
Linker-Free Synthesis of Antimicrobial Peptides Using a Novel Cleavage Reagent: Characterisation of the Molecular and Ionic Composition by nanoESI-HR MS
 , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
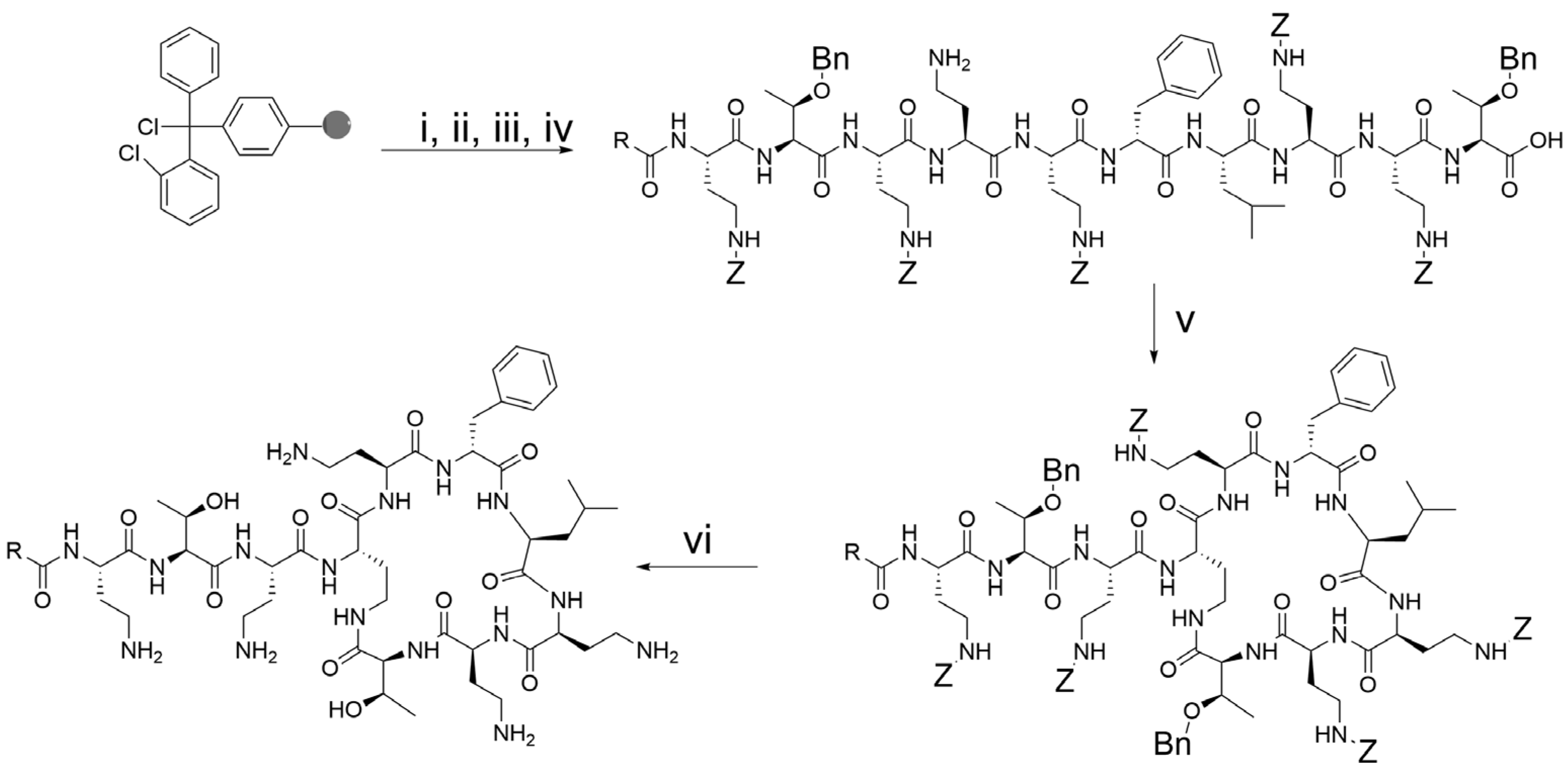
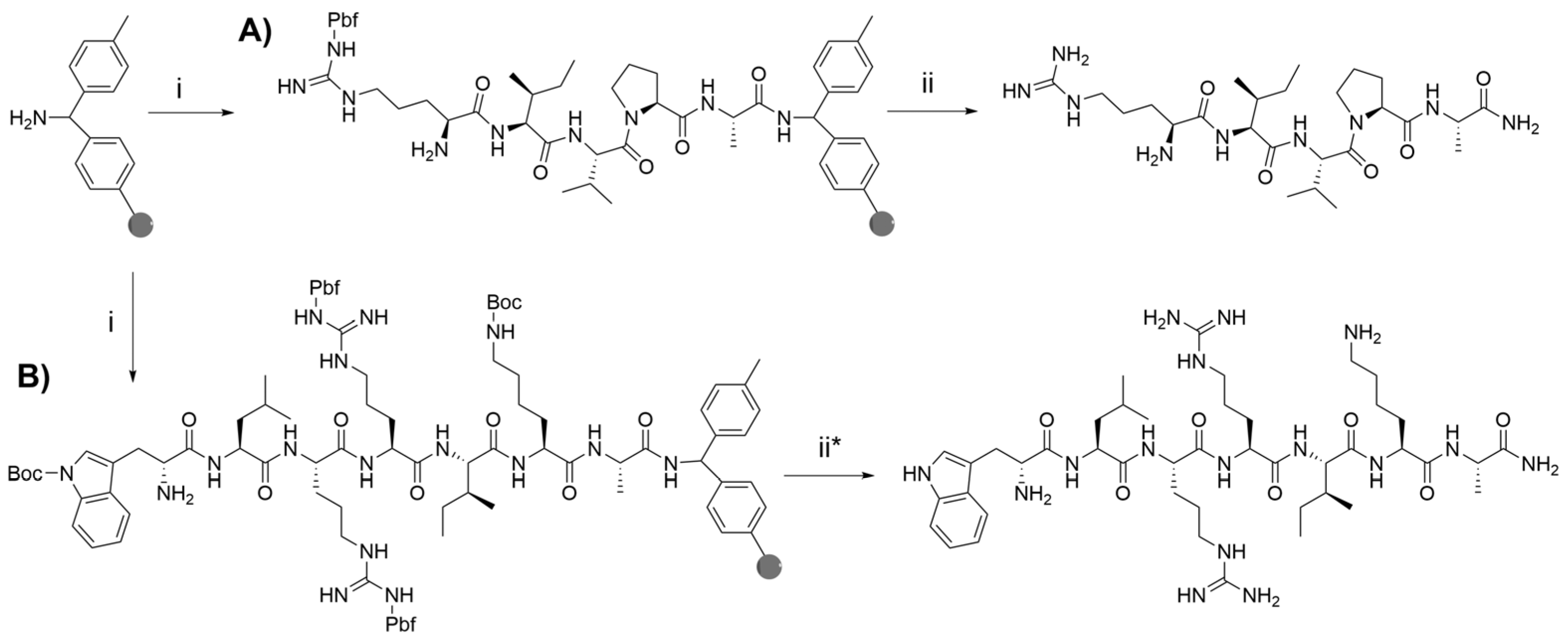
2.2. Peptide Synthesis and Purification
2.3. Peptide Counterion Exchange
2.4. CHNS Elemental Analysis
2.5. Chloride Determination by Titration
2.6. Nano-Electrospray Ionisation High-Resolution Mass Spectrometry
2.7. Electrospray Ionisation Mass Spectrometry
2.8. Antibacterial Susceptibility Testing
3. Results
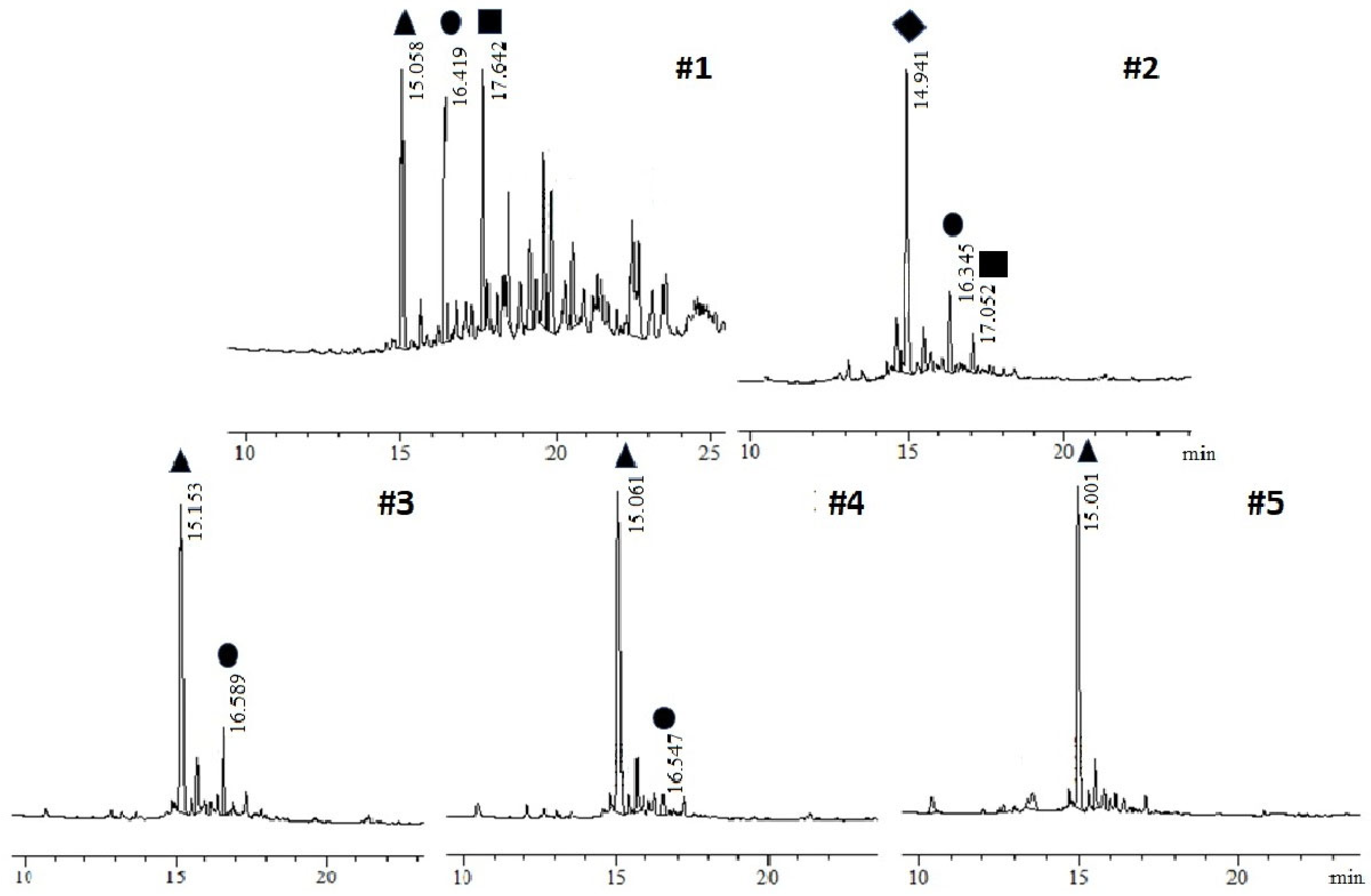
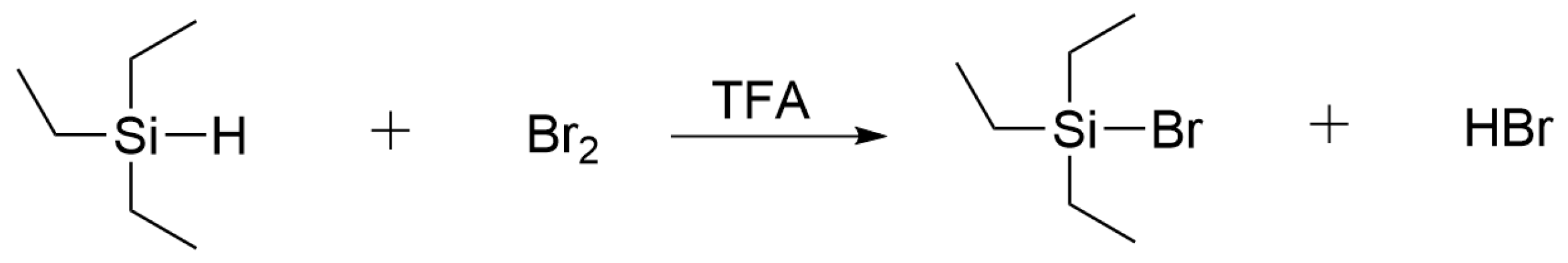
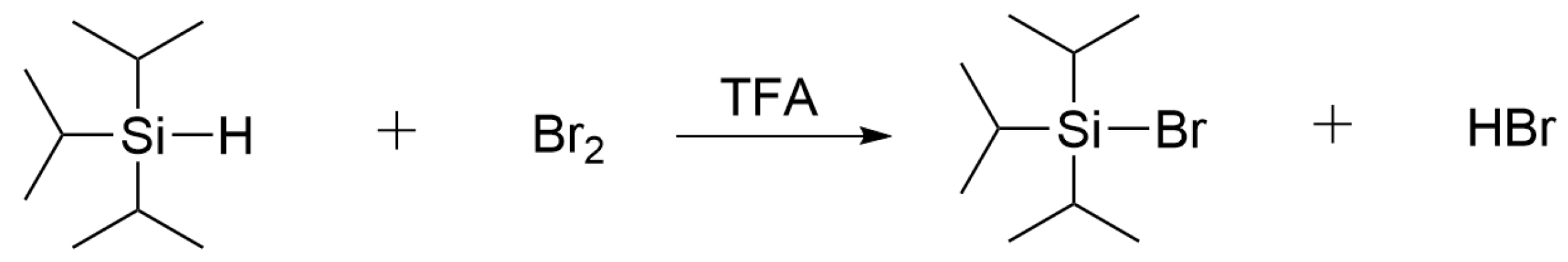
3.1. Novel Cleavage Reagent
3.2. CHNS Elemental Analysis
3.3. Chloride Determination by Titration
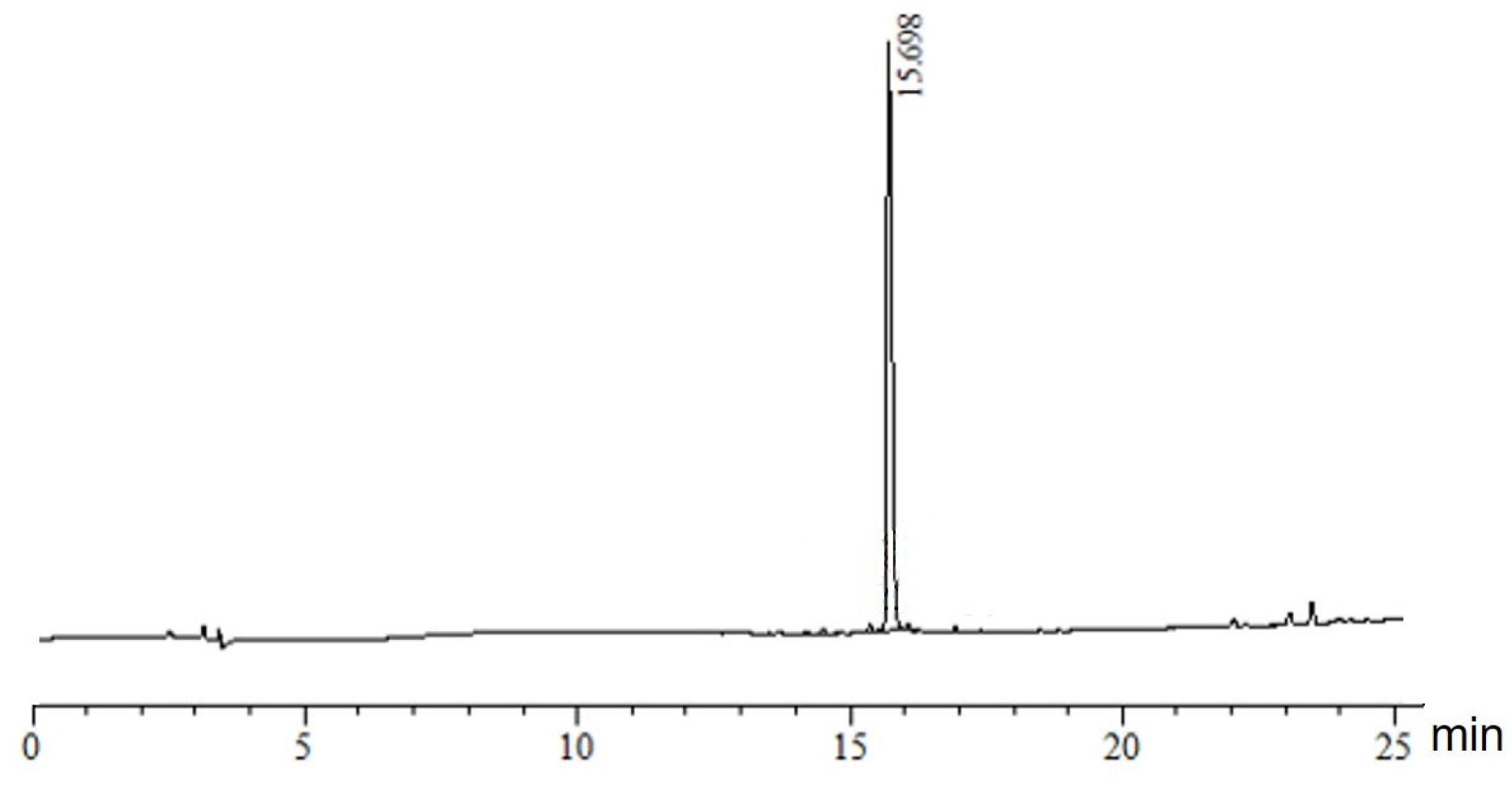
