
Lights and Shadows on the Cancer Multi-Target Inhibitor Rigosertib (ON-01910.Na)


Abstract
:1. Introduction
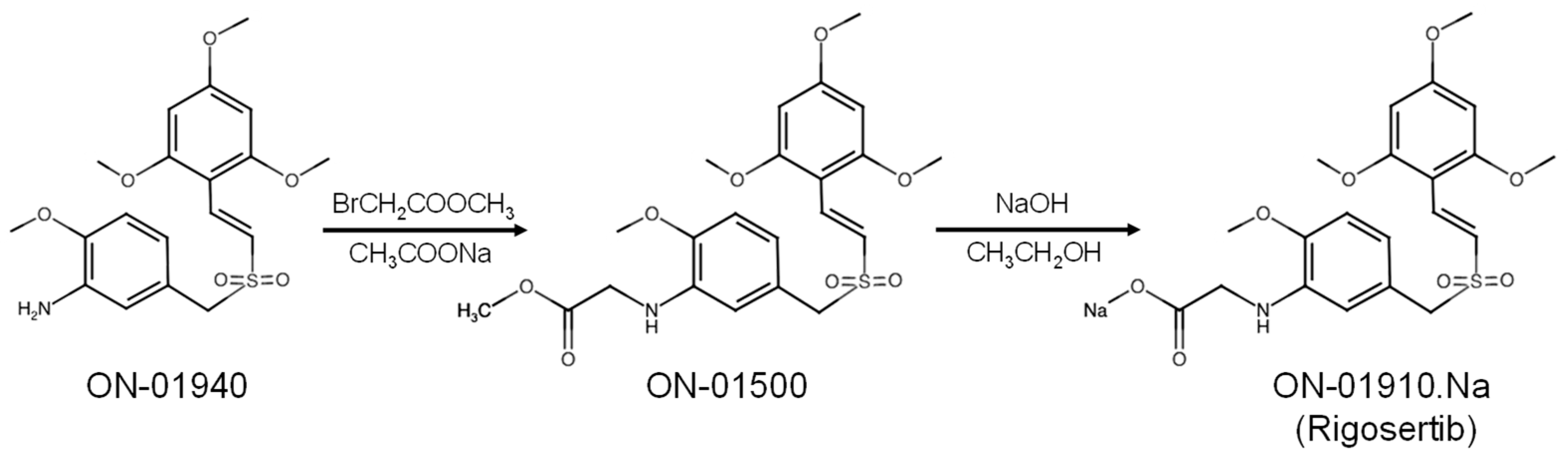
2. Development of Rigosertib (ON-01910.Na)
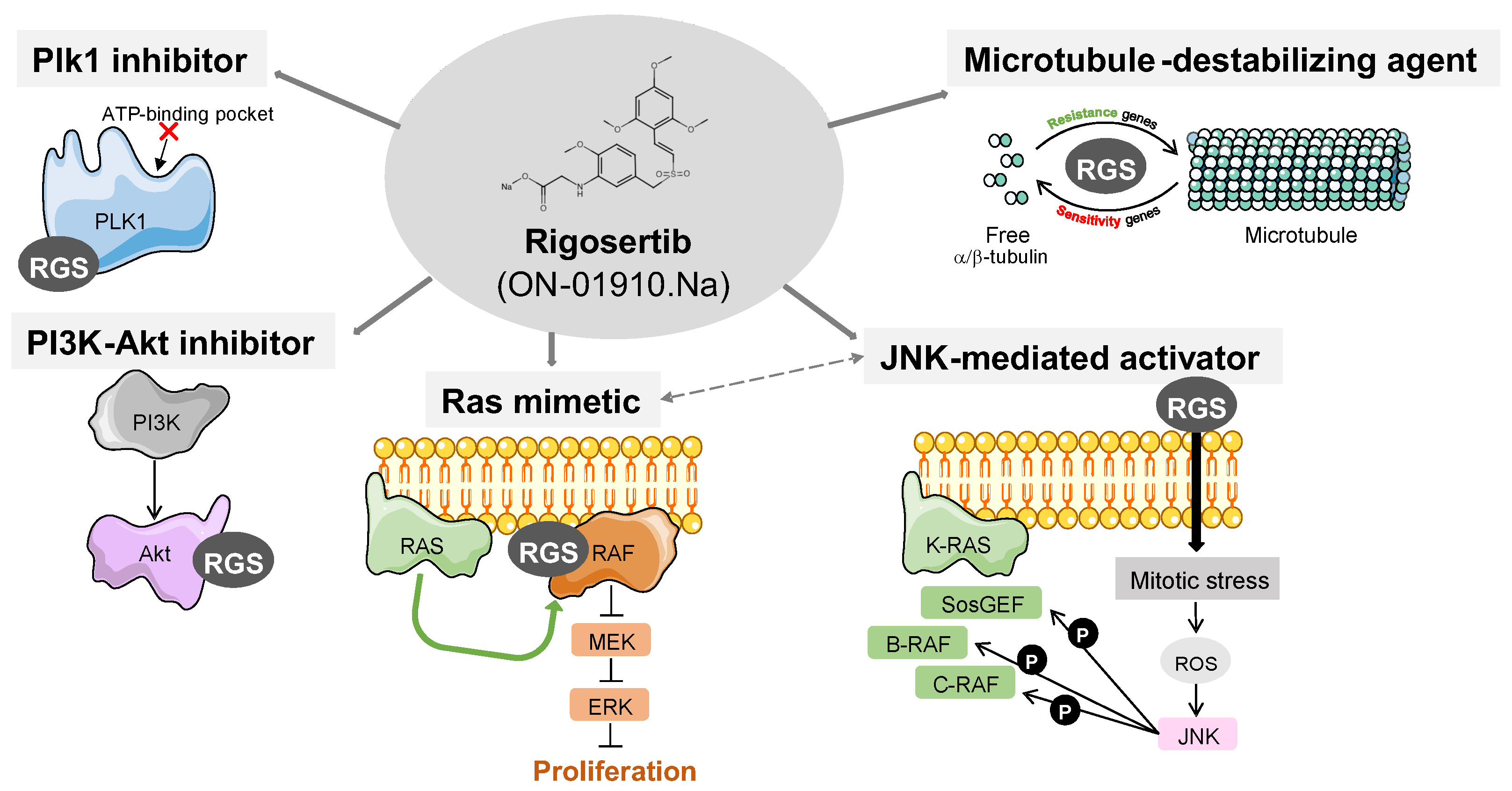
3. Described Targets for Rigosertib
3.1. Rigosertib as a Plk1 Inhibitor
3.2. Rigosertib as a PI3K–Akt Pathway Inhibitor
3.3. Rigosertib as a Ras Mimetic and Ras–Raf–MEK Axis Inhbitor
3.4. Rigosertib as a Tubulin Polymerization Destabilizer
4. Rigosertib Clinical Trials for Cancer Therapy

{kind=link}
{kind=link}
{kind=link}
| Phase | NCT Number | Treatment | Disease | Participants | References |
|---|---|---|---|---|---|
| Phase I | NCT01048619 | ON-01910.Na | Myelodysplastic Syndrome | 36 | [55] |
| NCT01168011 | ON-01910.Na | Solid Tumor | 68 | ||
| NCT01125891 | Gemcitabine or ON-01910.Na | Malignant Neoplasms Solid Tumors | 39 | [60] | |
| NCT00854646 | ON-01910.Na | Acute Myelocytic Leukemia Acute Lymphocytic Leukemia Chronic Myelocytic Leukemia Chronic Lymphocytic Leukemia Myelodysplastic Syndromes | 22 | [61] | |
| NCT01538537 | ON-01910.Na | Advanced Cancer Solid Tumors Cancer Neoplasms | 29 | [53] | |
| NCT01538563 | ON-01910.Na | Solid Tumors Advanced Cancer Neoplasms | 42 | - | |
| NCT01165905 | Gemcitabine or ON-01910.Na | Solid Tumor | 10 | - | |
| NCT00861783 | Irinotecan + ON-01910.Na Oxaliplatin + ON-01910.Na | Hepatoma Advanced Solid Tumor | 16 | - | |
| NCT00861328 | Irinotecan + ON-01910.Na Oxaliplatin + ON-01910.Na | Advanced Solid Tumors | 18 | - | |
| NCT00861510 | ON-01910.Na | Lymphoma Mantle-cell Leukemia Lymphocytic Chronic B-Cell Leukemia Hairy Cell Waldenstrom Macroglobulinemia Multiple Myeloma | 16 | [62] | |
| NCT00533416 | ON-01910.Na | Myelodysplastic Syndrome (MDS) | 14 | [63] | |
| Phase I/II | NCT01167166 | ON-01910.Na | Acute Myelocytic Leukemia Acute Lymphocytic Leukemia Myeloproliferative Disease Chronic Myeloid Leukemia | 30 | - |
| NCT00854945 | ON-01910.Na | Myelodysplastic Syndromes Acute Myeloid Leukemia | 36 | - | |
| NCT01926587 | Azacitidine + ON-01910.Na | Myelodysplastic Syndromes Acute Myeloid Leukemia Chronic Myelomonocytic Leukemia | 45 | [64] | |
| NCT04263090 | Nivolumab + ON-01910.Na | Non-small Cell Lung Cancer Adenocarcinoma Stage IV | 20 | - | |
| NCT03786237 | ON-01910.Na | Epidermolysis Bullosa Dystrophica Squamous Cell Carcinoma | 12 | - | |
| Phase II | NCT01807546 | ON-01910.Na | Head and Neck Squamous Cell Carcinoma Anal Squamous Cell Carcinoma Lung Squamous Cell Carcinoma Cervical Squamous Cell Carcinoma Esophageal Squamous Cell Carcinoma Skin Squamous Cell Carcinoma Penile Squamous Cell Carcinoma | 64 | - |
| NCT01904682 | ON-01910.Na | Myelodysplastic Syndromes | 45 | - | |
| NCT01584531 | ON-01910.Na | Myelodysplastic Syndromes MDS Trisomy 8 | 82 | - | |
| NCT00906334 | ON-01910.Na | Myelodysplastic Syndromes | 14 | [65] | |
| Phase III | NCT01241500 | ON-01910.Na | Myelodysplastic Syndromes MDS RAEB Chronic Myelomonocytic Leukemia | 299 | - |
