
Cancer Resistance to Immunotherapy: Comprehensive Insights with Future Perspectives


Abstract
:1. Introduction
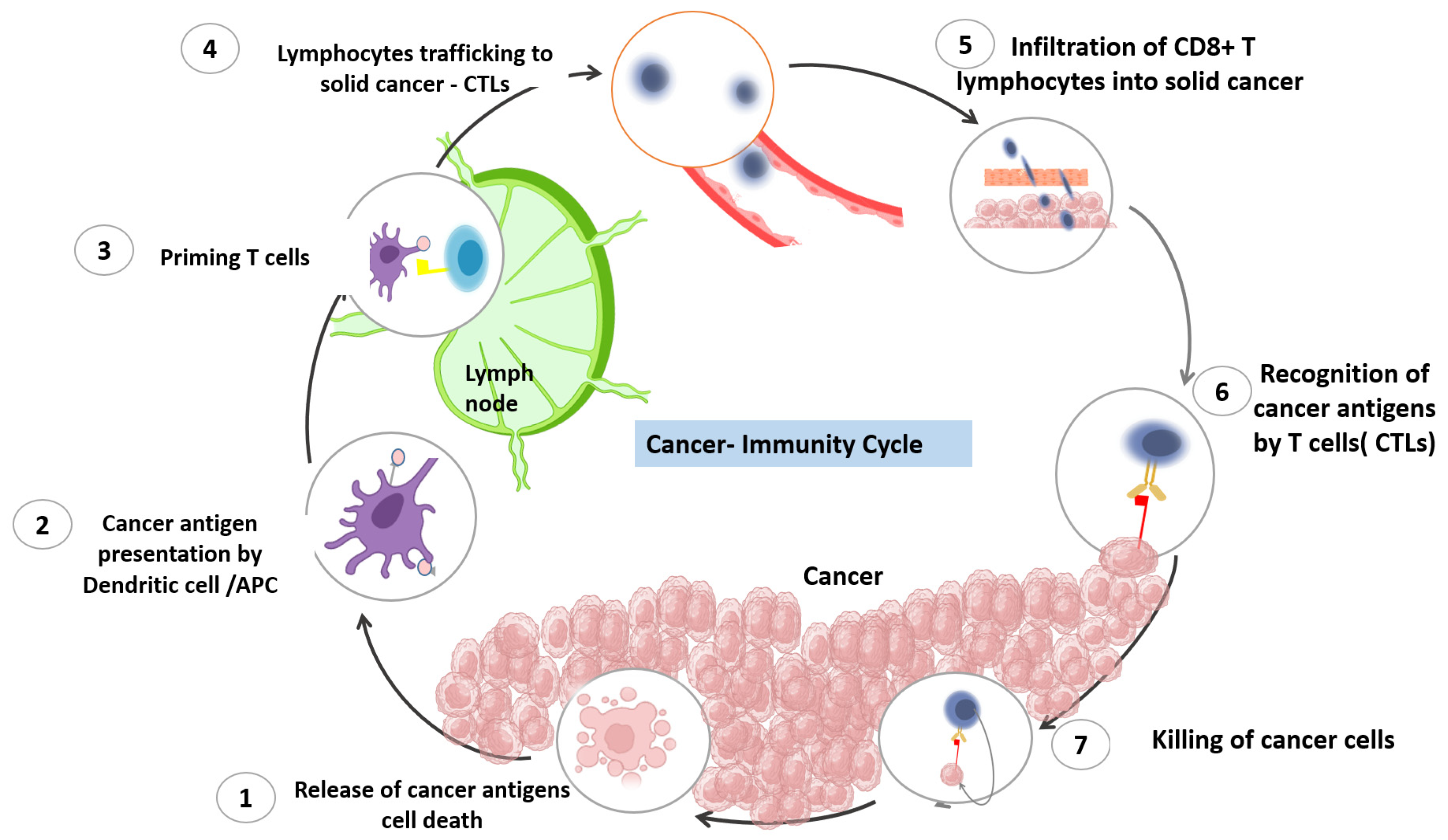
2. The Cancer Immune Cycle
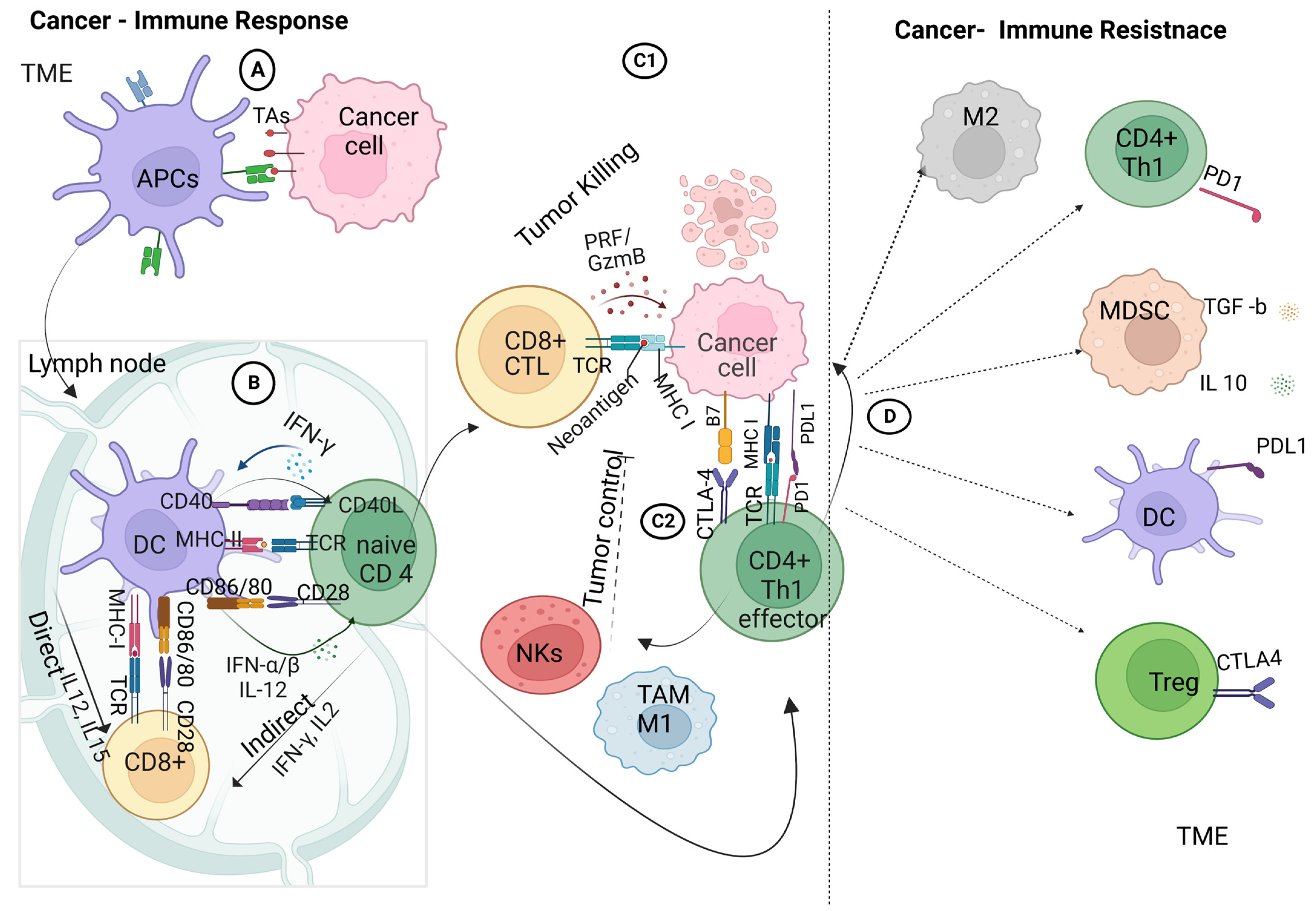
3. Innate and Adaptive Immune Responses in the Tumor Microenvironment (TME)
4. Resistance to Cancer Immunotherapy
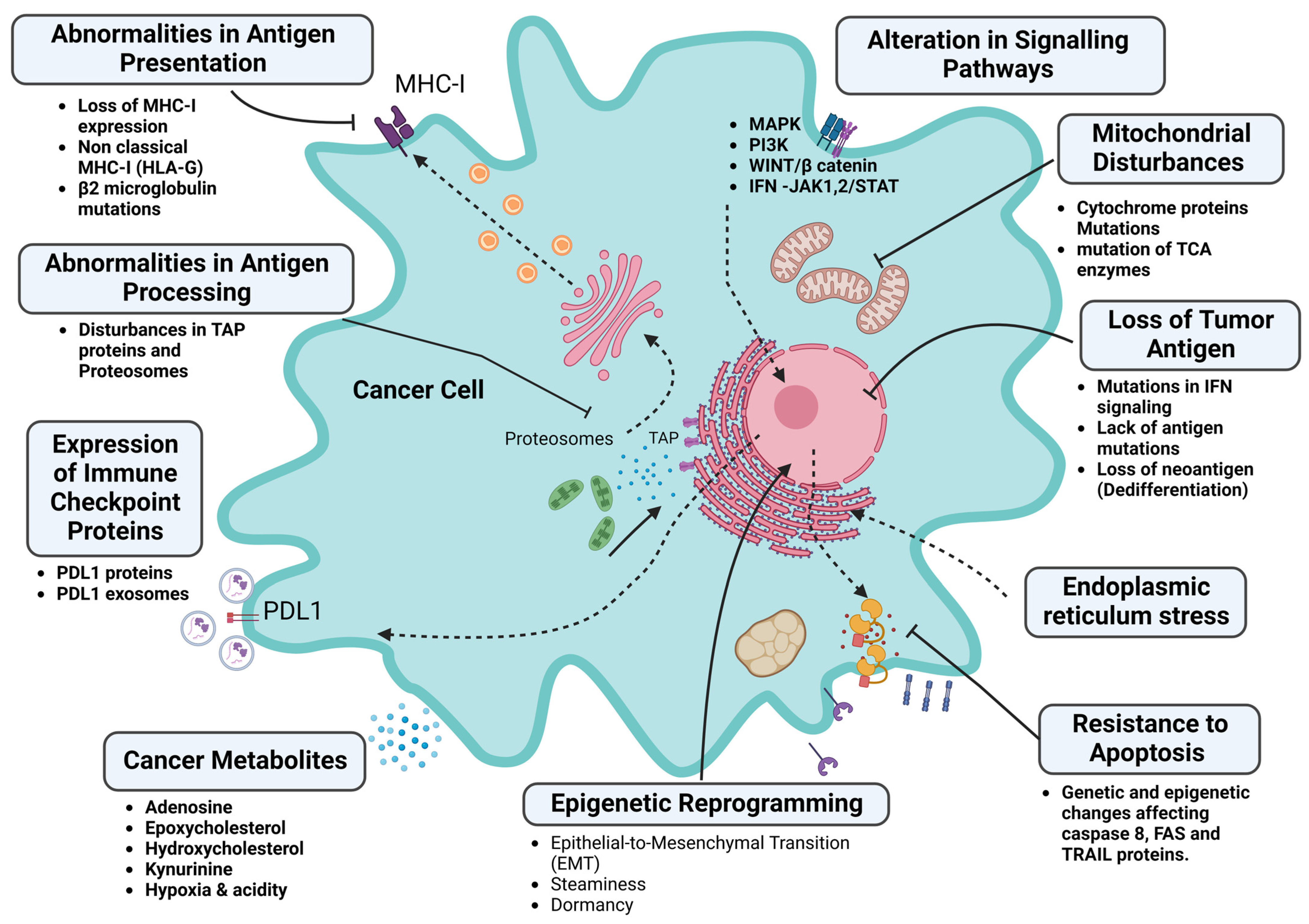
5. Tumor Factors (Intrinsic) Associated with Immunotherapy Resistance
5.1. Disorders of the Antigenic Determinants of Tumor Cells
5.2. Factors Modulating the Function of the Immune System