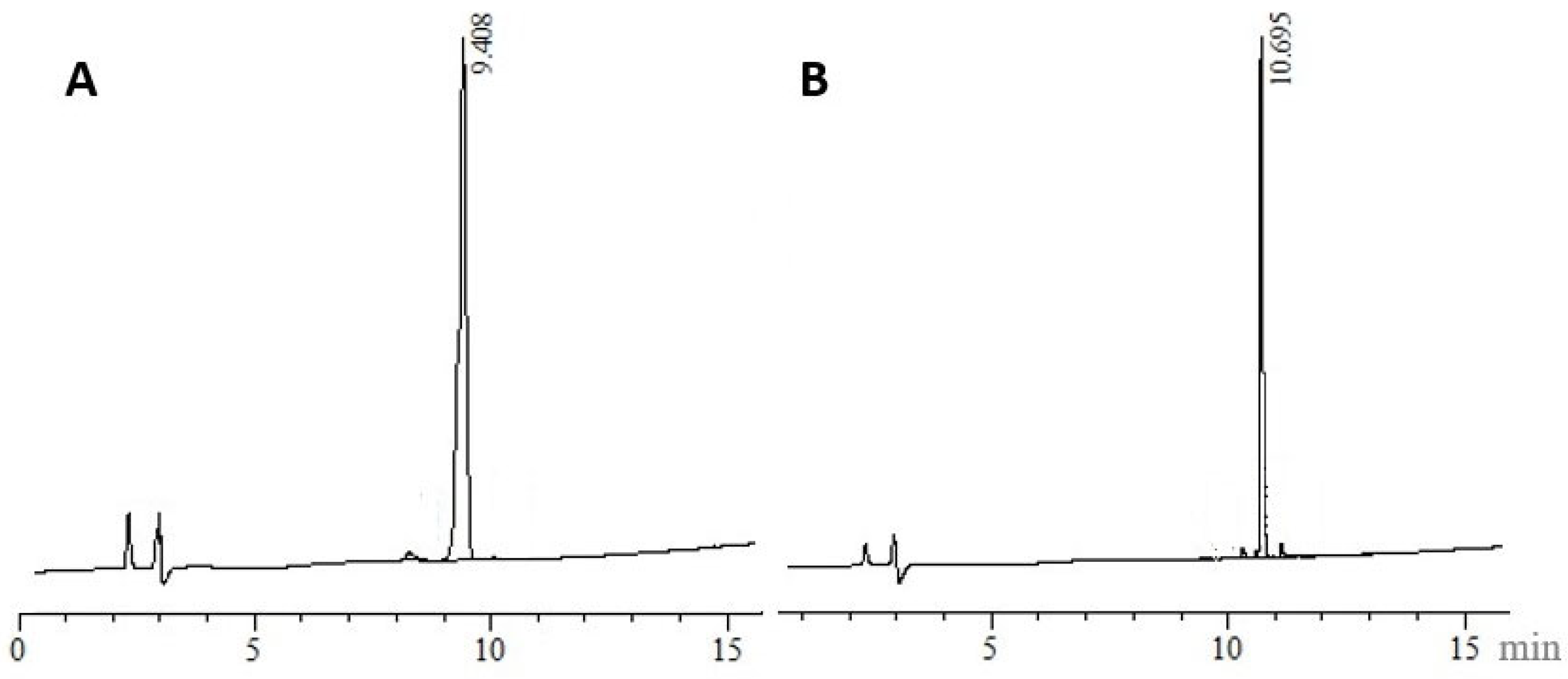
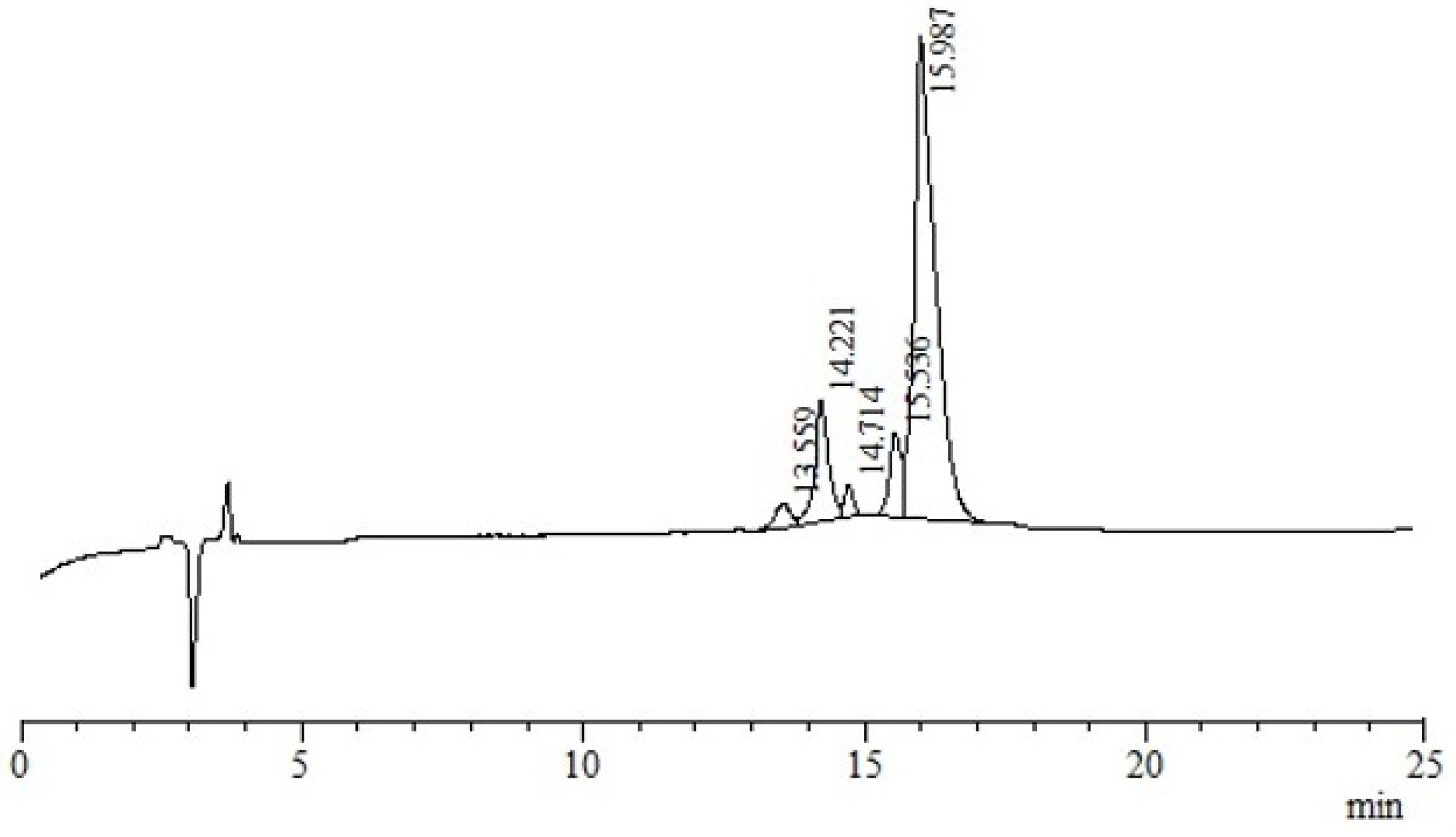
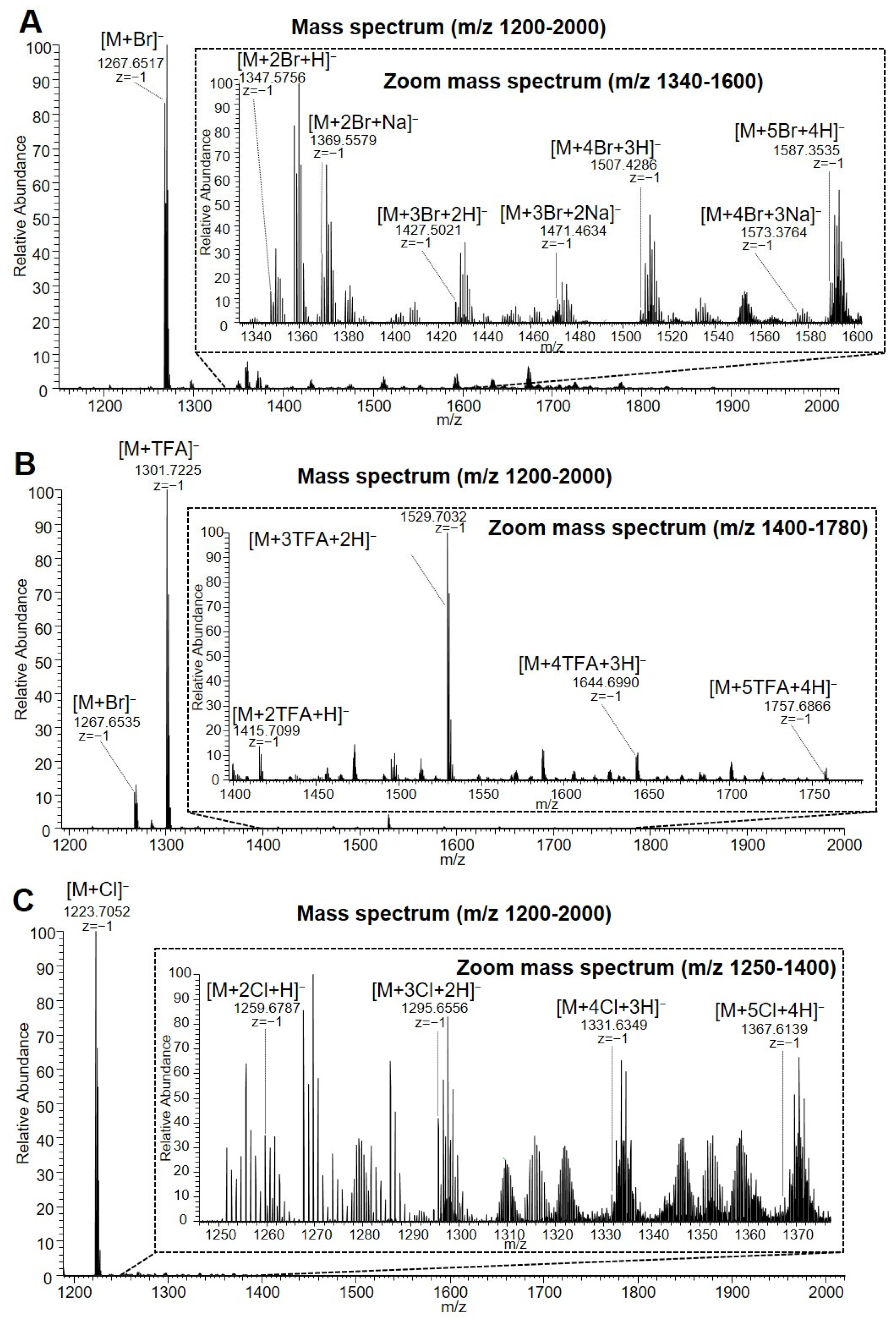
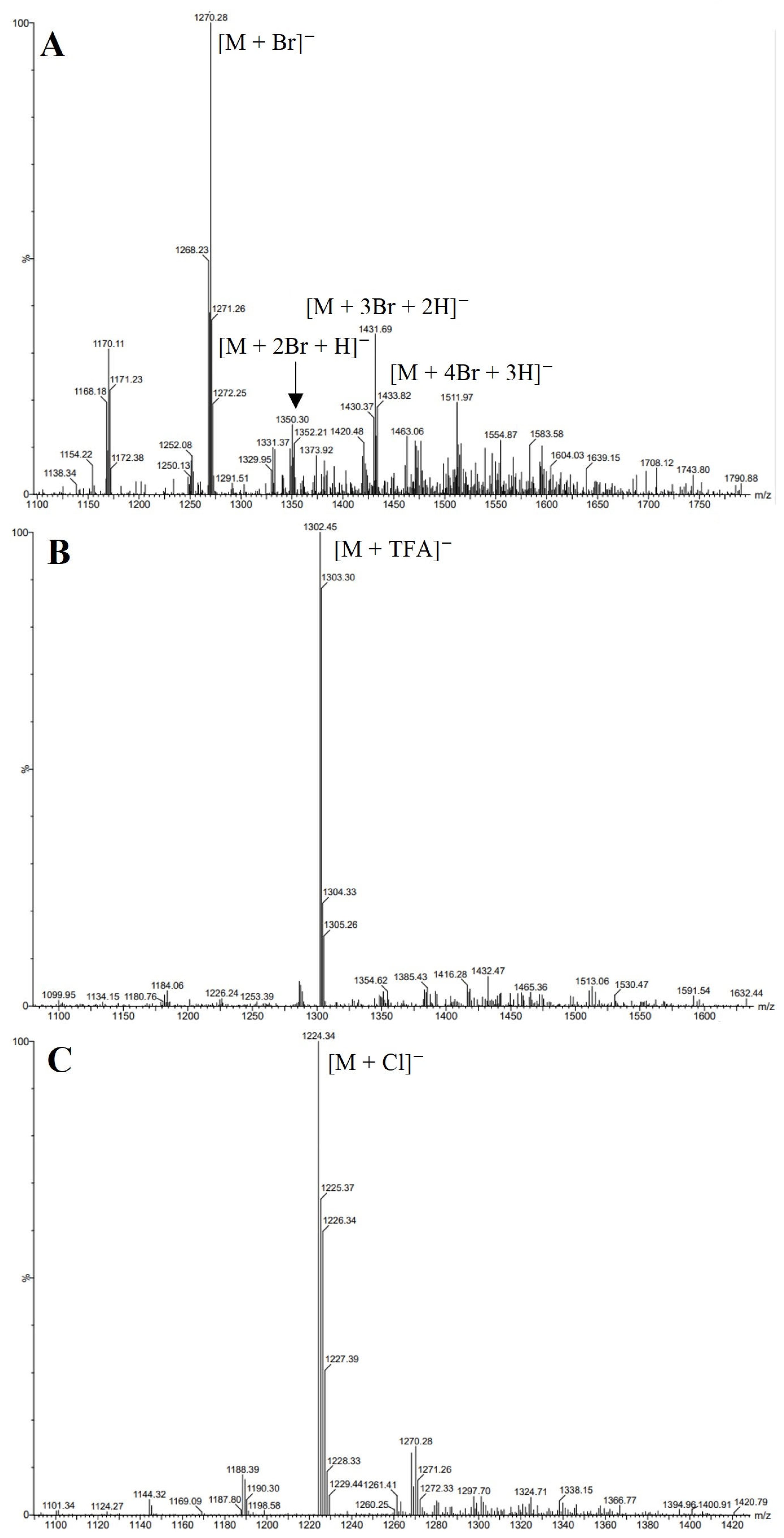
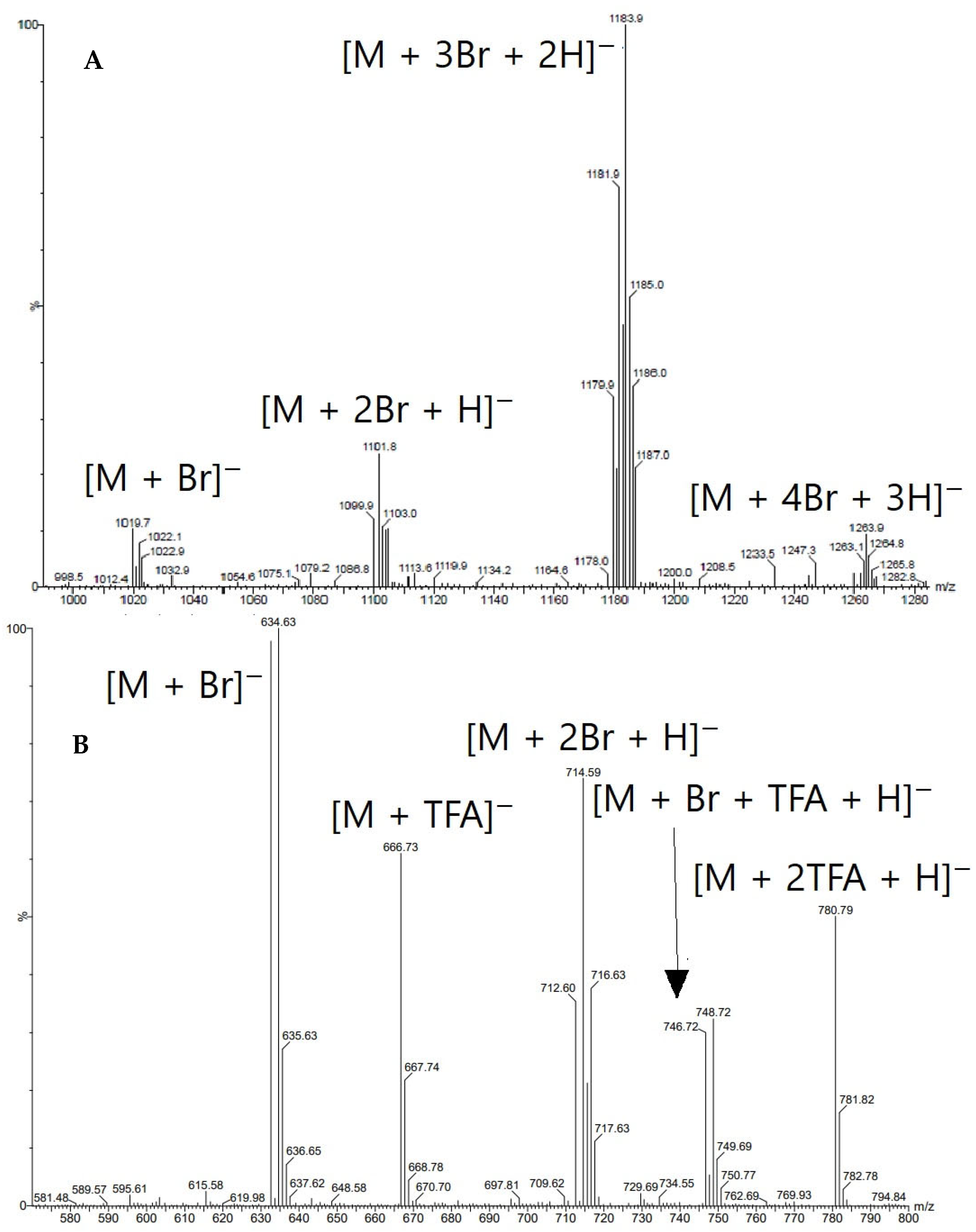
3.4. Electrospray Ionisation Mass Spectrometry (nanoESI-MS and ESI-MS)
3.5. Antimicrobial Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jadhav, K.B.; Woolcock, K.J.; Muttenthaler, M. Anhydrous Hydrogen Fluoride Cleavage in Boc Solid Phase Peptide Synthesis. Methods Mol. Biol. 2020, 2103, 41–57. [Google Scholar]
- Albericio, F.; Isidro-llobet, A.; Mercedes, A. Amino Acid Protecting Groups. Chem. Rev. 2009, 109, 2455–2504. [Google Scholar]
- Ben-Ishai, D.; Berger, A. Cleavage of N-Carbobenzoxy Groups by Dry Hydrogen Bromide and Hydrogen Chloride. J. Org. Chem. 1952, 17, 1564–1570. [Google Scholar] [CrossRef]
