
Clinical Relevance of Targeted Therapy and Immune-Checkpoint Inhibition in Lung Cancer
 , , ,
, , ,  ,
,  ,
,  , and
, and 
Abstract
:1. Introduction
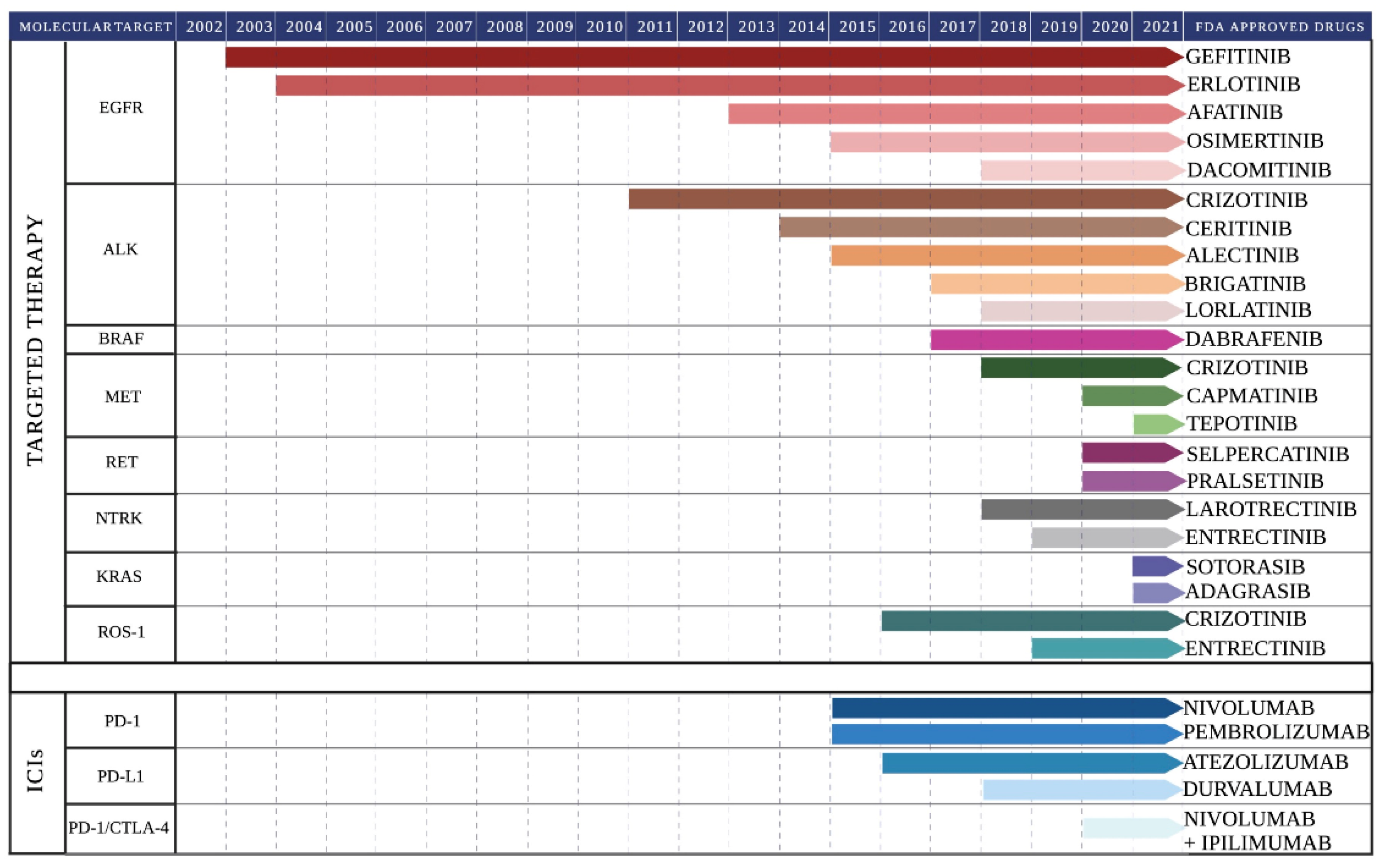
2. Molecular Landscape of Lung Cancer and Targeted Therapy
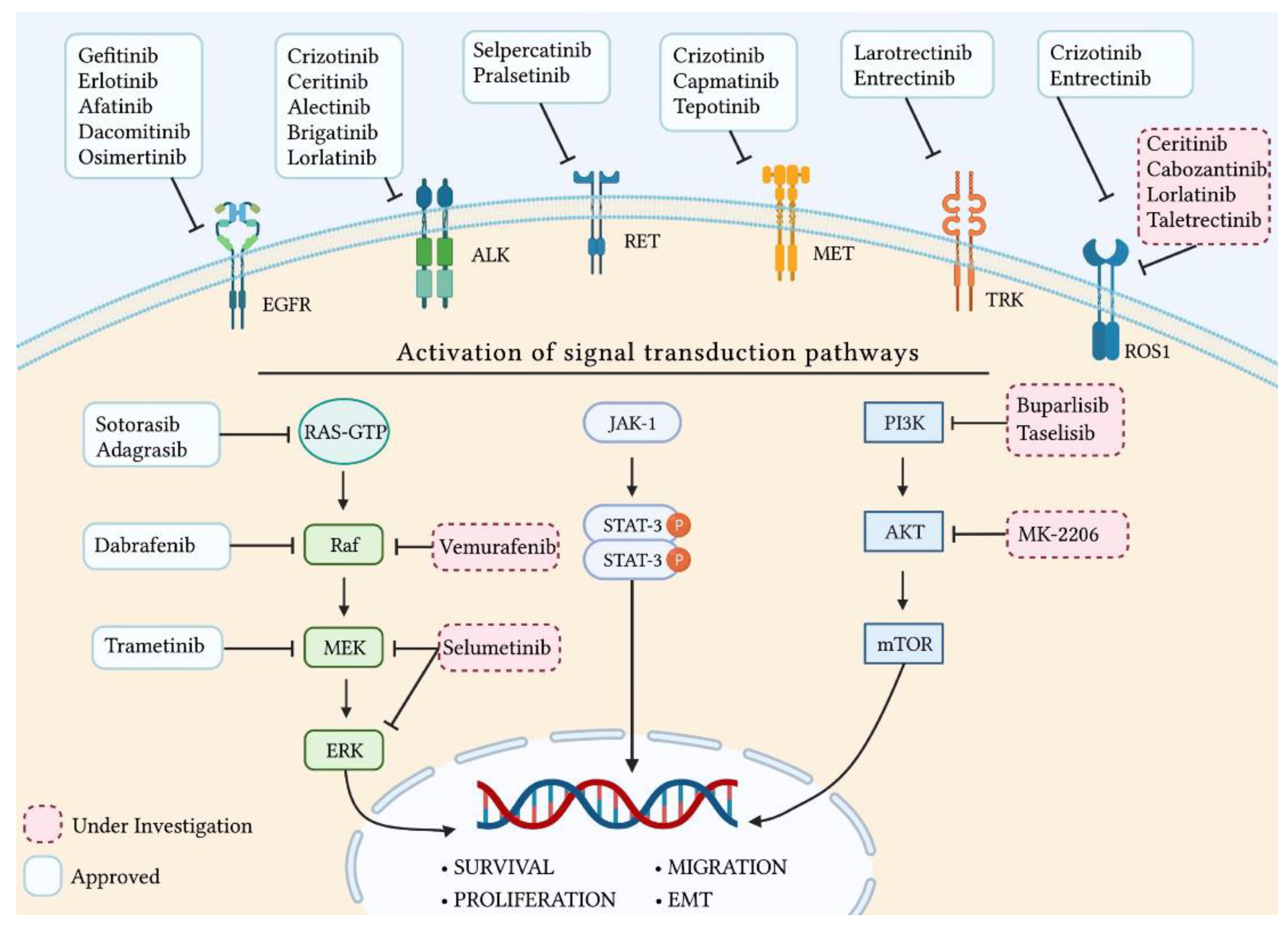
2.1. Epidermal Growth Factor Receptor
2.2. Anaplastic Lymphoma Kinase
2.3. B-Raf Proto-Oncogene
2.4. Rearrangement during Transfection
2.5. Hepatocyte Growth Factor Receptor
2.6. Neurotrophic Tropomyosin Receptor Tyrosine Kinases
2.7. Kirsten Rat Sarcoma Viral Oncogene Homolog
2.8. Proto-Oncogene Tyrosine-Protein Kinase ROS
2.9. Phosphoinositide 3-Kinase
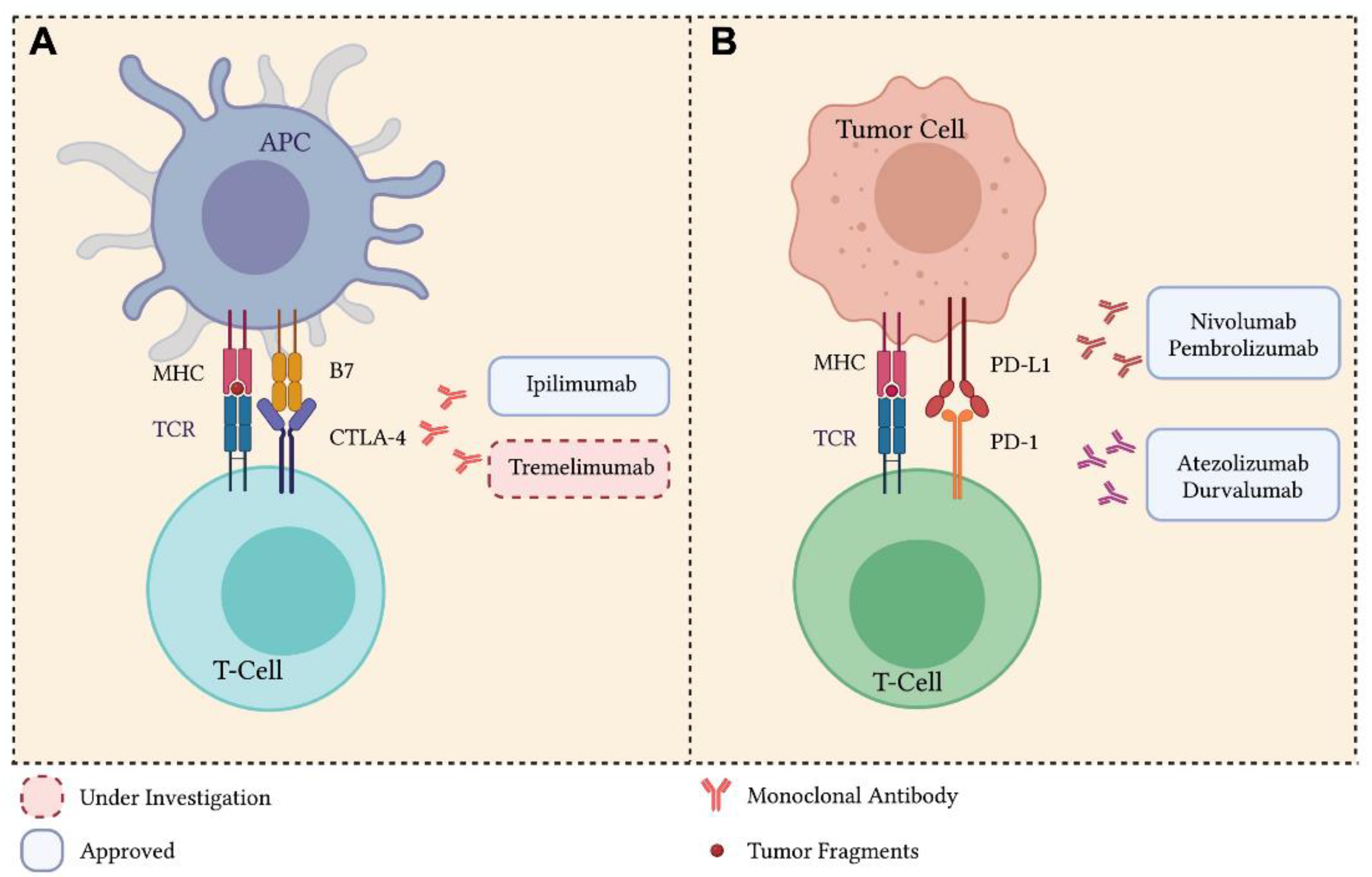
3. Immune Checkpoint Inhibitors
3.1. Cytotoxic T Lymphocyte Antigen-4 Inhibitors
3.2. Programmed Death 1 and Programmed Death Ligand 1 Inhibitors
4. Combination Therapies Using Immune Checkpoint Inhibitors plus Chemo- and/or Targeted Therapies
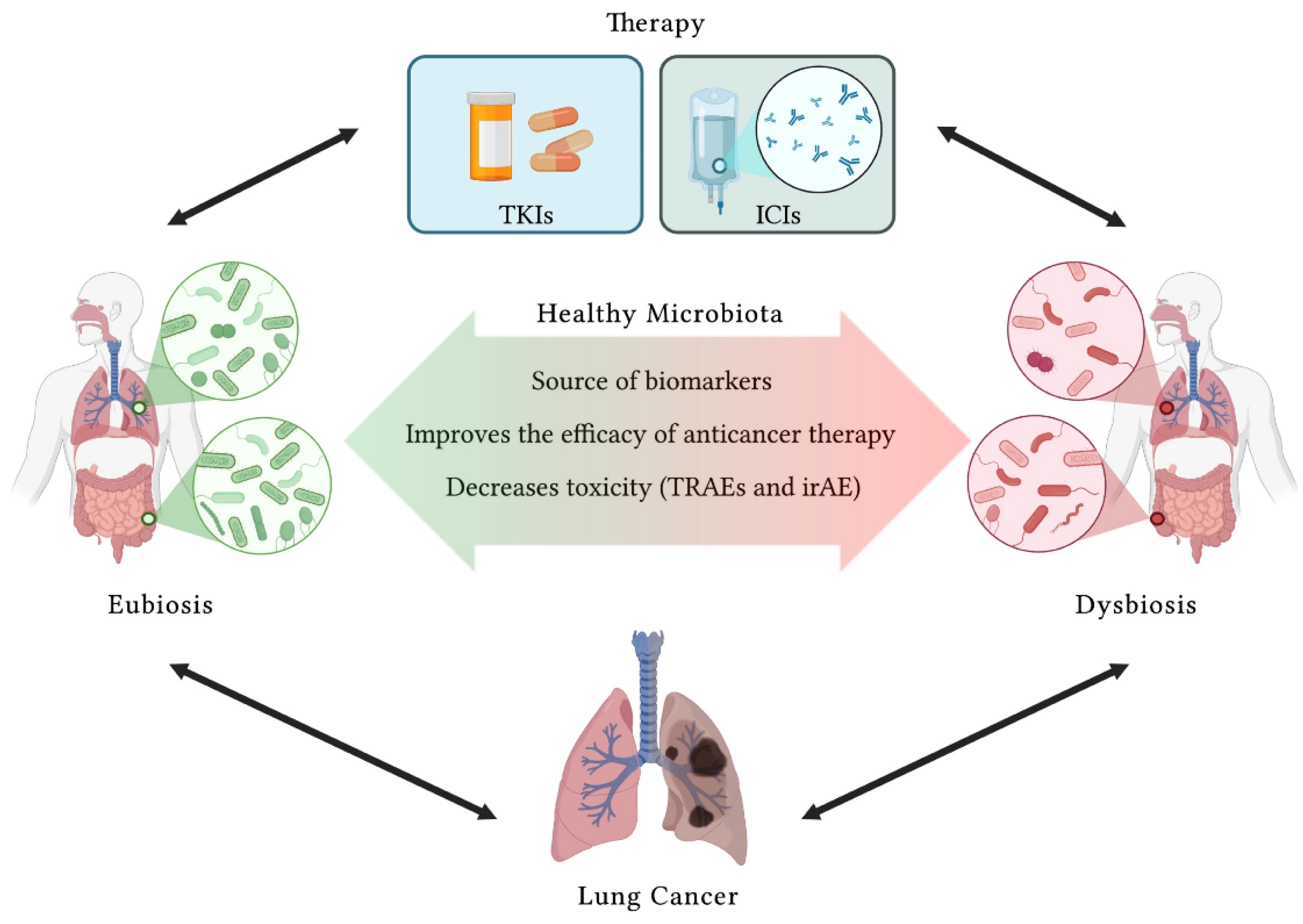
5. Microbiota Modulation in Lung Cancer
6. Main Concepts and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.M.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B.; et al. The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances Since the 2004 Classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef]
- Travis, W.D.; Brambilla, E.; Riely, G.J. New Pathologic Classification of Lung Cancer: Relevance for Clinical Practice and Clinical Trials. J. Clin. Oncol. 2013, 31, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Petersen, I. The morphological and molecular diagnosis of lung cancer. Dtsch. Arztebl. Int. 2011, 108, 525–535. [Google Scholar] [CrossRef]
- Travis, W.D.; Brambilla, E.; Noguchi, M.; Nicholson, A.G.; Geisinger, K.R.; Yatabe, Y.; Beer, D.G.; Powell, C.A.; Riely, G.J.; Van Schil, P.E.; et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J. Thorac. Oncol. 2011, 6, 244–285. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Brambilla, E.; Burke, A.P.; Marx, A.; Nicholson, A.G. Introduction to The 2015 World Health Organization Classification of Tumors of the Lung, Pleura, Thymus, and Heart. J. Thorac. Oncol. 2015, 10, 1240–1242. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Zheng, M. Classification and Pathology of Lung Cancer. Surg. Oncol. Clin. N. Am. 2016, 25, 447–468. [Google Scholar] [CrossRef]
- Hutchinson, B.D.; Shroff, G.S.; Truong, M.T.; Ko, J.P. Spectrum of Lung Adenocarcinoma. Semin. Ultrasound CT MRI 2019, 40, 255–264. [Google Scholar] [CrossRef]
- Dela Cruz, C.S.; Tanoue, L.T.; Matthay, R.A. Lung Cancer: Epidemiology, Etiology, and Prevention. Clin. Chest. Med. 2011, 32, 605–644. [Google Scholar] [CrossRef]
- Travis, W.D. Classification of Lung Cancer. Semin. Roentgenol. 2011, 46, 178–186. [Google Scholar] [CrossRef]
- Byers, L.A.; Rudin, C.M. Small cell lung cancer: Where do we go from here? Cancer 2015, 121, 664–672. [Google Scholar] [CrossRef]
- Travis, W.D. Lung Cancer Pathology. Clin. Chest. Med. 2020, 41, 67–85. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.G.; Haines, C.; Perkel, R.; Enck, R.E. Lung cancer: Diagnosis and management. Am. Fam. Physician 2007, 75, 56–63. [Google Scholar]
- Wadowska, K.; Bil-Lula, I.; Trembecki, Ł.; Śliwińska-Mossoń, M. Genetic Markers in Lung Cancer Diagnosis: A Review. Int. J. Mol. Sci. 2020, 21, 4569. [Google Scholar] [CrossRef]
- Rossi, G.; Pelosi, G.; Barbareschi, M.; Graziano, P.; Cavazza, A.; Papotti, M. Subtyping Non–Small Cell Lung Cancer. Int. J. Surg. Pathol. 2013, 21, 326–336. [Google Scholar] [CrossRef] [PubMed]
- van Meerbeeck, J.P.; Fennell, D.A.; De Ruysscher, D.K. Small-cell lung cancer. Lancet 2011, 378, 1741–1755. [Google Scholar] [CrossRef] [PubMed]
- Gridelli, C.; Rossi, A.; Carbone, D.P.; Guarize, J.; Karachaliou, N.; Mok, T.; Petrella, F.; Spaggiari, L.; Rosell, R. Non-small-cell lung cancer. Nat. Rev. Dis. Prim. 2015, 1, 15009. [Google Scholar] [CrossRef]
- Jemal, A.; Ma, J.; Rosenberg, P.S.; Siegel, R.; Anderson, W.F. Increasing Lung Cancer Death Rates Among Young Women in Southern and Midwestern States. J. Clin. Oncol. 2012, 30, 2739–2744. [Google Scholar] [CrossRef]
- Haddadin, S.; Perry, M.C. History of Small-Cell Lung Cancer. Clin. Lung Cancer 2011, 12, 87–93. [Google Scholar] [CrossRef]
- de Sousa, V.M.L.; Carvalho, L. Heterogeneity in Lung Cancer. Pathobiology 2018, 85, 96–107. [Google Scholar] [CrossRef]
- Alberg, A.J.; Brock, M.V.; Samet, J.M. Epidemiology of Lung Cancer: Looking to the Future. J. Clin. Oncol. 2005, 23, 3175–3185. [Google Scholar] [CrossRef]
- Malhotra, J.; Malvezzi, M.; Negri, E.; La Vecchia, C.; Boffetta, P. Risk factors for lung cancer worldwide. Eur. Respir. J. 2016, 48, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Falzone, L.; Marconi, A.; Loreto, C.; Franco, S.; Spandidos, D.A.; Libra, M. Occupational exposure to carcinogens: Benzene, pesticides and fibers (Review). Mol. Med. Rep. 2016, 14, 4467–4474. [Google Scholar] [CrossRef] [PubMed]
- Delva, F.; Andujar, P.; Lacourt, A.; Brochard, P.; Pairon, J.C. Occupational risk factors for lung cancer. Rev. Mal. Respir. 2016, 33, 444–459. [Google Scholar] [CrossRef] [PubMed]
- Falzone, L.; Bordonaro, R.; Libra, M. SnapShot: Cancer chemotherapy. Cell 2023, 186, 1816. [Google Scholar] [CrossRef]
- Kalemkerian, G.P.; Narula, N.; Kennedy, E.B.; Biermann, W.A.; Donington, J.; Leighl, N.B.; Lew, M.; Pantelas, J.; Ramalingam, S.S.; Reck, M.; et al. Molecular Testing Guideline for the Selection of Patients With Lung Cancer for Treatment With Targeted Tyrosine Kinase Inhibitors: American Society of Clinical Oncology Endorsement of the College of American Pathologists/International Association for the Study of Lung Cancer/Association for Molecular Pathology Clinical Practice Guideline Update. J. Clin. Oncol. 2018, 36, 911–919. [Google Scholar] [PubMed]
- Lim, S.W.; Ahn, M.J. Current status of immune checkpoint inhibitors in treatment of non-small cell lung cancer. Korean J. Intern. Med. 2019, 34, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, C.; Chen, G.; Hu, Q.; Gu, Z. Delivery Strategies for Immune Checkpoint Blockade. Adv. Healthc. Mater. 2018, 7, e1800424. [Google Scholar] [CrossRef]
- Seetharamu, N.; Budman, D.R.; Sullivan, K.M. Immune checkpoint inhibitors in lung cancer: Past, present and future. Futur. Oncol. 2016, 12, 1151–1163. [Google Scholar] [CrossRef]
- Ruiz-Cordero, R.; Devine, W.P. Targeted Therapy and Checkpoint Immunotherapy in Lung Cancer. Surg. Pathol. Clin. 2020, 13, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Carrot-Zhang, J.; Yao, X.; Devarakonda, S.; Deshpande, A.; Damrauer, J.S.; Silva, T.C.; Wong, C.K.; Choi, H.Y.; Felau, I.; Robertson, A.G.; et al. Whole-genome characterization of lung adenocarcinomas lacking the RTK/RAS/RAF pathway. Cell. Rep. 2021, 34, 108784. [Google Scholar] [CrossRef]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef] [PubMed]
- Lindeman, N.I.; Cagle, P.T.; Aisner, D.L.; Arcila, M.E.; Beasley, M.B.; Bernicker, E.H.; Colasacco, C.; Dacic, S.; Hirsch, F.R.; Kerr, K.; et al. Updated Molecular Testing Guideline for the Selection of Lung Cancer Patients for Treatment With Targeted Tyrosine Kinase Inhibitors: Guideline From the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. Arch. Pathol. Lab. Med. 2018, 142, 321–346. [Google Scholar] [PubMed]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef]
- Goffin, J.R.; Zbuk, K. Epidermal growth factor receptor: Pathway, therapies, and pipeline. Clin. Ther. 2013, 35, 1282–1303. [Google Scholar] [CrossRef]
- Wang, Z. ErbB Receptors and Cancer. Methods Mol. Biol. 2017, 1652, 3–35. [Google Scholar]
- Soo, R.A.; Kubo, A.; Ando, M.; Kawaguchi, T.; Ahn, M.J.; Ou, S.I. Association Between Environmental Tobacco Smoke Exposure and the Occurrence of EGFR Mutations and ALK Rearrangements in Never-smokers With Non–Small-cell Lung Cancer: Analyses From a Prospective Multinational ETS Registry. Clin. Lung Cancer 2017, 18, 535–542. [Google Scholar] [CrossRef]
- Gandhi, J.; Zhang, J.; Xie, Y.; Soh, J.; Shigematsu, H.; Zhang, W.; Yamamoto, H.; Peyton, M.; Girard, L.; Lockwood, W.W.; et al. Alterations in genes of the EGFR signaling pathway and their relationship to EGFR tyrosine kinase inhibitor sensitivity in lung cancer cell lines. PLoS ONE 2009, 4, e4576. [Google Scholar] [CrossRef]
- Cho, J.; Chen, L.; Sangji, N.; Okabe, T.; Yonesaka, K.; Francis, J.M.; Flavin, R.J.; Johnson, W.; Kwon, J.; Yu, S.; et al. Cetuximab response of lung cancer-derived EGF receptor mutants is associated with asymmetric dimerization. Cancer Res. 2013, 73, 6770–6779. [Google Scholar] [CrossRef]
- Yang, J.C.; Sequist, L.V.; Geater, S.L.; Tsai, C.M.; Mok, T.S.; Schuler, M.; Yamamoto, N.; Yu, C.J.; Ou, S.H.; Zhou, C.; et al. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: A combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol. 2015, 16, 830–838. [Google Scholar] [CrossRef]
