
Profiling Docetaxel in Plasma and Urine Samples from a Pediatric Cancer Patient Using Ultrasound-Assisted Dispersive Liquid–Liquid Microextraction Combined with LC–MS/MS
 , , and
, , and
Abstract
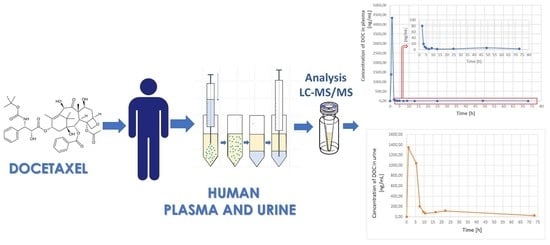
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Sample Collection
2.3. LC–MS/MS Analysis
2.4. Preparation of Plasma and Urine Standards
2.5. DLLME Procedure
2.6. Validation of Analytical Methods
2.7. Application for DOC Profiling in Real Plasma and Urine Samples
3. Results and Discussion
3.1. Optimization of UA-DLLME-LC–MS/MS Conditions
3.2. Method Validation
3.2.1. Selectivity
3.2.2. Linearity
3.2.3. Limit of Detection and Limit of Quantification
3.2.4. Accuracy and Precision
3.2.5. Carry Over and Recovery Results
3.2.6. Stability Study
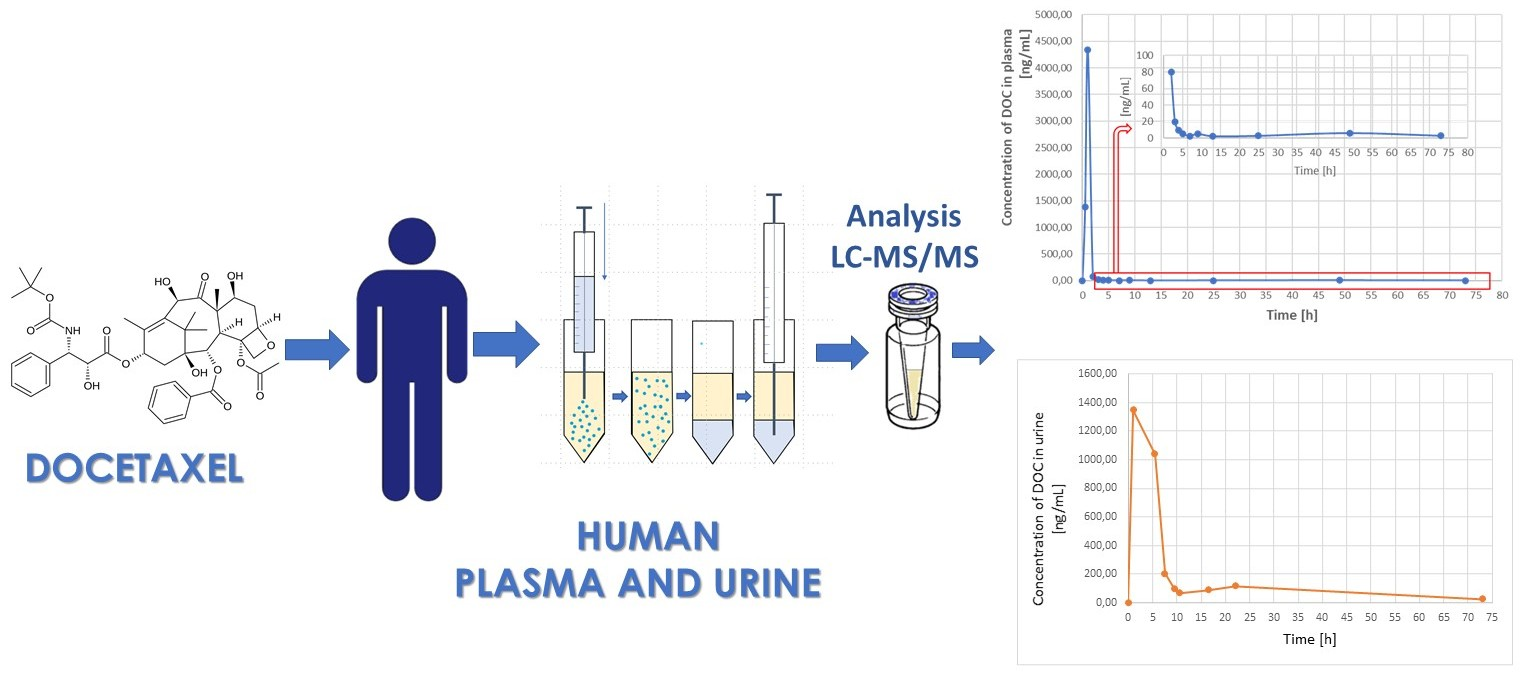
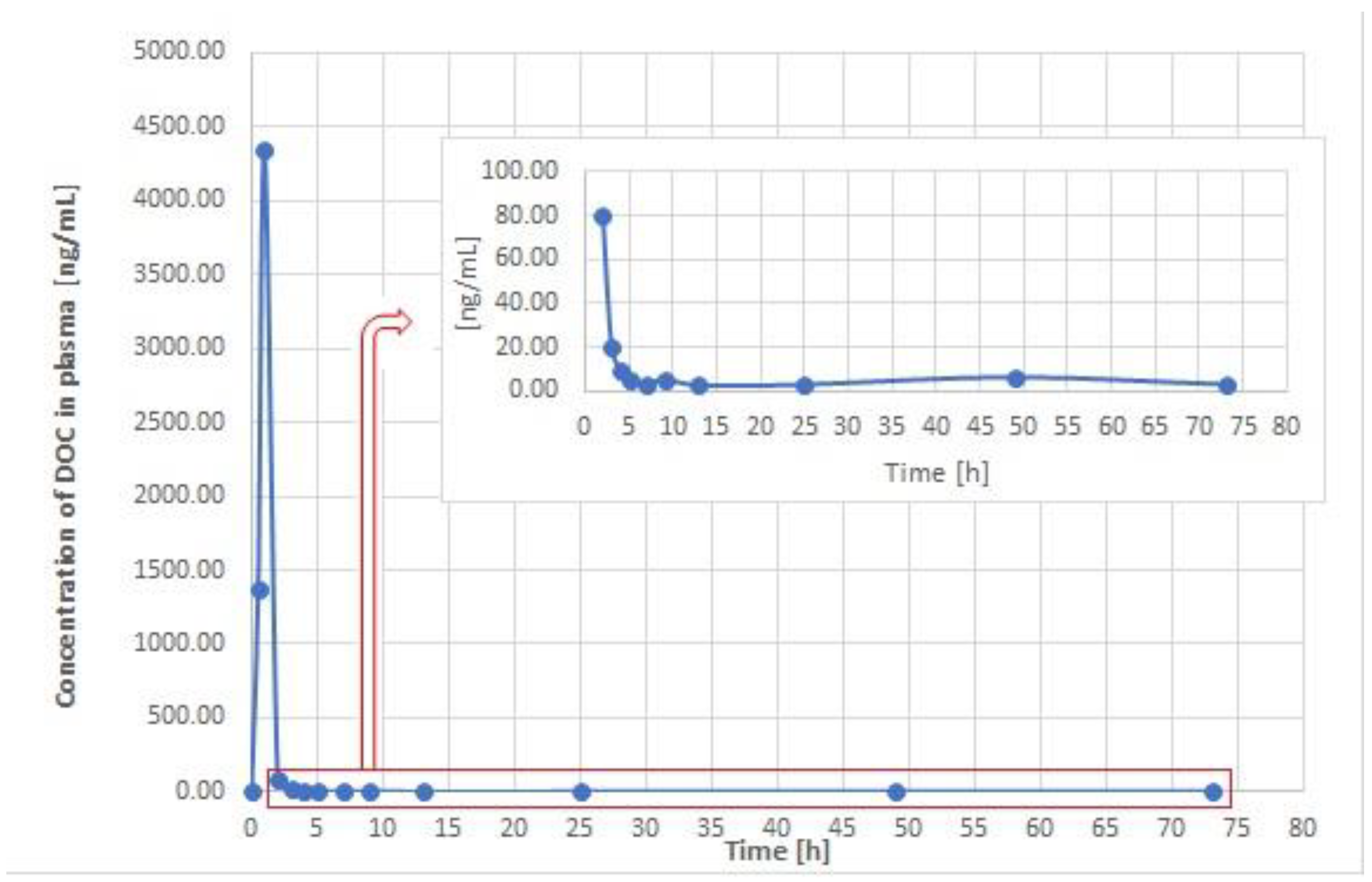
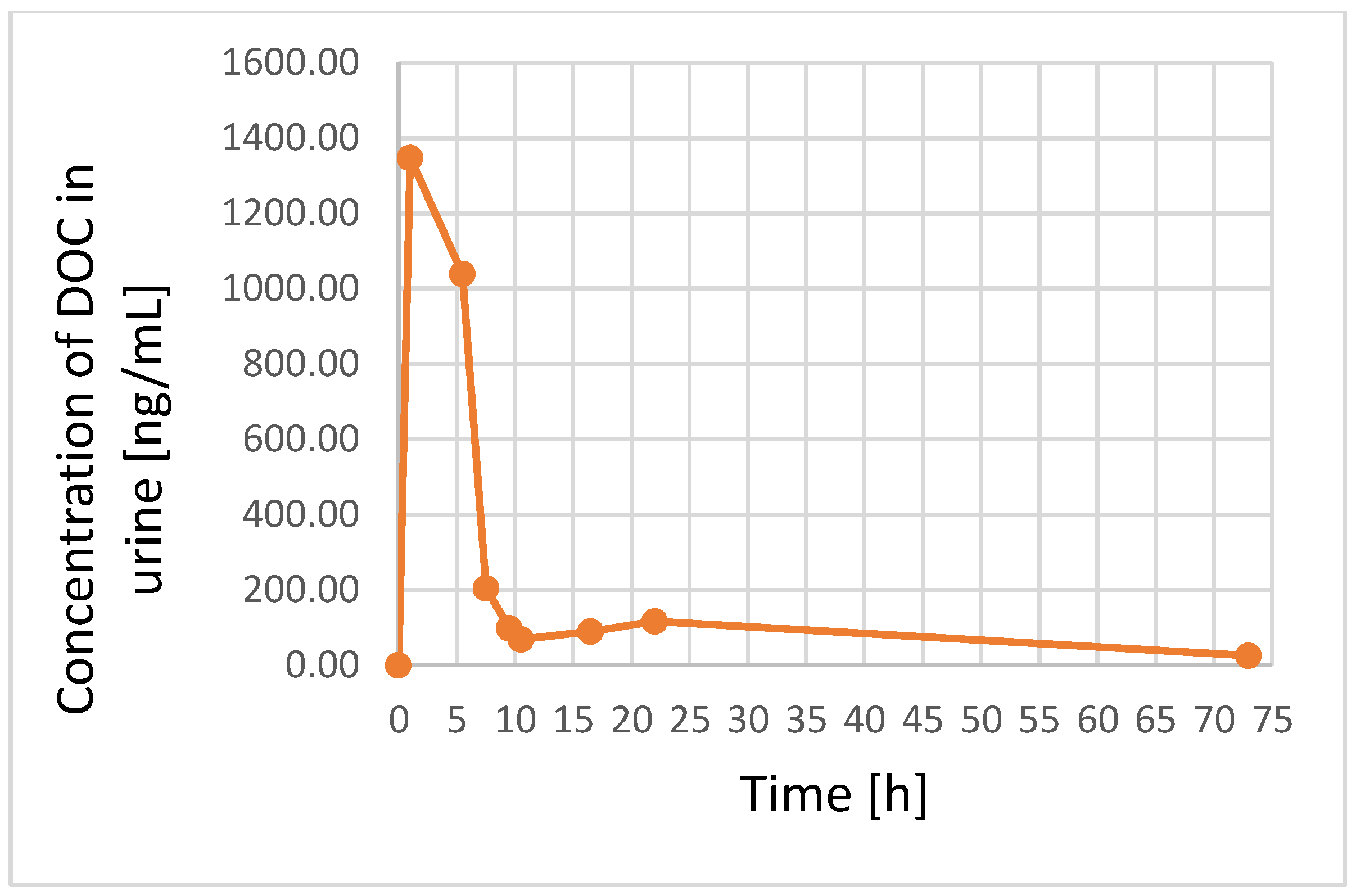
3.3. Application to Real Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Da Silva, G.H.; Fernandes, M.A.; Trevizan, L.N.F.; De Lima, F.T.; Eloy, J.; Chorilli, M. A Critical Review of Properties and Analytical Methods for the Determination of Docetaxel in Biological and Pharmaceutical Matrices. Crit. Rev. Anal. Chem. 2018, 48, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Pétronille, R.; Bernard, D.; Guillaume, L.G.; Victoire, V.; Thomas, F.; Jérôme, C.; Eloïse, D.C.; Gérard, P.; Sylvie, S.; Audrey, S.; et al. Comparative analysis of docetaxel: Physical and chemical characterisation of Taxotère® and generics. Eur. J. Cancer 2020, 135, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.; Ajani, J.; Scotté, F.; Winther, D.; Martin, M.; Aapro, M.S.; von Minckwitz, G. Docetaxel-related side effects and their management. Eur. J. Oncol. Nurs. 2009, 13, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Song, B.S.; Seo, J.; Kim, D.H.; Lim, J.S.; Yoo, J.Y.; Lee, J.A. Gemcitabine and docetaxel for the treatment of children and adolescents with recurrent or refractory osteosarcoma: Korea Cancer Center Hospital experience. Pediatr. Blood Cancer 2014, 61, 1376–1381. [Google Scholar] [CrossRef]
