


Dual-Drug Delivery by Anisotropic and Uniform Hybrid Nanostructures: A Comparative Study of the Function and Substrate–Drug Interaction Properties

Abstract
:1. Introduction
2. Uniform Structures
2.1. Solubility and Encapsulation
2.2. Release and Interactions
2.3. Internalization and Cytotoxicity Data
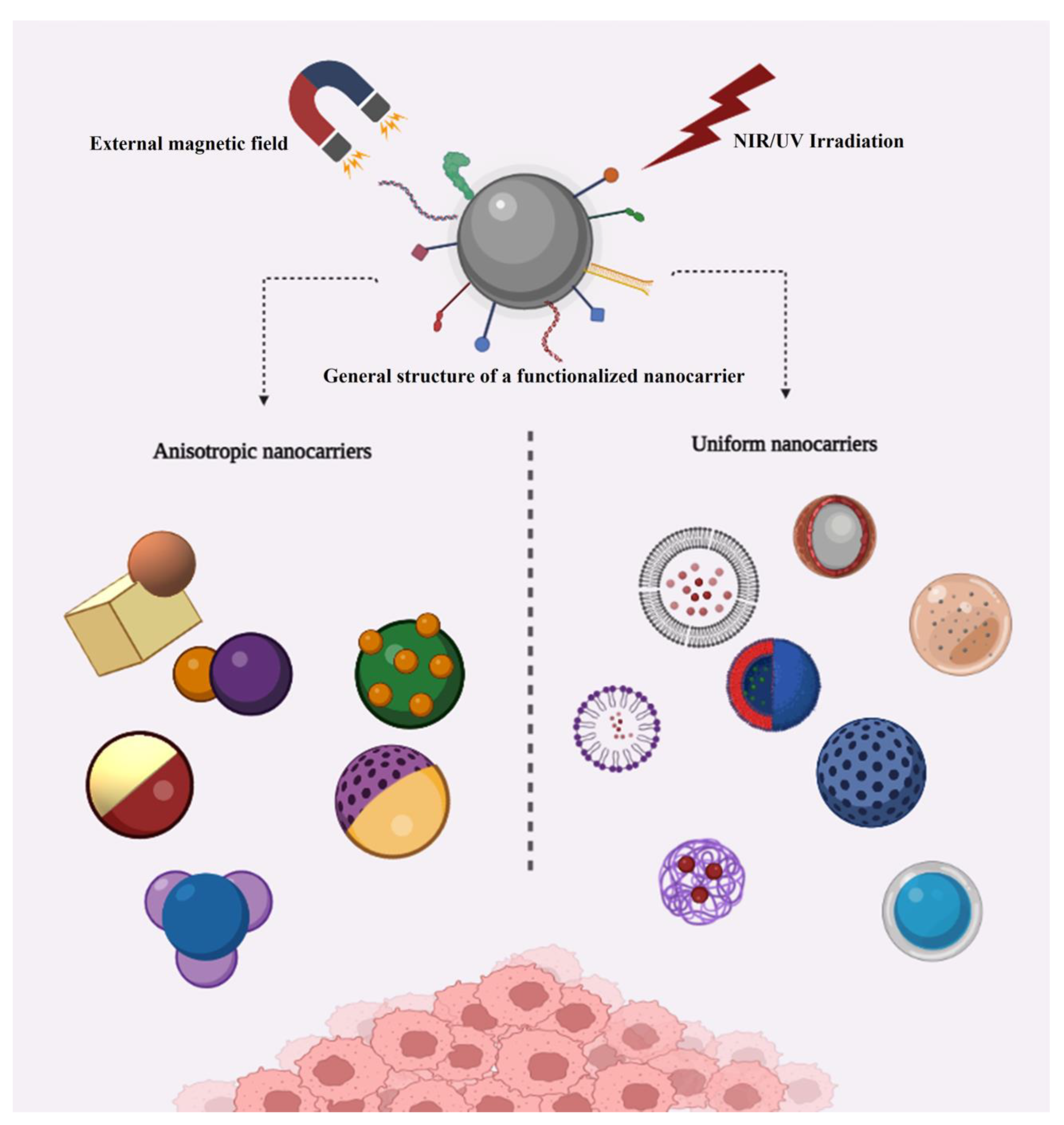
3. Anisotropic Structures
3.1. Solubility and Encapsulation
3.2. Anisotropic Structures Release and Interaction Data
3.3. Internalization and Cytotoxicity Evaluations
4. Discussion
5. Challenges and Outlook
6. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khosravian, P.; Shafiee Ardestani, M.; Khoobi, M.; Ostad, S.N.; Dorkoosh, F.A.; Akbari Javar, H.A.M. Mesoporous Silica Nanoparticles Functionalized with Folic Acid/Methionine for Active Targeted Delivery of Docetaxel. Dove Press 2016, 9, 7315–7330. [Google Scholar] [CrossRef] [Green Version]
- Quan, Q.; Xie, J.; Gao, H.; Yang, M.; Zhang, F.; Liu, G.; Lin, X.; Wang, A.; Eden, H.S.; Lee, S.; et al. HSA Coated Iron Oxide Nanoparticles as Drug Delivery Vehicles for Cancer Therapy. Mol. Pharm. 2011, 8, 1669–1676. [Google Scholar] [CrossRef] [Green Version]
- Hussein, H.A.; Abdullah, M.A. Novel Drug Delivery Systems Based on Silver Nanoparticles, Hyaluronic Acid, Lipid Nanoparticles and Liposomes for Cancer Treatment. Appl. Nanosci. 2021, 12, 3071–3096. [Google Scholar] [CrossRef]
- Ahangari, A.; Salouti, M.; Heidari, Z.; Kazemizadeh, A.R.; Safari, A.A. Development of Gentamicin-Gold Nanospheres for Antimicrobial Drug Delivery to Staphylococcal Infected Foci. Drug Deliv. 2013, 20, 34–39. [Google Scholar] [CrossRef]
- Ali, I.; Alsehli, M.; Scotti, L.; Scotti, M.T.; Tsai, S.T.; Yu, R.S.; Fa Hsieh, M.; Chen, J.C. Progress in Polymeric Nano-Medicines for Theranostic Cancer Treatment. Polymers 2020, 12, 598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, C.G.; Peters, G.J.; Ossendorp, F.; Cruz, L.J. The Potential of Multi-Compound Nanoparticles to Bypass Drug Resistance in Cancer. Cancer Chemother. Pharmacol. 2017, 80, 881–894. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Wang, B.; Wang, Y.; Lou, D. Dual Drug Release from Core-Shell Nanoparticles with Distinct Release Profiles. J. Pharm. Sci. 2014, 103, 3205–3216. [Google Scholar] [CrossRef]
- Li, X.; Wu, M.; Pan, L.; Shi, J. Tumor Vascular-Targeted Co-Delivery of Anti-Angiogenesis and Chemotherapeutic Agents by Mesoporous Silica Nanoparticle-Based Drug Delivery System for Synergetic Therapy of Tumor. Int. J. Nanomed. 2015, 11, 93–105. [Google Scholar] [CrossRef] [Green Version]
- Parhi, P.; Mohanty, C.; Sahoo, S.K. Nanotechnology-Based Combinational Drug Delivery: An Emerging Approach for Cancer Therapy. Drug Discov. Today 2012, 17, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, V.M.; Baril, P.; Costa, E.C.; De Melo-Diogo, D.; Foucher, F.; Queiroz, J.A.; Sousa, F.; Pichon, C.; Correia, I.J. Bioreducible Poly(2-Ethyl-2-Oxazoline)-PLA-PEI-SS Triblock Copolymer Micelles for Co-Delivery of DNA Minicircles and Doxorubicin. J. Control. Release 2015, 213, 175–191. [Google Scholar] [CrossRef]
- Tian, F.; Dahmani, F.Z.; Qiao, J.; Ni, J.; Xiong, H.; Liu, T.; Zhou, J.; Yao, J. Targeted Nanoplatform Chemotherapeutic and Antiangiogenic Drugs as a Tool to Reverse Multidrug Resistance in Breast Cancer. Acta Biomater. 2018, 75, 398–412. [Google Scholar] [CrossRef]
- Lin, J.; Liu, Z.; Zhu, Q.; Rong, X.; Liang, C.; Wang, J.; Ma, D.; Sun, J.; Wang, G. Colloids and Surfaces B: Biointerfaces Redox-Responsive Nanocarriers for Drug and Gene Co-Delivery Based on Chitosan Derivatives Modified Mesoporous Silica Nanoparticles. Colloids Surf. B Biointerfaces 2017, 155, 41–50. [Google Scholar] [CrossRef]
- Alven, S. Efficacy of Polymer-Based Nanocarriers for Co-Delivery of Curcumin and Selected Anticancer Drugs. Nanomaterials 2020, 10, 1556. [Google Scholar] [CrossRef] [PubMed]
- Naahidi, S.; Jafari, M.; Edalat, F.; Raymond, K.; Khademhosseini, A.; Chen, P. Biocompatibility of Engineered Nanoparticles for Drug Delivery. J. Control. Release 2013, 166, 182–194. [Google Scholar] [CrossRef]
- Tan, Y.; Li, S.; Pitt, B.R.; Huang, L. The Inhibitory Role of CpG Immunostimulatory Motifs in Cationic Lipid Vector-Mediated Transgene Expression in Vivo. Hum. Gene Ther. 1999, 10, 2153–2161. [Google Scholar] [CrossRef] [PubMed]
- Perrault, S.D.; Walkey, C.; Jennings, T.; Fischer, H.C.; Chan, W.C.W. Mediating Tumor Targeting Efficiency of Nanoparticles through Design. Nano Lett. 2009, 9, 1909–1915. [Google Scholar] [CrossRef]
- Vonarbourg, A.; Passirani, C.; Saulnier, P.; Benoit, J.-P. Parameters Influencing the Stealthiness of Colloidal Drug Delivery Systems. Biomaterials 2006, 27, 4356–4373. [Google Scholar] [CrossRef]
- Park, M.V.D.Z.; Neigh, A.M.; Vermeulen, J.P.; De La Fonteyne, L.J.J.; Verharen, H.W.; Briedé, J.J.; Loveren, H.V.; Jong, W.H.D. Biomaterials The Effect of Particle Size on the Cytotoxicity, in Fl Ammation, Developmental Toxicity and Genotoxicity of Silver Nanoparticles. Biomaterials 2011, 32, 9810–9817. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Zhang, C.; Liu, D.; Yan, J.; Liang, H. The Clinical Applications of Curcumin: Current State and the Future. Curr. Pharm. Des. 2013, 19, 2011–2031. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, G.; Jin, S.; Xu, L.; Zhao, C.X. Development of High-Drug-Loading Nanoparticles. Chempluschem 2020, 85, 2143–2157. [Google Scholar] [CrossRef]
- Bertrand, N.; Grenier, P.; Mahmoudi, M.; Lima, E.M.; Appel, E.A.; Dormont, F.; Lim, J.; Karnik, R.; Langer, R.; Farokhzad, O.C. Mechanistic Understanding of in vivo Protein Corona Formation on Polymeric Nanoparticles and Impact on Pharmacokinetics. Nat. Commun. 2017, 8, 777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yameen, B.; Choi, W.I.l.; Vilos, C.; Swami, A.; Shi, J.; Farokhzad, O.C. Insight into Nanoparticle Cellular Uptake and Intracellular Targeting. J. Control. Release 2014, 190, 485–499. [Google Scholar] [CrossRef] [Green Version]
- Tietjen, G.T.; Bracaglia, L.G.; Saltzman, W.M.; Pober, J.S. Focus on Fundamentals: Achieving Effective Nanoparticle Targeting. Trends Mol. Med. 2018, 24, 598–606. [Google Scholar] [CrossRef]
- Zhang, R.X.; Wong, H.L.; Xue, H.Y.; Eoh, J.Y.; Wu, X.Y. Nanomedicine of Synergistic Drug Combinations for Cancer Therapy–Strategies and Perspectives. J. Control. Release 2016, 240, 489–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haggag, Y.; Abu Ras, B.; El-Tanani, Y.; Tambuwala, M.M.; McCarron, P.; Isreb, M.; El-Tanani, M. Co-Delivery of a RanGTP Inhibitory Peptide and Doxorubicin Using Dual-Loaded Liposomal Carriers to Combat Chemotherapeutic Resistance in Breast Cancer Cells. Expert Opin. Drug Deliv. 2020, 17, 1655–1669. [Google Scholar] [CrossRef]
- Li, Y.; Thambi, T.; Lee, D.S. Co-Delivery of Drugs and Genes Using Polymeric Nanoparticles for Synergistic Cancer Therapeutic Effects. Adv. Healthc. Mater. 2018, 7, 1700886. [Google Scholar] [CrossRef]
- Birault, A.; Giret, S.; Théron, C.; Wong Chi Man, M.; Carcel, C.; Gallud, A.; Da Silva, A.; Durand, D.; Nguyen, C.; Bettache, N.; et al. Sequential Delivery of Synergistic Drugs by Silica Nanocarriers for Enhanced Tumour Treatment. J. Mater. Chem. B 2020, 8, 1472–1480. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Shen, X.; Geng, Y.; Chen, Z.; Li, L.; Li, S.; Yang, H.; Wu, C.; Zeng, H.; Liu, Y. Folate-Functionalized Magnetic-Mesoporous Silica Nanoparticles for Drug/Gene Codelivery to Potentiate the Antitumor Efficacy. ACS Appl. Mater. Interfaces 2016, 8, 13748–13758. [Google Scholar] [CrossRef]
- Rabanel, J.M.; Adibnia, V.; Tehrani, S.F.; Sanche, S.; Hildgen, P.; Banquy, X.; Ramassamy, C. Nanoparticle Heterogeneity: An Emerging Structural Parameter Influencing Particle Fate in Biological Media? Nanoscale 2019, 11, 383–406. [Google Scholar] [CrossRef]
- Fu, J.; An, D.; Song, Y.; Wang, C.; Qiu, M.; Zhang, H. Janus Nanoparticles for Cellular Delivery Chemotherapy: Recent Advances and Challenges. Coord. Chem. Rev. 2020, 422, 213467. [Google Scholar] [CrossRef]
- Le, T.C.; Zhai, J.; Chiu, W.H.; Tran, P.A.; Tran, N. Janus Particles: Recent Advances in the Biomedical Applications. Int. J. Nanomed. 2019, 14, 6749–6777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, Y.G.J.; Poh, K.C.W.; Loo, S.C.J. Hybrid Janus Microparticles Achieving Selective Encapsulation for Theranostic Applications via a Facile Solvent Emulsion Method. Macromol. Rapid Commun. 2019, 40, 1800801. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, K.; Lin, J.; Huang, P. Janus Nanoparticles in Cancer Diagnosis, Therapy and Theranostics. Biomater. Sci. 2019, 7, 1262–1275. [Google Scholar] [CrossRef]
- Vemula, V.R.; Lagishetty, V.; Lingala, S. Solubility Enhancement Techniques. Int. J. Pharm. Sci. Rev. Res. 2010, 5, 41–51. [Google Scholar]
- Loftsson, T.; Brewster, M.E. Pharmaceutical Applications of Cyclodextrins: Basic Science and Product Development. J. Pharm. Pharmacol. 2010, 62, 1607–1621. [Google Scholar] [CrossRef] [PubMed]
- McMorland, G.H.; Douglas, M.J.; Jeffery, W.K.; Ross, P.L.E.; Axelson, J.E.; Kim, J.H.K.; Gambling, D.R.; Robertson, K. Effect of PH-Adjustment of Bupivacaine on Onset and Duration of Epidural Analgesia in Parturients. Can. Anaesth. Soc. J. 1986, 33, 537–541. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Farajtabar, A.; Wang, N.; Liu, Z.; Fei, Z.; Zhao, H. Solubility of Chloroxine in Aqueous Co-Solvent Mixtures of N,N-Dimethylformamide, Dimethyl Sulfoxide, N-Methyl-2-Pyrrolidone and 1,4-Dioxane: Determination, Solvent Effect and Preferential Solvation Analysis. J. Chem. Thermodyn. 2019, 138, 288–296. [Google Scholar] [CrossRef]