- Gately, K.; O′Flaherty, J.; Cappuzzo, F.; Pirker, R.; Kerr, K.; O′Byrne, K. The role of the molecular footprint of EGFR in tailoring treatment decisions in NSCLC. J. Clin. Pathol. 2012, 65, 1–7. [Google Scholar] [CrossRef]
- Lee, C.K.; Davies, L.; Wu, Y.L.; Mitsudomi, T.; Inoue, A.; Rosell, R.; Zhou, C.; Nakagawa, K.; Thongprasert, S.; Fukuoka, M.; et al. Gefitinib or Erlotinib vs Chemotherapy for EGFR Mutation-Positive Lung Cancer: Individual Patient Data Meta-Analysis of Overall Survival. J. Natl. Cancer. Inst. 2017, 109, djw279. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.; Wu, Y.L.; Hirsh, V.; O’Byrne, K.; Yamamoto, N.; Mok, T.; Popat, S.; Sequist, L.V.; Massey, D.; Zazulina, V.; et al. First-Line Afatinib versus Chemotherapy in Patients with Non-Small Cell Lung Cancer and Common Epidermal Growth Factor Receptor Gene Mutations and Brain Metastases. J. Thorac. Oncol. 2016, 11, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Abdelgalil, A.A.; Al-Kahtani, H.M.; Al-Jenoobi, F.I. Erlotinib. Profiles Drug Subst. Excip. Relat. Methodol. 2020, 45, 93–117. [Google Scholar] [PubMed]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Burdett, S. Preoperative chemotherapy for non-small-cell lung cancer: A systematic review and meta-analysis of individual participant data. Lancet 2014, 383, 1561–1571. [Google Scholar]
- Xiong, L.; Lou, Y.; Bai, H.; Li, R.; Xia, J.; Fang, W.; Zhang, J.; Han-Zhang, H.; Lizaso, A.; Li, B.; et al. Efficacy of erlotinib as neoadjuvant regimen in EGFR-mutant locally advanced non-small cell lung cancer patients. J. Int. Med. Res. 2020, 48, 300060519887275. [Google Scholar] [CrossRef]
- Xie, H.; Wang, H.; Xu, L.; Li, M.; Peng, Y.; Cai, X.; Feng, Z.; Ren, W.; Peng, Z. Gefitinib Versus Adjuvant Chemotherapy in Patients With Stage II-IIIA Non-Small-Cell Lung Cancer Harboring Positive EGFR Mutations: A Single-Center Retrospective Study. Clin. Lung Cancer 2018, 19, 484–492. [Google Scholar] [CrossRef]
- Hosomi, Y.; Morita, S.; Sugawara, S.; Kato, T.; Fukuhara, T.; Gemma, A.; Takahashi, K.; Fujita, Y.; Harada, T.; Minato, K.; et al. Gefitinib Alone Versus Gefitinib Plus Chemotherapy for Non-Small-Cell Lung Cancer With Mutated Epidermal Growth Factor Receptor: NEJ009 Study. J. Clin. Oncol. 2020, 38, 115–123. [Google Scholar] [CrossRef]
- Yap, T.A.; Vidal, L.; Adam, J.; Stephens, P.; Spicer, J.; Shaw, H.; Ang, J.; Temple, G.; Bell, S.; Shahidi, M.; et al. Phase I trial of the irreversible EGFR and HER2 kinase inhibitor BIBW 2992 in patients with advanced solid tumors. J. Clin. Oncol. 2010, 28, 3965–3972. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Gale, C.M.; Lifshits, E.; Gonzales, A.J.; Shimamura, T.; Zhao, F.; Vincent, P.W.; Naumov, G.N.; Bradner, J.E.; et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007, 67, 11924–11932. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.; Yang, J.C.; Park, K.; Kim, J.H.; Bennouna, J.; Chen, Y.M.; Chouaid, C.; De Marinis, F.; Feng, J.F.; Grossi, F.; et al. Afatinib beyond progression in patients with non-small-cell lung cancer following chemotherapy, erlotinib/gefitinib and afatinib: Phase III randomized LUX-Lung 5 trial. Ann. Oncol. 2016, 27, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Felip, E.; Cobo, M.; Lu, S.; Syrigos, K.; Lee, K.H.; Göker, E.; Georgoulias, V.; Li, W.; Isla, D.; et al. Afatinib versus erlotinib as second-line treatment of patients with advanced squamous cell carcinoma of the lung (LUX-Lung 8): An open-label randomised controlled phase 3 trial. Lancet Oncol. 2015, 16, 897–907. [Google Scholar] [CrossRef]
- Felip, E.; Hirsh, V.; Popat, S.; Cobo, M.; Fülöp, A.; Dayen, C.; Trigo, J.M.; Gregg, R.; Waller, C.F.; Soria, J.C.; et al. Symptom and Quality of Life Improvement in LUX-Lung 8, an Open-Label Phase III Study of Second-Line Afatinib Versus Erlotinib in Patients With Advanced Squamous Cell Carcinoma of the Lung After First-Line Platinum-Based Chemotherapy. Clin. Lung Cancer 2018, 19, 74–83. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, S.H.; Ahn, J.S.; Ahn, M.J.; Park, K.; Sun, J.M. Efficacy and Safety of Afatinib for EGFR-mutant Non-small Cell Lung Cancer, Compared with Gefitinib or Erlotinib. Cancer Res. Treat. 2019, 51, 502–509. [Google Scholar] [CrossRef]
- Wu, Y.L.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Tsuji, F.; Linke, R.; Rosell, R.; Corral, J.; et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): A randomised, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 1454–1466. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Lee, M.; Linke, R.; Rosell, R.; Corral, J.; et al. Improvement in Overall Survival in a Randomized Study That Compared Dacomitinib With Gefitinib in Patients with Advanced Non-Small-Cell Lung Cancer and EGFR-Activating Mutations. J. Clin. Oncol. 2018, 36, 2244–2250. [Google Scholar] [CrossRef]
- Mok, T.S.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Chawla, A.; Rosell, R.; Corral, J.; Migliorino, M.R.; et al. Updated Overall Survival in a Randomized Study Comparing Dacomitinib with Gefitinib as First-Line Treatment in Patients with Advanced Non-Small-Cell Lung Cancer and EGFR-Activating Mutations. Drugs 2021, 81, 257–266. [Google Scholar] [CrossRef]
- Minari, R.; Bordi, P.; Tiseo, M. Third-generation epidermal growth factor receptor-tyrosine kinase inhibitors in T790M-positive non-small cell lung cancer: Review on emerged mechanisms of resistance. Transl. Lung Cancer Res. 2016, 5, 608–695. [Google Scholar] [CrossRef]
- Kuiper, J.L.; Heideman, D.A.M.; Thunnissen, E.; Paul, M.A.; van Wijk, A.W.; Postmus, P.E.; Smit, E.F. Incidence of T790M mutation in (sequential) rebiopsies in EGFR-mutated NSCLC-patients. Lung Cancer 2014, 85, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Chiarle, R.; Voena, C.; Ambrogio, C.; Piva, R.; Inghirami, G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat. Rev. Cancer 2008, 8, 11–23. [Google Scholar] [CrossRef]
- Leventaki, V.; Bhattacharyya, S.; Lim, M.S. Pathology and genetics of anaplastic large cell lymphoma. Semin. Diagn. Pathol. 2020, 37, 57–71. [Google Scholar] [CrossRef]
- Aygun, N. Biological and Genetic Features of Neuroblastoma and Their Clinical Importance. Curr. Pediatr. Rev. 2018, 14, 73–90. [Google Scholar] [CrossRef]
- Golding, B.; Luu, A.; Jones, R.; Viloria-Petit, A.M. The function and therapeutic targeting of anaplastic lymphoma kinase (ALK) in non-small cell lung cancer (NSCLC). Mol. Cancer 2018, 17, 52. [Google Scholar] [CrossRef]
- Hofman, P. ALK in Non-Small Cell Lung Cancer (NSCLC) Pathobiology, Epidemiology, Detection from Tumor Tissue and Algorithm Diagnosis in a Daily Practice. Cancers 2017, 9, 107. [Google Scholar] [CrossRef]
- Sabir, S.; Yeoh, S.; Jackson, G.; Bayliss, R. EML4-ALK Variants: Biological and Molecular Properties, and the Implications for Patients. Cancers 2017, 9, 118. [Google Scholar] [CrossRef]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Sanders, H.R.; Li, H.R.; Bruey, J.M.; Scheerle, J.A.; Meloni-Ehrig, A.M.; Kelly, J.C.; Novick, C.; Albitar, M. Exon scanning by reverse transcriptase-polymerase chain reaction for detection of known and novel EML4-ALK fusion variants in non-small cell lung cancer. Cancer Genet. 2011, 204, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Lin, J.; Liao, G.; Tian, Y.; Liang, Y.; Li, R.; Liu, M.; Yuan, Y. ALK Inhibitors in the Treatment of ALK Positive NSCLC. Front. Oncol. 2019, 8, 557. [Google Scholar] [CrossRef] [PubMed]
- Paik, J.; Dhillon, S. Alectinib: A Review in Advanced, ALK-Positive NSCLC. Drugs 2018, 78, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Tiseo, M.; Ahn, M.J.; Reckamp, K.L.; Hansen, K.H.; Kim, S.W.; Huber, R.M.; West, H.L.; Groen, H.J.M.; Hochmair, M.J.; et al. Brigatinib in Patients With Crizotinib-Refractory Anaplastic Lymphoma Kinase-Positive Non-Small-Cell Lung Cancer: A Randomized, Multicenter Phase II Trial. J. Clin. Oncol. 2017, 35, 2490–2498. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Tan, D.S.W.; Chiari, R.; Wu, Y.L.; Paz-Ares, L.; Wolf, J.; Geater, S.L.; Orlov, S.; Cortinovis, D.; Yu, C.J.; et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): A randomised, open-label, phase 3 study. Lancet 2017, 389, 917–929. [Google Scholar] [CrossRef]
- Patcas, A.; Chis, A.F.; Militaru, C.F.; Bordea, I.R.; Rajnoveanu, R.; Coza, O.F.; Hanna, R.; Tiberiu, T.; Todea, D.A. An insight into lung cancer: A comprehensive review exploring ALK TKI and mechanisms of resistance. Bosn. J. Basic Med. Sci. 2022, 22, 1–13. [Google Scholar] [CrossRef]
- Kwak, E.L.; Bang, Y.J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.H.; Dezube, B.J.; Jänne, P.A.; Costa, D.B.; et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef]
