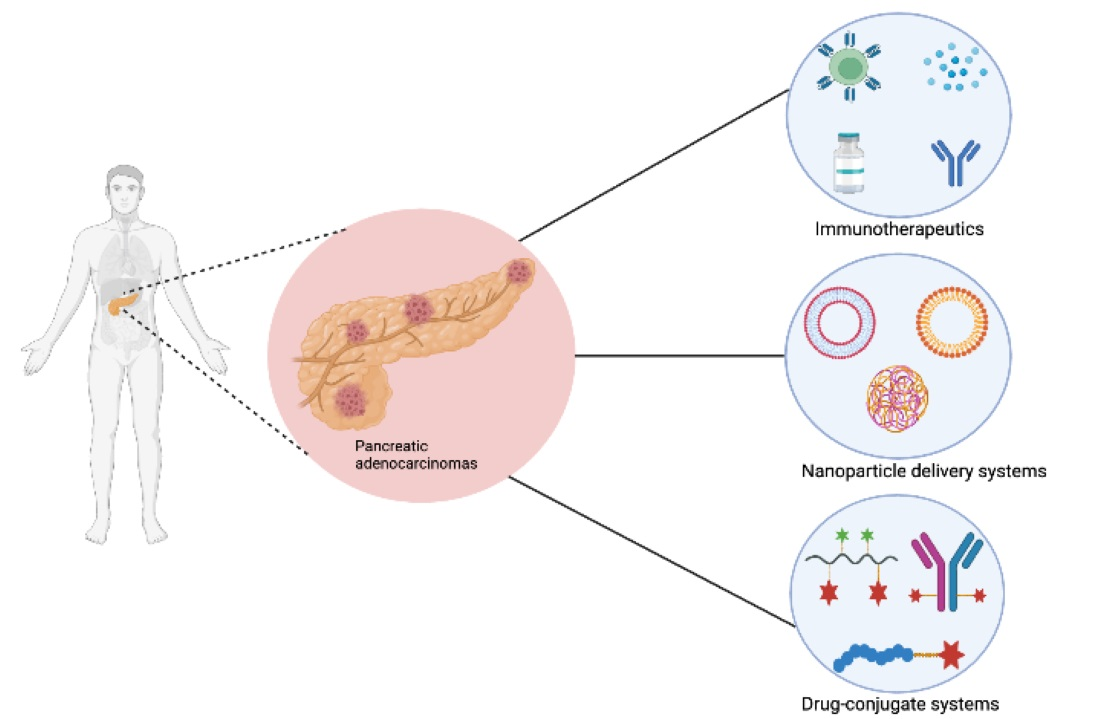
:
1. Introduction
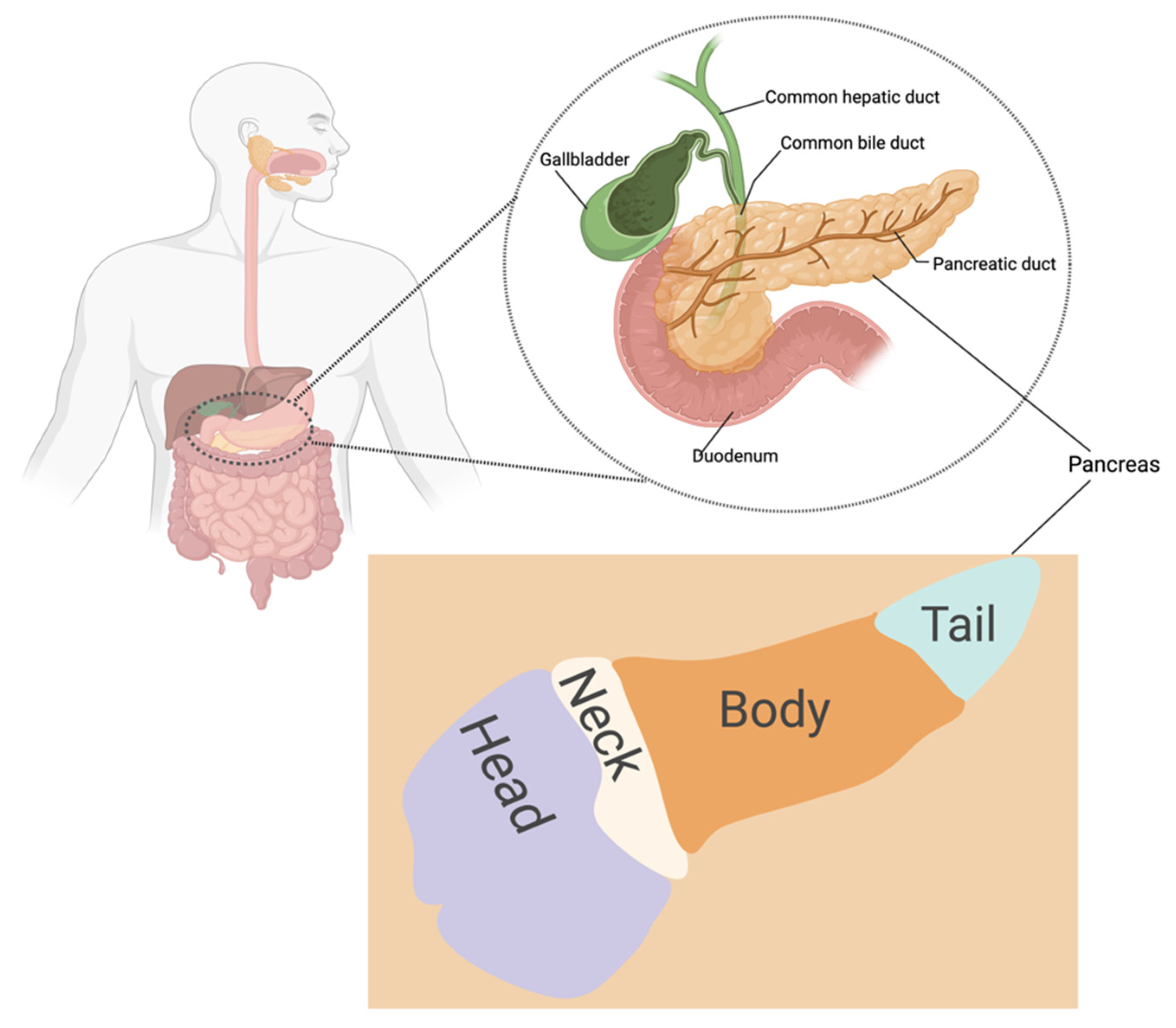
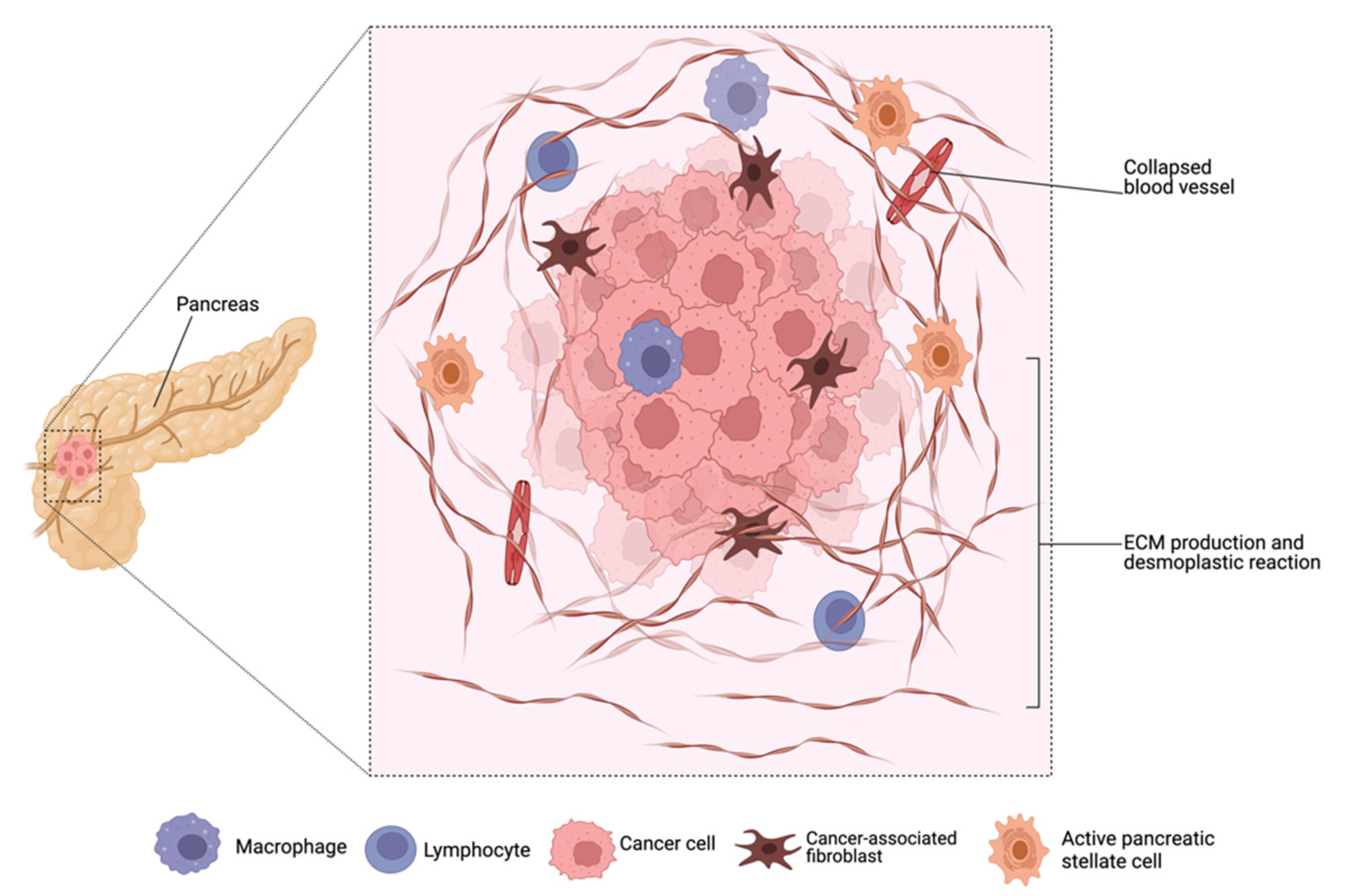
2. Peculiarities of Pancreatic Cancer
2.1. The Enhanced Permeability and Retention Effect in Pancreatic Cancer
2.2. Proteolytic Enzymes in Pancreatic Cancer
3. Recent Advances in Pancreatic Cancer Targeted Therapy and Limitations
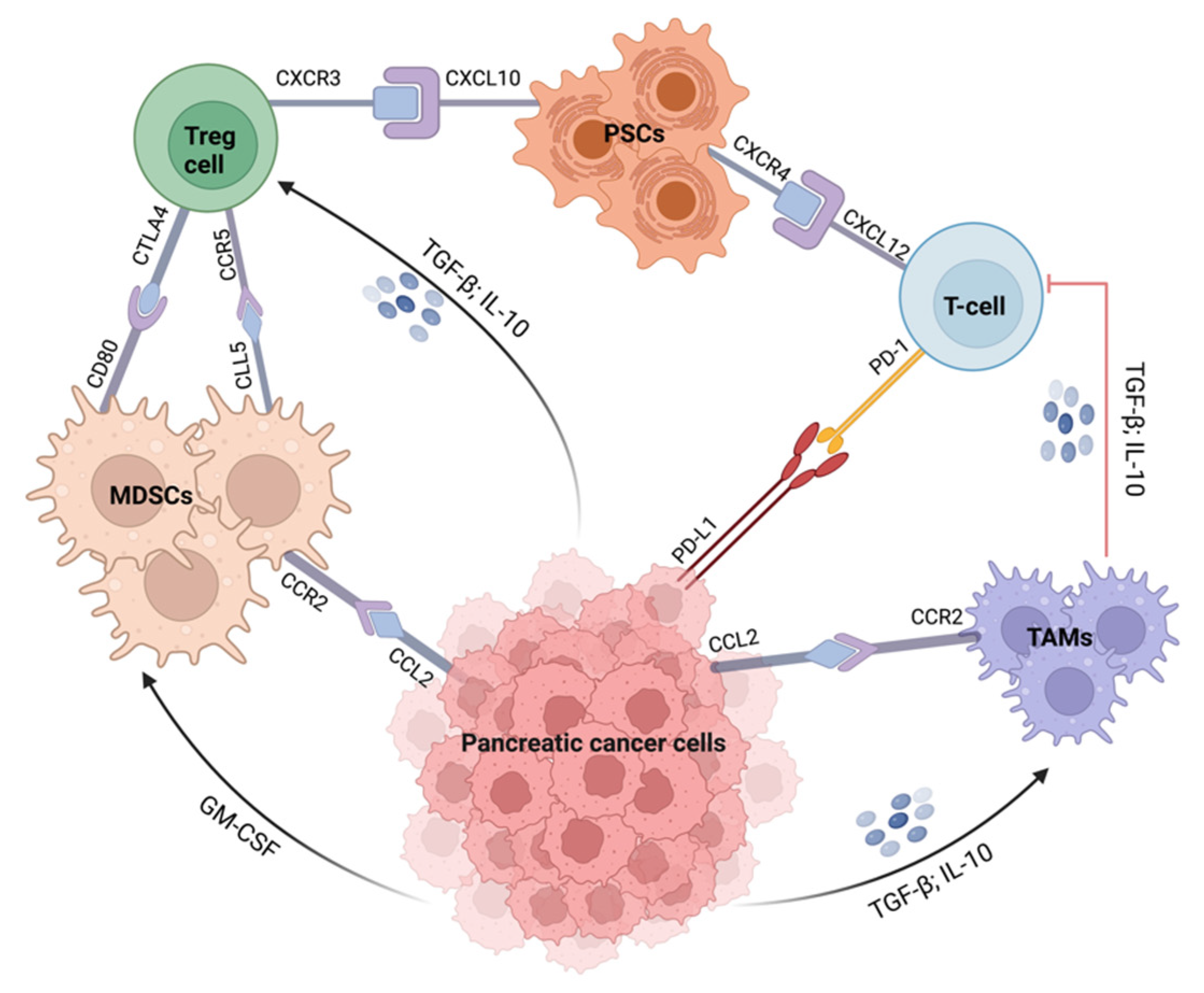
3.1. Immunotherapy Approaches for the Treatment of Pancreatic Cancer
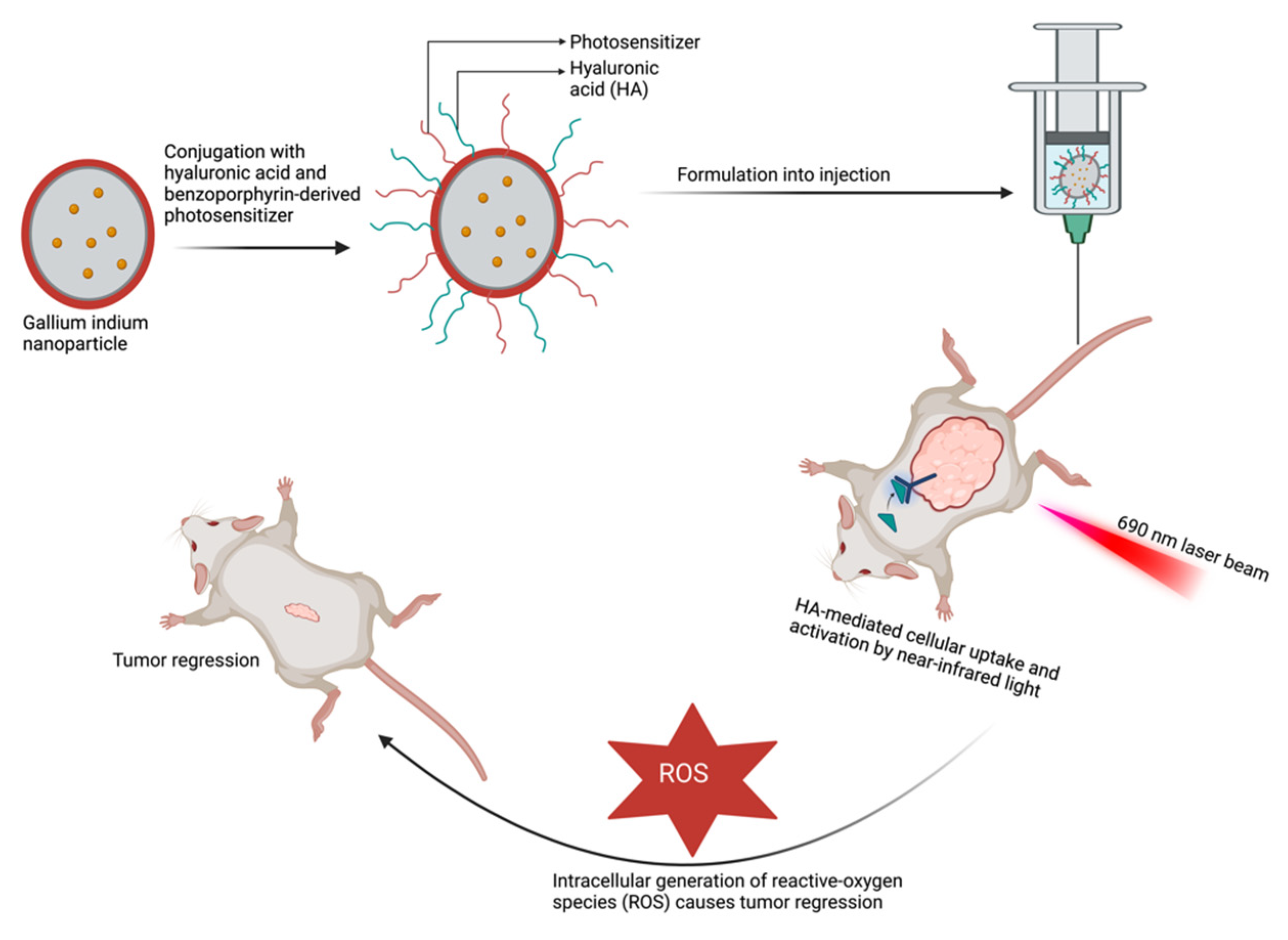
3.2. Photodynamic Therapy in Pancreatic Cancer
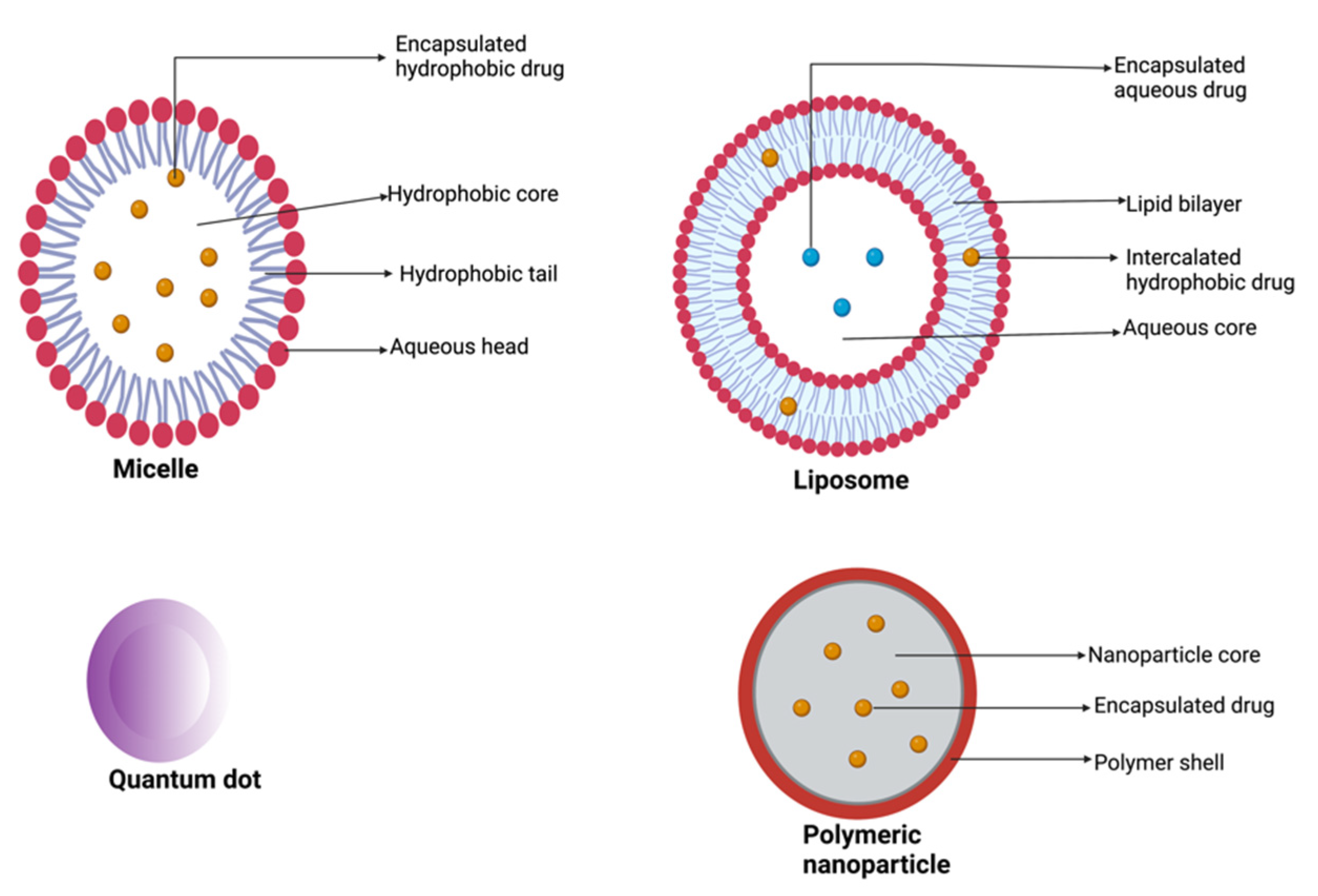
3.3. Nanootechnology in the Treatment of Pancreatic Cancer
3.3.1. Albumin-Based Nanoparticles for the Treatment of Pancreatic Cancer
3.3.2. Liposomal Nanoformulations for Pancreatic Cancer Treatment
3.3.3. Polymeric Nanoparticles for the Treatment of Pancreatic Adenocarcinoma
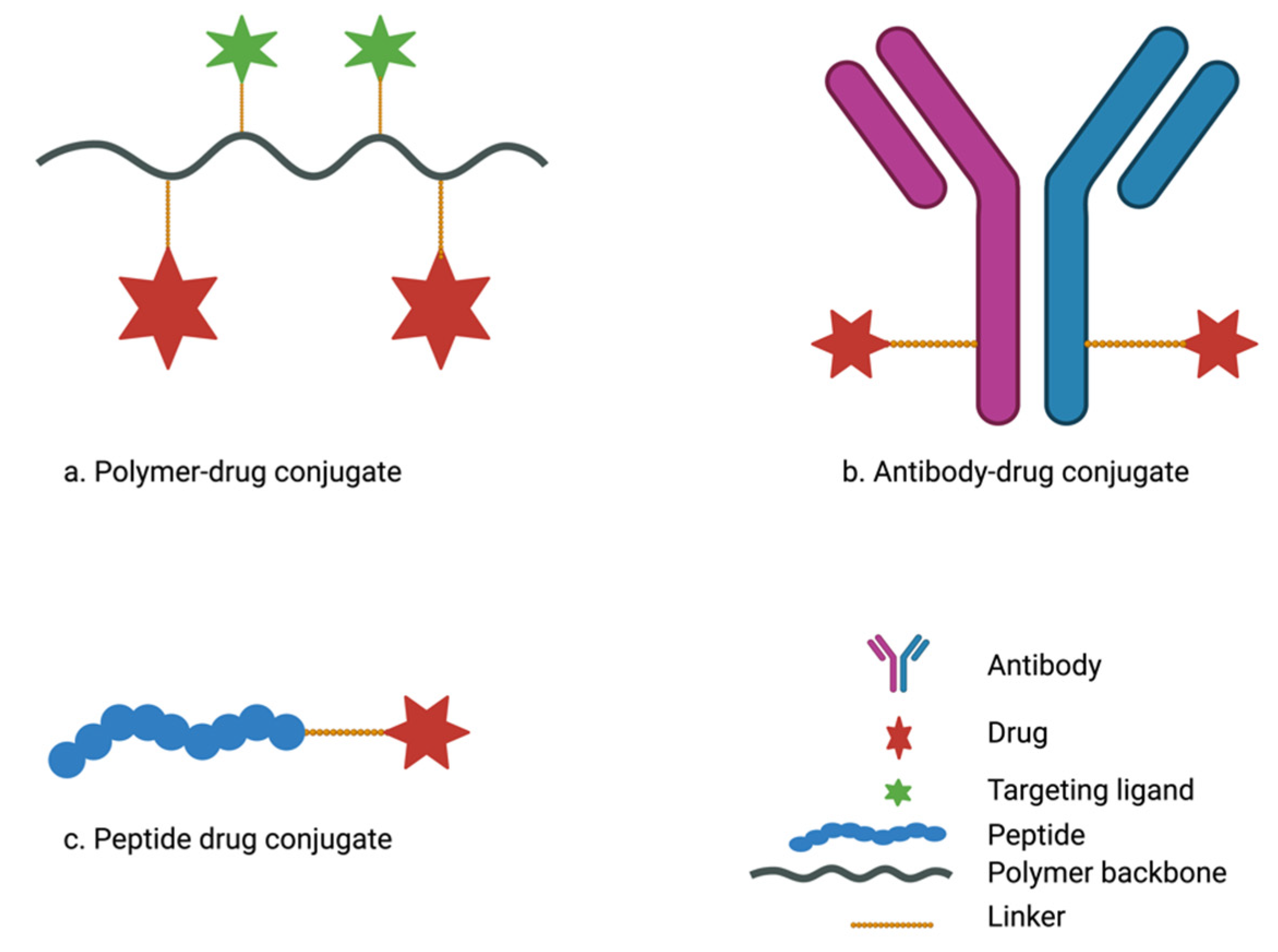
4. Drug-Conjugate Delivery Systems in Pancreatic Cancer Treatment
4.1. Polymer-Drug Conjugates for the Treatment of Pancreatic Cancer
4.2. Antibody-Drug Conjugates for Pancreatic Cancer Treatment
4.3. Peptide–Drug Conjugates for the Treatment of Pancreatic Cancer
5. Multistage Delivery Strategy
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ebelt, N.D.; Zamloot, V.; Manuel, E.R. Targeting desmoplasia in pancreatic cancer as an essential first step to effective therapy. Oncotarget 2020, 11, 3486–3488. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Qiu, Y.; Li, J.; Tang, X.; Wang, X.; Cun, X.; Xu, S.; Liu, Y.; Li, M.; Zhang, Z.; et al. Targeting cancer-associated fibroblasts by dual-responsive lipid-albumin nanoparticles to enhance drug perfusion for pancreatic tumor therapy. J. Control. Release 2020, 321, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Prakash, J. Nanomedicine Strategies to Enhance Tumor Drug Penetration in Pancreatic Cancer. Int. J. Nanomed. 2021, 16, 6313–6328. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yang, X.; Wang, X.; Zhao, X.; Zhang, Y.; Liu, S.; Anderson, G.J.; Kim, S.-J.; Li, Y.; Nie, G. Penetration Cascade of Size Switchable Nanosystem in Desmoplastic Stroma for Improved Pancreatic Cancer Therapy. ACS Nano 2021, 15, 14149–14161. [Google Scholar] [CrossRef]
- Kędzierska-Kapuza, K.; Witkowski, G.; Baumgart-Gryn, K.; Szylińska, A.; Durlik, M. Impact of COVID-19 on pancreatic cancer surgery: A high-volume Polish center experience. Adv. Clin. Exp. Med. 2022, 31, 389–398. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, Z.; Liu, H.; Fetse, J.P.; Jain, A.; Lin, C.-Y.; Cheng, K. Development of a tumor-responsive nanopolyplex targeting pancreatic cancer cells and stroma. ACS Appl. Mater. Interfaces 2019, 11, 45390–45403. [Google Scholar] [CrossRef]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic cancer stroma: An update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef]
- Xia, C.; Dong, X.; Li, H.; Cao, M.; Sun, D.; He, S.; Yang, F.; Yan, X.; Zhang, S.; Li, N.; et al. Cancer statistics in China and United States, 2022: Profiles, trends, and determinants. Chin. Med. J. 2022, 135, 584–590. [Google Scholar] [CrossRef]
- Ferlay, J.; Partensky, C.; Bray, F. More deaths from pancreatic cancer than breast cancer in the EU by 2017. Acta Oncol. Stockh. Swed. 2016, 55, 1158–1160. [Google Scholar] [CrossRef]
- Kokkinos, J.; Ignacio, R.M.; Sharbeen, G.; Boyer, C.; Gonzales-Aloy, E.; Goldstein, D.; McCarroll, J.A.; Phillips, P.A. Australian Pancreatic Cancer Genome Initiative. Targeting the undruggable in pancreatic cancer using nano-based gene silencing drugs. Biomaterials 2020, 240, 119742. [Google Scholar] [CrossRef]
- Bazeed, A.Y.; Day, C.M.; Garg, S. Pancreatic Cancer: Challenges and Opportunities in Locoregional Therapies. Cancers 2022, 14, 4257. [Google Scholar] [CrossRef]
- Kirkegård, J.; Bojesen, A.B.; Nielsen, M.F.; Mortensen, F.V. Trends in pancreatic cancer incidence, characteristics, and outcomes in Denmark 1980–2019: A nationwide cohort study. Cancer Epidemiol. 2022, 80, 102230. [Google Scholar] [CrossRef]
- Hidalgo, M.; Cascinu, S.; Kleeff, J.; Labianca, R.; Löhr, J.-M.; Neoptolemos, J.; Real, F.X.; Van Laethem, J.-L.; Heinemann, V. Addressing the challenges of pancreatic cancer: Future directions for improving outcomes. Pancreatology 2015, 15, 8–18. [Google Scholar] [CrossRef]
- Elechalawar, C.K.; Hossen, N.; Shankarappa, P.; Peer, C.J.; Figg, W.D.; Robertson, J.D.; Bhattacharya, R.; Mukherjee, P. Targeting Pancreatic Cancer Cells and Stellate Cells Using Designer Nanotherapeutics in vitro. Int. J. Nanomed. 2020, 15, 991–1003. [Google Scholar] [CrossRef]
- Carvalho, T.M.A.; Di Molfetta, D.; Greco, M.R.; Koltai, T.; Alfarouk, K.O.; Reshkin, S.J.; Cardone, R.A. Tumor Microenvironment Features and Chemoresistance in Pancreatic Ductal Adenocarcinoma: Insights into Targeting Physicochemical Barriers and Metabolism as Therapeutic Approaches. Cancers 2021, 13, 6135. [Google Scholar] [CrossRef]
- Pontious, C.; Kaul, S.; Hong, M.; Hart, P.A.; Krishna, S.G.; Lara, L.F.; Conwell, D.L.; Cruz-Monserrate, Z. Cathepsin E expression and activity: Role in the detection and treatment of pancreatic cancer. Pancreatology 2019, 19, 951–956. [Google Scholar] [CrossRef]
- Cannon, A.; Thompson, C.; Hall, B.R.; Jain, M.; Kumar, S.; Batra, S.K. Desmoplasia in pancreatic ductal adenocarcinoma: Insight into pathological function and therapeutic potential. Genes Cancer 2018, 9, 78–86. [Google Scholar] [CrossRef]
- Conte, M.; Cauda, V. Multimodal Therapies against Pancreatic Ductal Adenocarcinoma: A Review on Synergistic Approaches toward Ultimate Nanomedicine Treatments. Adv. Ther. 2022, 5, 2200079. [Google Scholar] [CrossRef]
- Badger, S.; Brant, J.; Jones, C.; McClements, J.; Loughrey, M.; Taylor, M.; Diamond, T.; McKie, L. The role of surgery for pancreatic cancer: A 12-year review of patient outcome. Ulst. Med. J. 2010, 79, 70–75. [Google Scholar]
- Tomasello, G.; Ghidini, M.; Costanzo, A.; Ghidini, A.; Russo, A.; Barni, S.; Passalacqua, R.; Petrelli, F. Outcome of head compared to body and tail pancreatic cancer: A systematic review and meta-analysis of 93 studies. J. Gastrointest. Oncol. 2019, 10, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Prim. 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Diab, M.; Azmi, A.; Mohammad, R.; Philip, P.A. Pharmacotherapeutic strategies for treating pancreatic cancer: Advances and challenges. Expert Opin. Pharmacother. 2019, 20, 535–546. [Google Scholar] [CrossRef]
- Lee, M.; Kwon, W.; Kim, H.; Byun, Y.; Han, Y.; Kang, J.S.; Choi, Y.J.; Jang, J.-Y. The Role of Location of Tumor in the Prognosis of the Pancreatic Cancer. Cancers 2020, 12, 2036. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zheng, Y.; Yang, F.; Zhu, L.; Zhu, X.-Q.; Wang, Z.-F.; Wu, X.-L.; Zhou, C.-H.; Yan, J.-Y.; Hu, B.-Y.; et al. The Molecular Biology of Pancreatic Adenocarcinoma: Translational Challenges and Clinical Perspectives. Signal Transduct. Target. Ther. 2021, 6, 249. [Google Scholar] [CrossRef]
- Silverman, D.T.; Hoover, R.N.; Brown, L.M.; Swanson, G.M.; Schiffman, M.; Greenberg, R.S.; Hayes, R.B.; Lillemoe, K.D.; Schoenberg, J.B.; Schwartz, A.G.; et al. Why do Black Americans have a higher risk of pancreatic cancer than White Americans? Epidemiology 2003, 14, 45–54. [Google Scholar] [CrossRef]