- Merrifield, R.B. Solid Phase Peptide Synthesis. I. The synthesis of a tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Sakakibara, S.; Shimonishi, Y.; Kishida, Y.; Okada, M.; Sugihara, H. Use of Anhydrous Hydrogen Fluoride in Peptide Synthesis. I. Behavior of Various Protective Groups in Anhydrous Hydrogen Fluoride. Bull. Chem. Soc. Jpn. 1967, 40, 2164–2167. [Google Scholar] [CrossRef]
- Tam, J.P.; Heath, W.F.; Merrifield, R.B. SN2 Deprotection of Synthetic Peptides with a Low Concentration of HF in Dimethyl Sulfide: Evidence and Application in Peptide Synthesis. J. Am. Chem. Soc. 1983, 105, 6442–6455. [Google Scholar] [CrossRef]
- Yajima, H.; Fujii, N.; Ogawa, H.; Kawatani, H. Trifluoromethanesulphonic Acid as a Deprotecting Reagent in Peptide Chemistry. J. Chem. Soc. Chem. Commun. 1974, 3, 107–108. [Google Scholar] [CrossRef]
- Tam, J.P.; Heath, W.F.; Merrifield, R.B. Mechanisms for the Removal of Benzyl Protecting Groups in Synthetic Peptides by Trifluoromethanesulfonic Acid Trifluoroacetic Acid-Dimethyl Sulfide. J. Am. Chem. Soc. 1986, 108, 5242–5251. [Google Scholar] [CrossRef]
- Yajima, H.; Fujii, N.; Funakoshi, S.; Watanabe, T.; Murayama, E.; Otaka, A. New Strategy for the Chemical Synthesis of Proteins. Tetrahedron 1988, 44, 805–819. [Google Scholar] [CrossRef]
- Hughes, J.L.; Leopold, E.J. Cleavage and Deprotection of Peptides from MBHA617 Resin with Bromotrimethylsilane. Tetrahedron Lett. 1993, 34, 7713–7716. [Google Scholar] [CrossRef]
- Jones, D.A. Hydrogenation of Protected Leucine Enkephalin from a Resin during Solid Phase Synthesis. Tetrahedron Lett. 1977, 33, 2853–2856. [Google Scholar] [CrossRef]
- Schlatter, J.M.; Mazur, R.H. Hydrogenation in Solid Phase Peptide Synthesis. I. Removal of Product from the Resin. Tetrahedron Lett. 1977, 18, 2851–2852. [Google Scholar] [CrossRef]