- Yoon, J.H.; Kwon, M.M.; Park, H.J.; Park, S.Y.; Lim, K.Y.; Joo, J.; Park, B.-K. A study of docetaxel and irinotecan in children and young adults with recurrent or refractory Ewing sarcoma family of tumors. BMC Cancer 2014, 14, 622. [Google Scholar] [CrossRef]
- Koo, J.; Knight-Perry, J.; Galambos, C.; Browne, L.P.; Cost, C.R. Pediatric Metastatic Cardiac Angiosarcoma Successfully Treated with Multimodal Therapy: Case Report and Review of Literature. J. Pediatr. Hematol. 2021, 43, e203–e206. [Google Scholar] [CrossRef]
- Kenmotsu, H.; Tanigawara, Y. Pharmacokinetics, dynamics and toxicity of docetaxel: Why the Japanese dose differs from the Western dose. Cancer Sci. 2015, 106, 497–504. [Google Scholar] [CrossRef]
- Imran, M.; Saleem, S.; Chaudhuri, A.; Ali, J.; Baboota, S. Docetaxel: An update on its molecular mechanisms, therapeutic trajectory and nanotechnology in the treatment of breast, lung and prostate cancer. J. Drug Deliv. Sci. Technol. 2020, 60, 101959. [Google Scholar] [CrossRef]
- Muth, M.; Ojara, F.W.; Kloft, C.; Joerger, M. Role of TDM-based dose adjustments for taxane anticancer drugs. Br. J. Clin. Pharmacol. 2020, 87, 306–316. [Google Scholar] [CrossRef]
- Ma, Y.; Lin, Q.; Yang, Y.; Liang, W.; Salamone, S.J.; Li, Y.; Lin, Y.; Zhao, H.; Zhao, Y.; Fang, W.; et al. Clinical pharmacokinetics and drug exposure-toxicity correlation study of docetaxel based chemotherapy in Chinese head and neck cancer patients. Ann. Transl. Med. 2020, 8, 236. [Google Scholar] [CrossRef]