- Dizaj, S.M.; Vazifehasl, Z.; Salatin, S.; Adibkia, K.; Javadzadeh, Y. Nanosizing of Drugs: Effect on Dissolution Rate. Res. Pharm. Sci. 2015, 10, 95–108. [Google Scholar]
- Kalepu, S.; Manthina, M.; Padavala, V. Oral Lipid-Based Drug Delivery Systems–an Overview. Acta Pharm. Sin. B 2013, 3, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Hasan, M.; Belhaj, N.; Benachour, H.; Barberi-Heyob, M.; Kahn, C.J.F.; Jabbari, E.; Linder, M.; Arab-Tehrany, E. Liposome Encapsulation of Curcumin: Physico-Chemical Characterizations and Effects on MCF7 Cancer Cell Proliferation. Int. J. Pharm. 2014, 461, 519–528. [Google Scholar] [CrossRef]
- Ting, J.M.; Porter, W.W.; Mecca, J.M.; Bates, F.S.; Reineke, T.M. Advances in Polymer Design for Enhancing Oral Drug Solubility and Delivery. Bioconjugate Chem. 2018, 29, 939–952. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Li, Z.; Wu, F.; Chen, M.; Wang, R.; Zhu, H.; Li, Q.; Yuan, Y. Improving Solubility and Bioavailability of Breviscapine with Mesoporous Silica Nanoparticles Prepared Using Ultrasound-Assisted Solution-Enhanced Dispersion by Supercritical Fluids Method. Int. J. Nanomed. 2020, 15, 1661–1675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalepu, S.; Nekkanti, V. Insoluble Drug Delivery Strategies: Review of Recent Advances and Business Prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veronese, F.M.; Schiavon, O.; Pasut, G.; Mendichi, R.; Andersson, L.; Tsirk, A.; Ford, J.; Wu, G.; Kneller, S.; Davies, J.; et al. PEG-Doxorubicin Conjugates: Influence of Polymer Structure on Drug Release, in vitro Cytotoxicity, Biodistribution, and Antitumor Activity. Bioconjug. Chem. 2005, 16, 775–784. [Google Scholar] [CrossRef]
- Banerjee, S.S.; Aher, N.; Patil, R.; Khandare, J. Poly(Ethylene Glycol)-Prodrug Conjugates: Concept, Design, and Applications. J. Drug Deliv. 2012, 2012, 103973. [Google Scholar] [CrossRef] [Green Version]
- Kolate, A.; Baradia, D.; Patil, S.; Vhora, I.; Kore, G.; Misra, A. PEG–A Versatile Conjugating Ligand for Drugs and Drug Delivery Systems. J. Control. Release 2014, 192, 67–81. [Google Scholar] [CrossRef]
- Ayubi, M.; Karimi, M.; Abdpour, S.; Rostamizadeh, K.; Parsa, M.; Zamani, M.; Saedi, A. Magnetic Nanoparticles Decorated with PEGylated Curcumin as Dual Targeted Drug Delivery: Synthesis, Toxicity and Biocompatibility Study. Mater. Sci. Eng. C 2019, 104, 109810. [Google Scholar] [CrossRef]
- Kratz, F.; Warnecke, A. Finding the Optimal Balance: Challenges of Improving Conventional Cancer Chemotherapy Using Suitable Combinations with Nano-Sized Drug Delivery Systems. J. Control. Release 2012, 164, 221–235. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, G.; Yang, H. Drug Delivery Systems for Differential Release in Combination Therapy. Expert Opin. Drug Deliv. 2011, 8, 171–190. [Google Scholar] [CrossRef]
- Liu, D.; Bimbo, L.M.; Mäkilä, E.; Villanova, F.; Kaasalainen, M.; Herranz-Blanco, B.; Caramella, C.M.; Lehto, V.P.; Salonen, J.; Herzig, K.H.; et al. Co-Delivery of a Hydrophobic Small Molecule and a Hydrophilic Peptide by Porous Silicon Nanoparticles. J. Control. Release 2013, 170, 268–278. [Google Scholar] [CrossRef]
- Kim, D.H.; Martin, D.C. Sustained Release of Dexamethasone from Hydrophilic Matrices Using PLGA Nanoparticles for Neural Drug Delivery. Biomaterials 2006, 27, 3031–3037. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Zhu, Q. Recent Advances of PLGA Micro/Nanoparticles for the Delivery of Biomacromolecular Therapeutics. Mater. Sci. Eng. C 2018, 92, 1041–1060. [Google Scholar] [CrossRef] [PubMed]
- Español, L.; Larrea, A.; Andreu, V.; Mendoza, G.; Arruebo, M.; Sebastian, V.; Aurora-Prado, M.S.; Kedor-Hackmann, E.R.M.; Santoro, M.I.R.M.; Santamaria, J. Dual Encapsulation of Hydrophobic and Hydrophilic Drugs in PLGA Nanoparticles by a Single-Step Method: Drug Delivery and Cytotoxicity Assays. RSC Adv. 2016, 6, 111060–111069. [Google Scholar] [CrossRef] [Green Version]
- Chuah, A.M.; Jacob, B.; Jie, Z.; Ramesh, S.; Mandal, S.; Puthan, J.K.; Deshpande, P.; Vaidyanathan, V.V.; Gelling, R.W.; Patel, G.; et al. Enhanced Bioavailability and Bioefficacy of an Amorphous Solid Dispersion of Curcumin. Food Chem. 2014, 156, 227–233. [Google Scholar] [CrossRef]
- Laha, D.; Pal, K.; Chowdhuri, A.R.; Parida, P.K.; Sahu, S.K.; Jana, K.; Karmakar, P. Fabrication of Curcumin-Loaded Folic Acid-Tagged Metal Organic Framework for Triple Negative Breast Cancer Therapy in in Vitro and in Vivo Systems. New J. Chem. 2019, 43, 217–229. [Google Scholar] [CrossRef]
- Pourjavadi, A.; Asgari, S.; Hosseini, S.H. Graphene Oxide Functionalized with Oxygen-Rich Polymers as a PH-Sensitive Carrier for Co-Delivery of Hydrophobic and Hydrophilic Drugs. J. Drug Deliv. Sci. Technol. 2020, 56, 101542. [Google Scholar] [CrossRef]
- Guo, W.; Song, Y.; Song, W.; Liu, Y.; Liu, Z.; Zhang, D.; Tang, Z.; Bai, O. Co-Delivery of Doxorubicin and Curcumin with Polypeptide Nanocarrier for Synergistic Lymphoma Therapy. Sci. Rep. 2020, 10, 7832. [Google Scholar] [CrossRef]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the First Proteasome Inhibitor Anticancer Drug: Current Status and Future Perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253. [Google Scholar] [CrossRef] [Green Version]
- Scott, K.; Hayden, P.J.; Howman, A.; Wheatley, K.; Coyne, I. Bortezomib for the Treatment of Multiple Myeloma. Cochrane Database Syst. Rev. 2013, 2013, CD010816. [Google Scholar] [CrossRef] [Green Version]
- Agyin, J.K.; Santhamma, B.; Nair, H.B.; Roy, S.S.; Tekmal, R.R. BU-32: A Novel Proteasome Inhibitor for Breast Cancer. Breast Cancer Res. 2009, 11, R74. [Google Scholar] [CrossRef] [Green Version]
- Medel, S.; Syrova, Z.; Kovacik, L.; Hrdy, J.; Hornacek, M.; Jager, E.; Hruby, M.; Lund, R.; Cmarko, D.; Stepanek, P.; et al. Curcumin-Bortezomib Loaded Polymeric Nanoparticles for Synergistic Cancer Therapy. Eur. Polym. J. 2017, 93, 116–131. [Google Scholar] [CrossRef]
- Manzano, M.; Vallet-Regí, M. Mesoporous Silica Nanoparticles for Drug Delivery. Adv. Funct. Mater. 2020, 30, 3–5. [Google Scholar] [CrossRef]
- Hosnedlova, B.; Kepinska, M.; Fernandez, C.; Peng, Q.; Ruttkay-Nedecky, B.; Milnerowicz, H.; Kizek, R. Carbon Nanomaterials for Targeted Cancer Therapy Drugs: A Critical Review. Chem. Rec. 2019, 19, 502–522. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zou, D.; Zhu, H.; Zhang, J. Mesoporous Metal–Organic Frameworks: Synthetic Strategies and Emerging Applications. Small 2018, 14, 1801454. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gu, F.X.; Chan, J.M.; Wang, A.Z.; Langer, R.S.; Farokhzad, O.C. Nanoparticles in Medicine: Therapeutic Applications and Developments. Clin. Pharmacol. Ther. 2008, 83, 761–769. [Google Scholar] [CrossRef]
- Rigby, S.; Fairhead, M.; van der Walle, C. Engineering Silica Particles as Oral Drug Delivery Vehicles. Curr. Pharm. Des. 2008, 14, 1821–1831. [Google Scholar] [CrossRef]
- Alyassin, Y.; Sayed, E.G.; Mehta, P.; Ruparelia, K.; Arshad, M.S.; Rasekh, M.; Shepherd, J.; Kucuk, I.; Wilson, P.B.; Singh, N.; et al. Application of Mesoporous Silica Nanoparticles as Drug Delivery Carriers for Chemotherapeutic Agents. Drug Discov. Today 2020, 25, 1513–1520. [Google Scholar] [CrossRef]
- Tian, B.; Liu, S.; Wu, S.; Lu, W.; Wang, D.; Jin, L.; Hu, B.; Li, K.; Wang, Z.; Quan, Z. PH-Responsive Poly (Acrylic Acid)-Gated Mesoporous Silica and Its Application in Oral Colon Targeted Drug Delivery for Doxorubicin. Colloids Surf. B Biointerfaces 2017, 154, 287–296. [Google Scholar] [CrossRef]
- Kumar, B.; Kulanthaivel, S.; Mondal, A.; Mishra, S.; Banerjee, B.; Bhaumik, A.; Banerjee, I.; Giri, S. Mesoporous Silica Nanoparticle Based Enzyme Responsive System for Colon Specific Drug Delivery through Guar Gum Capping. Colloids Surf. B Biointerfaces 2017, 150, 352–361. [Google Scholar] [CrossRef]
- Cui, Y.; Xu, Q.; Chow, P.K.H.; Wang, D.; Wang, C.H. Transferrin-Conjugated Magnetic Silica PLGA Nanoparticles Loaded with Doxorubicin and Paclitaxel for Brain Glioma Treatment. Biomaterials 2013, 34, 8511–8520. [Google Scholar] [CrossRef]
- Hsiang, Y.H.; Hertzberg, R.; Hecht, S.; Liu, L.F. Camptothecin Induces Protein-Linked DNA Breaks via Mammalian DNA Topoisomerase I. J. Biol. Chem. 1985, 260, 14873–14878. [Google Scholar] [CrossRef] [PubMed]
- Wall, M.E.; Wani, M.C.; Cook, C.E.; Palmer, K.H.; McPhail, A.T.; Sim, G.A. Plant Antitumor Agents. I. The Isolation and Structure of Camptothecin, a Novel Alkaloidal Leukemia and Tumor Inhibitor from Camptotheca Acuminata. J. Am. Chem. Soc. 1966, 88, 3888–3890. [Google Scholar] [CrossRef]
- Lu, J.; Liu, C.; Wang, P.; Ghazwani, M.; Xu, J.; Huang, Y.; Ma, X.; Zhang, P.; Li, S. The Self-Assembling Camptothecin-Tocopherol Prodrug: An Effective Approach for Formulating Camptothecin. Biomaterials 2015, 62, 176–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Edo, G.; Fornaguera, C.; Borrós, S.; Sánchez-García, D. Glycyrrhetinic Acid-Functionalized Mesoporous Silica Nanoparticles for the Co-Delivery of Dox/Cpt-Peg for Targeting Hepg2 Cells. Pharmaceutics 2020, 12, 1048. [Google Scholar] [CrossRef]
- Eytan, G.D. Mechanism of Multidrug Resistance in Relation to Passive Membrane Permeation. Biomed. Pharmacother. 2005, 59, 90–97. [Google Scholar] [CrossRef]
- Choudhury, H.; Pandey, M.; Yin, T.H.; Kaur, T.; Jia, G.W.; Tan, S.Q.L.; Weijie, H.; Yang, E.K.S.; Keat, C.G.; Bhattamishra, S.K.; et al. Rising Horizon in Circumventing Multidrug Resistance in Chemotherapy with Nanotechnology. Mater. Sci. Eng. C 2019, 101, 596–613. [Google Scholar] [CrossRef] [PubMed]