- Herremans, K.M.; Riner, A.N.; Winn, R.A.; Trevino, J.G. Diversity and Inclusion in Pancreatic Cancer Clinical Trials. Gastroenterology 2021, 161, 1741–1746.e3. [Google Scholar] [CrossRef]
- Moniri, M.R.; Dai, L.-J.; Warnock, G.L. The challenge of pancreatic cancer therapy and novel treatment strategy using engineered mesenchymal stem cells. Cancer Gene Ther. 2014, 21, 12–23. [Google Scholar] [CrossRef]
- Whatcott, C.J.; Diep, C.H.; Jiang, P.; Watanabe, A.; LoBello, J.; Sima, C.; Hostetter, G.; Shepard, H.M.; Von Hoff, D.D.; Han, H. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancerfibrosis in Pancreatic Metastases. Clin. Cancer Res. 2015, 21, 3561–3568. [Google Scholar] [CrossRef]
- Dókus, L.E.; Lajkó, E.; Ranđelović, I.; Mező, D.; Schlosser, G.; Kőhidai, L.; Tóvári, J.; Mező, G. Phage Display-Based Homing Peptide-Daunomycin Conjugates for Selective Drug Targeting to PANC-1 Pancreatic Cancer. Pharmaceutics 2020, 12, 576. [Google Scholar] [CrossRef]
- Tarannum, M.; Vivero-Escoto, J.L. Nanoparticle-based therapeutic strategies targeting major clinical challenges in pancreatic cancer treatment. Adv. Drug Deliv. Rev. 2022, 187, 114357. [Google Scholar] [CrossRef]
- Zhu, H.; Li, T.; Du, Y.; Li, M. Pancreatic cancer: Challenges and opportunities. BMC Med. 2018, 16, 214. [Google Scholar] [CrossRef]
- Manrai, M.; Tilak, T.V.S.V.G.K.; Dawra, S.; Srivastava, S.; Singh, A. Current and emerging therapeutic strategies in pancreatic cancer: Challenges and opportunities. World J. Gastroenterol. 2021, 27, 6572–6589. [Google Scholar] [CrossRef]
- Zhang, L.; Sanagapalli, S.; Stoita, A. Challenges in diagnosis of pancreatic cancer. World J. Gastroenterol. 2018, 24, 2047–2060. [Google Scholar] [CrossRef]
- Li, J.; Peng, L.; Chen, Q.; Ye, Z.; Zhao, T.; Hou, S.; Gu, J.; Hang, Q. Integrin β1 in Pancreatic Cancer: Expressions, Functions, and Clinical Implications. Cancers 2022, 14, 3377. [Google Scholar] [CrossRef]
- Liu, L.; Kshirsagar, P.G.; Gautam, S.K.; Gulati, M.; Wafa, E.I.; Christiansen, J.C.; White, B.M.; Mallapragada, S.K.; Wannemuehler, M.J.; Kumar, S.; et al. Nanocarriers for pancreatic cancer imaging, treatments, and immunotherapies. Theranostics 2022, 12, 1030–1060. [Google Scholar] [CrossRef]
- Khan, I.U.; Serra, C.A.; Anton, N.; Vandamme, T. Microfluidics: A focus on improved cancer targeted drug delivery systems. J. Control. Release 2013, 172, 1065–1074. [Google Scholar] [CrossRef]
- Merika, E.E.; Syrigos, K.N.; Saif, M.W. Desmoplasia in pancreatic cancer. Can we fight it? Gastroenterol. Res. Pract. 2012, 2012, 781765. [Google Scholar] [CrossRef]
- Adesina, S.K.; Holly, A.; Kramer-Marek, G.; Capala, J.; Akala, E.O. Polylactide-Based Paclitaxel-Loaded Nanoparticles Fabricated by Dispersion Polymerization: Characterization, Evaluation in Cancer Cell Lines, and Preliminary Biodistribution Studies. J. Pharm. Sci. 2014, 103, 2546–2555. [Google Scholar] [CrossRef]
- Khare, V.; Alam, N.; Saneja, A.; Dubey, R.D.; Gupta, P.N. Targeted Drug Delivery Systems for Pancreatic Cancer. J. Biomed. Nanotechnol. 2014, 10, 3462–3482. [Google Scholar] [CrossRef]
- Longnecker, D.S. Anatomy and Histology of the Pancreas (version 1.0). Pancreapedia Exocrine Pancreas Knowl. Base 2014. [Google Scholar] [CrossRef]
- Cesmebasi, A.; Malefant, J.; Patel, S.D.; Du Plessis, M.; Renna, S.; Tubbs, R.S.; Loukas, M. The surgical anatomy of the lymphatic system of the pancreas. Clin. Anat. 2015, 28, 527–537. [Google Scholar] [CrossRef] [PubMed]
- van Erning, F.N.; Mackay, T.M.; van der Geest, L.G.; Groot Koerkamp, B.; van Laarhoven, H.W.; Bonsing, B.A.; Wilmink, J.W.; van Santvoort, H.C.; de Vos-Geelen, J.; van Eijck, C.H.J.; et al. Association of the location of pancreatic ductal adenocarcinoma (head, body, tail) with tumor stage, treatment, and survival: A population-based analysis. Acta Oncol. 2018, 57, 1655–1662. [Google Scholar] [CrossRef] [PubMed]
- Artinyan, A.; Soriano, P.A.; Prendergast, C.; Low, T.; Ellenhorn, J.D.; Kim, J. The anatomic location of pancreatic cancer is a prognostic factor for survival. HPB 2008, 10, 371–376. [Google Scholar] [CrossRef]
- Jiang, S.; Fagman, J.B.; Ma, Y.; Liu, J.; Vihav, C.; Engstrom, C.; Liu, B.; Chen, C. A comprehensive review of pancreatic cancer and its therapeutic challenges. Aging 2022, 14, 7635–7649. [Google Scholar] [CrossRef]
- Masugi, Y. The Desmoplastic Stroma of Pancreatic Cancer: Multilayered Levels of Heterogeneity, Clinical Significance, and Therapeutic Opportunities. Cancers 2022, 14, 3293. [Google Scholar] [CrossRef]
- Falasca, M.; Kim, M.; Casari, I. Pancreatic cancer: Current research and future directions. Biochim. et Biophys. Acta (BBA)-Rev. Cancer 2016, 1865, 123–132. [Google Scholar] [CrossRef]
- Han, H.; Hou, Y.; Chen, X.; Zhang, P.; Kang, M.; Jin, Q.; Ji, J.; Gao, M. Metformin-Induced Stromal Depletion to Enhance the Penetration of Gemcitabine-Loaded Magnetic Nanoparticles for Pancreatic Cancer Targeted Therapy. J. Am. Chem. Soc. 2020, 142, 4944–4954. [Google Scholar] [CrossRef]
- Dimastromatteo, J.; Houghton, J.L.; Lewis, J.S.; Kelly, K.A. Challenges of Pancreatic Cancer. Cancer J. 2015, 21, 188–193. [Google Scholar] [CrossRef]
- Schnittert, J.; Bansal, R.; Prakash, J. Targeting Pancreatic Stellate Cells in Cancer. Trends Cancer 2019, 5, 128–142. [Google Scholar] [CrossRef]
- Miao, L.; Liu, Q.; Lin, C.M.; Luo, C.; Wang, Y.; Liu, L.; Yin, W.; Hu, S.; Kim, W.Y.; Huang, L. Targeting Tumor-Associated Fibroblasts for Therapeutic Delivery in Desmoplastic TumorsIn Situ Generation of Tumor-Suppressive Fibroblasts. Cancer Res. 2017, 77, 719–731. [Google Scholar] [CrossRef]
- Whittle, M.C.; Hingorani, S.R. Fibroblasts in Pancreatic Ductal Adenocarcinoma: Biological Mechanisms and Therapeutic Targets. Gastroenterology 2019, 156, 2085–2096. [Google Scholar] [CrossRef]
- Norton, J.; Foster, D.; Chinta, M.; Titan, A.; Longaker, M. Pancreatic Cancer Associated Fibroblasts (CAF): Under-Explored Target for Pancreatic Cancer Treatment. Cancers 2020, 12, 1347. [Google Scholar] [CrossRef]
- Nandi, T.; Pradyuth, S.; Singh, A.K.; Chitkara, D.; Mittal, A. Therapeutic agents for targeting desmoplasia: Current status and emerging trends. Drug Discov. Today 2020, 25, 2046–2055. [Google Scholar] [CrossRef]
- Polani, F.; Grierson, P.M.; Lim, K.-H. Stroma-targeting strategies in pancreatic cancer: Past lessons, challenges and prospects. World J. Gastroenterol. 2021, 27, 2105–2121. [Google Scholar] [CrossRef]
- Sivapalan, L.; Kocher, H.; Ross-Adams, H.; Chelala, C. Molecular profiling of ctDNA in pancreatic cancer: Opportunities and challenges for clinical application. Pancreatology 2021, 21, 363–378. [Google Scholar] [CrossRef]
- Chitkara, D.; Mittal, A.; Behrman, S.W.; Kumar, N.; Mahato, R.I. Self-assembling, amphiphilic polymer–gemcitabine conjugate shows enhanced antitumor efficacy against human pancreatic adenocarcinoma. Bioconjugate Chem. 2013, 24, 1161–1173. [Google Scholar] [CrossRef]