- Engels, F.K.; Loos, W.J.; van der Bol, J.M.; de Bruijn, P.; Mathijssen, R.H.; Verweij, J.; Mathot, R.A. Therapeutic Drug Monitoring for the Individualization of Docetaxel Dosing: A Randomized Pharmacokinetic Study. Clin. Cancer Res. 2011, 17, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Emami, J.; Kazemi, M.; Hasanzadeh, F.; Minaiyan, M.; Mirian, M.; Lavasanifar, A. Development of a RP-HPLC method for analysis of docetaxel in tumor-bearing mice plasma and tissues following injection of docetaxel-loaded pH responsive targeting polymeric micelles. Res. Pharm. Sci. 2020, 15, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ziaei, E.; Emami, J.; Kazemi, M.; Rezazadeh, M. Simultaneous Determination of Docetaxel and Celecoxib in Porous Microparticles and Rat Plasma by Liquid-Liquid Extraction and HPLC with UV Detection: In vitro and in vivo Validation and Application. J. Pharm. Pharm. Sci. 2020, 23, 289–303. [Google Scholar] [CrossRef]
- Kim, D.W.; Yousaf, A.M.; Li, D.X.; Kim, J.O.; Yong, C.S.; Cho, K.H.; Choi, H.-G. Development of RP-HPLC method for simultaneous determination of docetaxel and curcumin in rat plasma: Validation and stability. Asian J. Pharm. Sci. 2017, 12, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Kharkar, P.B.; Talkar, S.S.; Patravale, V.B. A Rapid and Sensitive Bio Analytical RP-HPLC Method for Detection of Docetaxel: Development and Validation. Indian J. Pharm. Educ. Res. 2017, 51, s729–s734. [Google Scholar] [CrossRef]
- Xu, Q.; Zhang, N.; Yin, X.; Wang, M.; Shen, Y.; Xu, S.; Zhang, L.; Gu, Z. Development and validation of a nylon6 nanofibers mat-based SPE coupled with HPLC method for the determination of docetaxel in rabbit plasma and its application to the relative bioavailability study. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2010, 878, 2403–2408. [Google Scholar] [CrossRef] [PubMed]
- Corona, G.; Elia, C.; Casetta, B.; Frustaci, S.; Toffoli, G. High-throughput plasma docetaxel quantification by liquid chromatography–tandem mass spectrometry. Clin. Chim. Acta 2011, 412, 358–364. [Google Scholar] [CrossRef]