- Lv, S.; Tang, Z.; Li, M.; Lin, J.; Song, W.; Liu, H.; Huang, Y.; Zhang, Y.; Chen, X. Co-Delivery of Doxorubicin and Paclitaxel by PEG-Polypeptide Nanovehicle for the Treatment of Non-Small Cell Lung Cancer. Biomaterials 2014, 35, 6118–6129. [Google Scholar] [CrossRef]
- Glück, S. Nab-Paclitaxel for the Treatment of Aggressive Metastatic Breast Cancer. Clin. Breast Cancer 2014, 14, 221–227. [Google Scholar] [CrossRef]
- Fu, L.; Liang, Y.; Deng, L.; Ding, Y.; Chen, L.; Ye, Y.; Yang, X.; Pan, Q. Characterization of Tetrandrine, a Potent Inhibitor of P-Glycoprotein- Mediated Multidrug Resistance. Cancer Chemother. Pharmacol. 2004, 53, 349–356. [Google Scholar] [CrossRef]
- Callaghan, R.; Luk, F.; Bebawy, M. Special Section on Transporters in Toxicity and Disease—Minireview Inhibition of the Multidrug Resistance P-Glycoprotein: Time for a Change of Strategy ? DRUG Metab. Dispos. 2014, 42, 623–631. [Google Scholar] [CrossRef] [Green Version]
- Jia, L.; Li, Z.; Shen, J.; Zheng, D.; Tian, X.; Guo, H.; Chang, P. Multifunctional Mesoporous Silica Nanoparticles Mediated Co-Delivery of Paclitaxel and Tetrandrine for Overcoming Multidrug Resistance. Int. J. Pharm. 2015, 489, 318–330. [Google Scholar] [CrossRef]
- Kreuter, J. Nanoparticles-a Historical Perspective. Int. J. Pharm. 2007, 331, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, A.S. The Origins and Evolution of “Controlled” Drug Delivery Systems. J. Control. Release 2008, 132, 153–163. [Google Scholar] [CrossRef]
- Lu, X.Y.; Wu, D.C.; Li, Z.J.; Chen, G.Q. Polymer Nanoparticles. Prog. Mol. Biol. Transl. Sci. 2011, 104, 299–323. [Google Scholar] [PubMed]
- Peddi, S.; Pan, X.; MacKay, J.A. Intracellular Delivery of Rapamycin from FKBP Elastin-like Polypeptides Is Consistent with Macropinocytosis. Front. Pharmacol. 2018, 9, 1184. [Google Scholar] [CrossRef]
- MacDonald, A.S. A Worldwide, Phase III, Randomized, Controlled, Safety and Efficacy Study of a Sirolimus/Cyclosporine Regimen for Prevention of Acute Rejection in Recipients of Primary Mismatched Renal Allografts. Transplantation 2001, 71, 271–280. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, J.; Sun, W.; Xie, Q.R.; Xia, W.; Gu, H. Delivering Hydrophilic and Hydrophobic Chemotherapeutics Simultaneously by Magnetic Mesoporous Silica Nanoparticles to Inhibit Cancer Cells. Int. J. Nanomed. 2012, 7, 999–1013. [Google Scholar] [CrossRef] [Green Version]
- Orido, T.; Fujino, H.; Kawashima, T.; Murayama, T. Decrease in Uptake of Arachidonic Acid by Indomethacin in LS174T Human Colon Cancer Cells; a Novel Cyclooxygenase-2-Inhibition-Independent Effect. Arch. Biochem. Biophys. 2010, 494, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Horibe, S.; Tanahashi, T.; Kawauchi, S.; Mizuno, S.; Rikitake, Y. Preventative Effects of Sodium Alginate on Indomethacin-Induced Small-Intestinal Injury in Mice. Int. J. Med. Sci. 2016, 13, 653–663. [Google Scholar] [CrossRef] [Green Version]
- Akhgari, A.; Heshmati, Z.; Afrasiabi Garekani, H.; Sadeghi, F.; Sabbagh, A.; Sharif Makhmalzadeh, B.; Nokhodchi, A. Indomethacin Electrospun Nanofibers for Colonic Drug Delivery: In Vitro Dissolution Studies. Colloids Surf. B Biointerfaces 2017, 152, 29–35. [Google Scholar] [CrossRef] [Green Version]
- Kovalainen, M.; Mönkäre, J.; Mäkilä, E.; Salonen, J.; Lehto, V.P.; Herzig, K.H.; Järvinen, K. Mesoporous Silicon (PSi) for Sustained Peptide Delivery: Effect of PSi Microparticle Surface Chemistry on Peptide YY3-36 Release. Pharm. Res. 2012, 29, 837–846. [Google Scholar] [CrossRef]
- Lim, E.B.; Vy, T.A.; Lee, S.W. Comparative Release Kinetics of Small Drugs (Ibuprofen and Acetaminophen) from Multifunctional Mesoporous Silica Nanoparticles. J. Mater. Chem. B 2020, 8, 2096–2106. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, L.; Tang, C.; Yin, C. Co-Delivery of Doxorubicin and Survivin ShRNA-Expressing Plasmid Via Microenvironment-Responsive Dendritic Mesoporous Silica Nanoparticles for Synergistic Cancer Therapy. Pharm. Res. 2017, 34, 2829–2841. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Yin, Q.; Chen, L.; Zhang, Z.; Li, Y. Co-Delivery of Paclitaxel and Survivin ShRNA by Pluronic P85-PEI/TPGS Complex Nanoparticles to Overcome Drug Resistance in Lung Cancer. Biomaterials 2012, 33, 8613–8624. [Google Scholar] [CrossRef]
- Shen, J.; Meng, Q.; Sui, H.; Yin, Q.; Zhang, Z.; Yu, H.; Li, Y. iRGD Conjugated TPGS Mediates Codelivery of Paclitaxel and Survivin shRNA for the Reversal of Lung Cancer Resistance. Mol. Pharmaceutics 2014, 11, 2579–2591. [Google Scholar] [CrossRef]
- Bahreyni, A.; Ramezani, M.; Alibolandi, M.; Hassanzadeh, P.; Abnous, K.; Taghdisi, S.M. High Affinity of AS1411 toward Copper; Its Application in a Sensitive Aptasensor for Copper Detection. Anal. Biochem. 2019, 575, 1–9. [Google Scholar] [CrossRef]
- Ghahremani, F.; Shahbazi-Gahrouei, D.; Kefayat, A.; Motaghi, H.; Mehrgardi, M.A.; Javanmard, S.H. AS1411 Aptamer Conjugated Gold Nanoclusters as a Targeted Radiosensitizer for Megavoltage Radiation Therapy of 4T1 Breast Cancer Cells. RSC Adv. 2018, 8, 4249–4258. [Google Scholar] [CrossRef] [Green Version]
- Babaei, M.; Abnous, K.; Mohammad, S.; Taghavi, S. European Journal of Pharmaceutics and Biopharmaceutics Targeted Rod-Shaped Mesoporous Silica Nanoparticles for the Co-Delivery of Camptothecin and Survivin ShRNA in to Colon Adenocarcinoma in vitro and in vivo. Eur. J. Pharm. Biopharm. 2020, 156, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Li, W.; Wu, L.; Huang, W.; Zhang, F. β-Cyclodextrin-Modified Mesoporous Silica Nanoparticles with Photo-Responsive Gatekeepers for Controlled Release of Hexaconazole. Coatings 2021, 11, 1489. [Google Scholar] [CrossRef]
- Beňová, E.; Hornebecq, V.; Zeleňák, V.; Huntošová, V.; Almáši, M.; Máčajová, M.; Bergé-Lefranc, D. PH-Responsive Mesoporous Silica Drug Delivery System, Its Biocompatibility and Co-Adsorption/Co-Release of 5-Fluorouracil and Naproxen. Appl. Surf. Sci. 2021, 561, 150011. [Google Scholar] [CrossRef]
- Aznar, E.; Sancenoín, F.; Marcos, M.D.; Martiínez-Maínez, R.; Stroeve, P.; Cano, J.; Amoroís, P. Delivery Modulation in Silica Mesoporous Supports via Alkyl Chain Pore Outlet Decoration. Langmuir 2012, 28, 2986–2996. [Google Scholar] [CrossRef]
- El Sayed, S.; Giménez, C.; Aznar, E.; Martínez-Máñez, R.; Sancenón, F.; Licchelli, M. Highly Selective and Sensitive Detection of Glutathione Using Mesoporous Silica Nanoparticles Capped with Disulfide-Containing Oligo(Ethylene Glycol) Chains. Org. Biomol. Chem. 2015, 13, 1017–1021. [Google Scholar] [CrossRef] [PubMed]
- Llinàs, M.C.; Martínez-Edo, G.; Cascante, A.; Porcar, I.; Borrós, S.; Sánchez-García, D. Preparation of a Mesoporous Silica-Based Nano-Vehicle for Dual DOX/CPT Ph-Triggered Delivery. Drug Deliv. 2018, 25, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Shan, H.; Li, K.; Zhao, D.; Chi, C.; Tan, Q.; Wang, X.; Yu, J.; Piao, M. Locally Controlled Release of Methotrexate and Alendronate by Thermo-Sensitive Hydrogels for Synergistic Inhibition of Osteosarcoma Progression. Front. Pharmacol. 2020, 11, 573. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Qi, M.; Guo, C.; Yu, Y.; Wang, B.; Fang, L.; Liu, M.; Wang, Z.; Fan, X.; Chen, D. Novel Dual Mitochondrial and CD44 Receptor Targeting Nanoparticles for Redox Stimuli-Triggered Release. Nanoscale Res. Lett. 2018, 13, 32. [Google Scholar] [CrossRef] [Green Version]
- Schanté, C.E.; Zuber, G.; Herlin, C.; Vandamme, T.F. Chemical Modifications of Hyaluronic Acid for the Synthesis of Derivatives for a Broad Range of Biomedical Applications. Carbohydr. Polym. 2011, 85, 469–489. [Google Scholar] [CrossRef]
- Lee, T.; Lim, E.K.; Lee, J.; Kang, B.; Choi, J.; Park, H.S.; Suh, J.S.; Huh, Y.M.; Haam, S. Efficient CD44-Targeted Magnetic Resonance Imaging (MRI) of Breast Cancer Cells Using Hyaluronic Acid (HA)-Modified MnFe2O4 Nanocrystals. Nanoscale Res. Lett. 2013, 8, 149. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Zou, S.; Guo, C.; Wang, K.; Zhao, F.; Fan, H.; Yin, J.; Chen, D. Multifunctional Redox-Responsive and CD44 Receptor Targeting Polymer-Drug Nanomedicine Based Curcumin and Alendronate: Synthesis, Characterization and in vitro Evaluation. Artif. Cells Nanomed. Biotechnol. 2018, 46, 168–177. [Google Scholar] [CrossRef] [Green Version]
- Hadinoto, K.; Sundaresan, A.; Cheow, W.S. Lipid-Polymer Hybrid Nanoparticles as a New Generation Therapeutic Delivery Platform: A Review. Eur. J. Pharm. Biopharm. 2013, 85, 427–443. [Google Scholar] [CrossRef]
- Shah, S.; Famta, P.; Raghuvanshi, R.S.; Singh, S.B.; Srivastava, S. Lipid Polymer Hybrid Nanocarriers: Insights into Synthesis Aspects, Characterization, Release Mechanisms, Surface Functionalization and Potential Implications. Colloids Interface Sci. Commun. 2022, 46, 100570. [Google Scholar] [CrossRef]
- Teixeira, M.C.; Carbone, C.; Souto, E.B. Beyond Liposomes: Recent Advances on Lipid Based Nanostructures for Poorly Soluble/Poorly Permeable Drug Delivery. Prog. Lipid Res. 2017, 68, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mendozza, M.; Caselli, L.; Salvatore, A.; Montis, C.; Berti, D. Nanoparticles and Organized Lipid Assemblies: From Interaction to Design of Hybrid Soft Devices. Soft Matter 2019, 15, 8951–8970. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.K.; Wang, C.; Cheung, H.Y.; Yang, M.; Fong, W.F. Triptolide Induces Bcl-2 Cleavage and Mitochondria Dependent Apoptosis in P53-Deficient HL-60 Cells. Cancer Lett. 2006, 241, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Nakazato, T.; Sagawa, M.; Kizaki, M. Triptolide Induces Apoptotic Cell Death of Multiple Myeloma Cells via Transcriptional Repression of Mcl-1. Int. J. Oncol. 2014, 44, 1131–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Yang, Z.; Wang, X.; Zhang, X.; Wang, M.; Wang, Y.; Mei, Q.; Wang, Z. Triptolide Inhibits Ovarian Cancer Cell Invasion by Repression of Matrix Metalloproteinase 7 and 19 and Upregulation of E-Cadherin. Exp. Mol. Med. 2012, 44, 633–641. [Google Scholar] [CrossRef] [Green Version]