| NCT01928537 | ON-01910.Na | Myelodysplastic Syndromes Refractory Anemia with Excess Blasts Chronic Myelomonocytic Leukemia Cytopenia | 67 | - | |
| NCT02562443 | Best supportive care (BSC) + ON-01910.Na Vs. Physician’s choice (PC) + ON-01910.Na | Myelodysplastic Syndromes MDS Refractory Anemia with Excess Blasts RAEB | 372 | [7] | |
| NCT01360853 | Gemcitabine + ON-01910.Na | Metastatic Pancreatic Adenocarcinoma | 160 | [8,56,66] |
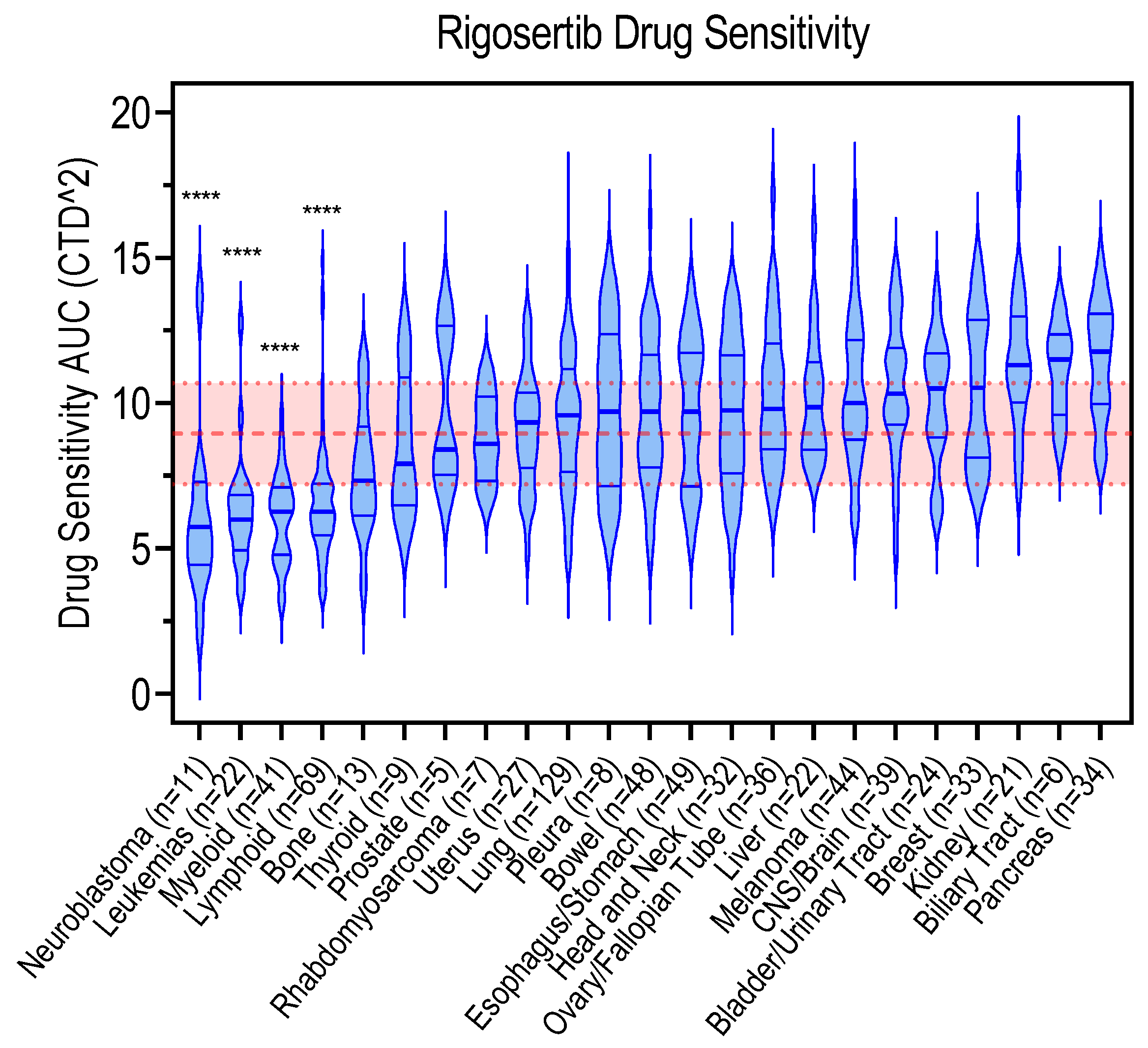
5. Genetic Biomarkers Associated with Rigosertib Response
6. Conclusions, Current Challenges, and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gumireddy, K.; Reddy, M.V.R.; Cosenza, S.C.; Nathan, R.B.; Baker, S.J.; Papathi, N.; Jiang, J.; Holland, J.; Reddy, E.P. ON01910, a Non-ATP-Competitive Small Molecule Inhibitor of Plk1, Is a Potent Anticancer Agent. Cancer Cell 2005, 7, 275–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, R.T.; Keysar, S.B.; Bowles, D.W.; Glogowska, M.J.; Astling, D.P.; Morton, J.J.; Le, P.; Umpierrez, A.; Eagles-Soukup, J.; Gan, G.N.; et al. The Dual Pathway Inhibitor Rigosertib Is Effective in Direct Patient Tumor Xenografts of Head and Neck Squamous Cell Carcinomas. Mol. Cancer Ther. 2013, 12, 1994–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, A.; Khudaynazar, N.; Tantravahi, R.V.; Gillum, A.M.; Hoffman, B.S. ON 01910.Na (Rigosertib) Inhibits PI3K/Akt Pathway and Activates Oxidative Stress Signals in Head and Neck Cancer Cell Lines. Oncotarget 2016, 7, 79388–79400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athuluri-Divakar, S.K.; Vasquez-Del Carpio, R.; Dutta, K.; Baker, S.J.; Cosenza, S.C.; Basu, I.; Gupta, Y.K.; Reddy, M.V.R.; Ueno, L.; Hart, J.R.; et al. A Small Molecule RAS-Mimetic Disrupts RAS Association with Effector Proteins to Block Signaling. Cell 2016, 165, 643–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jost, M.; Chen, Y.; Gilbert, L.A.; Horlbeck, M.A.; Krenning, L.; Menchon, G.; Rai, A.; Cho, M.Y.; Stern, J.J.; Prota, A.E.; et al. Combined CRISPRi/a-Based Chemical Genetic Screens Reveal That Rigosertib Is a Microtubule-Destabilizing Agent. Mol. Cell 2017, 68, 210–223.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritt, D.A.; Abreu-Blanco, M.T.; Bindu, L.; Durrant, D.E.; Zhou, M.; Specht, S.I.; Stephen, A.G.; Holderfield, M.; Morrison, D.K. Inhibition of Ras/Raf/MEK/ERK Pathway Signaling by a Stress-Induced Phospho-Regulatory Circuit. Mol. Cell 2016, 64, 875–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Manero, G.; Fenaux, P.; Al-Kali, A.; Baer, M.R.; Sekeres, M.A.; Roboz, G.J.; Gaidano, G.; Scott, B.L.; Greenberg, P.; Platzbecker, U.; et al. Rigosertib versus Best Supportive Care for Patients with High-Risk Myelodysplastic Syndromes after Failure of Hypomethylating Drugs (ONTIME): A Randomised, Controlled, Phase 3 Trial. Lancet Oncol. 2016, 17, 496–508. [Google Scholar] [CrossRef]
- O’Neil, B.H.; Scott, A.J.; Ma, W.W.; Cohen, S.J.; Aisner, D.L.; Menter, A.R.; Tejani, M.A.; Cho, J.K.; Granfortuna, J.; Coveler, L.; et al. A Phase II/III Randomized Study to Compare the Efficacy and Safety of Rigosertib plus Gemcitabine versus Gemcitabine Alone in Patients with Previously Untreated Metastatic Pancreatic Cancer. Ann. Oncol. 2015, 26, 1923–1929. [Google Scholar] [CrossRef]