5.3. Factors Associated with Resistance to the Effector Immune Response
5.4. Epigenetic Regulation of Cancer Resistance to Immunotherapy
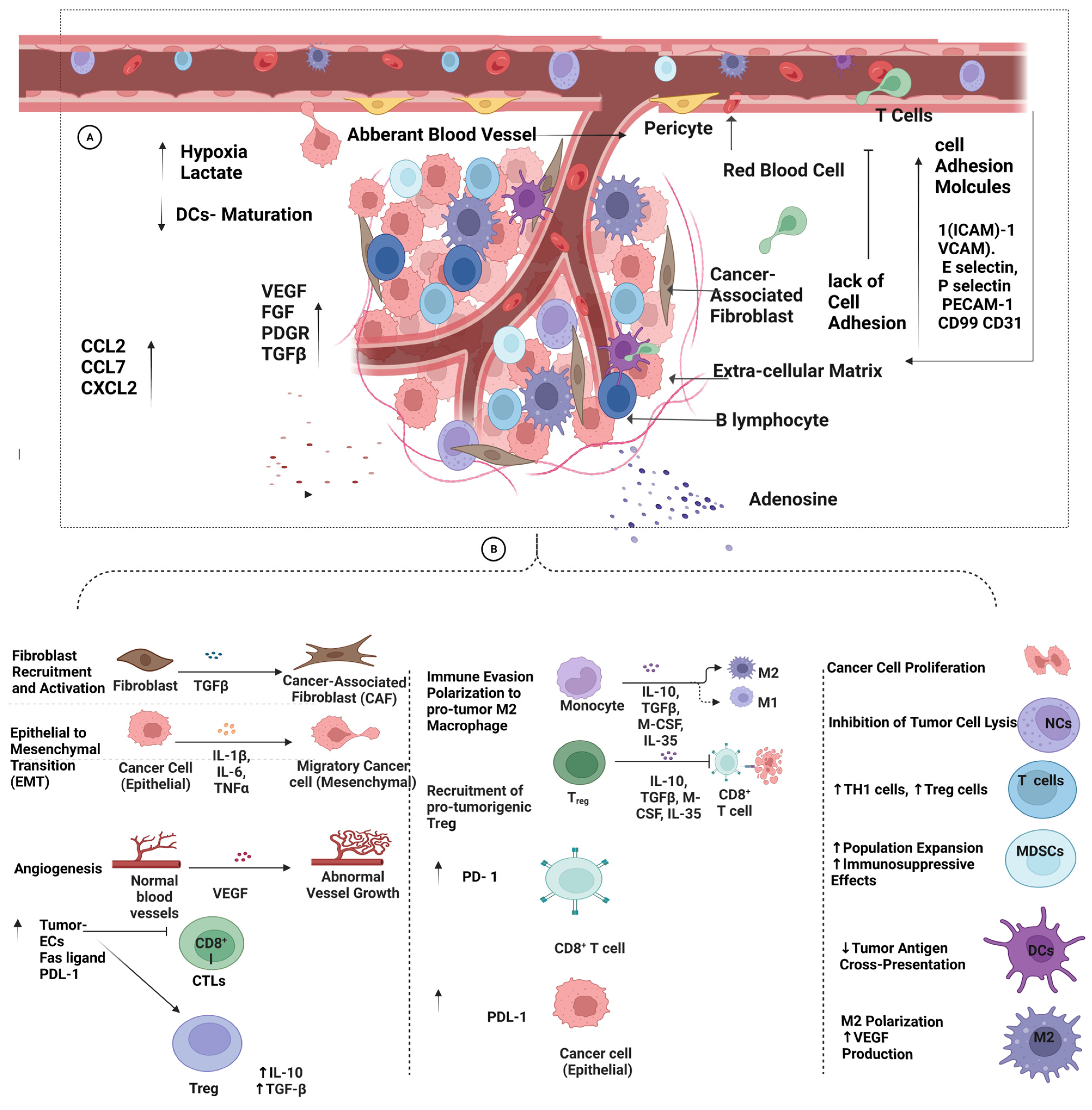
6. Extrinsic Factors Contributing to Tumor Resistance against Immunotherapy
6.1. Tumor-Infiltrating Lymphocytes
6.2. Chemokines
6.3. Vascular-Tumor Microenvironment
6.4. Immuno-Metabolic Products
6.5. Tumor-Associated Macrophages (TAMs)
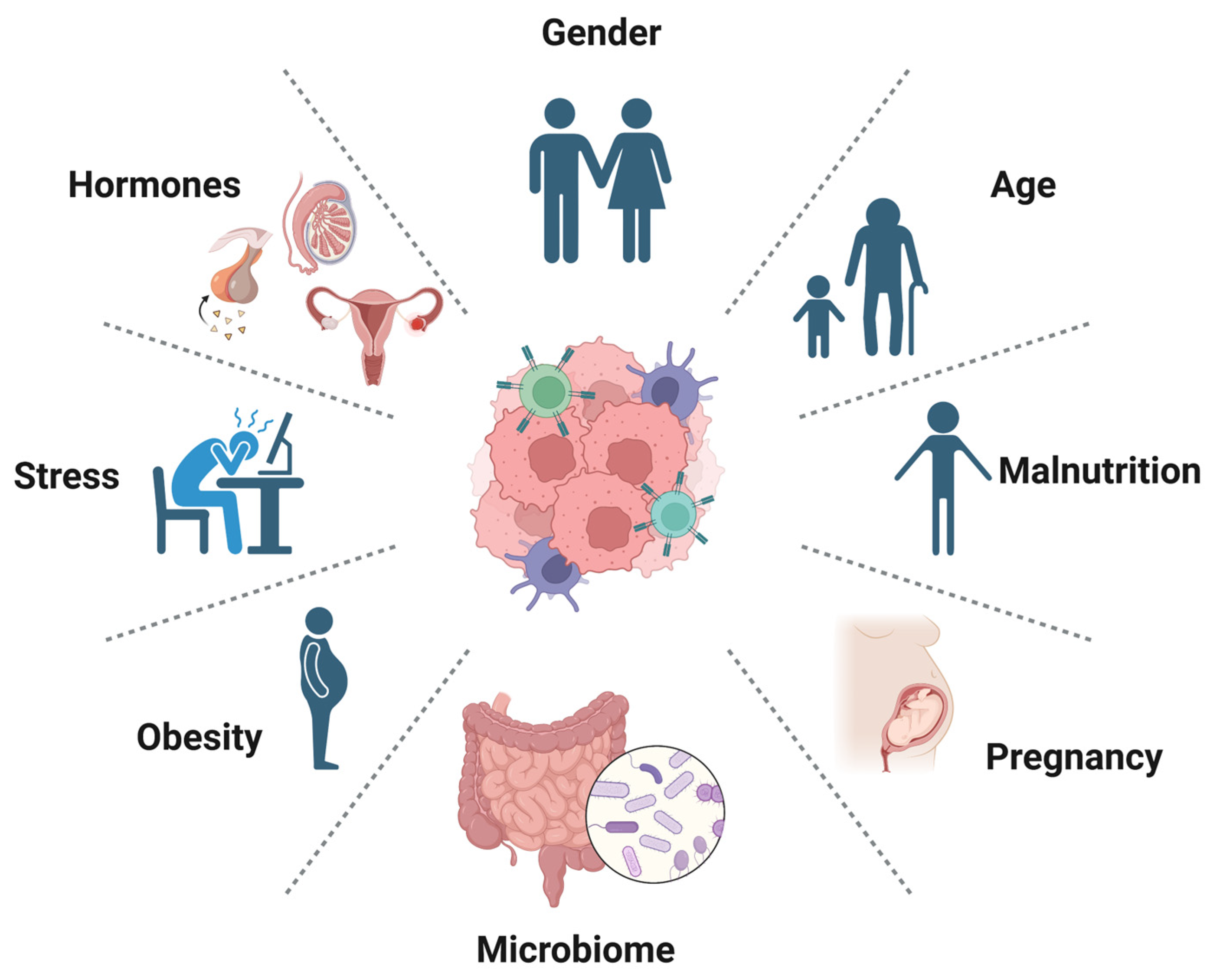
7. Host Factors Associated with Immunotherapy Resistance