- Qu, J.; Yu, S.S.; Du, D.; Wang, Y.D. Bioactive Constituents from Toxic Seed Plants in China. RSC Adv. 2013, 3, 10078–10102. [Google Scholar] [CrossRef]
- Zhou, Z.L.; Yang, Y.X.; Ding, J.; Li, Y.C.; Miao, Z.H. Triptolide: Structural Modifications, Structure-Activity Relationships, Bioactivities, Clinical Development and Mechanisms. Nat. Prod. Rep. 2012, 29, 457–475. [Google Scholar] [CrossRef]
- Meng, C.; Zhu, H.; Song, H.; Wang, Z.; Huang, G.; Li, D.; Ma, Z.; Ma, J.; Qin, Q.; Sun, X.; et al. Targets and Molecular Mechanisms of Triptolide in Cancer Therapy. Chin. J. Cancer Res. 2014, 26, 622–626. [Google Scholar] [CrossRef]
- Wu, B.; Lu, S.T.; Zhang, L.J.; Zhuo, R.X.; Xu, H.B.; Huang, S.W. Codelivery of Doxorubicin and Triptolide with Reduction-Sensitive Lipid–Polymer Hybrid Nanoparticles for in vitro and in vivo Synergistic Cancer Treatment. Int. J. Nanomed. 2017, 12, 1853–1862. [Google Scholar] [CrossRef] [Green Version]
- Tao, X.; Tao, T.; Wen, Y.; Yi, J.; He, L.; Huang, Z.; Nie, Y.; Yao, X.; Wang, Y.; He, C.; et al. Novel Delivery of Mitoxantrone with Hydrophobically Modified Pullulan Nanoparticles to Inhibit Bladder Cancer Cell and the Effect of Nano-Drug Size on Inhibition Efficiency. Nanoscale Res. Lett. 2018, 13, 345. [Google Scholar] [CrossRef] [Green Version]
- Kilic, M.A.; Ozlu, E.; Calis, S.; Blazkova, I.; Nguyen, H.V.; Dostalova, S.; Kopel, P.; Stanisavljevic, M.; Mosca, L.; Falvo, E.; et al. RGD-Modified Apoferritin Nanoparticles for Efficient Drug Delivery to Tumors. ACS Nano 2013, 7, 4830–4837. [Google Scholar]
- Kilic, M.A.; Ozlu, E.; Calis, S. A Novel Protein-Based Anticancer Drug Encapsulating Nanosphere: Apoferritin-Doxorubicin Complex. J. Biomed. Nanotechnol. 2012, 8, 508–514. [Google Scholar] [CrossRef]
- Bayón-Cordero, L.; Alkorta, I.; Arana, L. Application of Solid Lipid Nanoparticles to Improve the Efficiency of Anticancer Drugs. Nanomaterials 2019, 9, 474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geszke-Moritz, M.; Moritz, M. Solid Lipid Nanoparticles as Attractive Drug Vehicles: Composition, Properties and Therapeutic Strategies. Mater. Sci. Eng. C 2016, 68, 982–994. [Google Scholar] [CrossRef]
- Amer Ridha, A.; Kashanian, S.; Rafipour, R.; Hemati Azandaryani, A.; Zhaleh, H.; Mahdavian, E. A Promising Dual-Drug Targeted Delivery System in Cancer Therapy: Nanocomplexes of Folate–Apoferritin-Conjugated Cationic Solid Lipid Nanoparticles. Pharm. Dev. Technol. 2021, 26, 673–681. [Google Scholar] [CrossRef]
- Large, D.E.; Soucy, J.R.; Hebert, J.; Auguste, D.T. Advances in Receptor-Mediated, Tumor-Targeted Drug Delivery. Adv. Ther. 2019, 2, 1800091. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, N.; Wu, J.; Xu, X.; Kamaly, N.; Farokhzad, O.C. Cancer Nanotechnology: The Impact of Passive and Active Targeting in the Era of Modern Cancer Biology. Adv. Drug Deliv. Rev. 2014, 66, 2–25. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.; Park, C.; Yi, G.; Lee, D.; Koo, H. Active Targeting Strategies Using Biological Ligands for Nanoparticle Drug Delivery Systems. Cancers 2019, 11, 640. [Google Scholar] [CrossRef] [Green Version]
- Bae, Y.; Nishiyama, N.; Kataoka, K. In Vivo Antitumor Activity of the Folate-Conjugated PH-Sensitive Polymeric Micelle Selectively Releasing Adriamycin in the Intracellular Acidic Compartments. Bioconjug. Chem. 2007, 18, 1131–1139. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Thi, H.; Dang, L.H.; Vu-quang, H.; Tran, N.Q. Folate-Conjugated Chitosan-Pluronic P123 Nanogels: Synthesis and Characterizations towards Dual Drug Delivery. J. Nanomater. 2019, 2019, 1067821. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Li, B.; Qi, S.; Liu, Y.; Gai, Y.; Ye, P.; Yang, G.; Zhang, W.; Zhang, P.; He, X.; et al. Co-Delivery of Doxorubicin and Bmil SiRNA by Folate Receptor Targeted Liposomes Exhibits Enhanced Anti-Tumor Effects in vitro and in vivo. Theranostics 2014, 4, 1096–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, M.; Solanki, A.; Joshi, A.; Devkar, R.; Seshadri, S.; Thakore, S. β -Cyclodextrin Based Dual-Responsive Multifunctional Nanotheranostics for Cancer Cell Targeting and Dual Drug Delivery. Carbohydr. Polym. 2019, 206, 694–705. [Google Scholar] [CrossRef]
- Karaman, D.Ş.; Lähdeniemi, I.A.K.; Sahlgren, C.; Rosenholm, J.M.; Toivola, D.M. Targeted Modulation of Cell Differentiation in Distinct Regions of the Gastrointestinal Tract via Oral Administration of Differently PEG-PEI Functionalized Mesoporous Silica Nanoparticles. Int. J. Nanomed. 2016, 11, 299–313. [Google Scholar]
- Harrison, E.; Nicol, J.R.; Macias, M.; Burke, G.A.; Coulter, J.A.; Meenan, B.J.; Dixon, D. A Comparison of Gold Nanoparticle Surface Co-Functionalization Approaches Using Polyethylene Glycol (PEG) and the Effect on Stability, Non-Speci Fi c Protein Adsorption and Internalization. Mater. Sci. Eng. C 2016, 62, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Feiner-gracia, N.; Dols-perez, A.; Royo, M.; Solans, C.; Garcia-celma, M.J. Cell Penetrating Peptide Grafting of PLGA Nanoparticles to Enhance Cell Uptake. Eur. Polym. J. 2018, 108, 429–438. [Google Scholar] [CrossRef]
- Yang, X.; Hu, C.; Tong, F.; Liu, R.; Zhou, Y.; Qin, L.; Ouyang, L.; Gao, H. Tumor Microenvironment-Responsive Dual Drug Dimer-Loaded PEGylated Bilirubin Nanoparticles for Improved Drug Delivery and Enhanced Immune-Chemotherapy of Breast Cancer. Adv. Funct. Mater. 2019, 29, 1901896. [Google Scholar] [CrossRef]
- Nath, K.; Guo, L.; Nancolas, B.; Nelson, D.S.; Shestov, A.A.; Roman, J.; Zhou, R.; Leeper, D.B.; Halestrap, A.P.; Blair, I.A.; et al. Mechanism of Antineoplastic Activity of Lonidamine. Biochim. Biophys. Acta. 2016, 1866, 151–162. [Google Scholar] [CrossRef] [Green Version]
- Naumann, J.A.; Widen, J.C.; Jonart, L.A.; Ebadi, M.; Tang, J.; Gordon, D.J.; Harki, D.A.; Gordon, P.M. SN-38 Conjugated Gold Nanoparticles Activated by Ewing Sarcoma Specific MRNAs Exhibit in vitro and in vivo Efficacy. Bioconjugate Chem. 2018, 29, 1111–1118. [Google Scholar] [CrossRef]
- Bala, V.; Rao, S.; Boyd, B.J.; Prestidge, C.A. Prodrug and Nanomedicine Approaches for the Delivery of the Camptothecin Analogue SN38. J. Control. Release 2013, 172, 48–61. [Google Scholar] [CrossRef]
- Liao, J.; Song, Y.; Liu, C.; Li, D.; Zheng, H.; Lu, B. Polymer Chemistry. Polym. Chem. 2019, 10, 5879–5893. [Google Scholar] [CrossRef]
- Gao, N.; Xing, C.; Wang, H.; Feng, L.; Zeng, X.; Mei, L.; Peng, Z. PH-Responsive Dual Drug-Loaded Nanocarriers Based on Poly (2-Ethyl-2-Oxazoline) Modified Black Phosphorus Nanosheets for Cancer Chemo/Photothermal Therapy. Front. Pharmacol. 2019, 10, 270. [Google Scholar] [CrossRef] [PubMed]
- Romano, S.; Fonseca, N.; Simões, S.; Gonçalves, J.; Moreira, J.N. Nucleolin-Based Targeting Strategies for Cancer Therapy: From Targeted Drug Delivery to Cytotoxic Ligands. Drug Discov. Today 2019, 24, 1985–2001. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Sathiyaseelan, A.; Saravanakumar, K.; Mariadoss, A.V.A.; Wang, M.H. PH-Controlled Nucleolin Targeted Release of Dual Drug from Chitosan-Gold Based Aptamer Functionalized Nano Drug Delivery System for Improved Glioblastoma Treatment. Carbohydr. Polym. 2021, 262, 117907. [Google Scholar] [CrossRef]
- Wang, F.; Li, J.; Tang, X.; Huang, K.; Chen, L. Polyelectrolyte Three Layer Nanoparticles of Chitosan/Dextran Sulfate/Chitosan for Dual Drug Delivery. Colloids Surf. B Biointerfaces 2020, 190, 110925. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A Doxorubicin Delivery Platform Using Engineered Natural Membrane Vesicle Exosomes for Targeted Tumor Therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef]
- Peng, H.; Ji, W.; Zhao, R.; Yang, J.; Lu, Z.; Li, Y.; Zhang, X. Exosome: A Significant Nano-Scale Drug Delivery Carrier. J. Mater. Chem. B 2020, 8, 7591–7608. [Google Scholar] [CrossRef]
- Liang, G.; Zhu, Y.; Ali, D.J.; Tian, T.; Xu, H.; Si, K.; Sun, B.; Chen, B.; Xiao, Z. Engineered Exosomes for Targeted Co-Delivery of MiR-21 Inhibitor and Chemotherapeutics to Reverse Drug Resistance in Colon Cancer. J. Nanobiotechnology 2020, 18, 10. [Google Scholar] [CrossRef]
- Yin, Y.; Wu, X.; Yang, Z.; Zhao, J.; Wang, X.; Zhang, Q.; Yuan, M.; Xie, L.; Liu, H.; He, Q. The Potential Efficacy of R8-Modified Paclitaxel-Loaded Liposomes on Pulmonary Arterial Hypertension. Pharm. Res. 2013, 30, 2050–2062. [Google Scholar] [CrossRef]
- Yue, G.; Wang, C.; Liu, B.; Wu, M.; Huang, Y.; Guo, Y.; Ma, Q. Liposomes Co-Delivery System of Doxorubicin and Astragaloside IV Co-Modified by Folate Ligand and Octa-Arginine Polypeptide for Anti-Breast Cancer. RSC Adv. 2020, 10, 11573–11581. [Google Scholar] [CrossRef] [Green Version]
- Khoee, S.; Nouri, A. Preparation of Janus Nanoparticles and Its Application in Drug Delivery. Des. Dev. New Nanocarriers 2018, 2018, 145–180. [Google Scholar]
- Heo, D.N.; Yang, D.H.; Moon, H.J.; Lee, J.B.; Bae, M.S.; Lee, S.C.; Lee, W.J.; Sun, I.C.; Kwon, I.K. Gold Nanoparticles Surface-Functionalized with Paclitaxel Drug and Biotin Receptor as Theranostic Agents for Cancer Therapy. Biomaterials 2012, 33, 856–866. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Zhou, Y.; Zhang, Y.; Zhang, C.; Deng, X.; Dong, C.; Shuang, S. Facile Fabrication Route of Janus Gold-Mesoporous Silica Nanocarriers with Dual-Drug Delivery for Tumor Therapy. ACS Biomater. Sci. Eng. 2020, 6, 1573–1581. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, X.; Zhang, L.; Gao, Y.; Wang, C.; Hong, W.; Zhao, G.; Li, L.; Liu, R.; Wang, C. Amphiphilic Janus Nanoparticles for Imaging-Guided Synergistic Chemo-Photothermal Hepatocellular Carcinoma Therapy in the Second near-Infrared Window. Nanoscale 2021, 13, 3974–3982. [Google Scholar] [CrossRef] [PubMed]