- Jackson, A.E.; Johnstone, R.A.W. Rapid, Selective Removal of Benzyloxycarbonyl Groups from Peptides by Catalytic Transfer Hydrogenation. Synthesis 1976, 10, 685–687. [Google Scholar] [CrossRef]
- Gowda, D.C.; Abiraj, K. Heterogeneous Catalytic Transfer Hydrogenation in Peptide Synthesis. Int. J. Pept. Res. Ther. 2002, 9, 153–165. [Google Scholar] [CrossRef]
- Rabanal, F.; Cajal, Y. Recent Advances and Perspectives in the Design and Development of Polymyxins. Nat. Prod. Rep. 2017, 34, 886–908. [Google Scholar] [CrossRef] [PubMed]
- Segovia, R.; Solé, J.; Marqués, A.M.; Cajal, Y.; Rabanal, F. Unveiling the Membrane and Cell Wall Action of Antimicrobial Cyclic Lipopeptides: Modulation of the Spectrum of Activity. Pharmaceutics 2021, 13, 2180. [Google Scholar] [CrossRef]
- Grau-Campistany, A.; Strandberg, E.; Wadhwani, P.; Reichert, J.; Bürck, J.; Rabanal, F.; Ulrich, A.S. Hydrophobic Mismatch Demonstrated for Membranolytic Peptides, and Their Use as Molecular Rulers to Measure Bilayer Thickness in Native Cells. Sci. Rep. 2015, 5, 9388. [Google Scholar] [CrossRef]
- Roberts, K.D.; Zhu, Y.; Azad, M.A.; Han, M.L.; Wang, J.; Wang, L.; Yu, H.H.; Horne, A.S.; Pinson, J.A.; Rudd, D.; et al. Synthetic Lipopeptide Targeting Top-Priority Multidrug Resistant Gram-Negative Pathogens. Nat. Commun. 2022, 13, 1625. [Google Scholar] [CrossRef]
- Clausell, A.; Rabanal, F.; Garcia-Subirats, M.; Alsina, M.A.; Cajal, Y. Synthesis and Membrane Action of Polymyxin B Analogues. Luminescence 2005, 20, 117–123. [Google Scholar] [CrossRef]
- Lepak, A.J.; Wang, W.; Andes, D.R. Pharmacodynamic Evaluation of MRX-8, a Novel Polymyxin, in the Neutropenic Mouse Thigh and Lung Infection Models against Gram-Negative Pathogens. Antimicrob. Agents Chemother. 2020, 64, e01517-20. [Google Scholar] [CrossRef]
- Brown, P.; Abbott, E.; Abdulle, O.; Boakes, S.; Coleman, S.; Divall, N.; Duperchy, E.; Moss, S.; Rivers, D.; Simonovic, M.; et al. Design of Next Generation Polymyxins with Lower Toxicity: The Discovery of SPR206. ACS Infect. Dis. 2019, 5, 1645–1656. [Google Scholar] [CrossRef]
- Kudrimoti, M.; Curtis, A.; Azawi, S.; Worden, F.; Katz, S.; Adkins, D.; Bonomi, M.; Elder, J.; Sonis, S.T.; Straube, R.; et al. Dusquetide: A Novel Innate Defense Regulator 646 Demonstrating a Significant and Consistent Reduction in the Duration of Oral Mucositis in Preclinical Data and a Randomized, Placebo-Controlled Phase 2a Clinical Study. J. Biotechnol. 2016, 239, 115–125. [Google Scholar] [CrossRef] [PubMed]
- U.S. National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT03237325 (accessed on 11 February 2023).