- Kazandjian, D.; Blumenthal, G.M.; Chen, H.Y.; He, K.; Patel, M.; Justice, R.; Keegan, P.; Pazdur, R. FDA approval summary: Crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist 2014, 19, e5–e11. [Google Scholar] [CrossRef]
- Wu, Y.L.; Lu, S.; Lu, Y.; Zhou, J.; Shi, Y.K.; Sriuranpong, V.; Ho, J.C.M.; Ong, C.K.; Tsai, C.M.; Chung, C.H.; et al. Results of PROFILE 1029, a Phase III Comparison of First-Line Crizotinib versus Chemotherapy in East Asian Patients with ALK-Positive Advanced Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2018, 13, 1539–1548. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Crizotinib resistance: Implications for therapeutic strategies. Ann. Oncol. 2016, 27S, iii42–iii50. [Google Scholar] [CrossRef]
- Ling, Z.; Yunxia, L.; Shaohong, Z.; Chen, G.; Keke, N.; Youxin, J. Primary resistance to crizotinib treatment in a non-small cell lung cancer patient with an EML4-ALK rearrangement: A case report. Cancer Biol. Med. 2018, 15, 178. [Google Scholar] [CrossRef]
- Okada, K.; Araki, M.; Sakashita, T.; Ma, B.; Kanada, R.; Yanagitani, N.; Horiike, A.; Koike, S.; Oh-Hara, T.; Watanabe, K.; et al. Prediction of ALK mutations mediating ALK-TKIs resistance and drug re-purposing to overcome the resistance. EBioMedicine 2019, 41, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov. 2016, 6, 1118–1133. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Shaw, A.T.; Khan, T.M.; Mino-Kenudson, M.; Solomon, B.J.; Halmos, B.; Jessop, N.A.; Wain, J.C.; Yeo, A.T.; Benes, C.; et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci. Transl. Med. 2012, 4, 120ra17. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Akimoto, K.; Sato, H.; Manabe, R.; Kishino, Y.; Homma, T.; Kusumoto, S.; Yamaoka, T.; Tanaka, A.; Ohmori, T.; et al. Brigatinib and Alectinib for ALK Rearrangement-Positive Advanced Non-Small Cell Lung Cancer With or Without Central Nervous System Metastasis: A Systematic Review and Network Meta-Analysis. Cancers 2020, 12, 942. [Google Scholar] [CrossRef]
- Hida, T.; Nokihara, H.; Kondo, M.; Kim, Y.H.; Azuma, K.; Seto, T.; Takiguchi, Y.; Nishio, M.; Yoshioka, H.; Imamura, F.; et al. Alectinib versus crizotinib in patients with ALK-positive non-small-cell lung cancer (J-ALEX): An open-label, randomised phase 3 trial. Lancet 2017, 390, 29–39. [Google Scholar] [CrossRef]
- Nakagawa, K.; Hida, T.; Nokihara, H.; Morise, M.; Azuma, K.; Kim, Y.H.; Seto, T.; Takiguchi, Y.; Nishio, M.; Yoshioka, H.; et al. Final progression-free survival results from the J-ALEX study of alectinib versus crizotinib in ALK-positive non-small-cell lung cancer. Lung Cancer 2020, 139, 195–199. [Google Scholar] [CrossRef]
- Reckamp, K.; Lin, H.M.; Huang, J.; Proskorovsky, I.; Reichmann, W.; Krotneva, S.; Kerstein, D.; Huang, H.; Lee, J. Comparative efficacy of brigatinib versus ceritinib and alectinib in patients with crizotinib-refractory anaplastic lymphoma kinase-positive non-small cell lung cancer. Curr. Med. Res. Opin. 2019, 35, 569–576. [Google Scholar] [CrossRef]
- Camidge, D.R.; Kim, H.R.; Ahn, M.J.; Yang, J.C.; Han, J.Y.; Lee, J.S.; Hochmair, M.J.; Li, J.Y.; Chang, G.C.; Lee, K.H.; et al. Brigatinib versus Crizotinib in ALK-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2027–2039. [Google Scholar] [CrossRef]
- Lin, J.J.; Zhu, V.W.; Yoda, S.; Yeap, B.Y.; Schrock, A.B.; Dagogo-Jack, I.; Jessop, N.A.; Jiang, G.Y.; Le, L.P.; Gowen, K.; et al. Impact of EML4-ALK Variant on Resistance Mechanisms and Clinical Outcomes in ALK-Positive Lung Cancer. J. Clin. Oncol. 2018, 36, 1199–1206. [Google Scholar] [CrossRef]
- Yoda, S.; Lin, J.J.; Lawrence, M.S.; Burke, B.J.; Friboulet, L.; Langenbucher, A.; Dardaei, L.; Prutisto-Chang, K.; Dagogo-Jack, I.; Timofeevski, S.; et al. Sequential ALK Inhibitors Can Select for Lorlatinib-Resistant Compound ALK Mutations in ALK-Positive Lung Cancer. Cancer Discov. 2018, 8, 714–729. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.J.; Besse, B.; Bauer, T.M.; Felip, E.; Soo, R.A.; Camidge, D.R.; Chiari, R.; Bearz, A.; Lin, C.C.; Gadgeel, S.M.; et al. Lorlatinib in patients with ALK-positive non-small-cell lung cancer: Results from a global phase 2 study. Lancet Oncol. 2018, 19, 1654–1667. [Google Scholar] [CrossRef]
- Chen, J.; Ruiz-Garcia, A.; James, L.P.; Peltz, G.; Thurm, H.; Clancy, J.; Hibma, J. Lorlatinib Exposure-Response Analyses for Safety and Efficacy in a Phase I/II Trial to Support Benefit-Risk Assessment in Non-Small Cell Lung Cancer. Clin. Pharmacol. Ther. 2021, 110, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Bauer, T.M.; de Marinis, F.; Felip, E.; Goto, Y.; Liu, G.; Mazieres, J.; Kim, D.W.; Mok, T.; Polli, A.; et al. First-Line Lorlatinib or Crizotinib in Advanced ALK-Positive Lung Cancer. N. Engl. J. Med. 2020, 383, 2018–2029. [Google Scholar] [CrossRef]
- Gristina, V.; La Mantia, M.; Iacono, F.; Galvano, A.; Russo, A.; Bazan, V. The Emerging Therapeutic Landscape of ALK Inhibitors in Non-Small Cell Lung Cancer. Pharmaceuticals 2020, 13, 474. [Google Scholar] [CrossRef] [PubMed]
- Sheikine, Y.; Pavlick, D.; Klempner, S.J.; Trabucco, S.E.; Chung, J.H.; Rosenzweig, M.; Wang, K.; Velcheti, V.; Frampton, G.M.; Peled, N.; et al. BRAF in Lung Cancers: Analysis of Patient Cases Reveals Recurrent BRAF Mutations, Fusions, Kinase Duplications, and Concurrent Alterations. JCO Precis. Oncol. 2018, 2, 1–15. [Google Scholar] [CrossRef]
- Dankner, M.; Rose, A.A.N.; Rajkumar, S.; Siegel, P.M.; Watson, I.R. Classifying BRAF alterations in cancer: New rational therapeutic strategies for actionable mutations. Oncogene 2018, 37, 3183–3199. [Google Scholar] [CrossRef]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef]
- Degirmenci, U.; Wang, M.; Hu, J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells 2020, 9, 198. [Google Scholar] [CrossRef]
- Candido, S.; Rapisarda, V.; Marconi, A.; Malaponte, G.; Bevelacqua, V.; Gangemi, P.; Scalisi, A.; McCubrey, J.A.; Maestro, R.; Spandidos, D.A.; et al. Analysis of the B-RafV600E mutation in cutaneous melanoma patients with occupational sun exposure. Oncol. Rep. 2014, 31, 1079–1082. [Google Scholar] [CrossRef]
- Nam, S.K.; Yun, S.; Koh, J.; Kwak, Y.; Seo, A.N.; Park, K.U.; Kim, D.W.; Kang, S.B.; Kim, W.H.; Lee, H.S. BRAF, PIK3CA, and HER2 Oncogenic Alterations According to KRAS Mutation Status in Advanced Colorectal Cancers with Distant Metastasis. PLoS ONE 2016, 11, e0151865. [Google Scholar] [CrossRef] [PubMed]
- Nagayama, Y.; Mishima, H. Heterogenous nature of gene expression patterns in BRAF-like papillary thyroid carcinomas with BRAFV600E. Endocrine 2019, 66, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Villaruz, L.C.; Socinski, M.A.; Abberbock, S.; Berry, L.D.; Johnson, B.E.; Kwiatkowski, D.J.; Iafrate, A.J.; Varella-Garcia, M.; Franklin, W.A.; Camidge, D.R.; et al. Clinicopathologic features and outcomes of patients with lung adenocarcinomas harboring BRAF mutations in the Lung Cancer Mutation Consortium. Cancer 2015, 121, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. Mar. 2004, 116, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Zhang, H.; Ding, H.; Qian, J.; Lizaso, A.; Lin, J.; Han-Zhang, H.; Xiang, J.; Li, Y.; Zhu, H. The association between BRAF mutation class and clinical features in BRAF-mutant Chinese non-small cell lung cancer patients. J. Transl. Med. 2019, 17, 298. [Google Scholar] [CrossRef]
- Planchard, D.; Kim, T.M.; Mazieres, J.; Quoix, E.; Riely, G.; Barlesi, F.; Souquet, P.J.; Smit, E.F.; Groen, H.J.; Kelly, R.J.; et al. Dabrafenib in patients with BRAF(V600E)-positive advanced non-small-cell lung cancer: A single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 642–650. [Google Scholar] [CrossRef]
- Litvak, A.M.; Paik, P.K.; Woo, K.M.; Sima, C.S.; Hellmann, M.D.; Arcila, M.E.; Ladanyi, M.; Rudin, C.M.; Kris, M.G.; Riely, G.J. Clinical characteristics and course of 63 patients with BRAF mutant lung cancers. J. Thorac. Oncol. 2014, 9, 1669–1674. [Google Scholar] [CrossRef]
- Ding, X.; Zhang, Z.; Jiang, T.; Li, X.; Zhao, C.; Su, B.; Zhou, C. Clinicopathologic characteristics and outcomes of Chinese patients with non-small-cell lung cancer and BRAF mutation. Cancer Med. 2017, 6, 555–562. [Google Scholar] [CrossRef]
- Weart, T.C.; Miller, K.D.; Simone, C.B., 2nd. Spotlight on dabrafenib/trametinib in the treatment of non-small-cell lung cancer: Place in therapy. Cancer Manag. Res. 2018, 10, 647–652. [Google Scholar] [CrossRef]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef]