- Sawyers, C.L. Opportunities and Challenges in the Development of Kinase Inhibitor Therapy for Cancer. Genes Dev. 2003, 17, 2998–3010. [Google Scholar] [CrossRef] [Green Version]
- Reddy, E.; Reddy, M. Substituted Styryl Benzylsulfones for Treating Proliferative Disorders. U.S. Patent No. 6486210 B2, 22 November 2002. [Google Scholar]
- Reddy, E.; Reddy, M. Preparation of α,β-Unsaturated Sulfones for Treating Proliferative Disorders. U.S. Patent No. 6541475, 7 March 2002. [Google Scholar]
- Reddy, E.; Reddy, M. α,β-Unsaturated Sulfones for Treating Proliferative Disorders. U.S. Patent No. 6599932 B1, 29 July 2003. [Google Scholar]
- Reddy, M.V.R.; Venkatapuram, P.; Mallireddigari, M.R.; Pallela, V.R.; Cosenza, S.C.; Robell, K.A.; Akula, B.; Hoffman, B.S.; Reddy, E.P. Discovery of a Clinical Stage Multi-Kinase Inhibitor Sodium (E)-2-{2-Methoxy-5-[(2′,4′,6′-Trimethoxystyrylsulfonyl)Methyl]Phenylamino}acetate (ON 01910.Na): Synthesis, Structure–Activity Relationship, and Biological Activity. J. Med. Chem. 2011, 54, 6254–6276. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; He, Q.; Li, X.; Chang, C.-K.; Wu, L.-Y.; Zhang, Z.; Liu, L.; Shi, W.-H.; Zhu, Y.; Zhao, Y.-S.; et al. Rigosertib as a Selective Anti-Tumor Agent Can Ameliorate Multiple Dysregulated Signaling Transduction Pathways in High-Grade Myelodysplastic Syndrome. Sci. Rep. 2014, 4, 7310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archambault, V.; Normandin, K. Several Inhibitors of the Plk1 Polo-Box Domain Turn out to Be Non-Specific Protein Alkylators. Cell Cycle 2017, 16, 1220–1224. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Nie, C.; Chen, Y.; Li, J.; Xie, Y.; Tang, Z.; Gao, Y.; Ai, S.; Mao, Y.; Sun, Q.; et al. Therapeutic Targeting PLK1 by ON-01910.Na Is Effective in Local Treatment of Retinoblastoma. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2021, 28, 745–761. [Google Scholar] [CrossRef] [PubMed]
- de Cárcer, G.; Manning, G.; Malumbres, M. From Plk1 to Plk5. Cell Cycle 2011, 10, 2255–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiappa, M.; Petrella, S.; Damia, G.; Broggini, M.; Guffanti, F.; Ricci, F. Present and Future Perspective on PLK1 Inhibition in Cancer Treatment. Front. Oncol. 2022, 12, 903016. [Google Scholar] [CrossRef]
- Wachowicz, P.; Fernández-Miranda, G.; Marugán, C.; Escobar, B.; de Cárcer, G. Genetic Depletion of Polo-like Kinase 1 Leads to Embryonic Lethality Due to Mitotic Aberrancies. BioEssays 2016, 38, S96–S106. [Google Scholar] [CrossRef] [PubMed]
- Lénárt, P.; Petronczki, M.; Steegmaier, M.; Di Fiore, B.; Lipp, J.J.; Hoffmann, M.; Rettig, W.J.; Kraut, N.; Peters, J.-M. The Small-Molecule Inhibitor BI 2536 Reveals Novel Insights into Mitotic Roles of Polo-like Kinase 1. Curr. Biol. 2007, 17, 304–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haren, L.; Stearns, T.; Lüders, J. Plk1-Dependent Recruitment of γ-Tubulin Complexes to Mitotic Centrosomes Involves Multiple PCM Components. PLoS ONE 2009, 4, e5976. [Google Scholar] [CrossRef]