- Navarrete, A.; Martínez-Alcázar, M.P.; Durán, I.; Calvo, E.; Valenzuela, B.; Barbas, C.; García, A. Simultaneous online SPE–HPLC–MS/MS analysis of docetaxel, temsirolimus and sirolimus in whole blood and human plasma. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 921–922, 35–42. [Google Scholar] [CrossRef]
- Hendrikx, J.J.M.A.; Rosing, H.; Schinkel, A.H.; Schellens, J.H.M.; Beijnen, J.H. Combined quantification of paclitaxel, docetaxel and ritonavir in human feces and urine using LC-MS/MS. Biomed. Chromatogr. 2013, 28, 302–310. [Google Scholar] [CrossRef]
- da Silva, C.B.P.; Julio, I.P.; Donadel, G.E.; Martins, I. UPLC-MS/MS method for simultaneous determination of cyclophosphamide, docetaxel, doxorubicin and 5-fluorouracil in surface samples. J. Pharmacol. Toxicol. Methods 2016, 82, 68–73. [Google Scholar] [CrossRef]
- Shakalisava, Y.; Regan, F. CE separation approaches for combinations of anthracyclines and taxanes. Electrophoresis 2009, 30, 3110–3113. [Google Scholar] [CrossRef] [PubMed]
- Beumer, J.; Kozo, D.; Harney, R.L.; Baldasano, C.N.; Jarrah, J.; Christner, S.M.; Parise, R.; Baburina, I.; Courtney, J.B.; Salamone, S.J. An Automated Homogeneous Immunoassay for Quantitating Imatinib Concentrations in Plasma. Ther. Drug Monit. 2015, 37, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Fujikawa, A.; Ito, H.; Tanaka, N.; Furugen, A.; Miyamori, K.; Takahashi, N.; Ogura, J.; Kobayashi, M.; Yamada, T.; et al. A rapid and sensitive LC/ESI–MS/MS method for quantitative analysis of docetaxel in human plasma and its application to a pharmacokinetic study. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2012, 893–894, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Wang, H. Recent development and applications of dispersive liquid–liquid microextraction. J. Chromatogr. A 2013, 1295, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zuloaga, O.; Olivares, M.; Navarro, P.; Vallejo, A.; Prieto, A. Dispersive liquid–Liquid microextraction: Trends in the analysis of biological samples. Bioanalysis 2015, 7, 2211–2225. [Google Scholar] [CrossRef]
- Rykowska, I.; Ziemblińska, J.; Nowak, I. Modern approaches in dispersive liquid-liquid microextraction (DLLME) based on ionic liquids: A review. J. Mol. Liq. 2018, 259, 319–339. [Google Scholar] [CrossRef]
- Booth, B.P.; Simon, W.C. Analytical method validation. In New Drug Development; CRC Press: Boca Raton, FL, USA, 2004; pp. 138–159. [Google Scholar] [CrossRef]
- Available online: https://www.ich.org/page/quality-guidelines (accessed on 1 December 2022).
- Bien, E.; Stachowicz-Stencel, T.; Balcerska, A.; Godzinski, J.; Kazanowska, B.; Perek-Polnik, M.; Madziara, W.; Rybczynska, A.; Kurylak, A.; Zalewska-Szewczyk, B.; et al. Angiosarcoma in children—Still uncontrollable oncological problem. The report of the Polish Paediatric Rare Tumours Study. Eur. J. Cancer Care 2009, 18, 411–420. [Google Scholar] [CrossRef]
- Ostrowski, S.; Marcinkiewicz, A.; Kośmider, A.; Jaszewski, R. State of the art paper Sarcomas of the heart as a difficult interdisciplinary problem. Arch. Med. Sci. 2014, 1, 135–148. [Google Scholar] [CrossRef]
- Nakamura-Horigome, M.; Koyama, J.; Eizawa, T.; Kasai, H.; Kumazaki, S.; Tsutsui, H.; Koiwai, K.; Oguchi, K.; Kinoshita, O.; Ikeda, U. Successful Treatment of Primary Cardiac Angiosarcoma With Docetaxel and Radiotherapy. Angiology 2008, 59, 368–371. [Google Scholar] [CrossRef]
- Minichillo, S.; Pantaleo, M.A.; Nannini, M.; Coccolo, F.; Gatto, L.; Biasco, G.; Brandi, G. Efficacy of weekly docetaxel in locally advanced cardiac angiosarcoma. BMC Res. Notes 2015, 8, 325. [Google Scholar] [CrossRef]