- Lone, S.; Cheong, I.W. Fabrication of Polymeric Janus Particles by Droplet Microfluidics. RSC Adv. 2014, 4, 13322–13333. [Google Scholar] [CrossRef]
- Wurm, F.; Kilbinger, A.F.M. Polymeric Janus Particles. Angew. Chemie Int. Ed. 2009, 48, 8412–8421. [Google Scholar] [CrossRef]
- Xie, H.; She, Z.G.; Wang, S.; Sharma, G.; Smith, J.W. One-Step Fabrication of Polymeric Janus Nanoparticles for Drug Delivery. Langmuir 2012, 28, 4459–4463. [Google Scholar] [CrossRef] [Green Version]
- Winkler, J.S.; Barai, M.; Tomassone, M.S. Dual Drug-Loaded Biodegradable Janus Particles for Simultaneous Co-Delivery of Hydrophobic and Hydrophilic Compounds. Exp. Biol. Med. 2019, 244, 1162–1177. [Google Scholar] [CrossRef]
- Khoee, S.; Karimi, M.R. Dual-Drug Loaded Janus Graphene Oxide-Based Thermoresponsive Nanoparticles for Targeted Therapy. Polymer 2018, 142, 80–98. [Google Scholar] [CrossRef]
- Geppert, M.; Himly, M. Iron Oxide Nanoparticles in Bioimaging–An Immune Perspective. Front. Immunol. 2021, 12, 688927. [Google Scholar] [CrossRef]
- Dadfar, S.M.; Roemhild, K.; Drude, N.I.; von Stillfried, S.; Knüchel, R.; Kiessling, F.; Lammers, T. Iron Oxide Nanoparticles: Diagnostic, Therapeutic and Theranostic Applications. Adv. Drug Deliv. Rev. 2019, 138, 302–325. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Pauletti, G.M.; Wang, J.; Zhang, J.; Ewing, R.C.; Wang, Y.; Shi, D. Dual Surface-Functionalized Janus Nanocomposites of Polystyrene/Fe3O4@SiO2 for Simultaneous Tumor Cell Targeting and Stimulus-Induced Drug Release. Adv. Mater. 2013, 25, 3485–3489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafson, T.P.; Cao, Q.; Wang, S.T.; Berezin, M.Y. Design of Irreversible Optical Nanothermometers for Thermal Ablations. Chem. Commun. 2013, 49, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Borzenkov, M.; Chirico, G.; D’Alfonso, L.; Sironi, L.; Collini, M.; Cabrini, E.; Dacarro, G.; Milanese, C.; Pallavicini, P.; Taglietti, A.; et al. Thermal and Chemical Stability of Thiol Bonding on Gold Nanostars. Langmuir 2015, 31, 8081–8091. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, M.; Zhou, L.; Han, Q.; Chen, X.; Li, S.; Li, L.; Su, Z.; Wang, C. Dual Drug Delivery and Sequential Release by Amphiphilic Janus Nanoparticles for Liver Cancer Theranostics. Biomaterials 2018, 181, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhou, L.; Wei, Y.; El-Toni, A.M.; Zhang, F.; Zhao, D. Anisotropic Growth-Induced Synthesis of Dual-Compartment Janus Mesoporous Silica Nanoparticles for Bimodal Triggered Drugs Delivery. J. Am. Chem. Soc. 2014, 136, 15086–15092. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhou, L.; Wei, Y.; El-Toni, A.M.; Zhang, F.; Zhao, D. Anisotropic Encapsulation-Induced Synthesis of Asymmetric Single-Hole Mesoporous Nanocages. J. Am. Chem. Soc. 2015, 137, 5903–5906. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhang, Y.; Deng, W.; Hu, J. Antibacterial and Anticancer Activities of Asymmetric Lollipop-like Mesoporous Silica Nanoparticles Loaded with Curcumin and Gentamicin Sulfate. Colloids Surf. B Biointerfaces 2020, 186, 110744. [Google Scholar] [CrossRef]
- Zhao, D.; Wu, J.; Li, C.; Zhang, H.; Li, Z.; Luan, Y. Precise Ratiometric Loading of PTX and DOX Based on Redox-Sensitive Mixed Micelles for Cancer Therapy. Colloids Surf. B Biointerfaces 2017, 155, 51–60. [Google Scholar] [CrossRef]
- Sudha, T.; Bharali, D.J.; Yalcin, M.; Darwish, N.H.E.; Coskun, M.D.; Keating, K.A.; Lin, H.Y.; Davis, P.J.; Mousa, S.A. Targeted Delivery of Paclitaxel and Doxorubicin to Cancer Xenografts via the Nanoparticle of Nano-Diamino-Tetrac. Int. J. Nanomed. 2017, 12, 1305–1315. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Du, P.; Liu, P. PH-Responsive Intramolecular FRET-Based Self-Tracking Polymer Prodrug Nanoparticles for Real-Time Tumor Intracellular Drug Release Monitoring and Imaging. Int. J. Pharm. 2020, 588, 119723. [Google Scholar] [CrossRef]
- Abramczyk, H.; Brozek-Pluska, B. Raman Imaging in Biochemical and Biomedical Applications. Diagnosis and Treatment of Breast Cancer. Chem. Rev. 2013, 113, 5766–5781. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.; Ptolemy, A.S.; Niewczas, L.; Britz-McKibbin, P. Integrative Metabolomics for Characterizing Unknown Low-Abundance Metabolites by Capillary Electrophoresis-Mass Spectrometry with Computer Simulations. Anal. Chem. 2007, 79, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Hong, R.; Han, G.; Fernández, J.M.; Kim, B.J.; Forbes, N.S.; Rotello, V.M. Glutathione-Mediated Delivery and Release Using Monolayer Protected Nanoparticle Carriers. J. Am. Chem. Soc. 2006, 128, 1078–1079. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Yang, Y.; Chen, X.; Shao, Z. Intelligent Janus Nanoparticles for Intracellular Real-Time Monitoring of Dual Drug Release. Nanoscale 2016, 8, 6754–6760. [Google Scholar] [CrossRef]
- Huang, X.; Voit, B. Progress on Multi-Compartment Polymeric Capsules. Polym. Chem. 2013, 4, 435–443. [Google Scholar] [CrossRef]
- Lodge, T.P.; Rasdal, A.; Li, Z.; Hillmyer, M.A. Simultaneous, Segregated Storage of Two Agents in a Multicompartment Micelle. J. Am. Chem. Soc. 2005, 127, 17608–17609. [Google Scholar] [CrossRef]
- Sun, G.; Cui, H.; Lin, L.Y.; Lee, N.S.; Yang, C.; Neumann, W.L.; Freskos, J.N.; Shieh, J.J.; Dorshow, R.B.; Wooley, K.L. Multicompartment Polymer Nanostructures with Ratiometric Dual-Emission PH-Sensitivity. J. Am. Chem. Soc. 2011, 133, 8534–8543. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Song, H.; Zhang, J.; Li, P.; Li, C.; Wang, C.; Kong, D.; Zhao, Q. An Injectable, Thermosensitive and Multicompartment Hydrogel for Simultaneous Encapsulation and Independent Release of a Drug Cocktail as an Effective Combination Therapy Platform. J. Control. Release 2015, 203, 57–66. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, G.; Liu, G.; Hu, J.; Liu, S. Photo- and Thermo-Responsive Multicompartment Hydrogels for Synergistic Delivery of Gemcitabine and Doxorubicin. J. Control. Release 2017, 259, 149–159. [Google Scholar] [CrossRef]
- Tambasco, N.; Romoli, M.; Calabresi, P. Levodopa in Parkinson’s Disease: Current Status and Future Developments. Curr. Neuropharmacol. 2017, 16, 1239–1252. [Google Scholar] [CrossRef] [PubMed]
- Markham, C.H.; Diamond, S.G.; Treciokas, L.J. Carbidopa in Parkinson Disease and in Nausea and Vomiting of Levodopa. Arch. Neurol. 1974, 31, 128–133. [Google Scholar] [CrossRef]
- Seeberger, L.C.; Hauser, R.A. Levodopa/Carbidopa/Entacapone in Parkinson’s Disease. Expert Rev. Neurother. 2009, 9, 929–940. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, S.; Hitt, J.; Chang, S.W.L.; Lahann, J. Multicompartmental Microcylinders. Angew. Chemie Int. Ed. 2009, 48, 4589–4593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhaskar, S.; Pollock, K.M.; Yoshida, M.; Lahann, J. Towards Designer Microparticles: Simultaneous Control of Anisotropv, Shape and Size. Small 2010, 6, 404–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roh, K.H.; Martin, D.C.; Lahann, J. Biphasic Janus Particles with Nanoscale Anisotropy. Nat. Mater. 2005, 4, 759–763. [Google Scholar] [CrossRef]
- Parthipan, A.K.; Gupta, N.; Pandey, K.; Sharma, B.; Jacob, J.; Saha, S. One-Step Fabrication of Bicompartmental Microparticles as a Dual Drug Delivery System for Parkinson’s Disease Management. J. Mater. Sci. 2019, 54, 730–744. [Google Scholar] [CrossRef]
- Sun, L.; Wu, Q.; Peng, F.; Liu, L.; Gong, C. Strategies of Polymeric Nanoparticles for Enhanced Internalization in Cancer Therapy. Colloids Surf. B Biointerfaces 2015, 135, 56–72. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.; Walkey, C.; Chan, W.C.W. Polyethylene Glycol Backfilling Mitigates the Negative Impact of the Protein Corona on Nanoparticle Cell Targeting. Angew. Chemie Int. Ed. 2014, 53, 5093–5096. [Google Scholar] [CrossRef] [PubMed]
- Saw, P.E.; Park, J.; Lee, E.; Ahn, S.; Lee, J.; Kim, H.; Kim, J.; Choi, M.; Farokhzad, O.C.; Jon, S. Effect of PEG Pairing on the Efficiency of Cancer-Targeting Liposomes. Theranostics 2015, 5, 746–754. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Dai, R.; Wei, Q.; Li, W.; Zhu, G.; Chi, H.; Guo, Z.; Wang, L.; Cui, C.; Xu, J.; et al. Dual-Functionalized Janus Mesoporous Silica Nanoparticles with Active Targeting and Charge Reversal for Synergistic Tumor-Targeting Therapy. ACS Appl. Mater. Interfaces 2019, 11, 44582–44592. [Google Scholar] [CrossRef]
- Gao, P.; Sun, S.; Wang, Y.; Wei, Y.; Jiang, Y. Biodegradable T2-Phage-like Janus Nanoparticles for Actively-Targeted and Chemo-Photothermal Synergistic Therapy. Chem. Eng. J. 2022, 428, 131284. [Google Scholar] [CrossRef]
- Liu, C.; Li, C.Y.; Yuan, F. Mathematical Modeling of the Phoenix Rising Pathway. PLoS Comput. Biol. 2014, 10, e1003461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Tian, L.; Ma, J.; Gong, Y.; Zhang, Z.; Chen, Z.; Xu, B.; Xiong, H.; Li, C.; Huang, Q. Dying Tumor Cells Stimulate Proliferation of Living Tumor Cells via Caspase-Dependent Protein Kinase Cδ Activation in Pancreatic Ductal Adenocarcinoma. Mol. Oncol. 2015, 9, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Chandra, D.N.; Prasanth, G.K.; Singh, N.; Kumar, S.; Jithesh, O.; Sadasivan, C.; Sharma, S.; Singh, T.P.; Haridas, M. Identification of a Novel and Potent Inhibitor of Phospholipase A 2 in a Medicinal Plant: Crystal Structure at 1.93 Å and Surface Plasmon Resonance Analysis of Phospholipase A2 Complexed with Berberine. Biochim. Biophys. Acta Proteins Proteom. 2011, 1814, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Jia, Y.; Zheng, X.; Shao, D.; Zhao, Y.; Wang, Z.; Dawulieti, J.; Liu, W.; Sun, M.; Sun, W.; et al. Janus Nanocarrier-Based Co-Delivery of Doxorubicin and Berberine Weakens Chemotherapy-Exacerbated Hepatocellular Carcinoma Recurrence. Acta Biomater. 2019, 100, 352–364. [Google Scholar] [CrossRef]