- Boyd, L.N.; Andini, K.D.; Peters, G.J.; Kazemier, G.; Giovannetti, E. Heterogeneity and plasticity of cancer-associated fibroblasts in the pancreatic tumor microenvironment. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2022; Volume 82, pp. 184–196. [Google Scholar]
- Ren, B.; Cui, M.; Yang, G.; Wang, H.; Feng, M.; You, L.; Zhao, Y. Tumor microenvironment participates in metastasis of pancreatic cancer. Mol. Cancer 2018, 17, 108. [Google Scholar] [CrossRef]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—Clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef]
- Stine, Z.; Altman, B.; Hsieh, A.; Gouw, A.; Dang, C. Deregulation of the Cellular Energetics of Cancer Cells. In Pathobiology of Human Disease; Elsevier: Amsterdam, The Netherlands, 2014; pp. 444–455. [Google Scholar]
- Chen, X.; Zhou, W.; Liang, C.; Shi, S.; Yu, X.; Chen, Q.; Sun, T.; Lu, Y.; Zhang, Y.; Guo, Q.; et al. Codelivery Nanosystem Targeting the Deep Microenvironment of Pancreatic Cancer. Nano Lett. 2019, 19, 3527–3534. [Google Scholar] [CrossRef]
- Bannoura, S.F.; Uddin, H.; Nagasaka, M.; Fazili, F.; Al-Hallak, M.N.; Philip, P.A.; El-Rayes, B.; Azmi, A.S. Targeting KRAS in pancreatic cancer: New drugs on the horizon. Cancer Metastasis Rev. 2021, 40, 819–835. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, E.C.; Drezner, N.; Li, X.; Mishra-Kalyani, P.S.; Liu, Y.; Zhao, H.; Bi, Y.; Liu, J.; Rahman, A.; Wearne, E.; et al. FDA Approval Summary: Sotorasib for KRAS G12C-Mutated Metastatic NSCLC. Clin. Cancer Res. 2022, 28, 1482–1486. [Google Scholar] [CrossRef] [PubMed]
- Strickler, J.H.; Satake, H.; George, T.J.; Yaeger, R.; Hollebecque, A.; Garrido-Laguna, I.; Schuler, M.; Burns, T.F.; Coveler, A.L.; Falchook, G.S.; et al. Sotorasib in KRAS p. G12C–Mutated Advanced Pancreatic Cancer. N. Engl. J. Med. 2023, 388, 33–43. [Google Scholar] [CrossRef]
- Khawar, I.A.; Kim, J.H.; Kuh, H.-J. Improving drug delivery to solid tumors: Priming the tumor microenvironment. J. Control. Release 2015, 201, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Tsukigawa, K.; Fang, J. A Retrospective 30 Years After Discovery of the Enhanced Permeability and Retention Effect of Solid Tumors: Next-Generation Chemotherapeutics and Photodynamic Therapy-Problems, Solutions, and Prospects. Microcirculation 2016, 23, 173–182. [Google Scholar] [CrossRef]
- Ejigah, V.; Owoseni, O.; Bataille-Backer, P.; Ogundipe, O.D.; Fisusi, F.A.; Adesina, S.K. Approaches to Improve Macromolecule and Nanoparticle Accumulation in the Tumor Microenvironment by the Enhanced Permeability and Retention Effect. Polymers 2022, 14, 2601. [Google Scholar] [CrossRef]
- Greish, K. Enhanced permeability and retention effect for selective targeting of anticancer nanomedicine: Are we there yet? Drug Discov. Today Technol. 2012, 9, e161–e166. [Google Scholar] [CrossRef]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef]
- Natfji, A.A.; Ravishankar, D.; Osborn, H.M.I.; Greco, F. Parameters Affecting the Enhanced Permeability and Retention Effect: The Need for Patient Selection. J. Pharm. Sci. 2017, 106, 3179–3187. [Google Scholar] [CrossRef]
- Maeda, H.; Bharate, G.; Daruwalla, J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur. J. Pharm. Biopharm. 2009, 71, 409–419. [Google Scholar] [CrossRef]
- Kalyane, D.; Raval, N.; Maheshwari, R.; Tambe, V.; Kalia, K.; Tekade, R.K. Employment of enhanced permeability and retention effect (EPR): Nanoparticle-based precision tools for targeting of therapeutic and diagnostic agent in cancer. Mater. Sci. Eng. C 2019, 98, 1252–1276. [Google Scholar] [CrossRef]
- Rajora, A.K.; Ravishankar, D.; Osborn, H.M.I.; Greco, F. Impact of the Enhanced Permeability and Retention (EPR) Effect and Cathepsins Levels on the Activity of Polymer-Drug Conjugates. Polymers 2014, 6, 2186–2220. [Google Scholar] [CrossRef]
- Fang, J.; Islam, W.; Maeda, H. Exploiting the dynamics of the EPR effect and strategies to improve the therapeutic effects of nanomedicines by using EPR effect enhancers. Adv. Drug Deliv. Rev. 2020, 157, 142–160. [Google Scholar] [CrossRef]
- Edwards, P.; Kang, B.W.; Chau, I. Targeting the Stroma in the Management of Pancreatic Cancer. Front. Oncol. 2021, 11, 691185. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, J.; Meng, H. Transcytosis—An effective targeting strategy that is complementary to “EPR effect” for pancreatic cancer nano drug delivery. Theranostics 2019, 9, 8018–8025. [Google Scholar] [CrossRef]
- Islam, W.; Niidome, T.; Sawa, T. Enhanced Permeability and Retention Effect as a Ubiquitous and Epoch-Making Phenomenon for the Selective Drug Targeting of Solid Tumors. J. Pers. Med. 2022, 12, 1964. [Google Scholar] [CrossRef]
- Nel, A.; Ruoslahti, E.; Meng, H. New Insights into “Permeability” as in the Enhanced Permeability and Retention Effect of Cancer Nanotherapeutics. ACS Nano 2017, 11, 9567–9569. [Google Scholar] [CrossRef]
- Xie, Y.; Hang, Y.; Wang, Y.; Sleightholm, R.; Prajapati, D.R.; Bader, J.; Yu, A.; Tang, W.; Jaramillo, L.; Li, J.; et al. Stromal Modulation and Treatment of Metastatic Pancreatic Cancer with Local Intraperitoneal Triple miRNA/siRNA Nanotherapy. ACS Nano 2020, 14, 255–271. [Google Scholar] [CrossRef]
- Pandit, S.; Dutta, D.; Nie, S. Active transcytosis and new opportunities for cancer nanomedicine. Nat. Mater. 2020, 19, 478–480. [Google Scholar] [CrossRef]
- Zhou, Q.; Li, J.; Xiang, J.; Shao, S.; Zhou, Z.; Tang, J.; Shen, Y. Transcytosis-enabled active extravasation of tumor nanomedicine. Adv. Drug Deliv. Rev. 2022, 189, 114480. [Google Scholar] [CrossRef]
- Tanaka, H.Y.; Kano, M.R. Stromal barriers to nanomedicine penetration in the pancreatic tumor microenvironment. Cancer Sci. 2018, 109, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Wallrapp, C.; Hähnel, S.; Müller-Pillasch, F.; Burghardt, B.; Iwamura, T.; Ruthenbürger, M.; Lerch, M.M.; Adler, G.; Gress, T.M. A Novel Transmembrane Serine Protease (TMPRSS3) Overexpressed in Pancreatic Cancer1, 2. Cancer Res. 2000, 60, 2602–2606. [Google Scholar] [PubMed]
- Herszényi, L.; Barabás, L.; Hritz, I.; István, G.; Tulassay, Z. Impact of proteolytic enzymes in colorectal cancer development and progression. World J. Gastroenterol. 2014, 20, 13246–13257. [Google Scholar] [CrossRef] [PubMed]
- Uchima, Y.; Sawada, T.; Nishihara, T.; Maeda, K.; Ohira, M.; Hirakawa, K. Inhibition and Mechanism of Action of a Protease Inhibitor in Human Pancreatic Cancer Cells. Pancreas 2004, 29, 123–131. [Google Scholar] [CrossRef]
- Mótyán, J.A.; Tóth, F.; Tőzsér, J. Research applications of proteolytic enzymes in molecular biology. Biomolecules 2013, 3, 923–942. [Google Scholar] [CrossRef]
- Scott, C.J.; Taggart, C.C. Biologic protease inhibitors as novel therapeutic agents. Biochimie 2010, 92, 1681–1688. [Google Scholar] [CrossRef]
- Rudzińska, M.; Daglioglu, C.; Savvateeva, L.V.; Kaci, F.N.; Antoine, R.; Zamyatnin, A.A., Jr. Current status and perspectives of protease inhibitors and their combination with nanosized drug delivery systems for targeted cancer therapy. Drug Des. Dev. Ther. 2021, 15, 9–20. [Google Scholar] [CrossRef]
- Vandooren, J.; Opdenakker, G.; Loadman, P.M.; Edwards, D.R. Proteases in cancer drug delivery. Adv. Drug Deliv. Rev. 2016, 97, 144–155. [Google Scholar] [CrossRef]