- Peters, U.; Cherian, J.; Kim, J.H.; Kwok, B.H.; Kapoor, T.M. Probing Cell-Division Phenotype Space and Polo-like Kinase Function Using Small Molecules. Nat. Chem. Biol. 2006, 2, 618–626. [Google Scholar] [CrossRef]
- Oussenko, I.A.; Holland, J.F.; Reddy, E.P.; Ohnuma, T. Effect of ON 01910.Na, an Anticancer Mitotic Inhibitor, on Cell-Cycle Progression Correlates with RanGAP1 Hyperphosphorylation. Cancer Res. 2011, 71, 4968–4976. [Google Scholar] [CrossRef] [Green Version]
- Swaminathan, S.; Kiendl, F.; Körner, R.; Lupetti, R.; Hengst, L.; Melchior, F. RanGAP1*SUMO1 Is Phosphorylated at the Onset of Mitosis and Remains Associated with RanBP2 upon NPC Disassembly. J. Cell Biol. 2004, 164, 965–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atanasova, V.S.; Pourreyron, C.; Farshchian, M.; Lawler, M.; Brown, C.A.; Watt, S.A.; Wright, S.; Warkala, M.; Guttmann-Gruber, C.; Hofbauer, J.P.; et al. Identification of Rigosertib for the Treatment of Recessive Dystrophic Epidermolysis Bullosa–Associated Squamous Cell Carcinoma. Clin. Cancer Res. 2019, 25, 3384–3391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, Y.; Kang, Y.; Ham, Y.; Kim, M.-H.; Kim, S.-J.; Yoon, S.K.; Jang, S.K.; Park, J.B.; Cho, S.; Kim, J.H. PLK1-ELAVL1/HuR-MiR-122 Signaling Facilitates Hepatitis C Virus Proliferation. Proc. Natl. Acad. Sci. USA 2022, 119, e2214911119. [Google Scholar] [CrossRef] [PubMed]
- Vulin, M.; Jehanno, C.; Sethi, A.; Correia, A.L.; Obradović, M.M.S.; Couto, J.P.; Coissieux, M.-M.; Diepenbruck, M.; Preca, B.-T.; Volkmann, K.; et al. A High-Throughput Drug Screen Reveals Means to Differentiate Triple-Negative Breast Cancer. Oncogene 2022, 41, 4459–4473. [Google Scholar] [CrossRef] [PubMed]
- Hyoda, T.; Tsujioka, T.; Nakahara, T.; Suemori, S.; Okamoto, S.; Kataoka, M.; Tohyama, K. Rigosertib Induces Cell Death of a Myelodysplastic Syndrome-derived Cell Line by DNA Damage-induced G2/M Arrest. Cancer Sci. 2015, 106, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.G.; Shen, J.; Yoo, E.; Liu, R.; Yen, H.-Y.; Mehta, A.; Rajaei, A.; Yang, W.; Mhawech-Fauceglia, P.; DeMayo, F.J.; et al. Targeting the Glucose-Regulated Protein-78 Abrogates Pten-Null Driven AKT Activation and Endometrioid Tumorigenesis. Oncogene 2015, 34, 5418–5426. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Wang, M.; Wang, H.; Fang, L.; Gou, S. Targeting RAS-RAF Pathway Significantly Improves Antitumor Activity of Rigosertib-Derived Platinum(IV) Complexes and Overcomes Cisplatin Resistance. Eur. J. Med. Chem. 2020, 194, 112269. [Google Scholar] [CrossRef]
- Lu, T.; Laughton, C.A.; Wang, S.; Bradshaw, T.D. In Vitro Antitumor Mechanism of (E)-N-(2-Methoxy-5-(((2,4,6-Trimethoxystyryl)Sulfonyl)Methyl)Pyridin-3-Yl)Methanesulfonamide. Mol. Pharmacol. 2015, 87, 18–30. [Google Scholar] [CrossRef] [Green Version]