- Li, S.; Zhang, L.; Liang, X.; Wang, T.; Chen, X.; Liu, C.; Li, L.; Wang, C. Tailored Synthesis of Hollow MOF/Polydopamine Janus Nanoparticles for Synergistic Multi-Drug Chemo-Photothermal Therapy. Chem. Eng. J. 2019, 378, 122175. [Google Scholar] [CrossRef]
- Culp, M.B.; Soerjomataram, I.; Efstathiou, J.A.; Bray, F.; Catto, J. Recent Global Patterns in Prostate Cancer Incidence and Mortality Rates. Eur. Urol. 2019, 77, 38–52. [Google Scholar] [CrossRef]
- Tsakalozou, E.; Eckman, A.M.; Bae, Y. Combination Effects of Docetaxel and Doxorubicin in Hormone-Refractory Prostate Cancer Cells. Biochem. Res. Int. 2012, 2012, 832059. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Zhan, W.; Chen, Y.; Jha, R.K.; Chen, X. Docetaxel and Doxorubicin Codelivery by Nanocarriers for Synergistic Treatment of Prostate Cancer. Front. Pharmacol. 2019, 10, 1436. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Liu, S.; Shu, H.; Crawford, J.; Xing, Y.; Tao, F. Resveratrol Alleviates Temporomandibular Joint Inflammatory Pain by Recovering Disturbed Gut Microbiota. Brain. Behav. Immun. 2020, 87, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.C.; Zhou, X.P.; Zhu, Q.; Gong, S.; Qin, Z.H.; Reid, P.F.; Raymond, L.N.; Yin, Q.Z.; Jiang, X.H. Cobratoxin Inhibits Pain-Evoked Discharge of Neurons in Thalamic Parafascicular Nucleus in Rats: Involvement of Cholinergic and Serotonergic Systems. Toxicon 2009, 54, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yao, W.; Xie, X.; Gao, J.; Lu, X. PH-Sensitive Dual Drug Loaded Janus Nanoparticles by Oral Delivery for Multimodal Analgesia. J. Nanobiotechnology 2021, 19, 235. [Google Scholar] [CrossRef] [PubMed]
- Kooti, M.; Sedeh, A.N.; Motamedi, H.; Rezatofighi, S.E. Magnetic Graphene Oxide Inlaid with Silver Nanoparticles as Antibacterial and Drug Delivery Composite. Appl. Microbiol. Biotechnol. 2018, 102, 3607–3621. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Liu, G.; Cui, Y.; Jia, W.; Luo, Y.; Cheng, Z. A Hybrid System of Hydrogel/Frog Egg-like Microspheres Accelerates Wound Healing via Sustained Delivery of RCSPs. J. Appl. Polym. Sci. 2020, 137, 49521. [Google Scholar] [CrossRef]
- Ji, X.; Li, R.; Liu, G.; Jia, W.; Sun, M.; Liu, Y.; Luo, Y.; Cheng, Z. Phase Separation-Based Electrospun Janus Nanofibers Loaded with Rana Chensinensis Skin Peptides/Silver Nanoparticles for Wound Healing. Mater. Des. 2021, 207, 109864. [Google Scholar] [CrossRef]
- Wong, C.K.; Chen, F.; Walther, A.; Stenzel, M.H. Bioactive Patchy Nanoparticles with Compartmentalized Cargoes for Simultaneous and Trackable Delivery. Angew. Chemie Int. Ed. 2019, 58, 7335–7340. [Google Scholar] [CrossRef]
- Saeedi, M.; Eslamifar, M.; Khezri, K.; Dizaj, S.M. Applications of Nanotechnology in Drug Delivery to the Central Nervous System. Biomed. Pharmacother. 2019, 111, 666–675. [Google Scholar] [CrossRef]
- Zeinali, M.; Abbaspour-Ravasjani, S.; Ghorbani, M.; Babazadeh, A.; Soltanfam, T.; Santos, A.C.; Hamishehkar, H.; Hamblin, M.R. Nanovehicles for Co-Delivery of Anticancer Agents. Drug Discov. Today 2020, 25, 1416–1430. [Google Scholar] [CrossRef]
- Rizvi, S.A.A.; Saleh, A.M. Applications of Nanoparticle Systems in Drug Delivery Technology. Saudi Pharm. J. 2018, 26, 64–70. [Google Scholar] [CrossRef]
- Doermbach, K.; Pich, A. Facile Synthesis of Dumbbell-Shaped Multi-Compartment Nanoparticles. Nanoscale 2015, 7, 9169–9173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravaine, S.; Duguet, E. Current Opinion in Colloid & Interface Science Synthesis and Assembly of Patchy Particles: Recent Progress and Future Prospects. Curr. Opin. Colloid Interface Sci. 2017, 30, 45–53. [Google Scholar] [CrossRef]
- Wang, S.; Liu, K.; Wang, F.; Peng, F.; Tu, Y. The Application of Micro- and Nanomotors in Classified Drug Delivery. Chem. An Asian J. 2019, 14, 2336–2347. [Google Scholar] [CrossRef] [PubMed]
- Díez, P.; Lucena-Sánchez, E.; Escudero, A.; Llopis-Lorente, A.; Villalonga, R.; Martínez-Máñez, R. Ultrafast Directional Janus Pt-Mesoporous Silica Nanomotors for Smart Drug Delivery. ACS Nano 2021, 15, 4467–4480. [Google Scholar] [CrossRef]
- Ou, Z.; Luo, B.; Neophytou, A.; Chakrabarti, D. Synthesis and Self-Assembly of Janus and Triblock Patchy Particles. Front. Nanosci. 2019, 13, 61–85. [Google Scholar]
- Ye, Y.; Luan, J.; Wang, M.; Chen, Y.; Wilson, D.A.; Peng, F.; Tu, Y. Fabrication of Self-Propelled Micro- and Nanomotors Based on Janus Structures. Chem. A Eur. J. 2019, 25, 8663–8680. [Google Scholar] [CrossRef] [Green Version]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanocarrier | Cargo | Outcomes and Details | Ref. |
|---|---|---|---|
| PLGA NPs | diclofenac sodium (DS) and dexamethasone (DX) | Higher solubility and cell uptake for both drugs. Encapsulation efficiencies were (EE%) 67.7% for DX and 54.2% for DS (drug/polymer 1:10 ratio). Release rates were 80% for DS and 60% for DX. Cytotoxicity data revealed nearly 9% reduction in cell viability compared to the control group with 69.4% viability. | [53] |
| GO functionalized with poly(epichlorohydrin)-graft-hyperbranched polyglycerol (PCH-g-HPG) NPs | doxorubicin (DOX) and curcumin (CUR) | high dispersibility and biocompatibility particularly for CUR, drug loading efficiency (DLE) was highest at 85% for DOX, and at 80% for CUR. The release rate was nearly 90% for dox and 45% for CUR at pH 5. cytotoxicity on MCF-7 displayed nearly 20% cell viability for NPs loaded with both drugs compared to that of the 70% and 50% loaded with only CUR or DOX, respectively. | [56] |
| mPEG-b-P(Glu-co-Phe) | doxorubicin (DOX) and curcumin (CUR) | improved encapsulation and solubility of the CUR. Drug loading was calculated to be 9.7% for DOX and 8.1% for CUR, with DOX: CUR 1: 1.23 (molar ratio). DOX release after 60h at pH 5, 6.8, and 7.4 was reported 60%, 30%, 20%, respectively. Less cell viability (in most of the samples) and higher apoptotic rates in NPs with both drugs was observed compared to single-drug samples. | [57] |
| methoxy-poly (ethylene glycol)-block-poly lactic acid (mPEG-b-PLA) | curcumin (CUR) and bortezomib (BTZ) | enhanced solubility and cytotoxicity for both hydrophobic drugs specially CUR, 20–26% drug loading efficiency regarding the copolymer composition, higher cytotoxic effect of curcumin–BTZ complex loaded NPs for the MCF-7 cell line compared to the free curcumin–BTZ were observed. The 8k NPs were significantly more toxic compared to free complexes, and NPs with the shortest lactide chains were more cytotoxic. | [61] |
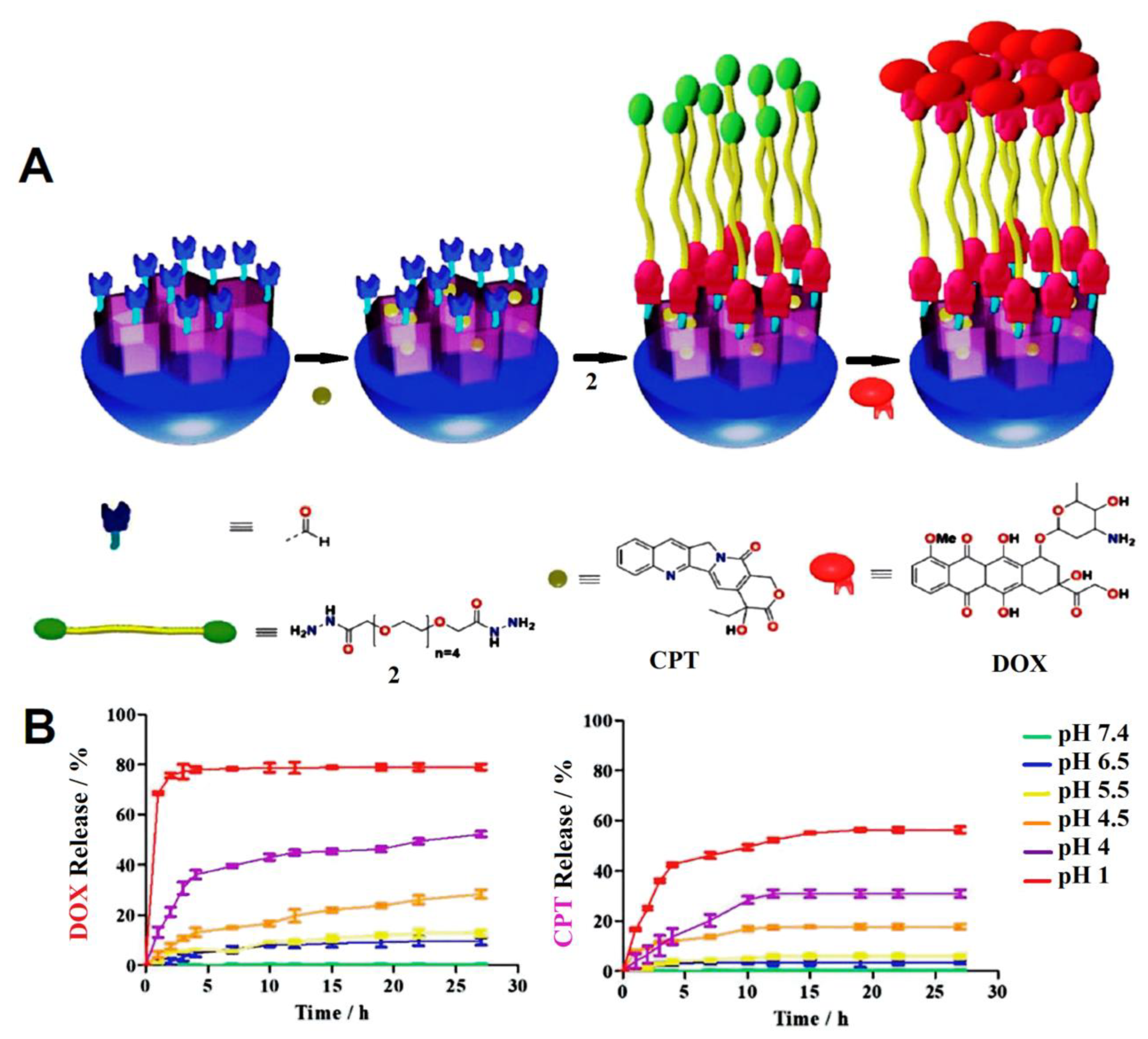
| mesoporous silica nanoparticle (MSNs) | doxorubicin and camptothecin (CPT) | sequential release, selective internalization. 25% DOX and 6.3 ± 3% CPT-PEG were loaded. No release at pH 7 was observed for both drugs while the release was 25% and 15% at pH 5.5 for DOX and CPT-PEG, respectively. HepG2 cells viability was 17% when using CPT-PEG@MSN-DOX compared to single-drug MSN-DOX cell viability at 47%. Selectivity of cytotoxic effect towards HepG2 cells was observed when NPs were decoreated with glycyrrhetinic acid (GA). | [74] |
| mesoporous silica nanoparticle (MSNs) | paclitaxel (PTX) and MDR reversal agent tetrandrine (TET) | pH-responsive release property, markedly increased intracellular accumulation of NPs. The drug loading efficiency was 7.23% for PTX and 1.21% for TET in NPs loaded with both drugs. pH-responsive release where release rates were 7.5% for PTX and 16.5% for TET after 72 h at pH 7, while release percentages of PTX and TET were 53.9% and 67.9%, respectively. More cell deaths and proliferation inhibition in the combination of two drugs using NPs than free drugs indicating better endocytosis and uptake of the drugs using MSNs. | [81] |
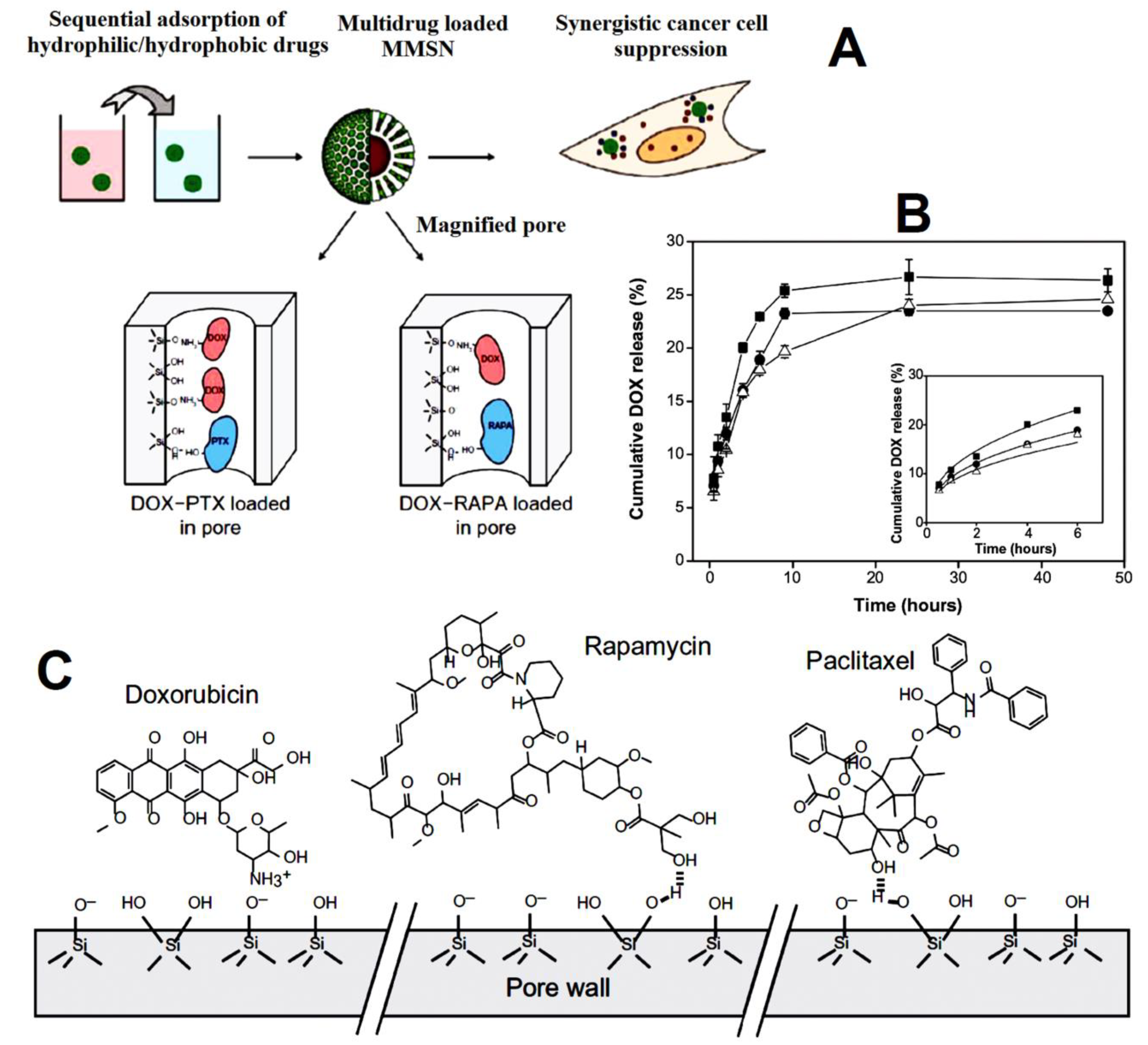
| magnetic mesoporous silica nanoparticles | doxorubicin (DOX) and hydrophobic drugs paclitaxel (PTX) and rapamycin (RAPA) | enhanced internalization, tumor cell apoptosis, and growth inhibition as compared to single-drug loaded MMSNs. The slower release was observed for DOX from DOX-PTXMMSNs (23%) and DOX-RAPA-MMSNs (19%) compared to the single drug (25%). Apoptotic ratios in cells were 30.72% for DOX-PTX-MMSNs and 47.30% for DOX-RAPA-MMSNs greater than the apoptotic ratios induced by each single-drug-loaded MMSNs. | [87] |
| thermally hydrocarbon sized-Psi (THCPSi) and thermally oxidized-PSi (TOPSi). | indomethacin (IMC), and peptide tyrosine tyrosine 3-36 (PYY3-36) | co-loading of therapeutics enhanced the loading capacity of the PSi NPs, accelerating the release rate and permeation. TOPSi-IMC-PYY showed a loading degree of 15.0 ± 0.1% for PYY3-36 and 13.7 ± 0.4% for IMC, and THCPSi-IMC-PYY with 18.4 ± 0.8% for PYY3-36 and 16.8 ± 0.8% for IMC. release rates of IMC and PYY3-36 from co-loaded PSi NPs were faster than single-drug-loaded ones at pH 7.4 and 5.5. Improvements in the cell viability rates were observed for the PSi NPs loaded with PYY3-36 compared to the plain PSi NPs. | [50] |
| FMSN@PDA@GO | ibuprofen (ibu) and acetaminophen (acet) | sequential release with individual kinetics for each drug. Loading amounts were 0.4 wt. % and 6.1 wt. % for MSNs-Ibu and FMSNs-Ibu, respectively. The cumulative release fraction was 90% at 36 h for Ibu and 15 h for Acet, respectively. Effective blocking layer of PDA @GO coating prevented rapid transmission of acetaminophen. | [92] |