- Patsouras, D.; Papaxoinis, K.; Kostakis, A.; Safioleas, M.C.; Lazaris, A.C.; Nicolopoulou-Stamati, P. Fibroblast activation protein and its prognostic significance in correlation with vascular endothelial growth factor in pancreatic adenocarcinoma. Mol. Med. Rep. 2015, 11, 4585–4590. [Google Scholar] [CrossRef]
- Cohen, S.J.; Alpaugh, R.K.; Palazzo, I.; Meropol, N.J.; Rogatko, A.; Xu, Z.; Hoffman, J.P.; Weiner, L.M.; Cheng, J.D. Fibroblast Activation Protein and Its Relationship to Clinical Outcome in Pancreatic Adenocarcinoma. Pancreas 2008, 37, 154–158. [Google Scholar] [CrossRef]
- Keane, F.M.; Yao, T.-W.; Seelk, S.; Gall, M.G.; Chowdhury, S.; Poplawski, S.E.; Lai, J.H.; Li, Y.; Wu, W.; Farrell, P.; et al. Quantitation of fibroblast activation protein (FAP)-specific protease activity in mouse, baboon and human fluids and organs. FEBS Open Bio 2014, 4, 43–54. [Google Scholar] [CrossRef]
- Akinboye, E.S.; Brennen, W.N.; Rosen, D.M.; Bakare, O.; Denmeade, S.R. Iterative design of emetine-based prodrug targeting fibroblast activation protein (FAP) and dipeptidyl peptidase IV DPPIV using a tandem enzymatic activation strategy. Prostate 2016, 76, 703–714. [Google Scholar] [CrossRef]
- Lo, A.; Li, C.-P.; Buza, E.L.; Blomberg, R.; Govindaraju, P.; Avery, D.; Monslow, J.; Hsiao, M.; Puré, E. Fibroblast activation protein augments progression and metastasis of pancreatic ductal adenocarcinoma. J. Clin. Investig. 2017, 2, e92232. [Google Scholar] [CrossRef]
- Zhao, L.; Chen, J.; Pang, Y.; Fu, K.; Shang, Q.; Wu, H.; Sun, L.; Lin, Q.; Chen, H. Fibroblast activation protein-based theranostics in cancer research: A state-of-the-art review. Theranostics 2022, 12, 1557–1569. [Google Scholar] [CrossRef]
- Huber, M.A.; Schubert, R.D.; Peter, R.U.; Kraut, N.; Park, J.E.; Rettig, W.J.; Garin-Chesa, P. Fibroblast Activation Protein: Differential Expression and Serine Protease Activity in Reactive Stromal Fibroblasts of Melanocytic Skin Tumors. J. Investig. Dermatol. 2003, 120, 182–188. [Google Scholar] [CrossRef]
- Liu, R.; Li, H.; Liu, L.; Yu, J.; Ren, X. Fibroblast activation protein: A potential therapeutic target in cancer. Cancer Biol. Ther. 2012, 13, 123–129. [Google Scholar] [CrossRef]
- Park, H.; Lee, Y.; Lee, H.; Kim, J.-W.; Hwang, J.-H.; Kim, J.; Yoon, Y.-S.; Han, H.-S.; Kim, H. The prognostic significance of cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. Tumor Biol. 2017, 39, 1010428317718403. [Google Scholar] [CrossRef]
- Lin, H.-J.; Liang, T.-L.; Chang, Y.-Y.; Liu, D.-Z.; Fan, J.-Y.; Roffler, S.R.; Lin, S.-Y. Development of Irinotecan Liposome Armed with Dual-Target Anti-Epidermal Growth Factor Receptor and Anti-Fibroblast Activation Protein-Specific Antibody for Pancreatic Cancer Treatment. Pharmaceutics 2022, 14, 1202. [Google Scholar] [CrossRef]
- Abd-Elgaliel, W.R.; Cruz-Monserrate, Z.; Wang, H.; Logsdon, C.D.; Tung, C.-H. Pancreatic cancer-associated Cathepsin E as a drug activator. J. Control. Release 2013, 167, 221–227. [Google Scholar] [CrossRef]
- Jones, L.; Ghaneh, P.; Humphreys, M.; Neoptolemos, J.P. The Matrix Metalloproteinases and Their Inhibitors in the Treatment of Pancreatic Cancer. Ann. N. Y. Acad. Sci. 1999, 880, 288–307. [Google Scholar] [CrossRef]
- Ghaneh, P.; Kawesha, A.; Evans, J.D.; Neoptolemos, J. Molecular prognostic markers in pancreatic cancer. J. Hepato-Biliary-Pancreatic Surg. 2002, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Zhu, H.-G. Caveolin-1 Regulating the Invasion and Expression of Matrix Metalloproteinase (MMPs) in Pancreatic Carcinoma Cells. J. Surg. Res. 2010, 159, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.S.; Haldar, M.K.; Nahire, R.R.; Katti, P.; Ambre, A.H.; Muhonen, W.W.; Shabb, J.B.; Padi, S.K.R.; Singh, R.K.; Borowicz, P.P.; et al. MMP-9 Responsive PEG Cleavable Nanovesicles for Efficient Delivery of Chemotherapeutics to Pancreatic Cancer. Mol. Pharm. 2014, 11, 2390–2399. [Google Scholar] [CrossRef] [PubMed]
- Niedergethmann, M.; Wostbrock, B.; Sturm, J.W.; Willeke, F.; Post, S.; Hildenbrand, R. Prognostic Impact of Cysteine Proteases Cathepsin B and Cathepsin L in Pancreatic Adenocarcinoma. Pancreas 2004, 29, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Valdepérez, D.; Jin, Q.; Yang, B.; Li, Z.; Wu, Y.; Pelaz, B.; Parak, W.J.; Ji, J. Dual Enzymatic Reaction-Assisted Gemcitabine Delivery Systems for Programmed Pancreatic Cancer Therapy. ACS Nano 2017, 11, 1281–1291. [Google Scholar] [CrossRef]
- Sulpizio, S.; Franceschini, N.; Piattelli, A.; Di Sebastiano, P.; Innocenti, P.; Selvaggi, F. Cathepsins and pancreatic cancer: The 2012 update. Pancreatology 2012, 12, 395–401. [Google Scholar] [CrossRef]
- Chu, E.; Sartorelli, A.C. Cancer chemotherapy. In Lange’s Basic and Clinical Pharmacology; McGraw Hill: New York, NY, USA, 2018; pp. 948–976. [Google Scholar]
- Liu, X.; Jiang, J.; Ji, Y.; Lu, J.; Chan, R.; Meng, H. Targeted drug delivery using iRGD peptide for solid cancer treatment. Mol. Syst. Des. Eng. 2017, 2, 370–379. [Google Scholar] [CrossRef]
- Arias, J.L. Drug Targeting Strategies in Cancer Treatment: An Overview. Mini-Rev. Med. Chem. 2011, 11, 1–17. [Google Scholar] [CrossRef]
- Young, K.; Hughes, D.J.; Cunningham, D.; Starling, N. Immunotherapy and pancreatic cancer: Unique challenges and potential opportunities. Ther. Adv. Med. Oncol. 2018, 10, 1758835918816281. [Google Scholar] [CrossRef]
- Fan, J.-Q.; Wang, M.-F.; Chen, H.-L.; Shang, D.; Das, J.K.; Song, J. Current advances and outlooks in immunotherapy for pancreatic ductal adenocarcinoma. Mol. Cancer 2020, 19, 32. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Beatty, G.L.; Dougan, S.K. Broadening the Impact of Immunotherapy to Pancreatic Cancer: Challenges and Opportunities. Gastroenterology 2019, 156, 2056–2072. [Google Scholar] [CrossRef]
- Bear, A.S.; Vonderheide, R.H.; O’Hara, M.H. Challenges and Opportunities for Pancreatic Cancer Immunotherapy. Cancer Cell 2020, 38, 788–802. [Google Scholar] [CrossRef]
- Wu, J.; Cai, J. Dilemma and Challenge of Immunotherapy for Pancreatic Cancer. Dig. Dis. Sci. 2020, 66, 359–368. [Google Scholar] [CrossRef]
- Martinez-Bosch, N.; Vinaixa, J.; Navarro, P. Immune Evasion in Pancreatic Cancer: From Mechanisms to Therapy. Cancers 2018, 10, 6. [Google Scholar] [CrossRef]
- Torphy, R.J.; Zhu, Y.; Schulick, R.D. Immunotherapy for pancreatic cancer: Barriers and breakthroughs. Ann. Gastroenterol. Surg. 2018, 2, 274–281. [Google Scholar] [CrossRef]
- Kamath, S.D.; Kalyan, A.; Kircher, S.; Nimeiri, H.; Fought, A.J.; Benson, A., III; Mulcahy, M. Ipilimumab and gemcitabine for advanced pancreatic cancer: A phase Ib study. Oncologist 2020, 25, e808–e815. [Google Scholar] [CrossRef]
- Wu, A.A.; Bever, K.M.; Ho, W.J.; Fertig, E.J.; Niu, N.; Zheng, L.; Parkinson, R.M.; Durham, J.N.; Onners, B.L.; Ferguson, A.K.; et al. A Phase II Study of Allogeneic GM-CSF–Transfected Pancreatic Tumor Vaccine (GVAX) with Ipilimumab as Maintenance Treatment for Metastatic Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 5129–5139. [Google Scholar] [CrossRef]
- Feng, M.; Xiong, G.; Cao, Z.; Yang, G.; Zheng, S.; Song, X.; You, L.; Zheng, L.; Zhang, T.; Zhao, Y. PD-1/PD-L1 and immunotherapy for pancreatic cancer. Cancer Lett. 2017, 407, 57–65. [Google Scholar] [CrossRef]