- Malacrida, A.; Rigolio, R.; Celio, L.; Damian, S.; Cavaletti, G.; Mazzaferro, V.; Miloso, M. In Vitro Evaluation of Rigosertib Antitumoral and Radiosensitizing Effects against Human Cholangiocarcinoma Cells. Int. J. Mol. Sci. 2021, 22, 8230. [Google Scholar] [CrossRef]
- Ruppenthal, S.; Kleiner, H.; Nolte, F.; Fabarius, A.; Hofmann, W.-K.; Nowak, D.; Seifarth, W. Increased Separase Activity and Occurrence of Centrosome Aberrations Concur with Transformation of MDS. PLoS ONE 2018, 13, e0191734. [Google Scholar] [CrossRef] [Green Version]
- Mielgo, A.; Seguin, L.; Huang, M.; Camargo, M.F.; Anand, S.; Franovic, A.; Weis, S.M.; Advani, S.J.; Murphy, E.A.; Cheresh, D.A. A MEK-Independent Role for CRAF in Mitosis and Tumor Progression. Nat. Med. 2011, 17, 1641–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, P.; Freese, K.; Mahli, A.; Thasler, W.E.; Hellerbrand, C.; Bosserhoff, A.K. Combined Effects of PLK1 and RAS in Hepatocellular Carcinoma Reveal Rigosertib as Promising Novel Therapeutic “Dual-Hit” Option. Oncotarget 2018, 9, 3605–3618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urasaki, Y.; Fiscus, R.R.; Le, T.T. Detection of the Cell Cycle-Regulated Negative Feedback Phosphorylation of Mitogen-Activated Protein Kinases in Breast Carcinoma Using Nanofluidic Proteomics. Sci. Rep. 2018, 8, 9991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galimov, E.R. The Role of P66shc in Oxidative Stress and Apoptosis. Acta Nat. 2010, 2, 44–51. [Google Scholar] [CrossRef]
- Günther, J.K.; Nikolajevic, A.; Ebner, S.; Troppmair, J.; Khalid, S. Rigosertib-Activated JNK1/2 Eliminate Tumor Cells through P66Shc Activation. Biology 2020, 9, 99. [Google Scholar] [CrossRef]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron Transfer between Cytochrome c and P66Shc Generates Reactive Oxygen Species That Trigger Mitochondrial Apoptosis. Cell 2005, 122, 221–233. [Google Scholar] [CrossRef]
- Rahmani, F.; Hashemzehi, M.; Avan, A.; Barneh, F.; Asgharzadeh, F.; Moradi Marjaneh, R.; Soleimani, A.; Parizadeh, M.; Ferns, G.A.; Ghayour Mobarhan, M.; et al. Rigosertib Elicits Potent Anti-Tumor Responses in Colorectal Cancer by Inhibiting Ras Signaling Pathway. Cell. Signal. 2021, 85, 110069. [Google Scholar] [CrossRef]
- Tong, A.W.; Stone, M.J. Prospects for CD40-Directed Experimental Therapy of Human Cancer. Cancer Gene Ther. 2003, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, C.; Saleh, N.; Yang, J.; Nebhan, C.A.; Vilgelm, A.E.; Reddy, E.P.; Roland, J.T.; Johnson, D.B.; Chen, S.-C.; Shattuck-Brandt, R.L.; et al. Novel Induction of CD40 Expression by Tumor Cells with RAS/RAF/PI3K Pathway Inhibition Augments Response to Checkpoint Blockade. Mol. Cancer 2021, 20, 85. [Google Scholar] [CrossRef]
- Lee, J.R.; Koretzky, G.A. Production of Reactive Oxygen Intermediates Following CD40 Ligation Correlates with C-Jun N-Terminal Kinase Activation and IL-6 Secretion in Murine B Lymphocytes. Eur. J. Immunol. 1998, 28, 4188–4197. [Google Scholar] [CrossRef]