- Mazieres, J.; Cropet, C.; Montané, L.; Barlesi, F.; Souquet, P.J.; Quantin, X.; Dubos-Arvis, C.; Otto, J.; Favier, L.; Avrillon, V.; et al. Vemurafenib in non-small-cell lung cancer patients with BRAFV600 and BRAFnonV600 mutations. Ann. Oncol. 2020, 31, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Khunger, A.; Khunger, M.; Velcheti, V. Dabrafenib in combination with trametinib in the treatment of patients with BRAF V600-positive advanced or metastatic non-small cell lung cancer: Clinical evidence and experience. Ther. Adv. Respir. Dis. 2018, 12, 175346661876761. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Smit, E.F.; Groen, H.J.M.; Mazieres, J.; Besse, B.; Helland, Å.; Giannone, V.; D’Amelio, A.M., Jr.; Zhang, P.; Mookerjee, B.; et al. Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: An open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1307–1316. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sasane, M.; Zhang, J.; Zhao, J.; Ricculli, M.L.; Yao, Z.; Redhu, S.; Signorovitch, J. Is time to progression associated with post-progression survival in previously treated metastatic non-small cell lung cancer with BRAF V600E mutation? A secondary analysis of phase II clinical trial data. BMJ Open 2018, 8, e021642. [Google Scholar] [CrossRef]
- Bustamante, J.G.; Otterson, G.A. Agents to treat BRAF-mutant lung cancer. Drugs Context 2019, 8, 212566. [Google Scholar] [CrossRef]
- Okimoto, R.A.; Lin, L.; Olivas, V.; Chan, E.; Markegard, E.; Rymar, A.; Neel, D.; Chen, X.; Hemmati, G.; Bollag, G.; et al. Preclinical efficacy of a RAF inhibitor that evades paradoxical MAPK pathway activation in protein kinase BRAF-mutant lung cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 13456–13461. [Google Scholar] [CrossRef]
- Miyauchi, S.; Shien, K.; Takeda, T.; Araki, K.; Nakata, K.; Miura, A.; Takahashi, Y.; Kurihara, E.; Ogoshi, Y.; Namba, K.; et al. Antitumor Effects of Pan-RAF Inhibitor LY3009120 Against Lung Cancer Cells Harboring Oncogenic BRAF Mutation. Anticancer Res. 2020, 40, 2667–2673. [Google Scholar] [CrossRef]
- Kohno, T.; Tabata, J.; Nakaoku, T. REToma: A cancer subtype with a shared driver oncogene. Carcinogenesis 2020, 41, 123–129. [Google Scholar] [CrossRef]
- Wang, R.; Hu, H.; Pan, Y.; Li, Y.; Ye, T.; Li, C.; Luo, X.; Wang, L.; Li, H.; Zhang, Y.; et al. RET fusions define a unique molecular and clinicopathologic subtype of non-small-cell lung cancer. J. Clin. Oncol. 2012, 30, 4352–4359. [Google Scholar] [CrossRef]
- Drilon, A.; Lin, J.J.; Filleron, T.; Ni, A.; Milia, J.; Bergagnini, I.; Hatzoglou, V.; Velcheti, V.; Offin, M.; Li, B.; et al. Frequency of Brain Metastases and Multikinase Inhibitor Outcomes in Patients With RET-Rearranged Lung Cancers. J. Thorac. Oncol. 2018, 13, 1595–1601. [Google Scholar] [CrossRef]
- Ferrara, R.; Auger, N.; Auclin, E.; Besse, B. Clinical and Translational Implications of RET Rearrangements in Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2018, 13, 27–45. [Google Scholar] [CrossRef]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, L. Clinical development of RET inhibitors in RET-rearranged non-small cell lung cancer: Update. Oncol. Rev. 2018, 12, 352. [Google Scholar] [CrossRef]
- Drilon, A.; Rekhtman, N.; Arcila, M.; Wang, L.; Ni, A.; Albano, M.; Van Voorthuysen, M.; Somwar, R.; Smith, R.S.; Montecalvo, J.; et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: An open-label, single-centre, phase 2, single-arm trial. Lancet Oncol. 2016, 17, 1653–1660. [Google Scholar] [CrossRef]
- Yoh, K.; Seto, T.; Satouchi, M.; Nishio, M.; Yamamoto, N.; Murakami, H.; Nogami, N.; Matsumoto, S.; Kohno, T.; Tsuta, K.; et al. Vandetanib in patients with previously treated RET-rearranged advanced non-small-cell lung cancer (LURET): An open-label, multicentre phase 2 trial. Lancet Respir. Med. 2017, 5, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Hida, T.; Velcheti, V.; Reckamp, K.L.; Nokihara, H.; Sachdev, P.; Kubota, T.; Nakada, T.; Dutcus, C.E.; Ren, M.; Tamura, T. A phase 2 study of lenvatinib in patients with RET fusion-positive lung adenocarcinoma. Lung Cancer 2019, 138, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Bradford, D.; Larkins, E.; Mushti, S.L.; Rodriguez, L.; Skinner, A.M.; Helms, W.S.; Price, L.S.L.; Zirkelbach, J.F.; Li, Y.; Liu, J.; et al. FDA Approval Summary: Selpercatinib for the Treatment of Lung and Thyroid Cancers with RET Gene Mutations or Fusions. Clin. Cancer Res. 2021, 27, 2130–2135. [Google Scholar] [CrossRef]
- Markham, A. Pralsetinib: First Approval. Drugs 2020, 80, 1865–1870. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors: A 2021 update. Pharmacol. Res. 2021, 165, 105463. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Oxnard, G.R.; Tan, D.S.W.; Loong, H.H.F.; Johnson, M.; Gainor, J.; McCoach, C.E.; Gautschi, O.; Besse, B.; Cho, B.C.; et al. Efficacy of Selpercatinib in RET Fusion-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Gainor, J.F.; Curigliano, G.; Kim, D.W.; Lee, D.H.; Besse, B.; Baik, C.S.; Doebele, R.C.; Cassier, P.A.; Lopes, G.; Tan, D.S.W.; et al. Pralsetinib for RET fusion-positive non-small-cell lung cancer (ARROW): A multi-cohort, open-label, phase 1/2 study. Lancet Oncol. 2021, 22, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Stinchcombe, T.E. Current management of RET rearranged non-small cell lung cancer. Ther. Adv. Med. Oncol. 2020, 12, 175883592092863. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. The human hepatocyte growth factor receptor gene: Complete structural organization and promoter characterization. Gene 1998, 215, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Skead, G.; Govender, D. Gene of the month: MET. J. Clin. Pathol. 2015, 68, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef]
- Cui, J.J. Targeting Receptor Tyrosine Kinase MET in Cancer: Small Molecule Inhibitors and Clinical Progress. J. Med. Chem. 2014, 57, 4427–4453. [Google Scholar] [CrossRef] [PubMed]
- Gelsomino, F.; Facchinetti, F.; Haspinger, E.R.; Garassino, M.C.; Trusolino, L.; De Braud, F.; Tiseo, M. Targeting the MET gene for the treatment of non-small-cell lung cancer. Crit. Rev. Oncol. Hematol. 2014, 89, 284–299. [Google Scholar] [CrossRef]
- Schrock, A.B.; Frampton, G.M.; Suh, J.; Chalmers, Z.R.; Rosenzweig, M.; Erlich, R.L.; Halmos, B.; Goldman, J.; Forde, P.; Leuenberger, K.; et al. Characterization of 298 Patients with Lung Cancer Harboring MET Exon 14 Skipping Alterations. J. Thorac. Oncol. 2016, 11, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Frampton, G.M.; Ali, S.M.; Rosenzweig, M.; Chmielecki, J.; Lu, X.; Bauer, T.M.; Akimov, M.; Bufill, J.A.; Lee, C.; Jentz, D.; et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015, 5, 850–859. [Google Scholar]
- Guo, B.; Cen, H.; Tan, X.; Liu, W.; Ke, Q. Prognostic Value of MET Gene Copy Number and Protein Expression in Patients with Surgically Resected Non-Small Cell Lung Cancer: A Meta-Analysis of Published Literatures. PLoS ONE 2014, 9, e99399. [Google Scholar] [CrossRef]
- Pyo, J.S.; Kang, G.; Cho, W.J.; Choi, S.B. Clinicopathological significance and concordance analysis of c-MET immunohistochemistry in non-small cell lung cancers: A meta-analysis. Pathol. Res. Pract. 2016, 212, 710–716. [Google Scholar] [CrossRef]
- Reungwetwattana, T.; Liang, Y.; Zhu, V.; Ou, S.H.I. The race to target MET exon 14 skipping alterations in non-small cell lung cancer: The Why, the How, the Who, the Unknown, and the Inevitable. Lung Cancer 2017, 103, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Paik, P.K.; Drilon, A.; Fan, P.D.; Yu, H.; Rekhtman, N.; Ginsberg, M.S.; Borsu, L.; Schultz, N.; Berger, M.F.; Rudin, C.M.; et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015, 5, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Cortot, A.B.; Kherrouche, Z.; Descarpentries, C.; Wislez, M.; Baldacci, S.; Furlan, A.; Tulasne, D. Exon 14 Deleted MET Receptor as a New Biomarker and Target in Cancers. JNCI J. Natl. Cancer Inst. 2017, 109, 5. [Google Scholar] [CrossRef] [PubMed]
- Salgia, R.; Sattler, M.; Scheele, J.; Stroh, C.; Felip, E. The promise of selective MET inhibitors in non-small cell lung cancer with MET exon 14 skipping. Cancer Treat. Rev. 2020, 87, 102022. [Google Scholar] [CrossRef]
- Drilon, A.; Clark, J.W.; Weiss, J.; Ou, S.I.; Camidge, D.R.; Solomon, B.J.; Otterson, G.A.; Villaruz, L.C.; Riely, G.J.; Heist, R.S.; et al. Antitumor activity of crizotinib in lung cancers harboring a MET exon 14 alteration. Nat. Med. 2020, 26, 47–51. [Google Scholar] [CrossRef]
- Dhillon, S. Capmatinib: First Approval. Drugs 2020, 80, 1125–1131. [Google Scholar] [CrossRef]
- Safi, D.; Abu Hejleh, T.; Furqan, M. Narrative review: Mesenchymal-epithelial transition inhibitors-meeting their target. Transl. Lung Cancer Res. 2021, 10, 462–474. [Google Scholar] [CrossRef]
- Wolf, J.; Seto, T.; Han, J.Y.; Reguart, N.; Garon, E.B.; Groen, H.J.M.; Tan, D.S.W.; Hida, T.; de Jonge, M.; Orlov, S.V.; et al. Capmatinib in MET Exon 14-Mutated or MET-Amplified Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 944–957. [Google Scholar] [CrossRef]