- Ribas, A.; Puzanov, I.; Dummer, R.; Schadendorf, D.; Hamid, O.; Robert, C.; Hodi, F.S.; Schachter, J.; Pavlick, A.C.; Lewis, K.D.; et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): A randomised, controlled, phase 2 trial. Lancet Oncol. 2015, 16, 908–918. [Google Scholar] [CrossRef]
- Weiss, G.J.; Blaydorn, L.; Beck, J.; Bornemann-Kolatzki, K.; Urnovitz, H.; Schütz, E.; Khemka, V. Phase Ib/II study of gemcitabine, nab-paclitaxel, and pembrolizumab in metastatic pancreatic adenocarcinoma. Investig. New Drugs 2018, 36, 96–102. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Hochster, H.S.; Kim, E.J.; George, B.; Kaylan, A.; Chiorean, E.G.; Waterhouse, D.M.; Guiterrez, M.; Parikh, A.; Jain, R.; et al. Open-label, Phase I Study of Nivolumab Combined with nab-Paclitaxel Plus Gemcitabine in Advanced Pancreatic CancerNivo Plus nab-Pac and Gem in Advanced Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 4814–4822. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Picozzi, V.J.; Ko, A.H.; Wainberg, Z.A.; Kindler, H.; Wang-Gillam, A.; Oberstein, P.E.; Morse, M.A.; Zeh, H.J.; Weekes, C.D.; et al. Results from a Phase IIb, Randomized, Multicenter Study of GVAX Pancreas and CRS-207 Compared with Chemotherapy in Adults with Previously Treated Metastatic Pancreatic Adenocarcinoma (ECLIPSE Study). Clin. Cancer Res. 2019, 25, 5493–5502. [Google Scholar] [CrossRef] [PubMed]
- Middleton, G.; Silcocks, P.; Cox, T.; Valle, J.; Wadsley, J.; Propper, D.; Coxon, F.; Ross, P.; Madhusudan, S.; Roques, T.; et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): An open-label, randomised, phase 3 trial. Lancet Oncol. 2014, 15, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Di Federico, A.; Mosca, M.; Pagani, R.; Carloni, R.; Frega, G.; De Giglio, A.; Rizzo, A.; Ricci, D.; Tavolari, S.; Di Marco, M.; et al. Immunotherapy in Pancreatic Cancer: Why Do We Keep Failing? A Focus on Tumor Immune Microenvironment, Predictive Biomarkers and Treatment Outcomes. Cancers 2022, 14, 2429. [Google Scholar] [CrossRef]
- Schizas, D.; Charalampakis, N.; Kole, C.; Economopoulou, P.; Koustas, E.; Gkotsis, E.; Ziogas, D.; Psyrri, A.; Karamouzis, M.V. Immunotherapy for pancreatic cancer: A 2020 update. Cancer Treat. Rev. 2020, 86, 102016. [Google Scholar] [CrossRef]
- Fan, B.-G.; Andrén-Sandberg, Å. Photodynamic Therapy for Pancreatic Cancer. Pancreas 2007, 34, 385–389. [Google Scholar] [CrossRef]
- Xie, Q.; Jia, L.; Liu, Y.-H.; Wei, C.-G. Synergetic anticancer effect of combined gemcitabine and photodynamic therapy on pancreatic cancer in vivo. World J. Gastroenterol. 2009, 15, 737–741. [Google Scholar] [CrossRef]
- Huggett, M.T.; Jermyn, M.; Gillams, A.; Illing, R.; Mosse, S.; Novelli, M.; Kent, E.; Bown, S.G.; Hasan, T.; Pogue, B.W.; et al. Phase I/II study of verteporfin photodynamic therapy in locally advanced pancreatic cancer. Br. J. Cancer 2014, 110, 1698–1704. [Google Scholar] [CrossRef]
- Lu, J.; Roy, B.; Anderson, M.; Leggett, C.L.; Levy, M.J.; Pogue, B.; Hasan, T.; Wang, K.K. Verteporfin- and sodium porfimer-mediated photodynamic therapy enhances pancreatic cancer cell death without activating stromal cells in the microenvironment. J. Biomed. Opt. 2019, 24, 118001. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Zhou, L.; Lu, J.; Jiang, B.; Liu, C.; Guo, J. Photodynamic therapy of pancreatic cancer: Where have we come from and where are we going? Photodiagn. Photodyn. Ther. 2020, 31, 101876. [Google Scholar] [CrossRef]
- Kim, M.M.; Darafsheh, A. Light Sources and Dosimetry Techniques for Photodynamic Therapy. Photochem. Photobiol. 2020, 96, 280–294. [Google Scholar] [CrossRef]
- Meng, Z.; Hou, W.; Zhou, H.; Zhou, L.; Chen, H.; Wu, C. Therapeutic Considerations and Conjugated Polymer-Based Photosensitizers for Photodynamic Therapy. Macromol. Rapid Commun. 2018, 39, 1700614. [Google Scholar] [CrossRef]
- Celli, J.P.; Solban, N.; Liang, A.; Pereira, S.P.; Hasan, T. Verteporfin-based photodynamic therapy overcomes gemcitabine insensitivity in a panel of pancreatic cancer cell lines. Lasers Surg. Med. 2011, 43, 565–574. [Google Scholar] [CrossRef]
- Yu, X.; Zhu, W.; Di, Y.; Gu, J.; Guo, Z.; Li, H.; Fu, D.; Jin, C. Triple-functional albumin-based nanoparticles for combined chemotherapy and photodynamic therapy of pancreatic cancer with lymphatic metastases. Int. J. Nanomed. 2017, 12, 6771–6785. [Google Scholar] [CrossRef]
- Yano, T.; Wang, K.K. Photodynamic Therapy for Gastrointestinal Cancer. Photochem. Photobiol. 2020, 96, 517–523. [Google Scholar] [CrossRef]
- Hafiz, S.S.; Xavierselvan, M.; Gokalp, S.; Labadini, D.; Barros, S.; Duong, J.; Foster, M.; Mallidi, S. Eutectic Gallium–Indium Nanoparticles for Photodynamic Therapy of Pancreatic Cancer. ACS Appl. Nano Mater. 2022, 5, 6125–6139. [Google Scholar] [CrossRef]
- Farokhzad, O.C.; Langer, R. Impact of Nanotechnology on Drug Delivery. ACS Nano 2009, 3, 16–20. [Google Scholar] [CrossRef]
- Attia, M.F.; Anton, N.; Wallyn, J.; Omran, Z.; Vandamme, T.F. An overview of active and passive targeting strategies to improve the nanocarriers efficiency to tumour sites. J. Pharm. Pharmacol. 2019, 71, 1185–1198. [Google Scholar] [CrossRef]
- Sun, R.; Xiang, J.; Zhou, Q.; Piao, Y.; Tang, J.; Shao, S.; Zhou, Z.; Bae, Y.H.; Shen, Y. The tumor EPR effect for cancer drug delivery: Current status, limitations, and alternatives. Adv. Drug Deliv. Rev. 2022, 191, 114614. [Google Scholar] [CrossRef]
- Adesina, S.K.; Akala, E.O. Nanotechnology Approaches for the Delivery of Exogenous siRNA for HIV Therapy. Mol. Pharm. 2015, 12, 4175–4187. [Google Scholar] [CrossRef]
- Alshawwa, S.Z.; Kassem, A.A.; Farid, R.M.; Mostafa, S.K.; Labib, G.S. Nanocarrier Drug Delivery Systems: Characterization, Limitations, Future Perspectives and Implementation of Artificial Intelligence. Pharmaceutics 2022, 14, 883. [Google Scholar] [CrossRef] [PubMed]
- Alshememry, A.K.; Alsaleh, N.B.; Alkhudair, N.; Alzhrani, R.; Alshamsan, A. Recent nanotechnology advancements to treat multidrug-resistance pancreatic cancer: Pre-clinical and clinical overview. Front. Pharmacol. 2022, 13, 933457. [Google Scholar] [CrossRef] [PubMed]
- Delplace, V.; Couvreur, P.; Nicolas, J. Recent trends in the design of anticancer polymer prodrug nanocarriers. Polym. Chem. 2014, 5, 1529–1544. [Google Scholar] [CrossRef]
- Bhattacharjee, S. Understanding the burst release phenomenon: Toward designing effective nanoparticulate drug-delivery systems. Ther. Deliv. 2021, 12, 21–36. [Google Scholar] [CrossRef]
- Li, S.-D.; Huang, L. Stealth nanoparticles: High density but sheddable PEG is a key for tumor targeting. J. Control. Release Off. J. Control. Release Soc. 2010, 145, 178–181. [Google Scholar] [CrossRef]
- Amoozgar, Z.; Yeo, Y. Recent advances in stealth coating of nanoparticle drug delivery systems. WIREs Nanomed. Nanobiotechnol. 2012, 4, 219–233. [Google Scholar] [CrossRef]
- Vllasaliu, D.; Fowler, R.; Stolnik, S. PEGylated nanomedicines: Recent progress and remaining concerns. Expert Opin. Drug Deliv. 2014, 11, 139–154. [Google Scholar] [CrossRef]
- Hoogenboezem, E.N.; Duvall, C.L. Harnessing albumin as a carrier for cancer therapies. Adv. Drug Deliv. Rev. 2018, 130, 73–89. [Google Scholar] [CrossRef]