- Konieczna, L.; Roszkowska, A.; Stachowicz-Stencel, T.; Synakiewicz, A.; Bączek, T. Bioanalysis of a panel of neurotransmitters and their metabolites in plasma samples obtained from pediatric patients with neuroblastoma and Wilms’ tumor. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018, 1074, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Judson, I.; Verweij, J.; Gelderblom, H.; Hartmann, J.T.; Schöffski, P.; Blay, J.-Y.; Kerst, J.M.; Sufliarsky, J.; Whelan, J.; Hohenberger, P.; et al. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: A randomised controlled phase 3 trial. Lancet Oncol. 2014, 15, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Pedrycz, A.; Kramkowska, A. Adriamycin—Efficacy and possible adverse effects. Curr. Probl. Psychiatry 2016, 17, 38–46. [Google Scholar] [CrossRef]
- Kim, J.H.; Park, H.S.; Heo, S.J.; Kim, S.K.; Han, J.W.; Shin, K.-H.; Kim, S.H.; Hur, H.; Kim, K.S.; Choi, Y.D.; et al. Differences in the Efficacies of Pazopanib and Gemcitabine/Docetaxel as Second-Line Treatments for Metastatic Soft Tissue Sarcoma. Oncology 2018, 96, 59–69. [Google Scholar] [CrossRef]
- Kuppens, I.E.L.M.; Van Maanen, M.J.; Rosing, H.; Schellens, J.H.M.; Beijnen, J.H. Quantitative analysis of docetaxel in human plasma using liquid chromatography coupled with tandem mass spectrometry. Biomed. Chromatogr. 2004, 19, 355–361. [Google Scholar] [CrossRef]
- Hirth, J.A.; Watkins, P.B.; Strawderman, M.; Schott, A.; Bruno, R.; Baker, L.H. The effect of an individual’s cytochrome CYP3A4 activity on docetaxel clearance. Clin. Cancer Res. 2000, 6, 1255–1258. [Google Scholar]
- Baker, S.D.; Sparreboom, A.; Verweij, J. Clinical pharmacokinetics of docetaxel: Recent developments. Clin. Pharmacokinet. 2006, 45, 235–252. [Google Scholar] [CrossRef]