| PEG@MSN nanorods | camptothecin and survivin shRNA-expressing plasmid (iSur-DNA) | higher targetability, uptake, and cytotoxicity owing to AS1411 aptamer. In total, 32% of encapsulation efficiency for CPT was reported. In total, 65% and 16% CPT was released from PEG@MSNR-CPT in citrate buffer (pH 5.4) and PBS (pH 7.4), respectively. The cytotoxicity of Apt-PEG@MSNR-CPT/Sur (20% cell viability) was significantly more than non-targeted NPs in the C26 cell line. | [98] |
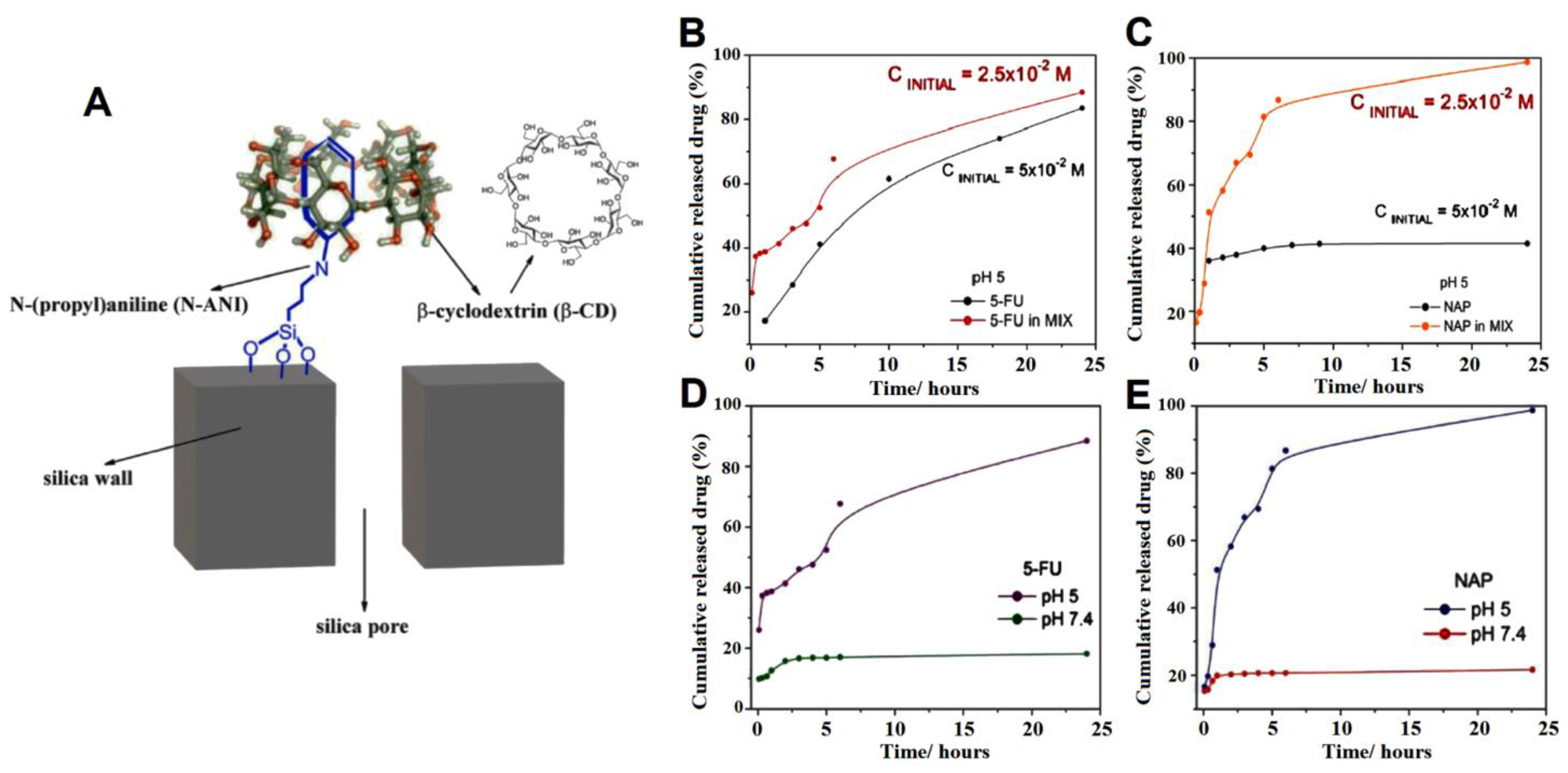
| MSN modified with N-(propyl)aniline and β-CD cap | 5-fluorouracil (5FU) and naproxen (NAP) | pH-responsive release in acidic condition controlled by CD, and more toxicity of drugs loaded in the nanoparticle. The total amount of drugs loaded in the SBA-15_N-ANI_5-FUN_ β-CD sample was 20 mg of 5FU and 62 mg of NAP per 1 g of solid. Less and slower release rates was detected in neutral pH due to the closed gatekeepers. The viability of U87 MG cells at pH = 6.3 was remarkedly reduced by the administration of SBA-15_N-ANI_5-FUN_β-CD (5FU + NAP 1:1) | [100] |
| MSN-hyd-PEG-hyd | doxorubicin (DOX) and camptothecin (CPT) | proper cytotoxicity of the dual delivery systems induced by pH-triggered release. Loading rates were 3.1% for CPT and 25% for DOX. Negligible release was detected at higher pH and physiological pH while nearly 10% of DOX and 5% of CPT were released in pH 5.5. | [103] |
| poly alendronate-hyaluronan-S-S-curcumin copolymer (ALN-oHA-S-S-CUR) | curcumin (CUR) and alendronate (ALN) | drug release under the reducing environments, high uptake and cytotoxicity of the active targeting redox-sensitive micelles on MDA-MB-231 cells owning CD44 receptors ALN-oHA-S-S-CUR micelles showed 52.58 ± 8.1 encapsulation efficiencies of CUR. Nearly 70% release from ALN-oHA-S-S-CUR micelles via 10mM GSH. The 40 µg/mL cur-loaded ALN-oHA-S-S-CUR micelles displayed the lowest cell viability after 48 h in MCF-7 cells and MDA-MB-231 cells | [108] |
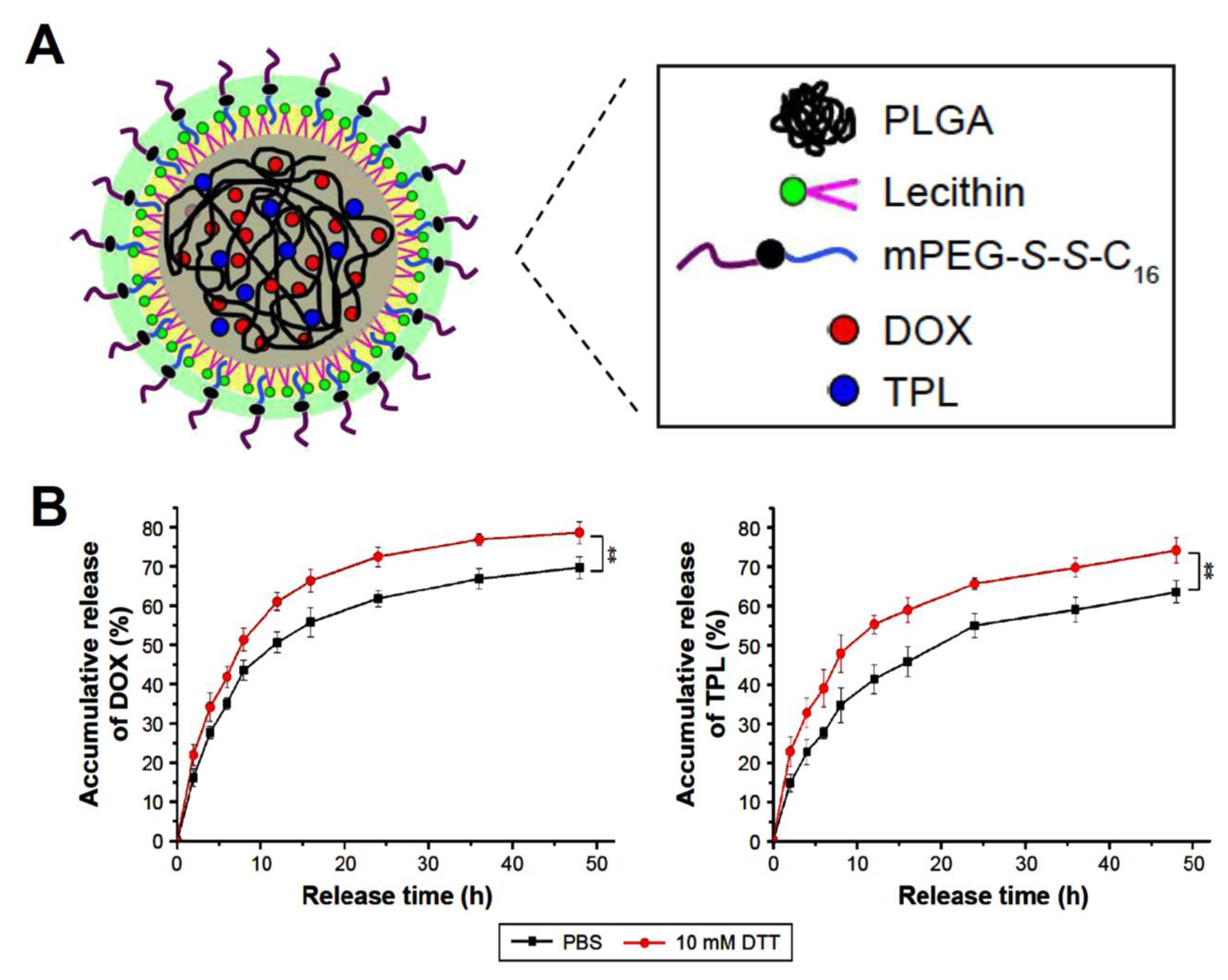
| lipid–polymer hybrid nanoparticles (LPNPs) | triptolide (TPL) and doxorubicin (DOX) | Encapsulation efficiencies were 75.5% for DOX and 58.3% for TPL in DOX/TPL-LPNPs. Roughly 78% of the DOX and 70% of TPL were released in the presence of 10 mM DTT. DOX/TPL-1/0.2-LPNPs demonstrated higher cytotoxicity than other samples with 20% cell viability and synergistic effect (CI < 1) at all concentrations. | [119] |
| protein–lipid nanocomplexes (DTPLNs) (Apoferritin (AFr)/cationic solid lipid nanoparticles (cSLN) ) | doxorubicin (DOX) and mitoxantrone (MTO) | At pH 6.8 DTPLNs released 75.99% of MTO and 94.52% of DOX. DTPLNs and TPNs displayed more growth inhibition compared to the free combination drugs with growth inhibition values of 31 ± 3.46, and 17 ± 3.42 (%) for MCF-7 cells, along with the greater cytotoxicity of the DTPLNs than other samples in folate receptor-positive cells due to the higher cellular uptake via FRs mediated endocytosis in PC-3 and MCF-7 lines. | [125] |
| liposomes (FA-DOX/siRNA-L) | doxorubicin (DOX) and Bmi1 siRNA | Significantly higher inhibitory and cytotoxicity efficacy than sole delivery with the help of targeting ligand. DOX encapsulation in FA-DOX/siRNA-L was 89.3% where the siRNA were fully encapsulated in the liposomes. FA-DOX/siRNA-L displayed improved cytotoxic features, where 90.5% of KB cells, 82% of HeLa, and 68% of Hep3B were killed, and enhanced tumor size reduction. | [131] |
| Nanocarriers based on β-Cyclodextrin modified with Maleic anhydride and NIPAM | doxorubicin (DOX) and curcumin (CUR) | nanoconjugates enhanced the endocytosis of poorly bioavailable drug curcumin, increased internalization using magnetic NPs. The drug entrapment efficiency of the nanoconjugates was 88% where the drug-loading content of curcumin and DOX was 45 wt% and 32 wt%, respectively. DOX release was Almost 60% at pH 5 and 37 °C vs. more than 80% at pH 5 and 40 °C, while curcumin release was less than 20% at pH 5 and 40 °C vs. 80% at pH 7.4 and 40 °C. Drug-loaded NPs displayed noticeable reduction in cell viability specifically in the presence of magnet, due to the enhanced uptake into the cell via endocytosis. | [132] |
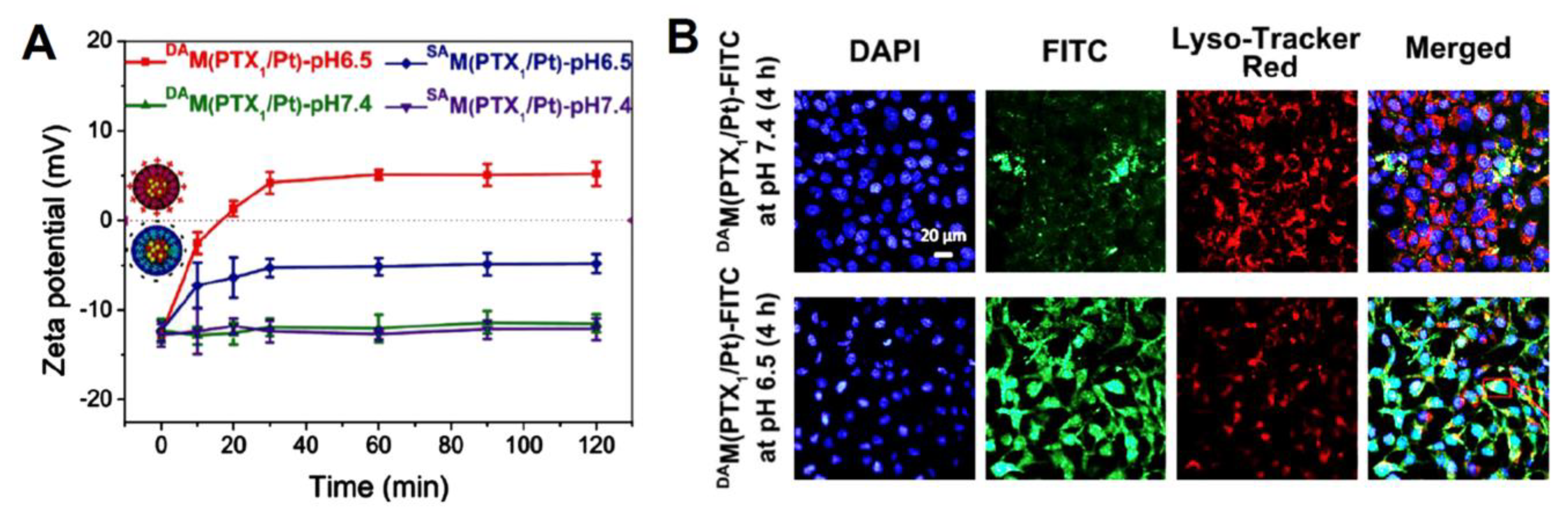
| poly(AMA-co-IMMA)-b-poly(OEGMA) (PAIPO) copolymers | paclitaxel (PTX) and cis-platinum (Pt (II)) | enhanced cellular uptake and apoptosis owing to the charge-conversion of micelles surface. The drug loading content (μmol/mg prodrug) in DAM (PTX1/Pt) was 0.101 for PTX and 0.112 for Pt (II). Approximately 82% of PTX and 88% of Pt (II)were released after 72 h in NaASc 5 mM + GSH 10 mM at pH 5.0. Cell-viability data of the HeLa and Skov-3 cell lines demonstrated the highest synergic effect in DAM(PTXn/Pt), exhibiting the lowest IC50 = 0.37–0.79 for HeLa, and 0.41–0.78 for Skov-3. | [140] |
| Black phosphorus nanosheets (BP @PDA-PEOz) | doxorubicin (DOX) and bortezomib (BTZ) | enhanced the cellular absorption of DOX and enhanced circulation time, possibly due to the charge reversal of PEOz.Higher release rates for both DOX and BTZ at 45% and 97% at pH 5 in the presence of NIR laser. Co-administration of DTX + BTZ being more cytotoxic than DOX or BTZ alone and DOX-loaded BP@PDAPEOz-BTZ with 808 nm laser irradiation (1.0 W cm−2) had the lowest rate of survival after 48 h. | [141] |
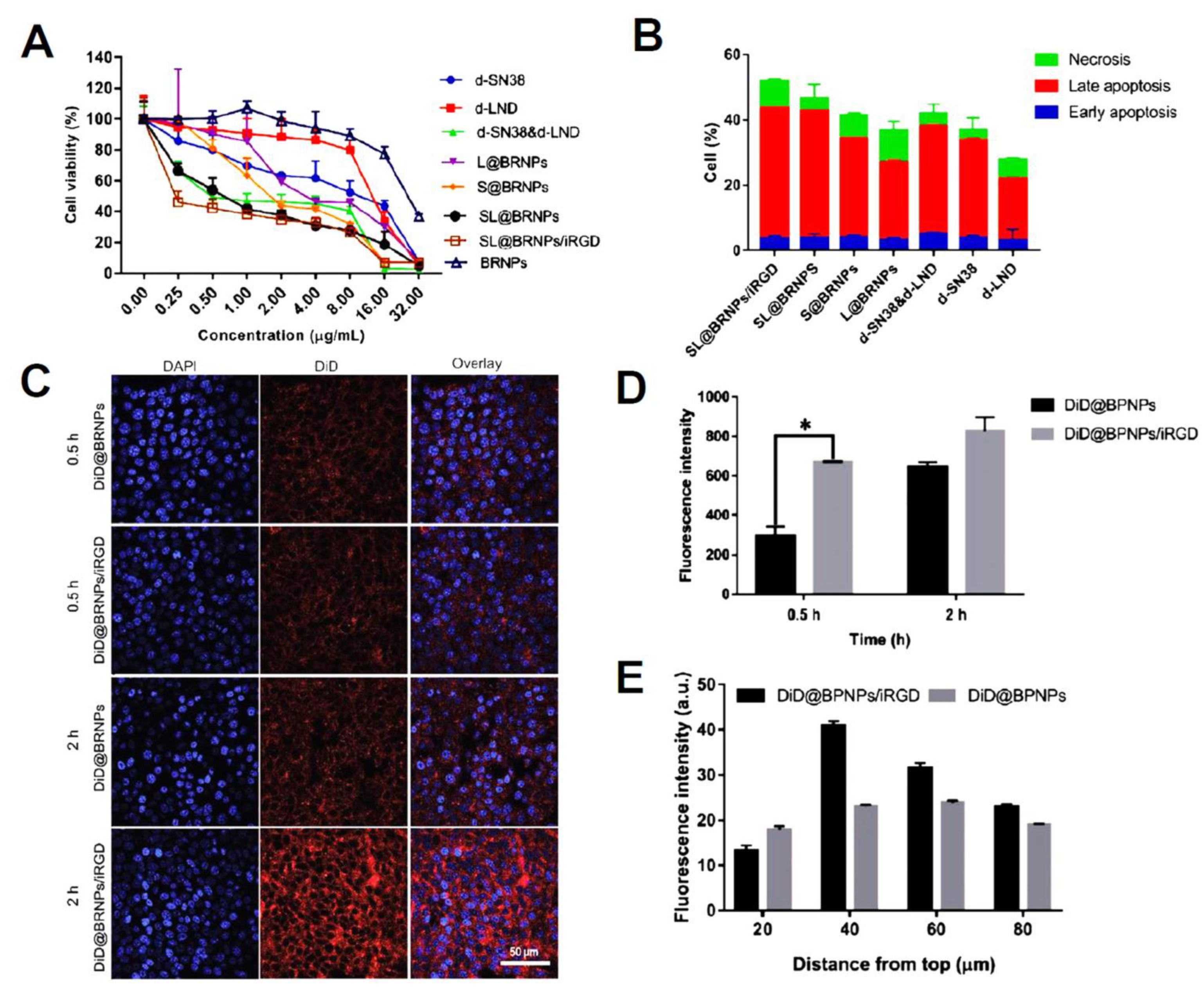
| PEGylated bilirubin nanoparticles (BRNPs) | dimer-7-ethyl-10 hydroxycamptothecin (d-SN38) and dimer-lonidamine (d-LND) | Encapsulation efficiencies of 95.33% and 92.34% for d-SN38 and d-LND were observed, respectively. The relative cumulative release of d-LND was 96.27% vs. 40.32% (100 × 103 M H2O2 vs. PBS), while that for d-SN38 was 67.39% vs. 25.95% (0.01 M H2O2 vs. PBS) indicating ROS-responsive release type. SL@BRNPs/iRGD groups cells apoptosis was at 54.2% (1.12-fold greater than SL@BRNPs), demonstrating the superior absorption of nanoparticles by iRGD and the subsequent increase in cell death. | [136] |
| Aptamer-Chitosan-AuNPs | 5-fluorouracil (5FU) and doxorubicin (DOX) | The optimum DEE(%) for 5FU was 74.59% and at the weight ratio of 0.125:1 (Dox: CS-Au-5FU NPs, the DEE of DOX was 83.67%. Nearly 90% of 5FU and 55% of DOX were released at pH 5.4 from Apt-Dox-CS-Au-5FU NPs after 120h. Higher cellular uptake in aptamer-functionalized NPs, in addition to enhanced proliferation inhibition induced by Apt-functionalized DOX-CS-Au-5FU NPs via less necrosis (7.03%) and a higher percentage of apoptotic cells (23.11%) compared to free drug and other groups. | [144] |
| chitosan/dextran sulfate/chitosan (CS/DEX/CS) NPs | 5-fluorouracil (5FU) and doxorubicin (DOX) | efficient internalization and uptake of PTX and 5FU in HepG2 cells, enhanced inhibition of cancer cells. the encapsulation efficiencies of PTX and 5FU in CS/DEX/CS NPs were 66.3% and 75.2%. Both medicines exhibited pH-sensitive release where at pH 5.67, 6.58, and 7.4, 5Fu release rates were 99.41%, 96.20%, and 87.00%, respectively, whereas PTX release was 51.90%, 40.00%, and 32.09%. the greatest cell inhibitory activity for CS-PTX/DEX/CS-5Fu and higher apoptosis rate of 39.55% for the dual drug-loaded nanoparticles in comparison with single-drug groups against HepG2 cells, indicating the synergic effect of drugs. | [145] |
| Exosomes | 5-fluorouracil (5FU) and miR-21i inhibitor | High cell uptake owing to the exosome fused with HER2 proteins, efficient synergic effect in cell killing compared to single drug. Exosomes’ loading capacity (LC) for 5-FU and miR-21i was around 3.1% and 0.5%, respectively. THLG-EXO penetrated Her2-mcherry-SGC-7901 cells more efficiently than SGC-7901 WT, and a significant reduction in tumor volume was seen in the group treated with the THLG-EXO/5FU/miR-21i compared to the THLG-EXO and THLG-EXO/5FU groups. | [148] |
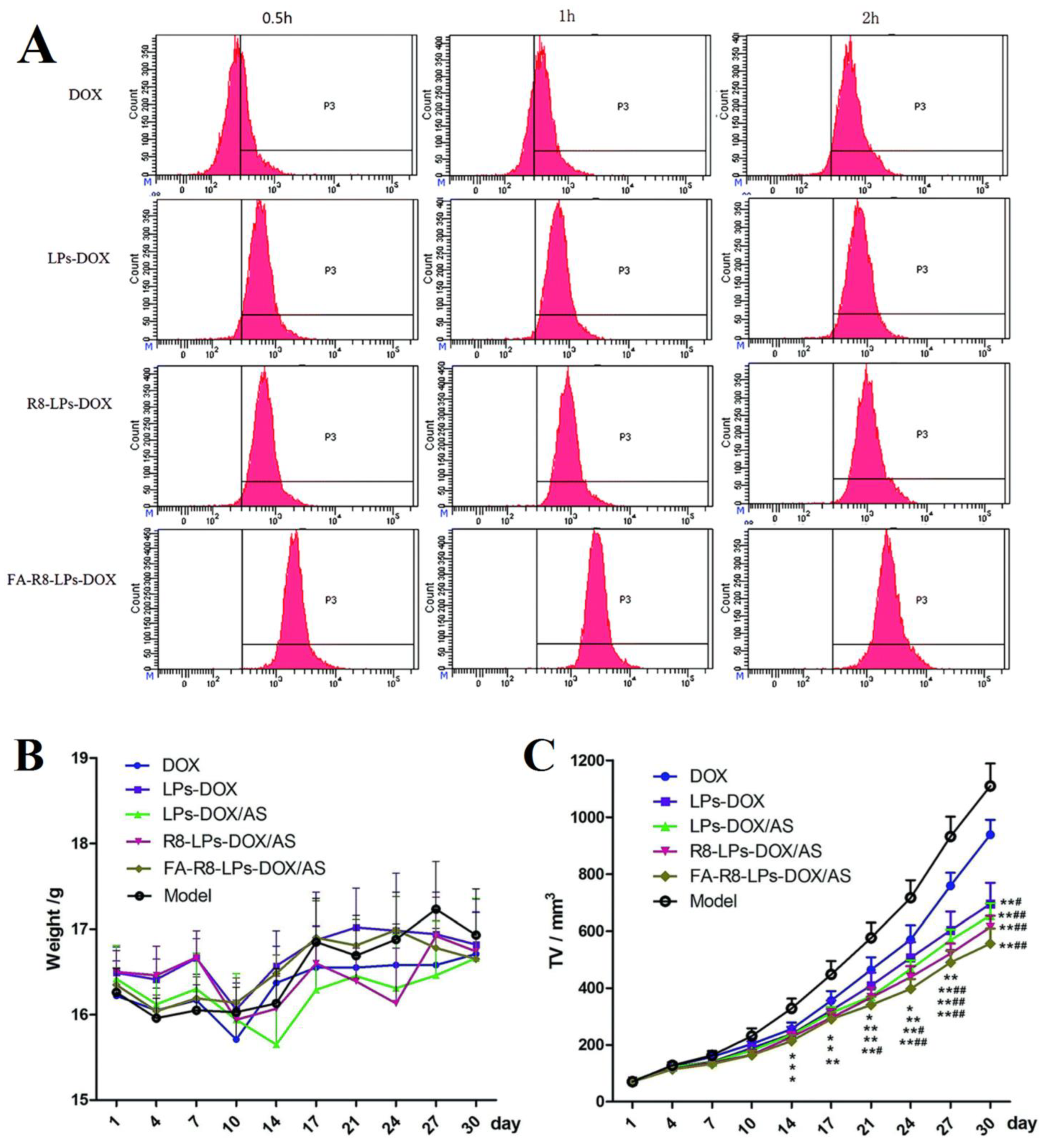
| FA-R8-functionalized liposome | astragaloside IV (ASIV) and doxorubicin (DOX) | FA-R8-LPs overcame DOX resistance, exhibited enhanced cytotoxicity. In total, 98.57% and 98.49% of EE% were obtained before freeze-drying. improved internalization and targetability of the final FA-R8-LPs-DOX/AS formulation indicating the optimal function of FA and R8). Tumor volume also was smallest after two weeks of treatment with FA-R8-LPs-DOX/AS. | [150] |
| Nanocarrier | Cargo | Outcomes and Details | Ref. |
|---|---|---|---|
| Janus Au-MSN NPs with preferential functionalized SH-β-CD on the surface of Au area | paclitaxel (PTX) and doxorubicin (DOX) | The loading efficiency of DOX and PTX on the PTX-AuMSN-DOX JNPs was approximately 58.05% and 95.23%, respectively. DOX was released at 22.31% at pH 7.4 and 73.48% at pH 5.0, while PTX showed cumulative releases of 67.55 and 62.75% in conditions with and without NIR laser, respectively. the dual-drug-loaded system presents high inhibition of tumor, and cytotoxicity with the CI value of 0.81 ± 0.01 indicating the synergistic effect of Janus nanoparticles. | [153] |
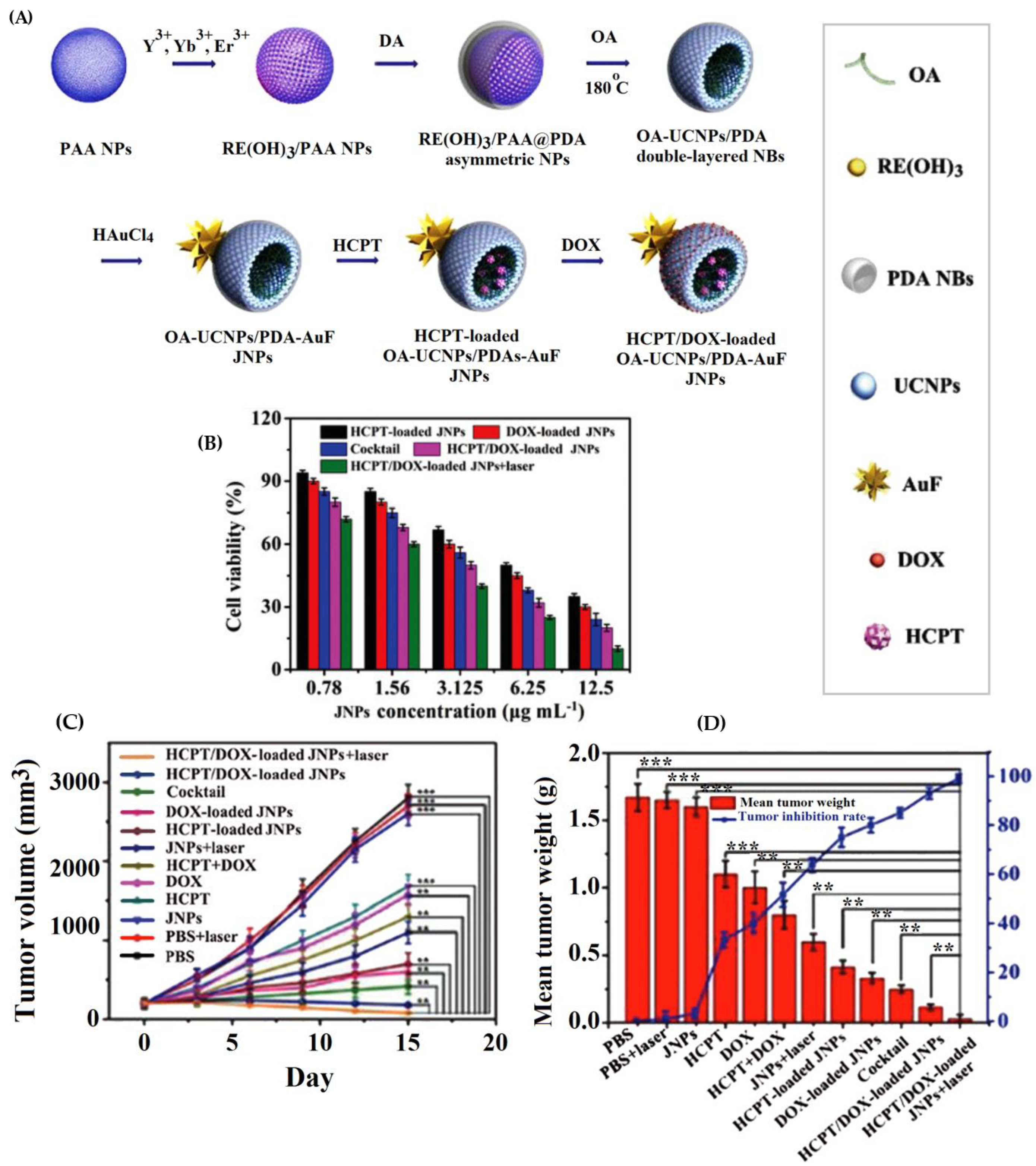
| oleic acid-NaYF4:Yb,Er/polydopamine Au nanoflower Janus NPs | hydroxycamptothecin (HCPT) and doxorubicin (DOX) | The loading efficiency (LE%) of the OA-UCNPs/PDA-AuF JNPs was 88% for DOX and 45% for HCPT. Drug release rates from drug-loaded OA-UCNPs/PDA-AuF JNPs within 104 h were 24% for HCPT and 26.5% for DOX at pH 7.4 vs. 87.1% for DOX and 60.5% for HCPT at pH 5.3. The HCPT/DOX-loaded OA-UCNPs/PDA-AuF JNPs (HCPT: DOX (1:1)) showed the lowest CI values (0.3805), best synergistic effect, and 52.0% apoptotic HepG2 cells using HCPT/DOX-loaded OA-UCNPs/PDA-AuF JNPs, higher than single-drug samples. | [154] |