- Hassanin, I.; Elzoghby, A. Albumin-based nanoparticles: A promising strategy to overcome cancer drug resistance. Cancer Drug Resist. 2020, 3, 93. [Google Scholar] [CrossRef]
- Kim, B.; Lee, C.; Lee, E.S.; Shin, B.S.; Youn, Y.S. Paclitaxel and curcumin co-bound albumin nanoparticles having antitumor potential to pancreatic cancer. Asian J. Pharm. Sci. 2016, 11, 708–714. [Google Scholar] [CrossRef]
- An, F.-F.; Zhang, X.-H. Strategies for Preparing Albumin-based Nanoparticles for Multifunctional Bioimaging and Drug Delivery. Theranostics 2017, 7, 3667–3689. [Google Scholar] [CrossRef]
- Cheng, Z.; Huang, Y.; Shen, Q.; Zhao, Y.; Wang, L.; Yu, J.; Lu, W. A camptothecin-based, albumin-binding prodrug enhances efficacy and safety in vivo. Eur. J. Med. Chem. 2021, 226, 113851. [Google Scholar] [CrossRef]
- Hirakawa, N.; Ishima, Y.; Kinoshita, R.; Nakano, R.; Chuang, V.T.G.; Ando, H.; Shimizu, T.; Okuhira, K.; Maruyama, T.; Otagiri, M.; et al. Reduction-Responsive and Multidrug Deliverable Albumin Nanoparticles: An Antitumor Drug to Abraxane against Human Pancreatic Tumor-Bearing Mice. ACS Appl. Bio Mater. 2021, 4, 4302–4309. [Google Scholar] [CrossRef]
- Tan, Y.L.; Ho, H.K. Navigating albumin-based nanoparticles through various drug delivery routes. Drug Discov. Today 2018, 23, 1108–1114. [Google Scholar] [CrossRef]
- Cho, H.; Jeon, S.I.; Ahn, C.-H.; Shim, M.K.; Kim, K. Emerging Albumin-Binding Anticancer Drugs for Tumor-Targeted Drug Delivery: Current Understandings and Clinical Translation. Pharmaceutics 2022, 14, 728. [Google Scholar] [CrossRef]
- Elzoghby, A.O.; Samy, W.M.; Elgindy, N.A. Albumin-based nanoparticles as potential controlled release drug delivery systems. J. Control. Release 2012, 157, 168–182. [Google Scholar] [CrossRef]
- Yu, X.; Jin, C. Application of albumin-based nanoparticles in the management of cancer. J. Mater. Sci. Mater. Med. 2016, 27, 4. [Google Scholar] [CrossRef]
- Goldstein, D.; El-Maraghi, R.H.; Hammel, P.; Heinemann, V.; Kunzmann, V.; Sastre, J.; Scheithauer, W.; Siena, S.; Tabernero, J.; Teixeira, L.; et al. nab-Paclitaxel Plus Gemcitabine for Metastatic Pancreatic Cancer: Long-Term Survival From a Phase III Trial. Gynecol. Oncol. 2015, 107, dju41313. [Google Scholar] [CrossRef]
- Feng, J.; Zhao, C.; Wang, L.; Qu, L.; Zhu, H.; Yang, Z.; An, G.; Tian, H.; Shou, C. Development of a novel albumin-based and maleimidopropionic acid-conjugated peptide with prolonged half-life and increased in vivo anti-tumor efficacy. Theranostics 2018, 8, 2094–2106. [Google Scholar] [CrossRef]
- Han, H.; Wang, J.; Chen, T.; Yin, L.; Jin, Q.; Ji, J. Enzyme-sensitive gemcitabine conjugated albumin nanoparticles as a versatile theranostic nanoplatform for pancreatic cancer treatment. J. Colloid Interface Sci. 2017, 507, 217–224. [Google Scholar] [CrossRef]
- Yue, C.; Liu, P.; Zheng, M.; Zhao, P.; Wang, Y.; Ma, Y.; Cai, L. IR-780 dye loaded tumor targeting theranostic nanoparticles for NIR imaging and photothermal therapy. Biomaterials 2013, 34, 6853–6861. [Google Scholar] [CrossRef] [PubMed]
- Arias, J.L. Liposomes in drug delivery: A patent review (2007–present). Expert Opin. Ther. Pat. 2013, 23, 1399–1414. [Google Scholar] [CrossRef] [PubMed]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975. [Google Scholar] [CrossRef] [PubMed]
- Ji, T.; Li, S.; Zhang, Y.; Lang, J.; Ding, Y.; Zhao, X.; Zhao, R.; Li, Y.; Shi, J.; Hao, J.; et al. An MMP-2 Responsive Liposome Integrating Antifibrosis and Chemotherapeutic Drugs for Enhanced Drug Perfusion and Efficacy in Pancreatic Cancer. ACS Appl. Mater. Interfaces 2016, 8, 3438–3445. [Google Scholar] [CrossRef] [PubMed]
- Raza, F.; Evans, L.; Motallebi, M.; Zafar, H.; Pereira-Silva, M.; Saleem, K.; Peixoto, D.; Rahdar, A.; Sharifi, E.; Veiga, F.; et al. Liposome-based diagnostic and therapeutic applications for pancreatic cancer. Acta Biomater. 2022, 157, 1–23. [Google Scholar] [CrossRef]
- Wang, X.; Liu, Y.; Xu, W.; Jia, L.; Chi, D.; Yu, J.; Wang, J.; He, Z.; Liu, X.; Wang, Y. Irinotecan and berberine co-delivery liposomes showed improved efficacy and reduced intestinal toxicity compared with Onivyde for pancreatic cancer. Drug Deliv. Transl. Res. 2021, 11, 2186–2197. [Google Scholar] [CrossRef]
- Ranjan, A.P.; Mukerjee, A.; Helson, L.; Gupta, R.; Vishwanatha, J.K. Efficacy of liposomal curcumin in a human pancreatic tumor xenograft model: Inhibition of tumor growth and angiogenesis. Anticancer. Res. 2013, 33, 3603–3609. [Google Scholar]
- Zinger, A.; Koren, L.; Adir, O.; Poley, M.; Alyan, M.; Yaari, Z.; Noor, N.; Krinsky, N.; Simon, A.; Gibori, H.; et al. Collagenase Nanoparticles Enhance the Penetration of Drugs into Pancreatic Tumors. ACS Nano 2019, 13, 11008–11021. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, F.; Jiang, X.; Zhao, X.; Wang, Y.; Kuai, Q.; Nie, G.; He, M.; Pan, Y.; Shi, W.; et al. Co-Delivery of Gemcitabine and Mcl-1 SiRNA via Cationic Liposome-Based System Enhances the Efficacy of Chemotherapy in Pancreatic Cancer. J. Biomed. Nanotechnol. 2019, 15, 966–978. [Google Scholar] [CrossRef]
- Passero, F.C., Jr.; Grapsa, D.; Syrigos, K.N.; Saif, M.W. The safety and efficacy of Onivyde (irinotecan liposome injection) for the treatment of metastatic pancreatic cancer following gemcitabine-based therapy. Expert Rev. Anticancer. Ther. 2016, 16, 697–703. [Google Scholar] [CrossRef]
- Wang-Gillam, A.; Li, C.-P.; Bodoky, G.; Dean, A.; Shan, Y.-S.; Jameson, G.; Macarulla, T.; Lee, K.-H.; Cunningham, D.; Blanc, J.F.; et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): A global, randomised, open-label, phase 3 trial. Lancet 2016, 387, 545–557. [Google Scholar] [CrossRef]
- Kaida, S.; Cabral, H.; Kumagai, M.; Kishimura, A.; Terada, Y.; Sekino, M.; Aoki, I.; Nishiyama, N.; Tani, T.; Kataoka, K. Visible Drug Delivery by Supramolecular Nanocarriers Directing to Single-Platformed Diagnosis and Therapy of Pancreatic Tumor ModelVisible DDS for Diagnosis and Therapy of Solid Tumors. Cancer Res. 2010, 70, 7031–7041. [Google Scholar] [CrossRef]
- Singh, A.P.; Biswas, A.; Shukla, A.; Maiti, P. Targeted therapy in chronic diseases using nanomaterial-based drug delivery vehicles. Signal Transduct. Target. Ther. 2019, 4, 33. [Google Scholar] [CrossRef]
- Srivastava, A.; Yadav, T.; Sharma, S.; Nayak, A.; Kumari, A.A.; Mishra, N. Polymers in drug delivery. J. Biosci. Med. 2015, 4, 69–84. [Google Scholar] [CrossRef]
- Wang, G.; Zhou, Z.; Zhao, Z.; Li, Q.; Wu, Y.; Yan, S.; Shen, Y.; Huang, P. Enzyme-Triggered Transcytosis of Dendrimer–Drug Conjugate for Deep Penetration into Pancreatic Tumors. ACS Nano 2020, 14, 4890–4904. [Google Scholar] [CrossRef]
- Wu, S.-T.; Fowler, A.; Garmon, C.B.; Fessler, A.B.; Ogle, J.D.; Grover, K.R.; Allen, B.C.; Williams, C.D.; Zhou, R.; Yazdanifar, M.; et al. Treatment of pancreatic ductal adenocarcinoma with tumor antigen specific-targeted delivery of paclitaxel loaded PLGA nanoparticles. BMC Cancer 2018, 18, 457. [Google Scholar] [CrossRef]
- Sun, J.; Wan, Z.; Chen, Y.; Xu, J.; Luo, Z.; Parise, R.A.; Diao, D.; Ren, P.; Beumer, J.H.; Lu, B.; et al. Triple drugs co-delivered by a small gemcitabine-based carrier for pancreatic cancer immunochemotherapy. Acta Biomater. 2020, 106, 289–300. [Google Scholar] [CrossRef]