- Kenmotsu, H.; Imamura, C.K.; Ono, A.; Omori, S.; Nakashima, K.; Wakuda, K.; Tanigawara, Y. The effects of advanced age and serum α1-acid glycoprotein on docetaxel unbound exposure and dose-limiting toxicity in cancer patients. Br. J. Clin. Pharmacol. 2017, 83, 2416–2425. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The Recovery of DOC [%] | ||||||||||||
| (v/v) | DChM:MeOH | DChM:EtOH | Chl:MeOH | Chl:EtOH | ||||||||
| AVG | SD | RSD [%] | AVG | SD | RSD [%] | AVG | SD | RSD [%] | AVG | SD | RSD [%] | |
| 4:6 | 75.09 | 6.65 | 8.86 | 72.53 | 4.52 | 6.23 | 79.86 | 5.99 | 7.50 | 95.00 | 4.89 | 5.15 |
| 3:7 | 78.19 | 4.88 | 6.24 | 71.63 | 5.99 | 8.36 | 88.08 | 6.13 | 6.96 | 93.63 | 5.21 | 5.56 |
| 2:8 | 69.67 | 7.05 | 10.12 | 74.67 | 4.41 | 5.91 | 82.42 | 5.69 | 6.90 | 93.69 | 5.75 | 6.14 |
| The Recovery of PAC [%] | ||||||||||||
| (v/v) | DChM:MeOH | DChM:EtOH | Chl:MeOH | Chl:EtOH | ||||||||
| AVG | SD | RSD [%] | AVG | SD | RSD [%] | AVG | SD | RSD [%] | AVG | SD | RSD [%] | |
| 4:6 | 77.86 | 7.11 | 9.13 | 75.20 | 6.66 | 8.86 | 82.80 | 6.04 | 7.29 | 98.50 | 4.36 | 4.43 |
| 3:7 | 81.07 | 5.06 | 6.24 | 74.28 | 5.87 | 7.90 | 91.38 | 7.06 | 7.73 | 97.08 | 5.77 | 5.94 |
| 2:8 | 72.24 | 6.45 | 8.93 | 77.43 | 5.05 | 6.52 | 85.46 | 6.23 | 7.29 | 97.14 | 6.02 | 6.20 |
| Parameters | Plasma Samples | Urine Samples |
|---|---|---|
| Linearity (ng/mL) | 2.5–2000 | 5–2000 |
| Equation parameter | ||
| Slope | 0.0013 ± 0.0000067 | 0.0025 ± 0.000013 |
| Intercept | −0.0007 ± 0.006 | −0.0063 ± 0.012 |
| Correlation coefficient (R2) | 0.9998 | 0.9997 |
| LOD (ng/mL) | 1 | 2.5 |
| Plasma Samples | Urine Samples | |||||||
|---|---|---|---|---|---|---|---|---|
| Concentration (ng/mL) | Concentration (ng/mL) | |||||||
| Spiked | Found (Mean ± SD) | Precision (RSD %) | Accuracy (%) | Spiked (ng/mL) | Found (Mean ± SD) | Precision (RSD %) | Accuracy (%) | |
| Intra-day (n = 6) | ||||||||
| LLOQ | 2.5 | 2.71 ± 0.25 | 9.42 | 108.21 | 5 | 4.92 ± 0.42 | 8.60 | 98.40 |