- Liu, J.; Yoshida, Y.; Yamashita, U. Suppressive Effect of Reactive Oxygen Species on CD40-Induced B Cell Activation. FEBS Lett. 2007, 581, 5043–5049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Du, P.; Jiang, D. Rigosertib Inhibits MEK1–ERK Pathway and Alleviates Lipopolysaccharide-induced Sepsis. Immun. Inflamm. Dis. 2021, 9, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, J.T.; Wan, X.; Hernandez, E.R.; Luo, R.; Lyons, G.C.; Wilson, K.M.; Gallardo, D.C.; Isanogle, K.A.; Robinson, C.M.; Mendoza, A.; et al. Rigosertib Induces Mitotic Arrest and Apoptosis in RAS-Mutated Rhabdomyosarcoma and Neuroblastoma. Mol. Cancer Ther. 2021, 20, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Radke, K.; Hansson, K.; Sjölund, J.; Wolska, M.; Karlsson, J.; Esfandyari, J.; Pietras, K.; Aaltonen, K.; Gisselsson, D.; Bexell, D. Anti-Tumor Effects of Rigosertib in High-Risk Neuroblastoma. Transl. Oncol. 2021, 14, 101149. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.J.; Cosenza, S.C.; Athuluri-Divakar, S.; Reddy, M.V.R.; Vasquez-Del Carpio, R.; Jain, R.; Aggarwal, A.K.; Reddy, E.P. A Contaminant Impurity, Not Rigosertib, Is a Tubulin Binding Agent. Mol. Cell 2020, 79, 180–190.e4. [Google Scholar] [CrossRef] [PubMed]
- Jost, M.; Chen, Y.; Gilbert, L.A.; Horlbeck, M.A.; Krenning, L.; Menchon, G.; Rai, A.; Cho, M.Y.; Stern, J.J.; Prota, A.E.; et al. Pharmaceutical-Grade Rigosertib Is a Microtubule-Destabilizing Agent. Mol. Cell 2020, 79, 191–198.e3. [Google Scholar] [CrossRef] [PubMed]
- Khachatryan, H.; Olszowy, B.; Barrero, C.A.; Gordon, J.; Perez-Leal, O. Identification of Inhibitors of Tubulin Polymerization Using a CRISPR-Edited Cell Line with Endogenous Fluorescent Tagging of β-Tubulin and Histone H1. Biomolecules 2023, 13, 249. [Google Scholar] [CrossRef]
- Jimeno, A.; Chan, A.; Cusatis, G.; Zhang, X.; Wheelhouse, J.; Solomon, A.; Chan, F.; Zhao, M.; Cosenza, S.C.; Reddy, M.R.; et al. Evaluation of the Novel Mitotic Modulator ON 01910.Na in Pancreatic Cancer and Preclinical Development of an Ex Vivo Predictive Assay. Oncogene 2009, 28, 610–618. [Google Scholar] [CrossRef] [Green Version]
- Navada, S.C.; Silverman, L.R. The Safety and Efficacy of Rigosertib in the Treatment of Myelodysplastic Syndromes. Expert Rev. Anticancer Ther. 2016, 16, 805–810. [Google Scholar] [CrossRef]
- Ohnuma, T.; Lehrer, D.; Ren, C.; Cho, S.Y.; Maniar, M.; Silverman, L.; Sung, M.; Gretz, H.F.; Benisovich, V.; Navada, S.; et al. Phase 1 Study of Intravenous Rigosertib (ON 01910.Na), a Novel Benzyl Styryl Sulfone Structure Producing G2/M Arrest and Apoptosis, in Adult Patients with Advanced Cancer. Am. J. Cancer Res. 2013, 3, 323–338. [Google Scholar]
- Silverman, L.R.; Greenberg, P.; Raza, A.; Olnes, M.J.; Holland, J.F.; Reddy, P.; Maniar, M.; Wilhelm, F. Clinical Activity and Safety of the Dual Pathway Inhibitor Rigosertib for Higher Risk Myelodysplastic Syndromes Following DNA Methyltransferase Inhibitor Therapy. Hematol. Oncol. 2015, 33, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Komrokji, R.S.; Raza, A.; Lancet, J.E.; Ren, C.; Taft, D.; Maniar, M.; Wilhelm, F.; List, A.F. Phase I Clinical Trial of Oral Rigosertib in Patients with Myelodysplastic Syndromes. Br. J. Haematol. 2013, 162, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Ottaiano, A.; Capozzi, M.; De Divitiis, C.; De Stefano, A.; Botti, G.; Avallone, A.; Tafuto, S. Gemcitabine Mono-Therapy versus Gemcitabine plus Targeted Therapy in Advanced Pancreatic Cancer: A Meta-Analysis of Randomized Phase III Trials. Acta Oncol. 2017, 56, 377–383. [Google Scholar] [CrossRef] [Green Version]