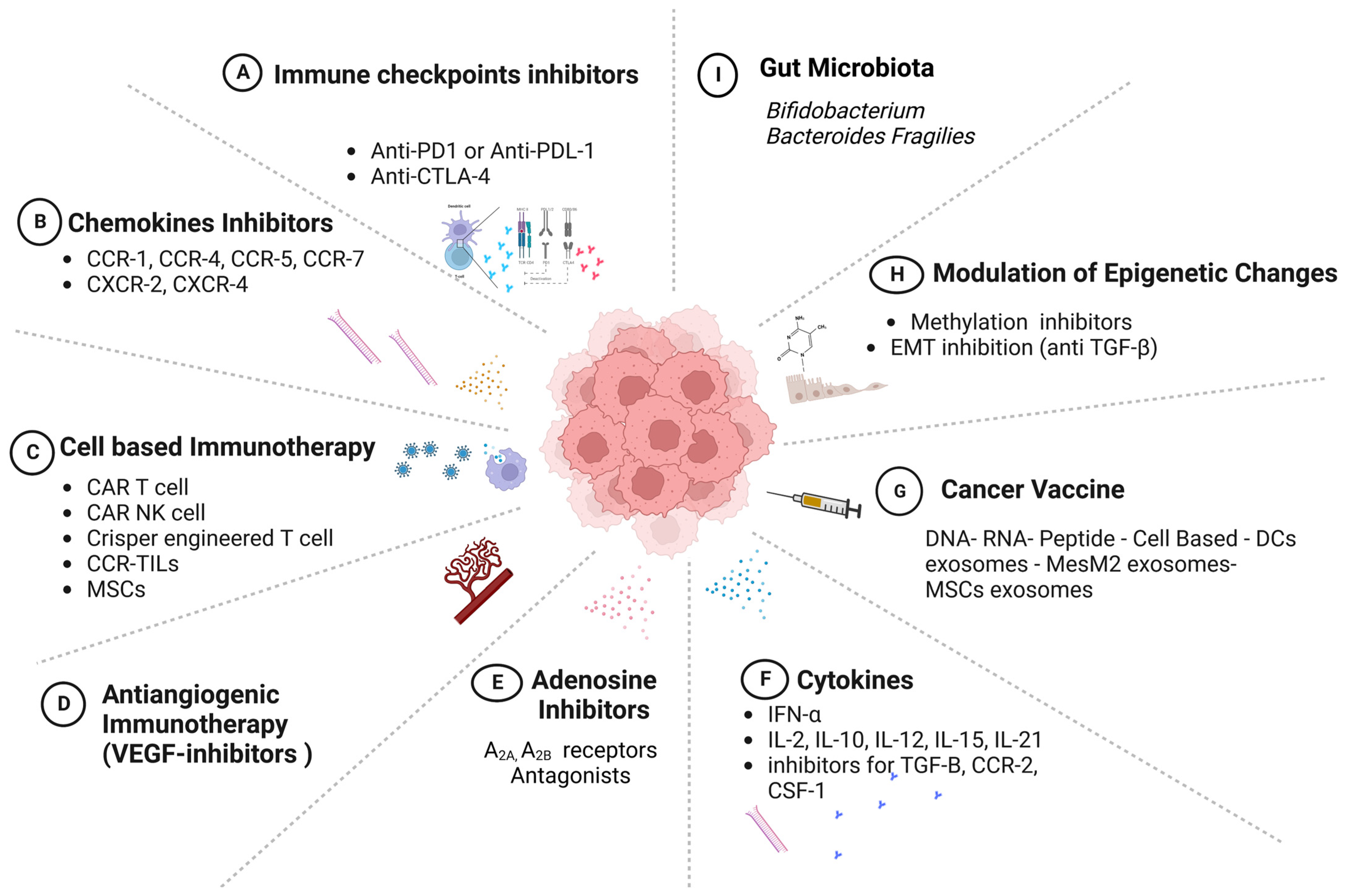
8. Models of Cancer Immunotherapy
8.1. Immune-Checkpoint Inhibitors
8.2. Chemokine Therapies
8.3. Adenosine-Targeting Therapies
8.4. Cell-Based Immunotherapy—Adoptive Cellular Transfer (ACT)
8.5. Vaccines
8.6. Exosomes
8.7. Epigenetic Modulators
8.8. Microbiota
9. Overcoming Cancer Resistance to Immunotherapy
10. Future Directions for Cancer Immunotherapy
11. Combination Therapies
12. Personalized Immunotherapy
13. Adoptive Cell Therapy
14. Overcoming Resistance
15. Novel Immunotherapy Agents