- Paik, P.K.; Felip, E.; Veillon, R.; Sakai, H.; Cortot, A.B.; Garassino, M.C.; Mazieres, J.; Viteri, S.; Senellart, H.; Van Meerbeeck, J.; et al. Tepotinib in Non-Small-Cell Lung Cancer with MET Exon 14 Skipping Mutations. N. Engl. J. Med. 2020, 383, 931–943. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.; Horton, A.; Burton, L.; Schmelzer, C.; Vandlen, R.; Rosenthal, A. Neurotrophin-4/5 is a mammalian-specific survival factor for distinct populations of sensory neurons. J. Neurosci. 1993, 13, 4961–4967. [Google Scholar] [CrossRef]
- Harada, T.; Yatabe, Y.; Takeshita, M.; Koga, T.; Yano, T.; Wang, Y.; Giaccone, G. Role and Relevance of TrkB Mutations and Expression in Non–Small Cell Lung Cancer. Clin. Cancer Res. 2011, 17, 2638–2645. [Google Scholar] [CrossRef]
- Marchetti, A.; Felicioni, L.; Pelosi, G.; Del Grammastro, M.; Fumagalli, C.; Sciarrotta, M.; Malatesta, S.; Chella, A.; Barassi, F.; Mucilli, F.; et al. Frequent mutations in the neurotrophic tyrosine receptor kinase gene family in large cell neuroendocrine carcinoma of the lung. Hum. Mutat. 2008, 29, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Dunn, D.B. Larotrectinib and Entrectinib: TRK Inhibitors for the Treatment of Pediatric and Adult Patients With NTRK Gene Fusion. J. Adv. Pract. Oncol. 2020, 11, 418–423. [Google Scholar]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in patients with TRK fusion-positive solid tumours: A pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020, 21, 531–540. [Google Scholar] [CrossRef]
- Filippi, R.; Depetris, I.; Satolli, M.A. Evaluating larotrectinib for the treatment of advanced solid tumors harboring an NTRK gene fusion. Expert Opin. Pharmacother. 2021, 22, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Fuse, M.J.; Okada, K.; Oh-hara, T.; Ogura, H.; Fujita, N.; Katayama, R. Mechanisms of Resistance to NTRK Inhibitors and Therapeutic Strategies in NTRK1-Rearranged Cancers. Mol. Cancer Ther. 2017, 16, 2130–2143. [Google Scholar] [CrossRef]
- Somwar, R.; Hofmann, N.E.; Smith, B.; Odintsov, I.; Vojnic, M.; Linkov, I.; Tam, A.; Khodos, I.; Mattar, M.S.; de Stanchina, E.; et al. NTRK kinase domain mutations in cancer variably impact sensitivity to type I and type II inhibitors. Commun. Biol. 2020, 3, 776. [Google Scholar] [CrossRef] [PubMed]
- Guin, S.; Theodorescu, D. The RAS-RAL axis in cancer: Evidence for mutation-specific selectivity in non-small cell lung cancer. Acta Pharmacol. Sin. 2015, 36, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Ramjaun, A.R.; Haiko, P.; Wang, Y.; Warne, P.H.; Nicke, B.; Nye, E.; Stamp, G.; Alitalo, K.; Downward, J. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell 2007, 129, 957–968. [Google Scholar] [CrossRef]
- Stokoe, D.; Macdonald, S.G.; Cadwallader, K.; Symons, M.; Hancock, J.F. Activation of Raf as a result of recruitment to the plasma membrane. Science 1994, 264, 1463–1467. [Google Scholar] [CrossRef]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Brandt, R.; Sell, T.; Lüthen, M.; Uhlitz, F.; Klinger, B.; Riemer, P.; Giesecke-Thiel, C.; Schulze, S.; El-Shimy, I.A.; Kunkel, D.; et al. Cell type-dependent differential activation of ERK by oncogenic KRAS in colon cancer and intestinal epithelium. Nat. Commun. 2019, 10, 2919. [Google Scholar] [CrossRef]
- Garrido, P.; Olmedo, M.E.; Gómez, A.; Paz Ares, L.; López-Ríos, F.; Rosa-Rosa, J.M.; Palacios, J. Treating KRAS-mutant NSCLC: Latest evidence and clinical consequences. Ther. Adv. Med. Oncol. 2017, 9, 589–597. [Google Scholar] [CrossRef]
- Kempf, E.; Rousseau, B.; Besse, B.; Paz-Ares, L. KRAS oncogene in lung cancer: Focus on molecularly driven clinical trials. Eur. Respir. Rev. 2016, 25, 71–76. [Google Scholar] [CrossRef]
- Westcott, P.M.K.; To, M.D. The genetics and biology of KRAS in lung cancer. Chin. J. Cancer 2013, 32, 63–70. [Google Scholar] [CrossRef]
- Feng, H.; Zhang, Y.; Bos, P.H.; Chambers, J.M.; Dupont, M.M.; Stockwell, B.R. K-Ras G12D Has a Potential Allosteric Small Molecule Binding Site. Biochemistry 2019, 58, 2542–2554. [Google Scholar] [CrossRef]
- Muñoz-Maldonado, C.; Zimmer, Y.; Medová, M. A Comparative Analysis of Individual RAS Mutations in Cancer Biology. Front. Oncol. 2019, 9, 1088. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Byers, L.A.; Diao, L.; Papadimitrakopoulou, V.A.; Tong, P.; Izzo, J.; Behrens, C.; Kadara, H.; Parra, E.R.; Canales, J.R.; et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015, 5, 860–877. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Zugazagoitia, J.; Herbertz, S.; John, W.; Paz-Ares, L.; Schmid-Bindert, G. KRAS-Mutant non-small cell lung cancer: From biology to therapy. Lung Cancer 2018, 124, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Veluswamy, R.; Mack, P.C.; Houldsworth, J.; Elkhouly, E.; Hirsch, F.R. KRAS G12C–Mutant Non–Small Cell Lung Cancer. J. Mol. Diagn. 2021, 23, 507–520. [Google Scholar] [CrossRef]
- Saleh, K.; Kordahi, M.; Felefly, T.; Kourie, H.R.; Khalife, N. KRAS-targeted therapies in advanced solid cancers: Drug the undruggable? Pharmacogenomics 2021, 22, 587–590. [Google Scholar] [CrossRef]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.A.; Rybkin, I.I.; Spira, A.I.; Riely, G.J.; Papadopoulos, K.P.; Sabari, J.K.; Johnson, M.L.; Heist, R.S.; Bazhenova, L.; Barve, M.; et al. KRYSTAL-1: Activity and Safety of Adagrasib (MRTX849) in Advanced/ Metastatic Non–Small-Cell Lung Cancer (NSCLC) Harboring KRAS G12C Mutation. Eur. J. Cancer 2020, 138, S1–S2. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- D’Angelo, A.; Sobhani, N.; Chapman, R.; Bagby, S.; Bortoletti, C.; Traversini, M.; Ferrari, K.; Voltolini, L.; Darlow, J.; Roviello, G. Focus on ROS1-Positive Non-Small Cell Lung Cancer (NSCLC): Crizotinib, Resistance Mechanisms and the Newer Generation of Targeted Therapies. Cancers 2020, 12, 3293. [Google Scholar] [CrossRef]
- Yoshida, A.; Kohno, T.; Tsuta, K.; Wakai, S.; Arai, Y.; Shimada, Y.; Asamura, H.; Furuta, K.; Shibata, T.; Tsuda, H. ROS1-rearranged lung cancer: A clinicopathologic and molecular study of 15 surgical cases. Am. J. Surg. Pathol. 2013, 37, 554–562. [Google Scholar] [CrossRef]
- Davies, K.D.; Doebele, R.C. Molecular Pathways: ROS1 Fusion Proteins in Cancer. Clin. Cancer Res. 2013, 19, 4040–4045. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.D.; Le, A.T.; Theodoro, M.F.; Skokan, M.C.; Aisner, D.L.; Berge, E.M.; Terracciano, L.M.; Cappuzzo, F.; Incarbone, M.; Roncalli, M.; et al. Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clin. Cancer Res. 2012, 18, 4570–4579. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Shaw, A.T. Recent Advances in Targeting ROS1 in Lung Cancer. J. Thorac. Oncol. 2017, 12, 1611–1625. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Ou, S.H.; Bang, Y.J.; Camidge, D.R.; Solomon, B.J.; Salgia, R.; Riely, G.J.; Varella-Garcia, M.; Shapiro, G.I.; Costa, D.B.; et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 371, 1963–1971. [Google Scholar] [CrossRef] [PubMed]
- Moro-Sibilot, D.; Cozic, N.; Pérol, M.; Mazières, J.; Otto, J.; Souquet, P.J.; Bahleda, R.; Wislez, M.; Zalcman, G.; Guibert, S.D.; et al. Crizotinib in c-MET- or ROS1-positive NSCLC: Results of the AcSé phase II trial. Ann. Oncol. 2019, 30, 1985–1991. [Google Scholar] [CrossRef]
- Lim, S.M.; Kim, H.R.; Lee, J.S.; Lee, K.H.; Lee, Y.G.; Min, Y.J.; Cho, E.K.; Lee, S.S.; Kim, B.S.; Choi, M.Y.; et al. Open-Label, Multicenter, Phase II Study of Ceritinib in Patients With Non-Small-Cell Lung Cancer Harboring ROS1 Rearrangement. J. Clin. Oncol. 2017, 35, 2613–2618. [Google Scholar] [CrossRef]
- Drilon, A.; Siena, S.; Dziadziuszko, R.; Barlesi, F.; Krebs, M.G.; Shaw, A.T.; de Braud, F.; Rolfo, C.; Ahn, M.J.; Wolf, J.; et al. Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: Integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020, 21, 261–270. [Google Scholar] [CrossRef]
- Davare, M.A.; Vellore, N.A.; Wagner, J.P.; Eide, C.A.; Goodman, J.R.; Drilon, A.; Deininger, M.W.; O’Hare, T.; Druker, B.J. Structural insight into selectivity and resistance profiles of ROS1 tyrosine kinase inhibitors. Proc. Natl. Acad. Sci. USA 2015, 112, E5381–E5390. [Google Scholar] [CrossRef]
- Pathak, D.; Choudhary, S.; Singh, P.K.; Singh, M.; Chadha, N.; Silakari, O. Pharmacophore-based designing of putative ROS-1 targeting agents for NSCLC. Mol. Divers. 2021, 25, 1091–1102. [Google Scholar] [CrossRef]
- Vanajothi, R.; Vedagiri, H.; Al-Ansari, M.M.; Al-Humaid, L.A.; Kumpati, P. Pharmacophore based virtual screening, molecular docking and molecular dynamic simulation studies for finding ROS1 kinase inhibitors as potential drug molecules. J. Biomol. Struct. Dyn. 2020, 17, 1–15. [Google Scholar] [CrossRef]