- Mohamed, M.F.; Brezden, A.; Mohammad, H.; Chmielewski, J.; Seleem, M.N. A Short D-Enantiomeric Antimicrobial Peptide with Potent Immunomodulatory and Antibiofilm Activity against Multidrug-Resistant Pseudomonas Aeruginosa and Acinetobacter Baumannii. Sci. Rep. 2017, 7, 6953. [Google Scholar] [CrossRef] [PubMed]
- Rabanal, F.; Rodriguez-Nuñez, M.; Cajal, Y. Method for the Deblocking/Deprotection of Compounds Obtained by Solid Phase Synthesis. WO Patent Application No. WO2009000950A1, 20 June 2008. [Google Scholar]
- Deans, D.R.; Eaborn, C. Organosilicon compounds. Part IX: The Reaction between Iodine and Trisubstituted Silanes. J. Chem. Soc. 1954, 3169–3173. [Google Scholar] [CrossRef]
- Sharma, S.K.; Wu, A.D.; Chandramouli, N.; Fotsch, C.; Kardash, G.; Bair, K.W. Solid-Phase Total Synthesis of Polymyxin B1. J. Pept. Res. 1999, 53, 501–506. [Google Scholar] [CrossRef]
- Ramesh, S.; Govender, T.; Kruger, H.G.; Albericio, F.; De La Torre, B.G. An Improved and Efficient Strategy for the Total Synthesis of a Colistin-like Peptide. Tetrahedron Lett. 2016, 57, 1885–1888. [Google Scholar] [CrossRef]
- Rink, H. Solid-Phase Synthesis of Protected Peptide Fragments Using a Trialkoxy Diphenyl-Methylester Resin. Tetrahedron Lett. 1987, 28, 3787–3790. [Google Scholar] [CrossRef]
- Wang, S.S.; Wang, B.S.; Hughes, J.L.; Leopold, E.J.; Wu, C.R.; Tam, J.P. Cleavage and Deprotection of Peptides on MBHA-Resin with Hydrogen Bromide. Int. J. Pept. Protein Res. 1992, 40, 344–349. [Google Scholar] [CrossRef]
- Choi, H.; Aldrich, J.V. Comparison of Methods for the Fmoc Solid-phase Synthesis and Cleavage of a Peptide Containing Both Tryptophan and Arginine. Int. J. Pept. Protein Res. 1993, 42, 58–63. [Google Scholar] [CrossRef]
- Rocheleau, M.-J. Analytical Methods for Determination of Counter-Ions in Pharmaceutical Salts. Curr. Pharm. Anal. 2008, 4, 25–32. [Google Scholar] [CrossRef]
- Patterson, R.K. Automated Pregl-Dumas Technique for Determining Total Carbon, Hydrogen, and Nitrogen in Atmospheric. Aerosols. Anal. Chem. 1973, 45, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Siu, K.W.M.; Guevremont, R.; Le Blanc, J.C.Y. Complexes of Silver(I) with Peptides and Proteins as Produced in Electrospray Mass Spectrometry. J. Am. Soc. Mass Spectrom. 1997, 8, 781–792. [Google Scholar] [CrossRef]
- Chabert, V.; Hologne, M.; Sénèque, O.; Crochet, A.; Walker, O.; Fromm, K.M. Model Peptide Studies of Ag+ Binding Sites from the Silver Resistance Protein SilE. Chem. Commun. 2017, 53, 6105–6108. [Google Scholar] [CrossRef]
- Bellina, B.; Compagnon, I.; MacAleese, L.; Chirot, F.; Lemoine, J.; Maître, P.; Broyer, M.; Antoine, R.; Kulesza, A.; Mitrić, R.; et al. Binding Motifs of Silver in Prion Octarepeat Model Peptides: A Joint Ion Mobility, IR and UV Spectroscopies, and Theoretical Approach. Phys. Chem. Chem. Phys. 2012, 14, 11433–11440. [Google Scholar] [CrossRef]
- Lentz, N.B.; Houk, R.S. Counter Ion Clusters. Am. Soc. Mass Spectrom. 2007, 18, 285–293. [Google Scholar] [CrossRef]
- Segovia, R. Novel Antimicrobial Peptides for Therapeutic Applications: Design, Synthesis, Characterization, and Evaluation of Their Biological and Biophysical Activity. Ph.D. Thesis, University of Barcelona, Barcelona, Spain, 2022. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acidic Mixture | HBr wt% | Volume Ratio | Time | Crude Purity | |
|---|---|---|---|---|---|
| #1 | TFA/TMSCl/TES/HBr 33% of AcOH | 1.8 | 64.2:25.8:5:5 | 2 h | 6% |
| #2 | TFA/TES/Br2 (TFA/HBr/Et3SiBr/TES) | 9.8 | 47.5:45:7.5 | 1 h | 0% |
| #3 | TFA/TES/Br2 (TFA/HBr/Et3SiBr/TES) | 2.8 | 82.5:15:2.5 | 30 min | 60% |
| #4 | TFA/TES/Br2 (TFA/HBr/Et3SiBr/TES) | 2.8 | 82.5:15:2.5 | 45 min | 75% |
| #5 | TFA/TES/Br2 (TFA/HBr/Et3SiBr/TES) | 2.8 | 82.5:15:2.5 | 90 min | 71% |
| Peptide | Sequence |
|---|---|
| Dusquetide | H-Arg-Ile-Val-Pro-Ala-NH2 |
| RR4 (8–14) | H-Trp-Leu-Arg-Arg-Ile-Lys-Ala-NH2 |
| Amount of Sample [mg] | Consumption of AgNO3 0.01 M [mL] | Cl Content [%] | Mean | Standard Deviation |
|---|---|---|---|---|
| 3.87 | 1.4012 | 12.69 | 12.70 | 0.00087 |
| 3.22 | 1.1674 | 12.67 | ||
| 3.59 | 1.3069 | 12.74 |
| Polymyxin Salts | MIC [μg·mL−1] Pseudomonas aeruginosa ATCC 27853 |
|---|---|
| Polymyxin B, sulfate salt, commercial | 1 |
| Polymyxin B3, trifluoroacetate salt, synthetic | 1 |
| Polymyxin B3, hydrochloride salt, synthetic | 1 |
| Polymyxin B3, hydrobromide salt, synthetic | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Segovia, R.; Díaz-Lobo, M.; Cajal, Y.; Vilaseca, M.; Rabanal, F. Linker-Free Synthesis of Antimicrobial Peptides Using a Novel Cleavage Reagent: Characterisation of the Molecular and Ionic Composition by nanoESI-HR MS. Pharmaceutics 2023, 15, 1310. https://doi.org/10.3390/pharmaceutics15041310
Segovia R, Díaz-Lobo M, Cajal Y, Vilaseca M, Rabanal F. Linker-Free Synthesis of Antimicrobial Peptides Using a Novel Cleavage Reagent: Characterisation of the Molecular and Ionic Composition by nanoESI-HR MS. Pharmaceutics. 2023; 15(4):1310. https://doi.org/10.3390/pharmaceutics15041310
Chicago/Turabian StyleSegovia, Roser, Mireia Díaz-Lobo, Yolanda Cajal, Marta Vilaseca, and Francesc Rabanal. 2023. "Linker-Free Synthesis of Antimicrobial Peptides Using a Novel Cleavage Reagent: Characterisation of the Molecular and Ionic Composition by nanoESI-HR MS" Pharmaceutics 15, no. 4: 1310. https://doi.org/10.3390/pharmaceutics15041310