| LQC | 50 | 45.44 ± 2.93 | 6.45 | 90.87 | 250 | 263.83 ± 16.49 | 6.25 | 105.53 |
| MQC | 750 | 745.37 ± 21.85 | 2.93 | 99.38 | 750 | 731.40 ± 33.47 | 4.58 | 97.52 |
| HQC | 1500 | 1460.54 ± 33.32 | 2.28 | 97.37 | 1500 | 1495.80 ± 59.11 | 3.95 | 99.72 |
| Inter-day (n = 6) | ||||||||
| LLOQ | 2.5 | 2.36 ± 0.26 | 11.09 | 94.27 | 5 | 4.84 ± 0.55 | 11.45 | 96.70 |
| LQC | 50 | 46.78 ± 2.42 | 5.18 | 93.56 | 250 | 245.54 ± 8.18 | 3.33 | 98.21 |
| MQC | 750 | 755.17 ± 25.03 | 3.31 | 100.69 | 750 | 738.34 ± 16.70 | 2.26 | 98.44 |
| HQC | 1500 | 1488.25 ± 39.26 | 2.64 | 99.22 | 1500 | 1495.47 ± 14.65 | 0.98 | 99.69 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maliszewska, O.; Roszkowska, A.; Lipiński, M.; Treder, N.; Olędzka, I.; Kowalski, P.; Bączek, T.; Bień, E.; Krawczyk, M.A.; Plenis, A. Profiling Docetaxel in Plasma and Urine Samples from a Pediatric Cancer Patient Using Ultrasound-Assisted Dispersive Liquid–Liquid Microextraction Combined with LC–MS/MS. Pharmaceutics 2023, 15, 1255. https://doi.org/10.3390/pharmaceutics15041255
Maliszewska O, Roszkowska A, Lipiński M, Treder N, Olędzka I, Kowalski P, Bączek T, Bień E, Krawczyk MA, Plenis A. Profiling Docetaxel in Plasma and Urine Samples from a Pediatric Cancer Patient Using Ultrasound-Assisted Dispersive Liquid–Liquid Microextraction Combined with LC–MS/MS. Pharmaceutics. 2023; 15(4):1255. https://doi.org/10.3390/pharmaceutics15041255
Chicago/Turabian StyleMaliszewska, Olga, Anna Roszkowska, Marcin Lipiński, Natalia Treder, Ilona Olędzka, Piotr Kowalski, Tomasz Bączek, Ewa Bień, Małgorzata Anna Krawczyk, and Alina Plenis. 2023. "Profiling Docetaxel in Plasma and Urine Samples from a Pediatric Cancer Patient Using Ultrasound-Assisted Dispersive Liquid–Liquid Microextraction Combined with LC–MS/MS" Pharmaceutics 15, no. 4: 1255. https://doi.org/10.3390/pharmaceutics15041255