- Pathak, D.; Chadha, N.; Silakari, O. Identification of non-resistant ROS-1 inhibitors using structure-based pharmacophore analysis. J. Mol. Graph. Model. 2016, 70, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.Y.; Li, Q.; Engstrom, L.D.; West, M.; Appleman, V.; Wong, K.A.; McTigue, M.; Deng, Y.L.; Liu, W.; Brooun, A.; et al. PF-06463922 is a potent and selective next-generation ROS1/ALK inhibitor capable of blocking crizotinib-resistant ROS1 mutations. Proc. Natl. Acad. Sci. USA 2015, 112, 3493–3498. [Google Scholar] [CrossRef]
- Ou, S.I.; Fujiwara, Y.; Shaw, A.T.; Yamamoto, N.; Nakagawa, K.; Fan, F.; Hao, Y.; Gao, Y.; Jänne, P.A.; Seto, T. Efficacy of Taletrectinib (AB-106/DS-6051b) in ROS1+ NSCLC: An Updated Pooled Analysis of U.S. and Japan Phase 1 Studies. JTO Clin. Res. Rep. 2020, 2, 100108. [Google Scholar] [CrossRef] [PubMed]
- Karakas, B.; Bachman, K.E.; Park, B.H. Mutation of the PIK3CA oncogene in human cancers. Br. J. Cancer 2006, 94, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.; Ghosh, P.; Kumavath, R. Strophanthidin Attenuates MAPK, PI3K/AKT/mTOR, and Wnt/β-Catenin Signaling Pathways in Human Cancers. Front. Oncol. 2020, 9, 1469. [Google Scholar] [CrossRef]
- Lu, H.; Qin, J.; Han, N.; Lei, L.; Xie, F.; Li, C. EGFR, KRAS, BRAF, PTEN, and PIK3CA mutation in plasma of small cell lung cancer patients. Onco Targets Ther. 2018, 11, 2217–2226. [Google Scholar] [CrossRef] [PubMed]
- Scheffler, M.; Bos, M.; Gardizi, M.; König, K.; Michels, S.; Fassunke, J.; Heydt, C.; Künstlinger, H.; Ihle, M.; Ueckeroth, F.; et al. PIK3CA mutations in non-small cell lung cancer (NSCLC): Genetic heterogeneity, prognostic impact and incidence of prior malignancies. Oncotarget 2015, 6, 1315–1326. [Google Scholar] [CrossRef]
- Li, S.; Li, L.; Zhu, Y.; Huang, C.; Qin, Y.; Liu, H.; Ren-Heidenreich, L.; Shi, B.; Ren, H.; Chu, X.; et al. Coexistence of EGFR with KRAS, or BRAF, or PIK3CA somatic mutations in lung cancer: A comprehensive mutation profiling from 5125 Chinese cohorts. Br. J. Cancer 2014, 110, 2812–2820. [Google Scholar] [CrossRef] [PubMed]
- Vansteenkiste, J.F.; Canon, J.L.; De Braud, F.; Grossi, F.; De Pas, T.; Gray, J.E.; Su, W.C.; Felip, E.; Yoshioka, H.; Gridelli, C.; et al. Safety and Efficacy of Buparlisib (BKM120) in Patients with PI3K Pathway-Activated Non-Small Cell Lung Cancer: Results from the Phase II BASALT-1 Study. J. Thorac. Oncol. 2015, 10, 1319–1327. [Google Scholar] [CrossRef]
- Lopez-Chavez, A.; Thomas, A.; Rajan, A.; Raffeld, M.; Morrow, B.; Kelly, R.; Carter, C.A.; Guha, U.; Killian, K.; Lau, C.C.; et al. Molecular profiling and targeted therapy for advanced thoracic malignancies: A biomarker-derived, multiarm, multihistology phase II basket trial. J. Clin. Oncol. 2015, 33, 1000–1007. [Google Scholar] [CrossRef]
- Langer, C.J.; Redman, M.W.; Wade, J.L., 3rd; Aggarwal, C.; Bradley, J.D.; Crawford., J.; Stella, P.J.; Knapp, M.H.; Miao, J.; Minichiello, K.; et al. SWOG S1400B (NCT02785913), a Phase II Study of GDC-0032 (Taselisib) for Previously Treated PI3K-Positive Patients with Stage IV Squamous Cell Lung Cancer (Lung-MAP Sub-Study). J. Thorac. Oncol. 2019, 14, 1839–1846. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Chan, T.A.; Kroemer, G.; Wolchok, J.D.; López-Soto, A. The hallmarks of successful anticancer immunotherapy. Sci. Transl. Med. 2018, 10, eaat7807. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.S.; Melucci, A.D.; Chacon, A.C.; Prieto, P.A. Adoptive T Cell Therapy for Solid Tumors: Pathway to Personalized Standard of Care. Cells 2021, 10, 808. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar]
- Jung, N.C.; Lee, J.H.; Chung, K.H.; Kwak, Y.S.; Lim, D.S. Dendritic Cell-Based Immunotherapy for Solid Tumors. Transl. Oncol. 2018, 11, 686–690. [Google Scholar] [CrossRef]
- Liu, Z.; Han, C.; Fu, Y.X. Targeting innate sensing in the tumor microenvironment to improve immunotherapy. Cell. Mol. Immunol. 2020, 17, 13–26. [Google Scholar]
- Shin, E.C. Cancer immunotherapy: Special issue of BMB Reports in 2021. BMB Rep. 2021, 54, 1. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Dong, Y.; Sun, Q.; Zhang, X. PD-1 and its ligands are important immune checkpoints in cancer. Oncotarget 2017, 8, 2171–2186. [Google Scholar]
- Shen, X.; Zhang, L.; Li, J.; Li, Y.; Wang, Y.; Xu, Z.X. Recent Findings in the Regulation of Programmed Death Ligand 1 Expression. Front. Immunol. 2019, 10, 1337. [Google Scholar]
- Wu, Y.; Chen, W.; Xu, Z.P.; Gu, W. PD-L1 Distribution and Perspective for Cancer Immunotherapy-Blockade, Knockdown, or Inhibition. Front. Immunol. 2019, 10, 2022. [Google Scholar] [CrossRef] [PubMed]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef]
- Zhou, X.; Yao, Z.; Yang, H.; Liang, N.; Zhang, X.; Zhang, F. Are immune-related adverse events associated with the efficacy of immune checkpoint inhibitors in patients with cancer? A systematic review and meta-analysis. BMC Med. 2020, 18, 87. [Google Scholar] [CrossRef]
- Twomey, J.D.; Zhang, B. Cancer Immunotherapy Update: FDA-Approved Checkpoint Inhibitors and Companion Diagnostics. AAPS J. 2021, 23, 39. [Google Scholar] [CrossRef]
- Zhou, F.; Qiao, M.; Zhou, C. The cutting-edge progress of immune-checkpoint blockade in lung cancer. Cell. Mol. Immunol. 2021, 18, 279–293. [Google Scholar] [PubMed]
- Teixidor, E.; Bosch-Barrera, J. The dark side of immunotherapy: Challenges facing the new hope in cancer treatment. Ann. Transl. Med. 2019, 7, S183. [Google Scholar] [CrossRef]
- Kim, J.M.; Chen, D.S. Immune escape to PD-L1/PD-1 blockade: Seven steps to success (or failure). Ann. Oncol. 2016, 27, 1492–1504. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Chen, C.; Gu, Y.; Lu, W.; Zhan, P.; Liu, H.; Lv, T.; Song, Y.; Zhang, F. Immune-Related Adverse Events Predict the Efficacy of Immune Checkpoint Inhibitors in Lung Cancer Patients: A Meta-Analysis. Front. Oncol. 2021, 11, 631949. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Ibarrondo, F.J. Comin-Anduix B and Escuin-Ordinas H: Tremelimumab: Research and clinical development. Onco Targets Ther. 2016, 9, 1767–1776. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Bondarenko, I.; Luft, A.; Serwatowski, P.; Barlesi, F.; Chacko, R.; Sebastian, M.; Neal, J.; Lu, H.; Cuillerot, J.M.; et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: Results from a randomized, double-blind, multicenter phase II study. J. Clin. Oncol. 2012, 30, 2046–2054. [Google Scholar] [CrossRef]
- Reck, M.; Bondarenko, I.; Luft, A.; Serwatowski, P.; Barlesi, F.; Chacko, R.; Sebastian, M.; Lu, H.; Cuillerot, J.M.; Lynch, T.J. Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: Results from a randomized, double-blind, multicenter phase 2 trial. Ann. Oncol. 2013, 24, 75–83. [Google Scholar] [CrossRef]
- Arriola, E.; Wheater, M.; Galea, I.; Cross, N.; Maishman, T.; Hamid, D.; Stanton, L.; Cave, J.; Geldart, T.; Mulatero, C.; et al. Outcome and Biomarker Analysis from a Multicenter Phase 2 Study of Ipilimumab in Combination with Carboplatin and Etoposide as First-Line Therapy for Extensive-Stage SCLC. J. Thorac. Oncol. 2016, 11, 1511–1521. [Google Scholar] [CrossRef]
- Reck, M.; Luft, A.; Szczesna, A.; Havel, L.; Kim, S.W.; Akerley, W.; Pietanza, M.C.; Wu, Y.L.; Zielinski, C.; Thomas, M.; et al. Phase III Randomized Trial of Ipilimumab Plus Etoposide and Platinum Versus Placebo Plus Etoposide and Platinum in Extensive-Stage Small-Cell Lung Cancer. J. Clin. Oncol. 2016, 34, 3740–3748. [Google Scholar] [CrossRef] [PubMed]
- Fehrenbacher, L.; Spira, A.; Ballinger, M.; Kowanetz, M.; Vansteenkiste, J.; Mazieres, J.; Park, K.; Smith, D.; Artal-Cortes, A.; Lewanski, C.; et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016, 387, 1837–1846. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Hotta, K.; Nishio, M.; Saito, H.; Okamoto, I.; Nakahara, Y.; Hayashi, H.; Hayama, M.; Laud, P.; Jiang, H.; Paz-Ares, L.; et al. First-line durvalumab plus platinum-etoposide in extensive-stage small-cell lung cancer: CASPIAN Japan subgroup analysis. Int. J. Clin. Oncol. 2021, 26, 1073–1082. [Google Scholar] [CrossRef]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef]