- Rees, M.G.; Seashore-Ludlow, B.; Cheah, J.H.; Adams, D.J.; Price, E.V.; Gill, S.; Javaid, S.; Coletti, M.E.; Jones, V.L.; Bodycombe, N.E.; et al. Correlating Chemical Sensitivity and Basal Gene Expression Reveals Mechanism of Action. Nat. Chem. Biol. 2016, 12, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Seashore-Ludlow, B.; Rees, M.G.; Cheah, J.H.; Cokol, M.; Price, E.V.; Coletti, M.E.; Jones, V.; Bodycombe, N.E.; Soule, C.K.; Gould, J.; et al. Harnessing Connectivity in a Large-Scale Small-Molecule Sensitivity Dataset. Cancer Discov. 2015, 5, 1210–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson, K.; Radke, K.; Aaltonen, K.; Saarela, J.; Mañas, A.; Sjölund, J.; Smith, E.M.; Pietras, K.; Påhlman, S.; Wennerberg, K.; et al. Therapeutic Targeting of KSP in Preclinical Models of High-Risk Neuroblastoma. Sci. Transl. Med. 2020, 12, eaba4434. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.W.; Messersmith, W.A.; Dy, G.K.; Weekes, C.D.; Whitworth, A.; Ren, C.; Maniar, M.; Wilhelm, F.; Eckhardt, S.G.; Adjei, A.A.; et al. Phase I Study of Rigosertib, an Inhibitor of the Phosphatidylinositol 3-Kinase and Polo-like Kinase 1 Pathways, Combined with Gemcitabine in Patients with Solid Tumors and Pancreatic Cancer. Clin. Cancer Res. 2012, 18, 2048–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navada, S.C.; Fruchtman, S.M.; Odchimar-Reissig, R.; Demakos, E.P.; Petrone, M.E.; Zbyszewski, P.S.; Holland, J.F.; Silverman, L.R. A Phase 1/2 Study of Rigosertib in Patients with Myelodysplastic Syndromes (MDS) and MDS Progressed to Acute Myeloid Leukemia. Leuk. Res. 2018, 64, 10–16. [Google Scholar] [CrossRef]
- Roschewski, M.; Farooqui, M.; Aue, G.; Wilhelm, F.; Wiestner, A. Phase I Study of ON 01910.Na (Rigosertib), a Multikinase PI3K Inhibitor in Relapsed/Refractory B-Cell Malignancies. Leukemia 2013, 27, 1920–1923. [Google Scholar] [CrossRef]
- Olnes, M.J.; Shenoy, A.; Weinstein, B.; Pfannes, L.; Loeliger, K.; Tucker, Z.; Tian, X.; Kwak, M.; Wilhelm, F.; Yong, A.S.M.; et al. Directed Therapy for Patients with Myelodysplastic Syndromes (MDS) by Suppression of Cyclin D1 with ON 01910.Na. Leuk. Res. 2012, 36, 982–989. [Google Scholar] [CrossRef] [Green Version]
- Navada, S.C.; Garcia-Manero, G.; OdchimarReissig, R.; Pemmaraju, N.; Alvarado, Y.; Ohanian, M.N.; John, R.B.; Demakos, E.P.; Zbyszewski, P.S.; Maniar, M.; et al. Rigosertib in Combination with Azacitidine in Patients with Myelodysplastic Syndromes or Acute Myeloid Leukemia: Results of a Phase 1 Study. Leuk. Res. 2020, 94, 106369. [Google Scholar] [CrossRef] [PubMed]