| PLGA Janus NPs | paclitaxel (PTX) and doxorubicin hydrocholoride (DOX. HCl) | one-step process of Janus synthesis using a fluidic nanoprecipitation system (FNPS). PTX and DOX in Janus particles each showed an encapsulation efficiency of 80% and 15%. The cumulative drug release was nearly 90% for PTX and more than 70% for DOX from Janus NPs after 120h. | [157] |
| PLGA/PCL Janus particles | curcumin (CUR) and quercetin (QCT) and acetaminophen (APAP) | The encapsulation efficiency (EE%) in Janus NPs was 93.11% for CUR and 92.03% for QCT, while it was 93.38% for CUR and 86.9% for QCT in PCL. The EE% of the APAP and NPX was reported as 54.9% and 93.98% in O/W with ethyl acetate, 21.04% and 91.88% in O/W with DCM + methanol, and 68.29% and 85.49% in W/O/W. Double emulsion method resulted in high encapsulation efficiency, compartmentalization, drug release control. | [158] |
| Janus and mixed structures of GO-based PCL/terpolymer NPs | quercetin (QCT) and 5-fluorouracil (5FU) | Encapsulation efficiency (EE%) of QCT and 5FU was 41.91% and 53.25% in Janus NPs, respectively, vs. 38.03% for QCT and 71.77% for 5FU in mixed NPs. More than 80% and nearly 80% of 5FU, and nearly 40% and 50% of QCT were released from m-(PCL-NGO-Terpolymer) and J-(PCL-NGO-Terpolymer) at 40 °C, respectively. The dual-loaded NPs were less toxic than the free mix of drugs when maintained at 37 °C but much more toxic when heated to 40 °C. The inhibitory effect of dual-drug-loaded nanoparticles on normal cells was dependent on nanoparticle morphology, with mixed nanoparticles showing the greatest degree of inhibition. | [159] |
| polystyrene/Fe3O4 @SiO2 Janus nanocomposites (SJNCs) | Fe3O4 and doxorubicin (DOX) | The loading efficiency of ≈2% (w/w) was calculated for DOX, with release rates of 25.1% (w/w) at pH 7.4, 47.1% (w/w) at pH 6.0, and 82.6% (w/w) at pH 5.0. The apparent cytotoxicity of DOX coupled to non-targeted Janus particles was 5-fold reduced when compared to free DOX, while FA-functionalized Janus particles (FA-SJNCs-DOX) were more toxic reducing the viability of breast cancer cells with an IC50 of 255.3 ± 55.1 μg/mL. Moreover, NPs were equipped with multimodal imaging and hyperthermia induced by Fe3O4. | [162] |
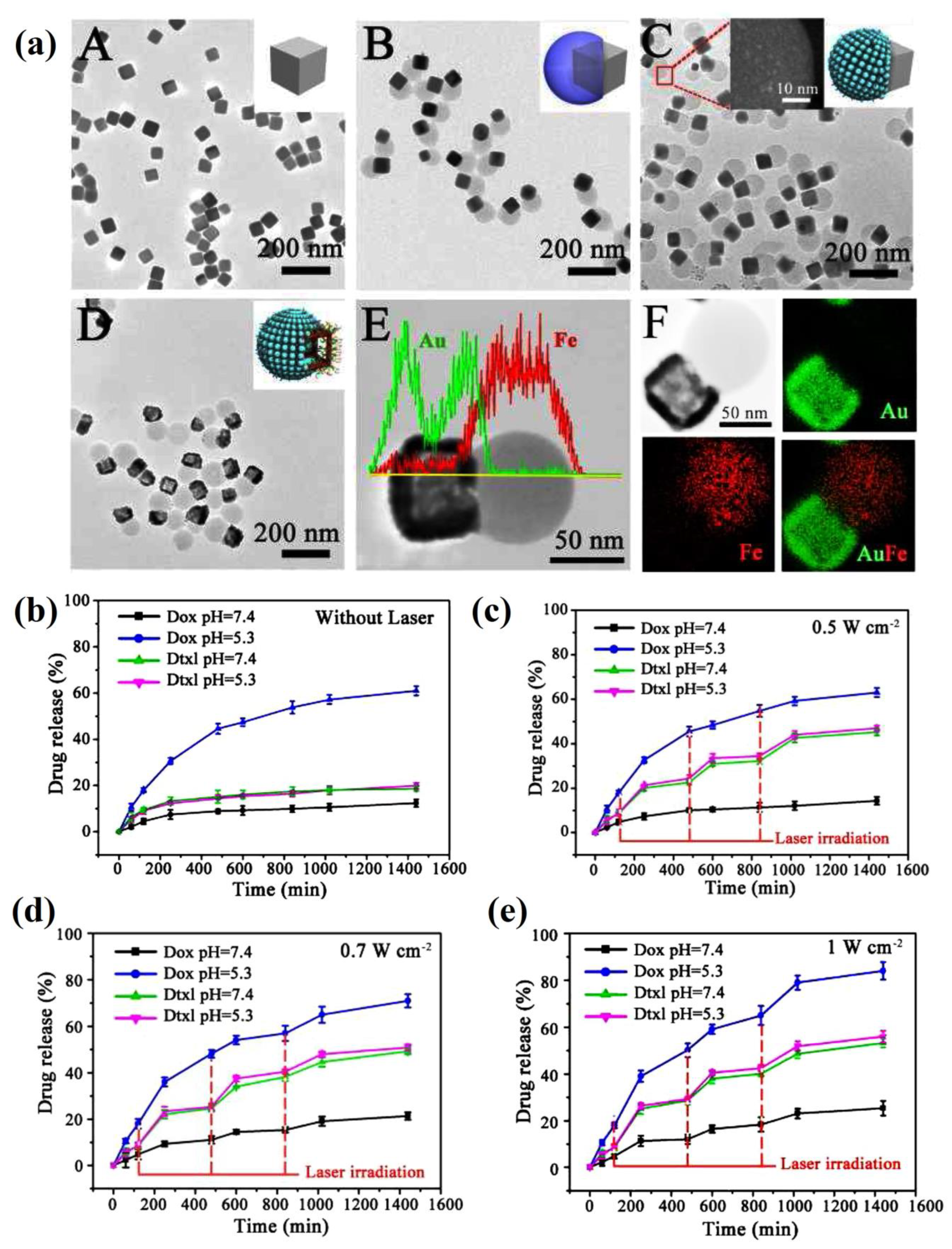
| PCL-AuNC/Fe(OH)3-PAA Janus nanoparticle | doxorubicin (DOX) and docetaxel (DOC) | The loading capacities of docetaxel (DOC) and DOX in PCL-AuNC/Fe(OH)3-PAA JNPs were about 5 wt. % and 20 wt. %, respectively. DOX release rate (60%) at an acidic pH vs. negligible release of DOX in neutral PBS, and it slightly enhanced using NIR laser, while DOC release was enhanced using NIR laser irradiation at 0.5 W cm−2 for 5 min up to nearly 60%. The dual-drug-loaded NPs displayed higher cytotoxicity compared with the cocktail (DOX loaded JNPs/DOC-loaded JNPs) and single-drug groups, and CI of 0.42, indicating the synergistic effect of the JNPs, specially in addition to the laser induction. | [165] |
| UCNP@SiO2@mSiO2&PMO cubic-spheres | paclitaxel (PTX) and doxorubicin (DOX) | Higher efficiency of cancer cell killing (more than 50%) compared to that of the single drugs (~25%) induced by NIR and UV-Vis irradiations. | [166] |
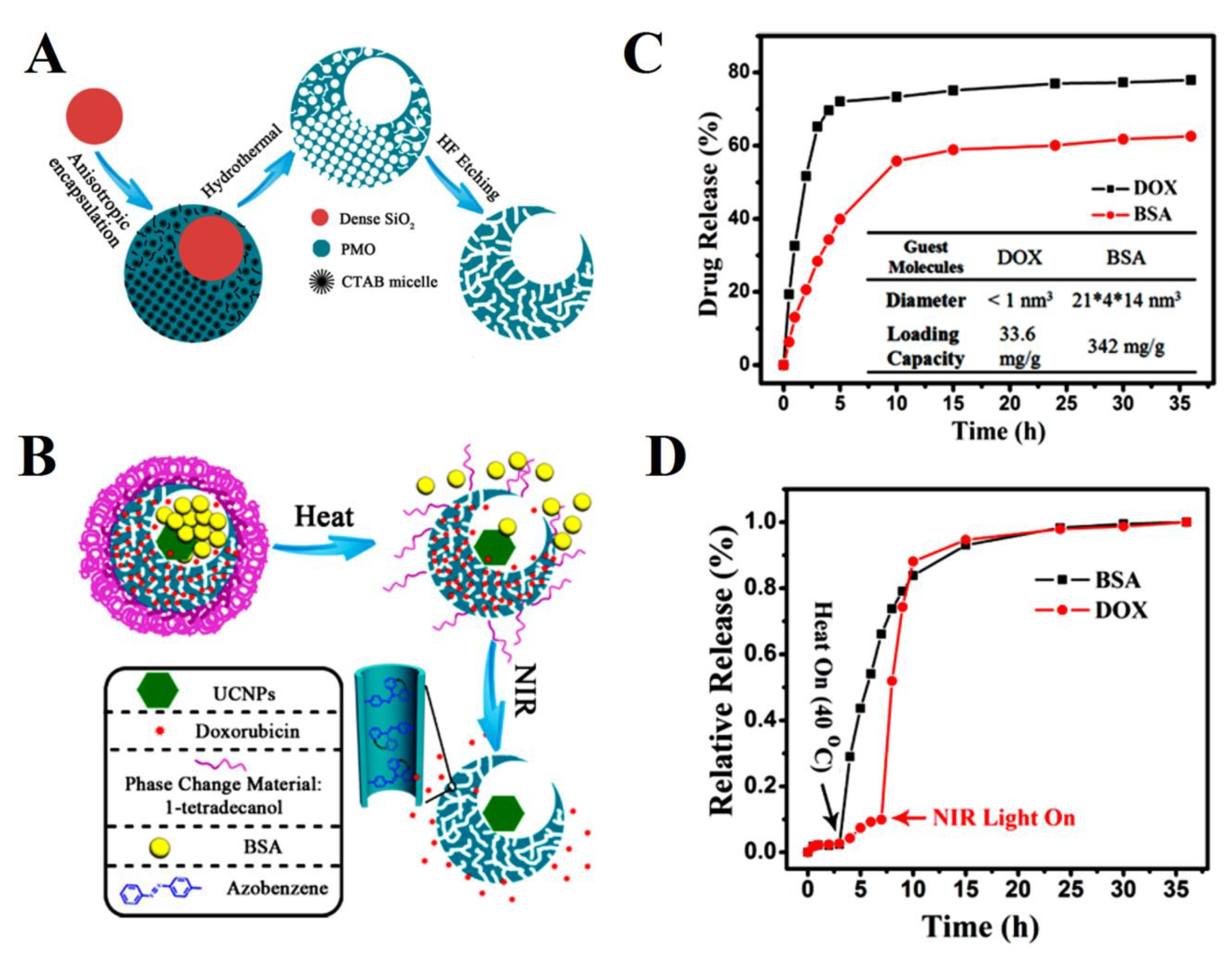
| mesoporous silica nanocages | bovine serum albumin (BSA) and doxorubicin (DOX) | Release control capabilities through the specific drug loading site, guest molecules size, and heat and NIR light. The loading capacities were 342 mg/g for BSA and 33.6 mg/g for DOX. Release rates of both DOX and BSA molecules from single-hole nanorattles under the stimuli of heat (up to 40 °C) and NIR light reached nearly 100% after 35 h. | [167] |
| asymmetric lollipop-like Fe3O4@SiO2&EPMO NPs | curcumin (CUR) and gentamicin (GS) | Higher loading capacity of 249 mg/g for GS than 25.8 mg/g for Cur IN Fe3O4@SiO2&EPMO NPs due to intermolecular hydrogen bonds formed between the OH and amino groups of GS and the silanols (Si−OH) of the hydrophilic mesoporous SiO2. The total GS released amount were 99.2 and 96.8%, while 63.0 and 50.3% for DOX from Fe3O4@SiO2-GS&EPMO and Fe3O4@SiO2-GS&EPMO-Cur, respectively. while only Cur and GS kill 48.2% and 19% of cells, respectively, Fe3O4@SiO2-GS&EPMO-Cur kill 89.6% of the cells, indicating the synergic effect of the dual-drug loaded NPs. | [168] |
| multicompartment hydrogel (MCH) | doxorubicin (DOX) and paclitaxel (PTX) | The drug loading amount was approximately 0.75% (w/w) for PTX and 4.2% (w/w) for DOX. PTX and DOX accumulative released amount from single-loaded MCH was 62.2% and 20.7%, respectively, while the data was around 64.9% for PTX and 23% for DOX, indicating that drugs were well segregated. MCH loaded with both drugs also had the lowest MCF-7 cell viability at only 10%, lowest tumor volume, and side effects compared to other groups, especially single-drug samples. | [179] |
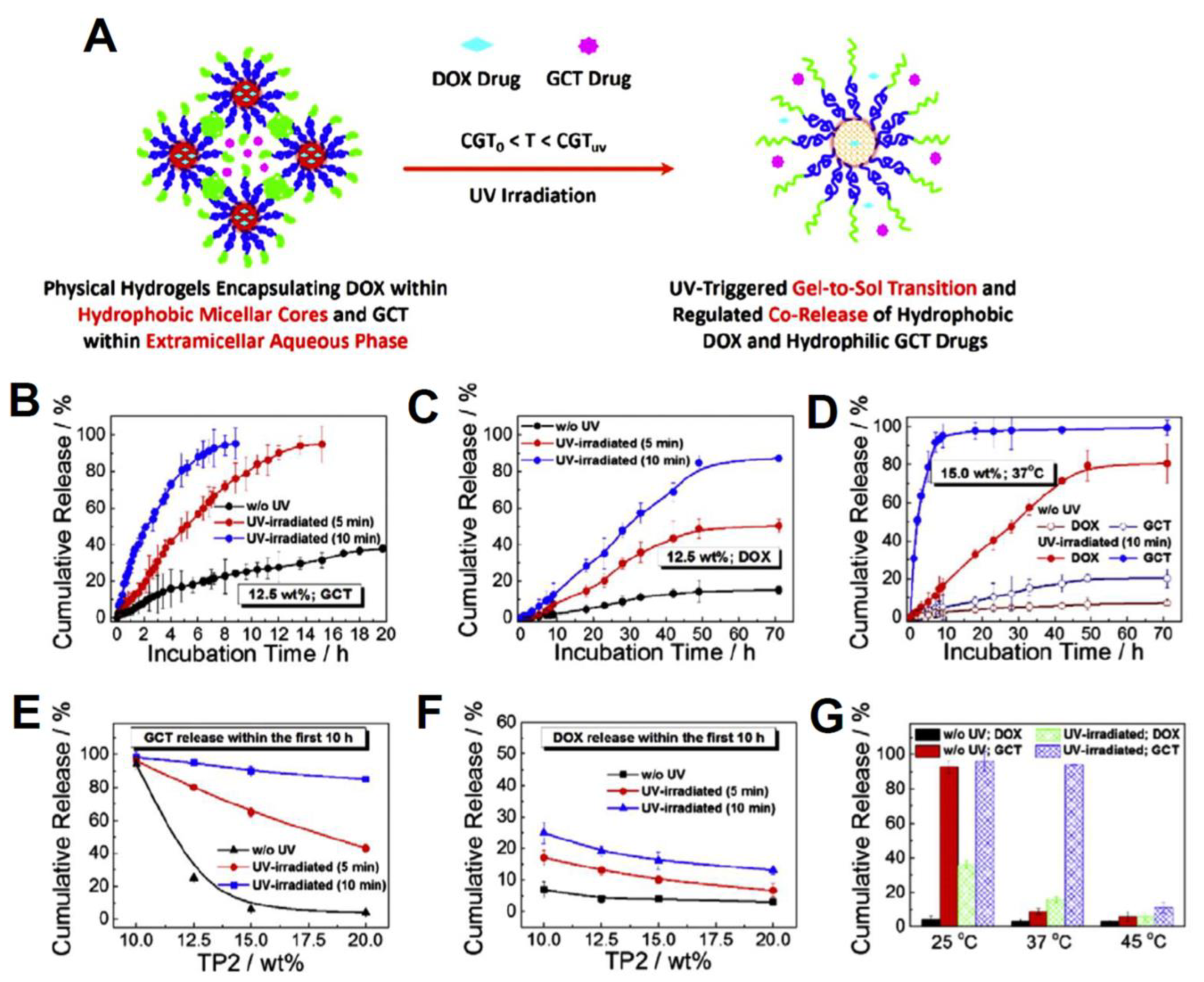
| PNIPAM-b-PNAM-b-PNBOC self-assembled hydrogels | gemcitabine (GCT) and doxorubicin (DOX) | Enhanced internalization, and release control and gel-to-sol transition vis UV light and NIR irradiation was observed. The loading efficiencies (LE) were calculated to be 80.5% for DOX and 100.0% for GCT. At 15 wt. % and 37 °C, less than 20% of GCT and 10% of DOX were released without UV, while more than 98% of GCT and nearly 80% of DOX were released with UV inductions, after 70 h. | [180] |
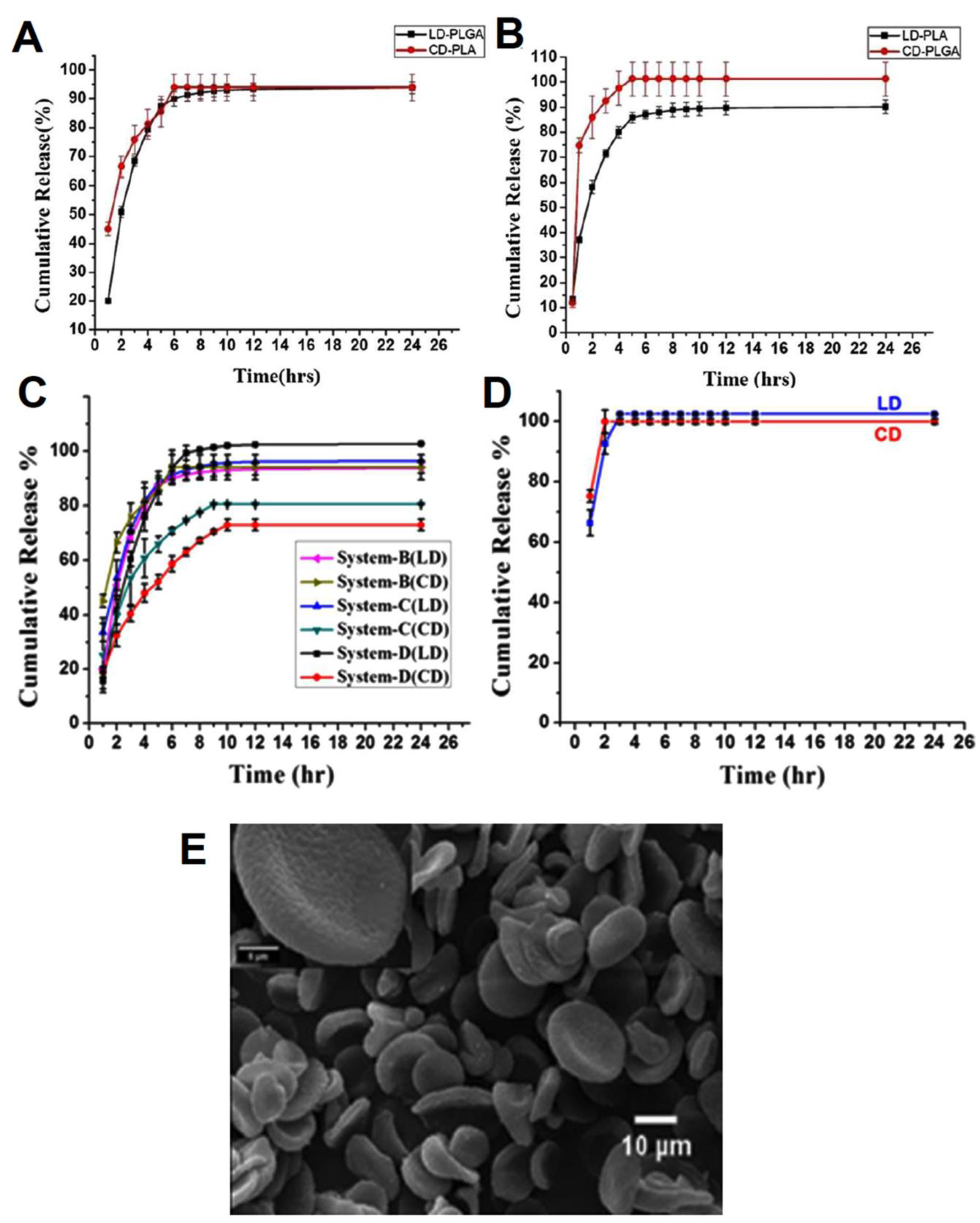
| PLGA/PLA nano discs | levodopa (LD) and carbidopa (CD) | Sustained release rate for both drugs in acidic conditions. The encapsulation efficiencies were 97% for LD and 78% for CD in system B (LD in PLGA and CD in PLA). The release profiles of system B illustrated nearly 90% of drugs were released after 24 h. | [187] |
| MSN Janus NPs | doxorubicin and berberine (BER) | Loading efficiencies were 58.81% for DOX and 54.17% for BER. the highest apoptotic efficiency of HA-MSN@DB at 48.10% among other systems, and a considerable growth prevention and tumor proliferation inhibition as well as enhanced cell internalization via CD44 receptor-mediated pathways, were observed. | [196] |
| H-ZIF-8/polydopamine Janus nanoparticles | doxorubicin hydrochloride (DOX.HCl) and 10-Hydroxycamptothecin (HCPT) | Loading capacity (LC) of H-ZIF-8/PDA-CD JNPs was 42.0 wt. % for DOX and 9.8 wt. % for HCPT. All release profiles at various pH conditions were enhanced after using a laser, where the release rate of DOX and HCPT reached nearly 80% and 60% at pH 5.3. cell viabilities and tumor volumes treated with dual-loaded JNPs plus laser were lower compared to other groups, highlighting the effective PTT and chemotherapy synergy. | [197] |
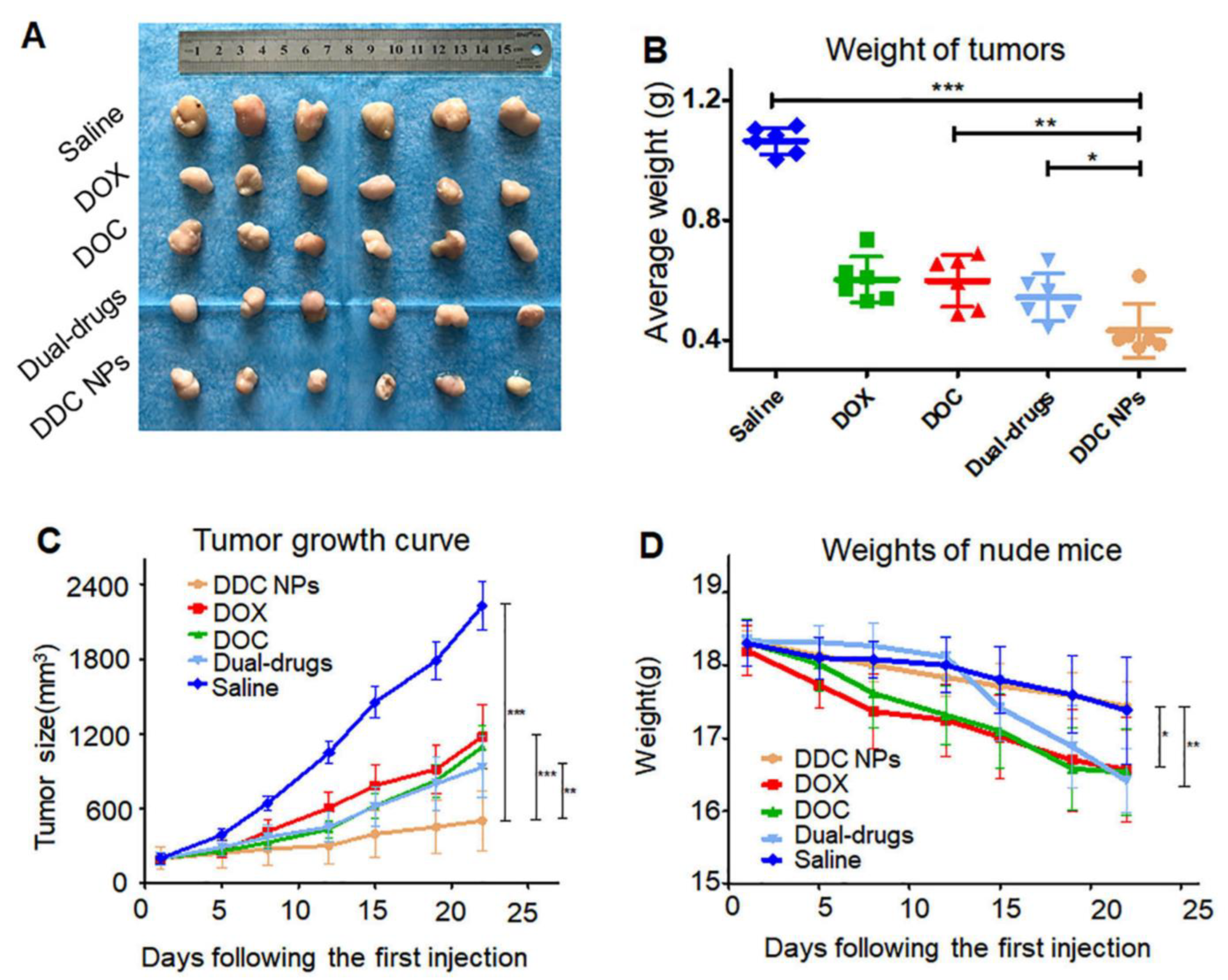
| self-assembled hyaluronic acid (HA) and cationic amphipathic starch (CSaSt) NPs | docetaxel (DOC) and doxorubicin (DOX) | The encapsulation efficiencies were 96.1 ± 2.3% for DOC and 91.4 ± 3.7% for DOX. approximately 70% of DOX was released within 12 h when HAase was added compared to 40% without HAase. DDC NPs attenuated the toxicity of DOC and DOX in vivo, with 20% vs. 80% mortality, and lower tumor growth in mice treated with the combined drug treatment. DDC NPs could also enhance the accumulation of drugs in tumors via HA-targeting agents. | [200] |
| chitosan/sodium alginate/PLGA Janus NPs | venom protein (αCT) and resveratrol (Res) | Improved bioavailability, cell entrance, and intestinal absorption. The entrapment efficiency (EE%) was 58.14% for αCT and 87.49% for Res. Compared with free αCT and Res, the JNPs have obvious sustained-release characteristics where the cumulative release of αCT in αCT-JNP and αCT/Res-JNP was 78.44% and 73.82%, respectively, compared to the cumulative release of 94.41% for Res in Res-JNP and 94.57% from αCT/ResJNP, respectively. | [203] |
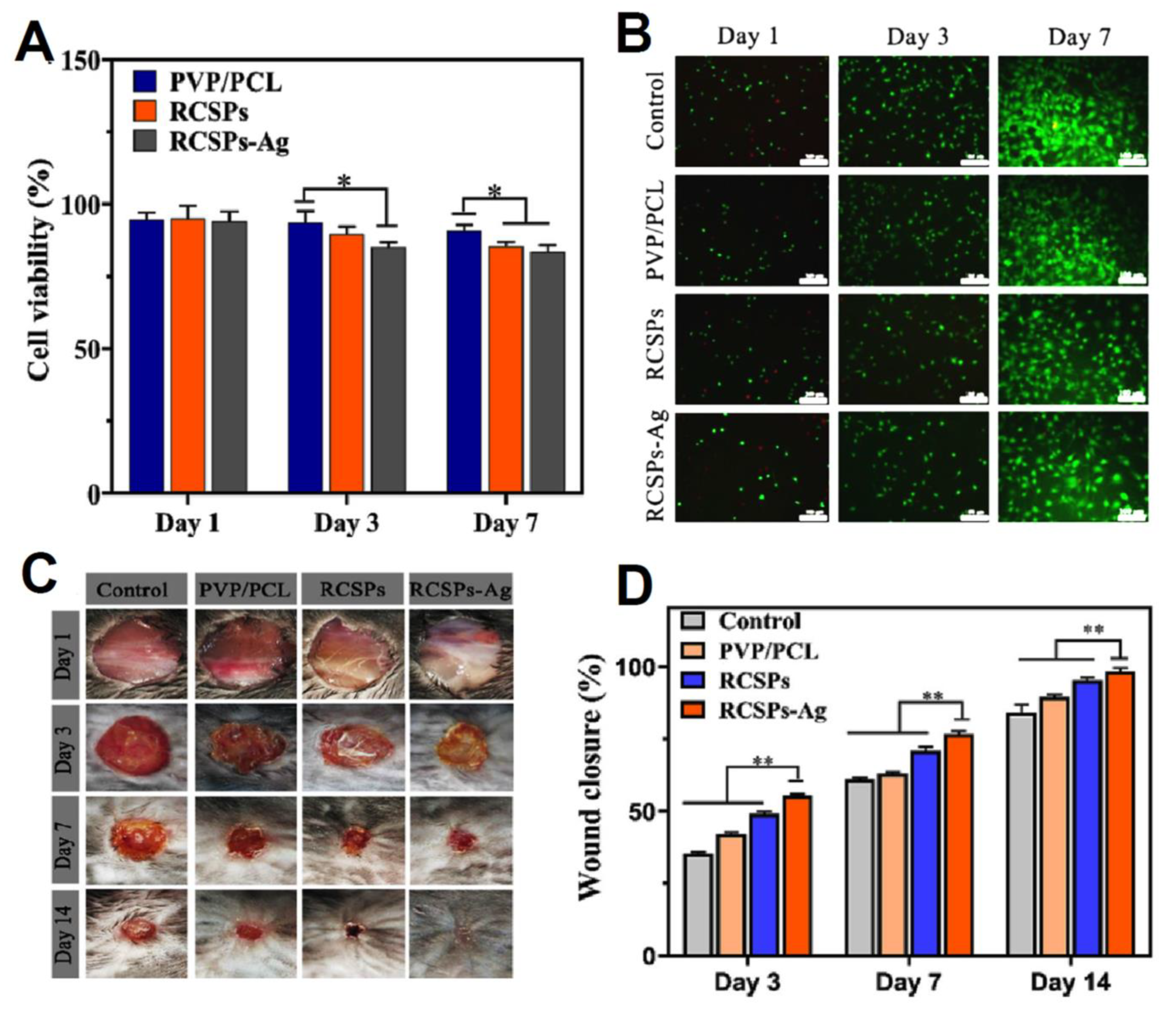
| PVP/PCL Janus nanofibers | Rana chensinensis skin peptides (RCSPs) and Ag NPs | Good wettability, mechanical, antibacterial, Biocompatibility, and wound healing properties. The cumulative release rate of RCSPs from RCSPs-Ag nanofibers reached 94.15% after 35 min. Lowest cell viability and highest wound closure for RCSPs-Ag nanofibers after 7 and 14 days, respectively. | [206] |
| assembled triblock terpolymers into multicompartment patchy NPs | doxorubicin (DOX) and Cyanine5 (Cy5) | FRET real-time monitoring capacity, simultaneous release and delivery to cancerous cells. The loading efficiencies of DOX and Cy5 were 49.27% and 18.76%, while the co-loading method decreased DOX loading efficiency and capacity by ~15% with negligible changes in the data of Cy5. | [207] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kargari Aghmiouni, D.; Khoee, S. Dual-Drug Delivery by Anisotropic and Uniform Hybrid Nanostructures: A Comparative Study of the Function and Substrate–Drug Interaction Properties. Pharmaceutics 2023, 15, 1214. https://doi.org/10.3390/pharmaceutics15041214
Kargari Aghmiouni D, Khoee S. Dual-Drug Delivery by Anisotropic and Uniform Hybrid Nanostructures: A Comparative Study of the Function and Substrate–Drug Interaction Properties. Pharmaceutics. 2023; 15(4):1214. https://doi.org/10.3390/pharmaceutics15041214
Chicago/Turabian StyleKargari Aghmiouni, Delaram, and Sepideh Khoee. 2023. "Dual-Drug Delivery by Anisotropic and Uniform Hybrid Nanostructures: A Comparative Study of the Function and Substrate–Drug Interaction Properties" Pharmaceutics 15, no. 4: 1214. https://doi.org/10.3390/pharmaceutics15041214