- Vokes, E.E.; Ready, N.; Felip, E.; Horn, L.; Burgio, M.A.; Antonia, S.J.; Arén Frontera, O.; Gettinger, S.; Holgado, E.; Spigel, D.; et al. Nivolumab versus docetaxel in previously treated advanced non-small-cell lung cancer (CheckMate 017 and CheckMate 057): 3-year update and outcomes in patients with liver metastases. Ann. Oncol. 2018, 29, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Lu, S.; Cheng, Y.; Zhou, C.; Wang, J.; Mok, T.; Zhang, L.; Tu, H.Y.; Wu, L.; Feng, J.; et al. Nivolumab Versus Docetaxel in a Predominantly Chinese Patient Population With Previously Treated Advanced NSCLC: CheckMate 078 Randomized Phase III Clinical Trial. J. Thorac. Oncol. 2019, 14, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Pai-Scherf, L.; Blumenthal, G.M.; Li, H.; Subramaniam, S.; Mishra-Kalyani, P.S.; He, K.; Zhao, H.; Yu, J.; Paciga, M.; Goldberg, K.B.; et al. FDA Approval Summary: Pembrolizumab for Treatment of Metastatic Non-Small Cell Lung Cancer: First-Line Therapy and Beyond. Oncologist 2017, 22, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.K.; Wu, Y.L.; Kudaba, I.; Kowalski, D.M.; Cho, B.C.; Turna, H.Z.; Castro, G., Jr.; Srimuninnimit, V.; Laktionov, K.K.; Bondarenko, I.; et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): A randomised, open-label, controlled, phase 3 trial. Lancet 2019, 393, 1819–1830. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Langer, C.J.; Paz-Ares, L.; Rodríguez-Abreu, D.; Halmos, B.; Garassino, M.C.; Houghton, B.; Kurata, T.; Cheng, Y.; Lin, J.; et al. Pembrolizumab plus chemotherapy versus chemotherapy alone in patients with advanced non-small cell lung cancer without tumor PD-L1 expression: A pooled analysis of 3 randomized controlled trials. Cancer 2020, 126, 4867–4877. [Google Scholar] [CrossRef]
- Theelen, W.S.M.E.; Peulen, H.M.U.; Lalezari, F.; van der Noort, V.; de Vries, J.F.; Aerts, J.G.J.V.; Dumoulin, D.W.; Bahce, I.; Niemeijer, A.N.; de Langen, A.J.; et al. Effect of Pembrolizumab After Stereotactic Body Radiotherapy vs Pembrolizumab Alone on Tumor Response in Patients With Advanced Non-Small Cell Lung Cancer: Results of the PEMBRO-RT Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1276–1282. [Google Scholar] [CrossRef]
- Welsh, J.; Menon, H.; Chen, D.; Verma, V.; Tang, C.; Altan, M.; Hess, K.; de Groot, P.; Nguyen, Q.N.; Varghese, R.; et al. Pembrolizumab with or without radiation therapy for metastatic non-small cell lung cancer: A randomized phase I/II trial. J. Immunother Cancer 2020, 8, e001001. [Google Scholar] [CrossRef]
- Theelen, W.S.M.E.; Chen, D.; Verma, V.; Hobbs, B.P.; Peulen, H.M.U.; Aerts, J.G.J.V.; Bahce, I.; Niemeijer, A.L.N.; Chang, J.Y.; de Groot, P.M.; et al. Pembrolizumab with or without radiotherapy for metastatic non-small-cell lung cancer: A pooled analysis of two randomised trials. Lancet Respir. Med. 2021, 9, 467–475. [Google Scholar] [CrossRef]
- Krishnamurthy, A.; Jimeno, A. Atezolizumab: A novel PD-L1 inhibitor in cancer therapy with a focus in bladder and non-small cell lung cancers. Drugs Today 2017, 53, 217. [Google Scholar] [CrossRef]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Kurata, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC. N. Engl. J. Med. 2018, 379, 2342–2350. [Google Scholar] [CrossRef]
- Faivre-Finn, C.; Vicente, D.; Kurata, T.; Planchard, D.; Paz-Ares, L.; Vansteenkiste, J.F.; Spigel, D.R.; Garassino, M.C.; Reck, M.; Senan, S.; et al. Four-Year Survival with Durvalumab After Chemoradiotherapy in Stage III NSCLC—An Update From the PACIFIC Trial. J. Thorac. Oncol. 2021, 16, 860–867. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Dvorkin, M.; Chen, Y.; Reinmuth, N.; Hotta, K.; Trukhin, D.; Statsenko, G.; Hochmair, M.J.; Özgüroğlu, M.; Ji, J.H.; et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): A randomised, controlled, open-label, phase 3 trial. Lancet 2019, 394, 1929–1939. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.E.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Kurata, T.; Chiappori, A.; Lee, K.H.; Cho, B.C.; et al. Three-Year Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC—Update from PACIFIC. J. Thorac. Oncol. 2020, 15, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Barlesi, F.; Vansteenkiste, J.; Spigel, D.; Ishii, H.; Garassino, M.; de Marinis, F.; Özgüroğlu, M.; Szczesna, A.; Polychronis, A.; Uslu, R.; et al. Avelumab versus docetaxel in patients with platinum-treated advanced non-small-cell lung cancer (JAVELIN Lung 200): An open-label, randomised, phase 3 study. Lancet. Oncol. 2018, 19, 1468–1479. [Google Scholar] [CrossRef] [PubMed]
- Pabla, S.; Conroy, J.M.; Nesline, M.K.; Glenn, S.T.; Papanicolau-Sengos, A.; Burgher, B.; Hagen, J.; Giamo, V.; Andreas, J.; Lenzo, F.L.; et al. Proliferative potential and resistance to immune checkpoint blockade in lung cancer patients. J. Immunother. Cancer 2019, 7, 27. [Google Scholar] [CrossRef]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. [Google Scholar] [CrossRef]
- Ready, N.E.; Ott, P.A.; Hellmann, M.D.; Zugazagoitia, J.; Hann, C.L.; de Braud, F.; Antonia, S.J.; Ascierto, P.A.; Moreno, V.; Atmaca, A.; et al. Nivolumab Monotherapy and Nivolumab Plus Ipilimumab in Recurrent Small Cell Lung Cancer: Results From the CheckMate 032 Randomized Cohort. J. Thorac. Oncol. 2020, 15, 426–435. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Ciuleanu, T.E.; Cobo, M.; Schenker, M.; Zurawski, B.; Menezes, J.; Richardet, E.; Bennouna, J.; Felip, E.; Juan-Vidal, O.; et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): An international, randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 198–211. [Google Scholar] [CrossRef]
- Boyer, M.; Şendur, M.A.N.; Rodríguez-Abreu, D.; Park, K.; Lee, D.H.; Çiçin, I.; Yumuk, P.F.; Orlandi, F.J.; Leal, T.A.; Molinier, O.; et al. Pembrolizumab Plus Ipilimumab or Placebo for Metastatic Non-Small-Cell Lung Cancer With PD-L1 Tumor Proportion Score ≥ 50%: Randomized, Double-Blind Phase III KEYNOTE-598 Study. J. Clin. Oncol. 2021, 39, 2327–2338. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Reinmuth, N.; Orlov, S.; Fischer, J.R.; Sugawara, S.; Mandziuk, S.; Marquez-Medina, D.; Novello, S.; Takeda, Y.; Soo, R.; et al. ARCTIC: Durvalumab with or without tremelimumab as third-line or later treatment of metastatic non-small-cell lung cancer. Ann. Oncol. 2020, 31, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Cho, B.C.; Reinmuth, N.; Lee, K.H.; Luft, A.; Ahn, M.J.; van den Heuvel, M.M.; Cobo, M.; Vicente, D.; Smolin, A.; et al. Durvalumab With or Without Tremelimumab vs Standard Chemotherapy in First-line Treatment of Metastatic Non-Small Cell Lung Cancer: The MYSTIC Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Onoi, K.; Chihara, Y.; Uchino, J.; Shimamoto, T.; Morimoto, Y.; Iwasaku, M.; Kaneko, Y.; Yamada, T.; Takayama, K. Immune Checkpoint Inhibitors for Lung Cancer Treatment: A Review. J. Clin. Med. 2020, 9, 1362. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Li, X.; Huang, Q.; Zheng, X.; Liu, M. Progress and Challenges of Predictive Biomarkers for Immune Checkpoint Blockade. Front. Oncol. 2021, 11, 617335. [Google Scholar] [CrossRef]
- Bai, R.; Lv, Z.; Xu, D.; Cui, J. Predictive biomarkers for cancer immunotherapy with immune checkpoint inhibitors. Biomark. Res. 2020, 8, 34. [Google Scholar] [CrossRef]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef]
- Herbst, R.S.; Baas, P.; Kim, D.W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.Y.; Molina, J.; Kim, J.H.; Arvis, C.D.; Ahn, M.J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef]
- Hong, L.; Negrao, M.V.; Dibaj, S.S.; Chen, R.; Reuben, A.; Bohac, J.M.; Liu, X.; Skoulidis, F.; Gay, C.M.; Cascone, T.; et al. Programmed Death-Ligand 1 Heterogeneity and Its Impact on Benefit From Immune Checkpoint Inhibitors in NSCLC. J. Thorac. Oncol. 2020, 15, 1449–1459. [Google Scholar] [CrossRef]
- Gaule, P.; Smithy, J.W.; Toki, M.; Rehman, J.; Patell-Socha, F.; Cougot, D.; Collin, P.; Morrill, P.; Neumeister, V.; Rimm, D.L. A Quantitative Comparison of Antibodies to Programmed Cell Death 1 Ligand 1. JAMA Oncol. 2017, 3, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Greillier, L.; Tomasini, P.; Barlesi, F. The clinical utility of tumor mutational burden in non-small cell lung cancer. Transl. Lung Cancer Res. 2018, 7, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhao, J.; Wang, G.; Zhang, F.; Zhang, Z.; Zhang, F.; Zhang, Y.; Dong, H.; Zhao, X.; Duan, J.; et al. Comutations in DNA Damage Response Pathways Serve as Potential Biomarkers for Immune Checkpoint Blockade. Cancer Res. 2018, 78, 6486–6496. [Google Scholar] [CrossRef] [PubMed]
- Gandara, D.R.; Paul, S.M.; Kowanetz, M.; Schleifman, E.; Zou, W.; Li, Y.; Rittmeyer, A.; Fehrenbacher, L.; Otto, G.; Malboeuf, C.; et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat. Med. 2018, 24, 1441–1448. [Google Scholar] [CrossRef]
- Garassino, M.; Rodriguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; Speranza, G.; Reck, M.; Hui, R.; Boyer, M.; Cristescu, R.; et al. OA04.06 Evaluation of TMB in KEYNOTE-189: Pembrolizumab Plus Chemotherapy vs Placebo Plus Chemotherapy for Nonsquamous NSCLC. J. Thorac. Oncol. 2019, 14, S216–S217. [Google Scholar] [CrossRef]
- Rackaityte, E.; Lynch, S.V. The human microbiome in the 21st century. Nat. Commun. 2020, 11, 5256. [Google Scholar] [CrossRef] [PubMed]