- Sun, I.-C.; Yoon, H.Y.; Lim, D.-K.; Kim, K. Recent Trends in In Situ Enzyme-Activatable Prodrugs for Targeted Cancer Therapy. Bioconjug. Chem. 2020, 31, 1012–1024. [Google Scholar] [CrossRef]
- Santoni, M.; Miccini, F.; Cimadamore, A.; Piva, F.; Massari, F.; Cheng, L.; Lopez-Beltran, A.; Montironi, R.; Battelli, N. An update on investigational therapies that target STAT3 for the treatment of cancer. Expert Opin. Investig. Drugs 2021, 30, 245–251. [Google Scholar] [CrossRef]
- Bimonte, S.; Barbieri, A.; Leongito, M.; Piccirillo, M.; Giudice, A.; Pivonello, C.; de Angelis, C.; Granata, V.; Palaia, R.; Izzo, F. Curcumin AntiCancer Studies in Pancreatic Cancer. Nutrients 2016, 8, 433. [Google Scholar] [CrossRef]
- Bagley, A.F.; Ludmir, E.B.; Maitra, A.; Minsky, B.D.; Smith, G.L.; Das, P.; Koong, A.C.; Holliday, E.B.; Taniguchi, C.M.; Katz, M.H.; et al. NBTXR3, a first-in-class radioenhancer for pancreatic ductal adenocarcinoma: Report of first patient experience. Clin. Transl. Radiat. Oncol. 2022, 33, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Bort, G.; Lux, F.; Dufort, S.; Crémillieux, Y.; Verry, C.; Tillement, O. EPR-mediated tumor targeting using ultrasmall-hybrid nanoparticles: From animal to human with theranostic AGuIX nanoparticles. Theranostics 2020, 10, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Song, Y.; He, J.; Zhang, M.; Liu, J.; Ni, P. Zwitterionic shielded polymeric prodrug with folate-targeting and pH responsiveness for drug delivery. J. Mater. Chem. B 2019, 7, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Vrettos, E.I.; Mező, G.; Tzakos, A.G. On the design principles of peptide–drug conjugates for targeted drug delivery to the malignant tumor site. Beilstein J. Org. Chem. 2018, 14, 930–954. [Google Scholar] [CrossRef]
- Wang, Y.; Cheetham, A.G.; Angacian, G.; Su, H.; Xie, L.; Cui, H. Peptide–drug conjugates as effective prodrug strategies for targeted delivery. Adv. Drug Deliv. Rev. 2017, 110–111, 112–126. [Google Scholar] [CrossRef]
- Alas, M.; Saghaeidehkordi, A.; Kaur, K. Peptide–Drug Conjugates with Different Linkers for Cancer Therapy. J. Med. Chem. 2020, 64, 216–232. [Google Scholar] [CrossRef]
- Chavda, V.P.; Solanki, H.K.; Davidson, M.; Apostolopoulos, V.; Bojarska, J. Peptide-Drug Conjugates: A New Hope for Cancer Management. Molecules 2022, 27, 7232. [Google Scholar] [CrossRef]
- Guo, X.; Wang, L.; Wei, X.; Zhou, S. Polymer-based drug delivery systems for cancer treatment. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 3525–3550. [Google Scholar] [CrossRef]
- Manzur, A.; Oluwasanmi, A.; Moss, D.; Curtis, A.; Hoskins, C. Nanotechnologies in Pancreatic Cancer Therapy. Pharmaceutics 2017, 9, 39. [Google Scholar] [CrossRef]
- Seifu, M.F.; Nath, L.K. Polymer-Drug Conjugates: Novel Carriers for Cancer Chemotherapy. Polym. Technol. Mater. 2019, 58, 158–171. [Google Scholar] [CrossRef]
- Mosiane, K.S.; Nweke, E.E.; Balogun, M.; Fru, P.N. Polyethyleneglycol-Betulinic Acid (PEG-BA) Polymer-Drug Conjugate Induces Apoptosis and Antioxidation in a Biological Model of Pancreatic Cancer. Polymers 2023, 15, 448. [Google Scholar] [CrossRef]
- Almawash, S.A.; Mondal, G.; Mahato, R.I. Coadministration of Polymeric Conjugates of Docetaxel and Cyclopamine Synergistically Inhibits Orthotopic Pancreatic Cancer Growth and Metastasis. Pharm. Res. 2018, 35, 17. [Google Scholar] [CrossRef]
- Arias-Pinilla, G.A.; Modjtahedi, H. Therapeutic Application of Monoclonal Antibodies in Pancreatic Cancer: Advances, Challenges and Future Opportunities. Cancers 2021, 13, 1781. [Google Scholar] [CrossRef]
- Tolcher, A.W. Antibody drug conjugates: Lessons from 20 years of clinical experience. Ann. Oncol. 2016, 27, 2168–2172. [Google Scholar] [CrossRef]
- Birrer, M.J.; Moore, K.N.; Betella, I.; Bates, R.C. Antibody-Drug Conjugate-Based Therapeutics: State of the Science. JNCI J. Natl. Cancer Inst. 2019, 111, 538–549. [Google Scholar] [CrossRef]
- Parslow, A.C.; Parakh, S.; Lee, F.-T.; Gan, H.K.; Scott, A.M. Antibody–drug conjugates for cancer therapy. Molecules 2020, 25, 4764. [Google Scholar] [CrossRef]
- Sorbara, M.; Cordelier, P.; Bery, N. Antibody-Based Approaches to Target Pancreatic Tumours. Antibodies 2022, 11, 47. [Google Scholar] [CrossRef]
- Nagaoka, K.; Bai, X.; Ogawa, K.; Dong, X.; Zhang, S.; Zhou, Y.; Carlson, R.I.; Jiang, Z.-G.; Fuller, S.; Lebowitz, M.S.; et al. Anti-tumor activity of antibody drug conjugate targeting aspartate-β-hydroxylase in pancreatic ductal adenocarcinoma. Cancer Lett. 2019, 449, 87–98. [Google Scholar] [CrossRef]
- Nishigaki, T.; Takahashi, T.; Serada, S.; Fujimoto, M.; Ohkawara, T.; Hara, H.; Sugase, T.; Otsuru, T.; Saito, Y.; Tsujii, S.; et al. Anti-glypican-1 antibody–drug conjugate is a potential therapy against pancreatic cancer. Br. J. Cancer 2020, 122, 1333–1341. [Google Scholar] [CrossRef]
- Huang, J.; Agoston, A.T.; Guo, P.; Moses, M.A. A Rationally Designed ICAM1 Antibody Drug Conjugate for Pancreatic Cancer. Adv. Sci. 2020, 7, 2002852. [Google Scholar] [CrossRef]
- Xu, J.; Li, X.; Du, Y. Antibody–Pattern Recognition Receptor Agonist Conjugates: A Promising Therapeutic Strategy for Cancer. Adv. Biol. 2022, 6, 2101065. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Guo, H.; Li, L.; Zhang, Y.; Cui, J. The promising role of antibody drug conjugate in cancer therapy: Combining targeting ability with cytotoxicity effectively. Cancer Med. 2021, 10, 4677–4696. [Google Scholar] [CrossRef] [PubMed]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Marei, H.E.; Cenciarelli, C.; Hasan, A. Potential of antibody–drug conjugates (ADCs) for cancer therapy. Cancer Cell Int. 2022, 22, 255. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, J.; Nilvebrant, J.; Nygren, P.; Lehmann, F. Progress and Future Directions with Peptide-Drug Conjugates for Targeted Cancer Therapy. Molecules 2021, 26, 6042. [Google Scholar] [CrossRef]
- Xu, L.; Xu, S.; Xiang, T.; Liu, H.; Chen, L.; Jiang, B.; Yao, J.; Zhu, H.; Hu, R.; Chen, Z. Multifunctional building elements for the construction of peptide drug conjugates. Eng. Regen. 2022, 3, 92–109. [Google Scholar] [CrossRef]
- Hoppenz, P.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide-Drug Conjugates and Their Targets in Advanced Cancer Therapies. Front. Chem. 2020, 8, 571. [Google Scholar] [CrossRef]
- Heh, E.; Allen, J.; Ramirez, F.; Lovasz, D.; Fernandez, L.; Hogg, T.; Riva, H.; Holland, N.; Chacon, J. Peptide Drug Conjugates and Their Role in Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 829. [Google Scholar] [CrossRef]
- Cooper, B.M.; Iegre, J.; Donovan, D.H.O.; Halvarsson, M.; Spring, D.R. Peptides as a platform for targeted therapeutics for cancer: Peptide–drug conjugates (PDCs). Chem. Soc. Rev. 2021, 50, 1480–1494. [Google Scholar] [CrossRef]
- Berillo, D.; Yeskendir, A.; Zharkinbekov, Z.; Raziyeva, K.; Saparov, A. Peptide-Based Drug Delivery Systems. Medicina 2021, 57, 1209. [Google Scholar] [CrossRef]
- Fu, C.; Yu, L.; Miao, Y.; Liu, X.; Yu, Z.; Wei, M. Peptide–drug conjugates (PDCs): A novel trend of research and development on targeted therapy, hype or hope? Acta Pharm. Sin. B 2022, 13, 498–516. [Google Scholar] [CrossRef]