- Seetharam, M.; Fan, A.C.; Tran, M.; Xu, L.; Renschler, J.P.; Felsher, D.W.; Sridhar, K.; Wilhelm, F.; Greenberg, P.L. Treatment of Higher Risk Myelodysplastic Syndrome Patients Unresponsive to Hypomethylating Agents with ON 01910.Na. Leuk. Res. 2012, 36, 98–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazha, A.; Sekeres, M.A.; Komrokji, R.; Steensma, D.P.; Kantarjian, H.; Roboz, G.; Fenaux, P.; Prebet, T.; Azarnia, N.; Zbyszewski, P.S.; et al. Validation of a Post-Hypomethylating Agent Failure Prognostic Model in Myelodysplastic Syndromes Patients Treated in a Randomized Controlled Phase III Trial of Rigosertib vs. Best Supportive Care. Blood Cancer J. 2017, 7, 644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Burgos, L.; Navarro-González, B.; García-Martín, S.; Sirozh, O.; Mota-Pino, J.; Fueyo-Marcos, E.; Tejero, H.; Antón, M.E.; Murga, M.; Al-Shahrour, F.; et al. Activation of the Integrated Stress Response Is a Vulnerability for Multidrug-resistant FBXW7-deficient Cells. EMBO Mol. Med. 2022, 14, e15855. [Google Scholar] [CrossRef] [PubMed]
- Saei, A.; Palafox, M.; Benoukraf, T.; Kumari, N.; Jaynes, P.W.; Iyengar, P.V.; Muñoz-Couselo, E.; Nuciforo, P.; Cortés, J.; Nötzel, C.; et al. Loss of USP28-Mediated BRAF Degradation Drives Resistance to RAF Cancer Therapies. J. Exp. Med. 2018, 215, 1913–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haase, D.; Germing, U.; Schanz, J.; Pfeilstöcker, M.; Nösslinger, T.; Hildebrandt, B.; Kundgen, A.; Lübbert, M.; Kunzmann, R.; Giagounidis, A.A.N.; et al. New Insights into the Prognostic Impact of the Karyotype in MDS and Correlation with Subtypes: Evidence from a Core Dataset of 2124 Patients. Blood 2007, 110, 4385–4395. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.; Kohlmann, A.; Dugas, M.; Kern, W.; Hiddemann, W.; Schnittger, S.; Haferlach, T. Genomic Gains and Losses Influence Expression Levels of Genes Located within the Affected Regions: A Study on Acute Myeloid Leukemias with Trisomy 8, 11, or 13, Monosomy 7, or Deletion 5q. Leukemia 2005, 19, 1224–1228. [Google Scholar] [CrossRef] [Green Version]
- Tanenbaum, M.E.; Medema, R.; Akhmanova, A. Regulation of Localization and Activity of the Microtubule Depolymerase MCAK. Bioarchitecture 2011, 1, 80–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hood, F.E.; Royle, S.J. Pulling It Together. Bioarchitecture 2011, 1, 105–109. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monfort-Vengut, A.; de Cárcer, G. Lights and Shadows on the Cancer Multi-Target Inhibitor Rigosertib (ON-01910.Na). Pharmaceutics 2023, 15, 1232. https://doi.org/10.3390/pharmaceutics15041232
Monfort-Vengut A, de Cárcer G. Lights and Shadows on the Cancer Multi-Target Inhibitor Rigosertib (ON-01910.Na). Pharmaceutics. 2023; 15(4):1232. https://doi.org/10.3390/pharmaceutics15041232
Chicago/Turabian StyleMonfort-Vengut, Ana, and Guillermo de Cárcer. 2023. "Lights and Shadows on the Cancer Multi-Target Inhibitor Rigosertib (ON-01910.Na)" Pharmaceutics 15, no. 4: 1232. https://doi.org/10.3390/pharmaceutics15041232