- Carbone, C.; Piro, G.; Di Noia, V.; D’Argento, E.; Vita, E.; Ferrara, M.G.; Pilotto, S.; Milella, M.; Cammarota, G.; Gasbarrini, A.; et al. Lung and Gut Microbiota as Potential Hidden Driver of Immunotherapy Efficacy in Lung Cancer. Mediat. Inflamm. 2019, 2019, 7652014. [Google Scholar] [CrossRef]
- Vivarelli, S.; Salemi, R.; Candido, S.; Falzone, L.; Santagati, M.; Stefani, S.; Torino, F.; Banna, G.L.; Tonini, G.; Libra, M. Gut Microbiota and Cancer: From Pathogenesis to Therapy. Cancers 2019, 11, 38. [Google Scholar] [CrossRef]
- Martins, D.; Mendes, F.; Schmitt, F. Microbiome: A Supportive or a Leading Actor in Lung Cancer? Pathobiology 2021, 88, 198–207. [Google Scholar] [CrossRef]
- Baruch, E.N.; Youngster, I.; Ben-Betzalel, G.; Ortenberg, R.; Lahat, A.; Katz, L.; Adler, K.; Dick-Necula, D.; Raskin, S.; Bloch, N.; et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2021, 371, 602–609. [Google Scholar] [CrossRef]
- Davar, D.; Dzutsev, A.K.; McCulloch, J.A.; Rodrigues, R.R.; Chauvin, J.M.; Morrison, R.M.; Deblasio, R.N.; Menna, C.; Ding, Q.; Pagliano, O.; et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 2021, 371, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Vivarelli, S.; Falzone, L.; Basile, M.; Nicolosi, D.; Genovese, C.; Libra, M.; Salmeri, M. Benefits of using probiotics as adjuvants in anticancer therapy (Review). World Acad. Sci. J. 2019, 1, 125–135. [Google Scholar] [CrossRef]
- Falzone, L.; Scandurra, G.; Lombardo, V.; Gattuso, G.; Lavoro, A.; Distefano, A.B.; Scibilia, G.; Scollo, P. A multidisciplinary approach remains the best strategy to improve and strengthen the management of ovarian cancer (Review). Int. J. Oncol. 2021, 59, 53. [Google Scholar] [CrossRef]
- Ramirez, R.A.; Cass, A.S.; Das, S.; Low, S.W.; Mehrad, M.; Rickman, O.B.; Scherer, P.M.; Thomas, K.E.; Gillaspie, E.A. A multidisciplinary approach to the work up and management of pulmonary carcinoid tumors and DIPNECH: A narrative review. Transl. Lung Cancer Res. 2022, 11, 2567–2587. [Google Scholar] [CrossRef]
- Kapoor, A.; Prabhash, K. Analysis of Outcomes With Addition of Immunotherapy to Chemoradiation Therapy for Non-Small Cell Lung Cancer. JAMA Oncol. 2022, 8, 168. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, R.A.; Lu, J.; Thomas, K.E.H. Quality of life for non-small cell lung cancer patients in the age of immunotherapy. Transl. Lung Cancer Res. 2018, 7, S149–S152. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.E.R.; Mansfield, A.S. Validating chemoimmunotherapy in small-cell lung cancer. Lancet Oncol 2022, 23, 692–693. [Google Scholar] [CrossRef]
- Pennell, N.A. Strategies and End Points in the Development of Novel Immunotherapy Trials for Patients With Unresectable, Locally Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2022, 40, 3353–3356. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Target | Trial Type | Agents | Side Effects | References |
|---|---|---|---|---|
| EGFR | Phase II/III | erlotinib | Rash, diarrhea, anorexia, fatigue, dyspnea, cough, nausea, vomiting | [46,48,54,56,62,63] |
| Phase II/III | gefitinib | Abdominal or stomach pain or tenderness, clay-colored stools, dark urine, diarrhea, severe Fever, headache, nausea and vomiting, weakness, yellow eyes or skin | [49,50,56,57,58,59,62,63] | |
| Phase III | afatinib | Diarrhea, paronychia, skin rush, stomatitis, pruritus | [53,54,55,56] | |
| Phase III | dacomitinib | Skin lesions, diarrhea, cough, fever, headache, nasal congestion, sore throat sores, ulcers, or white spots on the lips, tongue, or inside the mouth, weakness | [57,58,59] | |
| Phase III | osimertinib | Low blood cell counts, pain, diarrhea, tiredness, cough, mouth sores, dry skin, rash | [62,63] | |
| ALK | Phase II/III | crizotinib | Diarrhea, vomiting, nausea, vision disorder, headache, musculoskeletal pain, stomatitis, fatigue | [77,78,86,94] |
| Phase I/II/III | alectinib | Tiredness, constipation, muscle pain, anemia, swelling | [86,88] | |
| Phase II | brigatinib | Asthenia, blurred vision, cough, nausea, diarrhea, hyperglycemia, hypertension, myalgia, fatigue | [88] | |
| Phase I/II | ceritinib | Abdominal pain, diarrhea, hyperglycemia, increased serum ALT and AST, nausea, vomiting | [88] | |
| Phase I/II | erlotinib | Anorexia, eye infection, diarrhea, vomiting, skin rash, nausea | [88] | |
| Phase III | lorlatinib | Diarrhea, neuropathy, obesity, cough, edema, cognitive disorders | [92,93,94] | |
| BRAF | Phase II | vemurafenib | Alopecia, skin photosensitivity, hyperkeratosis, arthralgia, fatigue, rash, diarrhea, headache, skin papilloma | [110,111] |
| Phase II | dabrafenib | Fever, hyperglycemia, squamous cell carcinoma | [106,113,114] | |
| MEK | Phase II | trametinib | Rash, swelling (eye, face, tongue), cough, trouble breathing | [113,114] |
| RET | Phase I/II | selpercatinib | Bleeding, constipation, hair loss, hyperglycemia, difficulty in breathing, pneumonia, increased AST and ALT levels, hyponatremia, neutropenia | [130] |
| Phase I/II | pralsetinib | Bone pain, cramping, swelling, stomatitis, diarrhea, weakness | [131] | |
| MET | Phase I | crizotinib | Abdominal pain, diarrhea, hyperglycemia, increased serum ALT and AST, nausea, vomiting | [146] |
| Phase II | capmatinib | Edema, nausea, vomiting, high creatinine level, skin rush, stomach pain | [149] | |
| Phase II | tepotinib | Cough, anxiety, dark urine, sore throat, trouble breathing, fever, muscle and bone pain | [150] | |
| NTRK | Phase I/II | larotrectinib | Diarrhea, nausea, dizziness, vomiting, anemia, increased AST and ALT levels, cough, constipation, fatigue | [156] |
| Phase I/II | entrectinib | Vision disorders, diarrhea, nausea, edema, cognitive disorders, vomiting, dizziness, dyspnea, myalgia | [159,187] | |
| KRAS | Phase I/II | adagrasib | Nausea, diarrhea, vomiting, fatigue, increased ALT, hyponatremia | [177] |
| Phase I/II | sotorasib | Pneumonia, diarrhea, muscle and bone pain, nausea, fatigue, increased AST and ALT levels, respiratory failure, heart diseases | [176,178] | |
| ROS-1 | Phase I/II | crizotinib | Diarrhea, vomiting, nausea, vision disorder, headache, musculoskeletal pain, stomatitis, fatigue | [184,185] |
| Phase II | ceritinib | Abdominal pain, diarrhea, hyperglycemia, increased serum ALT and AST, nausea, vomiting | [186] | |
| Phase I/II | entrectinib | Vision disorders, diarrhea, nausea, edema, cognitive disorders, vomiting, dizziness, dyspnea, myalgia | [159,187] | |
| Phase I | taletrectinib | Increased AST and ALT levels, nausea, diarrhea | [193] | |
| PI3K | Phase I | buparlisib | Hyperglycemia, fatigue, rash, anxiety, depression, mood disorders | [199] |
| Phase II | MK-2206 | Hyperglycemia, thrombocytopenia, fatigue, rash, nausea | [200] | |
| Phase II | taselisib | Diarrhea, hyperglycemia, nausea, fatigue, headache, stomatitis, vomiting, rash | [201] |
| Molecular Target | Phase | Agents | Side Effects | References |
|---|---|---|---|---|
| CTLA-4 | Phase II/III | ipilimumab | Diarrhea, rash, pruritus, fatigue, nausea, vomiting, decreased appetite, abdominal pain, and colitis | [222,223,224,225,249,250,251] |
| Phase III | tremelimumab | Nausea, fatigue, muscle and bone pain, rash, diarrhea, neutropenia, hyponatremia | [252,253] | |
| PD1 | Phase III | nivolumab | Rash, weakness, muscle and bone pain, diarrhea, respiratory tract infection, fever | [227,230,232,249,250] |
| Phase II/III | pembrolizumab | Fatigue, muscle and bone pain, rash, diarrhea, fever, pruritus, abdominal pain, nausea | [234,235,236,237,251] | |
| PD-L1 | Phase II/III | atezolizumab | Fatigue, nausea, infection, fever, constipation, anemia, pneumonia | [226,240,241] |
| Phase II/III | durvalumab | Rash, diarrhea, pruritus, anemia, neutrophilia, fatigue, dyspnea, asthenia, loss of appetite | [243,244] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leone, G.M.; Candido, S.; Lavoro, A.; Vivarelli, S.; Gattuso, G.; Calina, D.; Libra, M.; Falzone, L. Clinical Relevance of Targeted Therapy and Immune-Checkpoint Inhibition in Lung Cancer. Pharmaceutics 2023, 15, 1252. https://doi.org/10.3390/pharmaceutics15041252
Leone GM, Candido S, Lavoro A, Vivarelli S, Gattuso G, Calina D, Libra M, Falzone L. Clinical Relevance of Targeted Therapy and Immune-Checkpoint Inhibition in Lung Cancer. Pharmaceutics. 2023; 15(4):1252. https://doi.org/10.3390/pharmaceutics15041252
Chicago/Turabian StyleLeone, Gian Marco, Saverio Candido, Alessandro Lavoro, Silvia Vivarelli, Giuseppe Gattuso, Daniela Calina, Massimo Libra, and Luca Falzone. 2023. "Clinical Relevance of Targeted Therapy and Immune-Checkpoint Inhibition in Lung Cancer" Pharmaceutics 15, no. 4: 1252. https://doi.org/10.3390/pharmaceutics15041252