- Moore, K.M.; Desai, A.; Delgado, B.D.L.; Trabulo, S.M.D.; Reader, C.; Brown, N.F.; Murray, E.R.; Brentnall, A.; Howard, P.; Masterson, L.; et al. Integrin αvβ6-specific therapy for pancreatic cancer developed from foot-and-mouth-disease virus. Theranostics 2020, 10, 2930–2942. [Google Scholar] [CrossRef]
- Worm, D.J.; Els-Heindl, S.; Beck-Sickinger, A.G. Targeting of peptide-binding receptors on cancer cells with peptide-drug conjugates. Pept. Sci. 2020, 112, e24171. [Google Scholar] [CrossRef]
- de Mendoza, T.H.; Mose, E.S.; Botta, G.P.; Braun, G.B.; Kotamraju, V.R.; French, R.P.; Suzuki, K.; Miyamura, N.; Teesalu, T.; Ruoslahti, E.; et al. Tumor-penetrating therapy for β5 integrin-rich pancreas cancer. Nat. Commun. 2021, 12, 1541. [Google Scholar] [CrossRef]
- Peng, Z.-H.; Kopeček, J. Enhancing Accumulation and Penetration of HPMA Copolymer–Doxorubicin Conjugates in 2D and 3D Prostate Cancer Cells via iRGD Conjugation with an MMP-2 Cleavable Spacer. J. Am. Chem. Soc. 2015, 137, 6726–6729. [Google Scholar] [CrossRef]
- Kang, S.; Lee, S.; Park, S. iRGD Peptide as a Tumor-Penetrating Enhancer for Tumor-Targeted Drug Delivery. Polymers 2020, 12, 1906. [Google Scholar] [CrossRef]
- Peng, Z.-H.; Jogdeo, C.M.; Li, J.; Xie, Y.; Wang, Y.; Sheinin, Y.M.; Kopeček, J.; Oupický, D. Tumor Microenvironment-Responsive Polymeric iRGD and Doxorubicin Conjugates Reduce Spontaneous Lung Metastasis in an Orthotopic Breast Cancer Model. Pharmaceutics 2022, 14, 1725. [Google Scholar] [CrossRef]
- Ragozin, E.; Hesin, A.; Bazylevich, A.; Tuchinsky, H.; Bovina, A.; Zahavi, T.S.; Oron-Herman, M.; Kostenich, G.; Firer, M.; Rubinek, T.; et al. New somatostatin-drug conjugates for effective targeting pancreatic cancer. Bioorg. Med. Chem. 2018, 26, 3825–3836. [Google Scholar] [CrossRef]
- Dean, A.; Gill, S.; McGregor, M.; Broadbridge, V.; Järveläinen, H.A.; Price, T. Dual αV-integrin and neuropilin-1 targeting peptide CEND-1 plus nab-paclitaxel and gemcitabine for the treatment of metastatic pancreatic ductal adenocarcinoma: A first-in-human, open-label, multicentre, phase 1 study. Lancet Gastroenterol. Hepatol. 2022, 7, 943–951. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Chen, B.; Dai, W.; He, B.; Zhang, H.; Wang, X.; Wang, Y.; Zhang, Q. Current Multistage Drug Delivery Systems Based on the Tumor Microenvironment. Theranostics 2017, 7, 538–558. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Hsiao, A.; Mann, A.P.; Landry, M.G.; Meric-Bernstam, F.; Ferrari, M. Nanomedicine in cancer therapy: Innovative trends and prospects. Cancer Sci. 2011, 102, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Sangai, T.; Hsiao, A.; Ferrati, S.; Bai, L.; Liu, X.; Meric-Bernstam, F.; Ferrari, M. Multistage delivery of chemotherapeutic nanoparticles for breast cancer treatment. Cancer Lett. 2013, 334, 245–252. [Google Scholar] [CrossRef]
- Stylianopoulos, T.; Jain, R.K. Design considerations for nanotherapeutics in oncology. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 1893–1907. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.; Brown, B.S.; Quattrocchi, N.; Evangelopoulos, M.; Ferrari, M.; Tasciotti, E. Multifunctional to multistage delivery systems: The evolution of nanoparticles for biomedical applications. Chin. Sci. Bull. 2012, 57, 3961–3971. [Google Scholar] [CrossRef] [PubMed]
- Stylianopoulos, T.; Wong, C.; Bawendi, M.G.; Jain, R.K.; Fukumura, D. Multistage nanoparticles for improved delivery into tumor tissue. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2012; Volume 508, pp. 109–130. [Google Scholar]
- Wong, C.; Stylianopoulos, T.; Cui, J.; Martin, J.; Chauhan, V.P.; Jiang, W.; Popović, Z.; Jain, R.K.; Bawendi, M.G.; Fukumura, D. Multistage nanoparticle delivery system for deep penetration into tumor tissue. Proc. Natl. Acad. Sci. USA 2011, 108, 2426–2431. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Yao, Z.; Ding, J.; Min, Q.; Jiang, L.-P.; Zhu, J.-J. Cascaded Aptamers-Governed Multistage Drug-Delivery System Based on Biodegradable Envelope-Type Nanovehicle for Targeted Therapy of HER2-Overexpressing Breast Cancer. ACS Appl. Mater. Interfaces 2018, 10, 34050–34059. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, X.; Qiu, L. The anti-tumor efficacy of curcumin when delivered by size/charge-changing multistage polymeric micelles based on amphiphilic poly(β-amino ester) derivates. Biomaterials 2014, 35, 3467–3479. [Google Scholar] [CrossRef]
- Serda, R.E.; Godin, B.; Blanco, E.; Chiappini, C.; Ferrari, M. Multi-stage delivery nano-particle systems for therapeutic applications. Biochim. Et Biophys. Acta (BBA)-Gen. Subj. 2011, 1810, 317–329. [Google Scholar] [CrossRef]
- Cun, X.; Chen, J.; Li, M.; He, X.; Tang, X.; Guo, R.; Deng, M.; Li, M.; Zhang, Z.; He, Q. Tumor-Associated Fibroblast-Targeted Regulation and Deep Tumor Delivery of Chemotherapeutic Drugs with a Multifunctional Size-Switchable Nanoparticle. ACS Appl. Mater. Interfaces 2019, 11, 39545–39559. [Google Scholar] [CrossRef]
- Li, H.J.; Du, J.Z.; Liu, J.; Du, X.J.; Shen, S.; Zhu, Y.H.; Wang, X.; Ye, X.; Nie, S.; Wang, J. Smart superstructures with ultrahigh pH-sensitivity for targeting acidic tumor microenvironment: Instantaneous size switching and improved tumor penetration. ACS Nano 2016, 10, 6753–6761. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
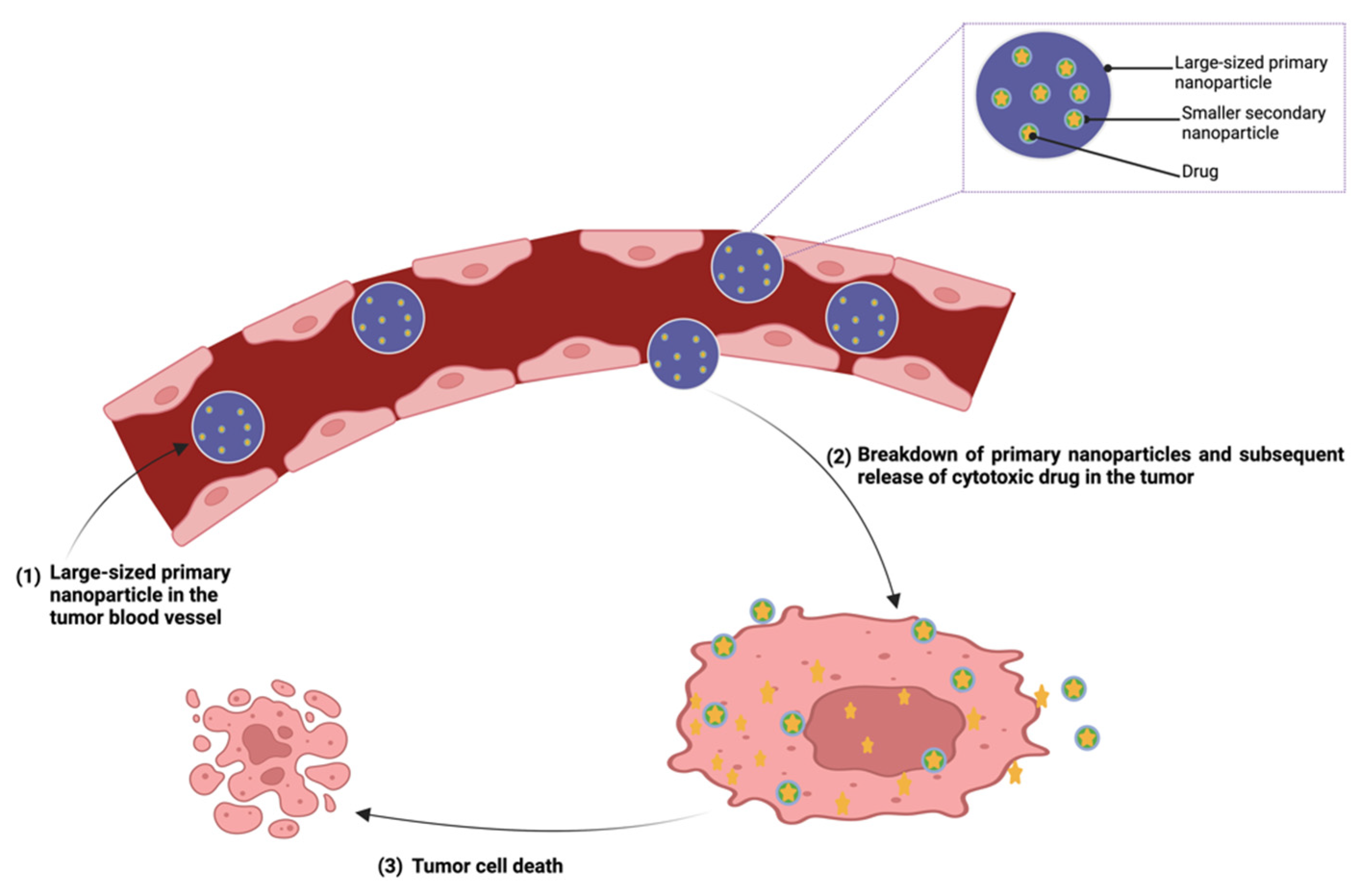{kind=link}
| Anticancer Agent | Molecular Target | Phase | Sponsor | ClinicalTrials.gov Identifier |
|---|---|---|---|---|
| Curcumin and doxorubicin (Imx-110) | Stat3/NF-kB/poly-tyrosine kinase/topoisomerase II | 1/2a | Immix Biopharma Australia Pty Ltd. | NCT03382340 |
| Inorganic hafnium oxide (NBTXR3) | Radiation | 1 | M.D. Anderson Cancer Center | NCT04484909 |
| AGuIX-NP (Theranostic agent) | EPR effect | 1/2 | Dana-Farber Cancer Institute | NCT04789486 |
| Anticancer Agent | Molecular Target | Phase | Sponsor | ClinicalTrials.gov Identifier |
|---|---|---|---|---|
| Monomethyl auristatin E (TORL-2-307-ADC) | Claudin 18.2 | 1 | TORL Biotherapeutics, LLC | NCT05156866 |
| Anthracycline PNU-159682 (SOT102) | Claudin 18.2 | 1/2 | SOTIO Biotech | NCT05525286 |
| Auristatin moiety (A166) | * HER2 | 1/2 | Klus Pharma Inc. | NCT03602079 |
| Monomethyl auristatin E (XB002) | Tissue factor | 1 | Exelixis | NCT04925284 |
| Duocarmycin analog (vobramitamab duocarmazine) | B7-homolog 3 | 1 | MacroGenics | NCT05293496 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olajubutu, O.; Ogundipe, O.D.; Adebayo, A.; Adesina, S.K. Drug Delivery Strategies for the Treatment of Pancreatic Cancer. Pharmaceutics 2023, 15, 1318. https://doi.org/10.3390/pharmaceutics15051318
Olajubutu O, Ogundipe OD, Adebayo A, Adesina SK. Drug Delivery Strategies for the Treatment of Pancreatic Cancer. Pharmaceutics. 2023; 15(5):1318. https://doi.org/10.3390/pharmaceutics15051318
Chicago/Turabian StyleOlajubutu, Oluwabukunmi, Omotola D. Ogundipe, Amusa Adebayo, and Simeon K. Adesina. 2023. "Drug Delivery Strategies for the Treatment of Pancreatic Cancer" Pharmaceutics 15, no. 5: 1318. https://doi.org/10.3390/pharmaceutics15051318