Author Contributions
Funding
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Cancer Epidemiol. 2021, 149, 778–789. [Google Scholar] [CrossRef]
- DeVita, V.T., Jr.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [Green Version]
- Wagner, E.; Elliot, K.; Enders, W.; Chobanuk, J.; Tsui, A. BOOK REVIEW: Chemotherapy and Biotherapy Guidelines, and Recommendations for Practice 4th Edition. Can. Oncol. Nurs. J. Rev. Can. De Soins Infirm. En Oncol. 2015, 25, 480. [Google Scholar]
- Almajali, B.; Al-Jamal, H.A.N.; Wan Taib, W.R.; Ismail, I.; Johan, M.F.; Doolaanea, A.A.; Ibrahim, W.N.; Tajudin, S.A. Thymoquinone Suppresses Cell Proliferation and Enhances Apoptosis of HL60 Leukemia Cells through Re-Expression of JAK/STAT Negative Regulators. Asian Pac. J. Cancer Prev. 2021, 22, 879–885. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Dobosz, P.; Dzieciątkowski, T. The Intriguing History of Cancer Immunotherapy. Front. Immunol. 2019, 10, 2965. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Liu, J.; Chen, Z.; Li, Y.; Zhao, W.; Wu, J.; Zhang, Z. PD-1/PD-L1 Checkpoint Inhibitors in Tumor Immunotherapy. Front. Pharmacol. 2021, 12, 731798. [Google Scholar] [CrossRef]
- Maggs, L.; Cattaneo, G.; Dal, A.E.; Moghaddam, A.S.; Ferrone, S. CAR T Cell-Based Immunotherapy for the Treatment of Glioblastoma. Front. Neurosci. 2021, 15, 662064. [Google Scholar] [CrossRef]
- van Elsas, M.J.; van Hall, T.; van der Burg, S.H. Future Challenges in Cancer Resistance to Immunotherapy. Cancers 2020, 12, 935. [Google Scholar] [CrossRef] [Green Version]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef] [Green Version]
- Vilgelm, A.E.; Richmond, A. Chemokines Modulate Immune Surveillance in Tumorigenesis, Metastasis, and Response to Immunotherapy. Front. Immunol. 2019, 10, 333. [Google Scholar] [CrossRef] [Green Version]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8(+) T cells in cancer and cancer immunotherapy. Br. J. Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef]
- Oh, D.Y.; Fong, L. Cytotoxic CD4(+) T cells in cancer: Expanding the immune effector toolbox. Immunity 2021, 54, 2701–2711. [Google Scholar] [CrossRef]
- Dallavalasa, S.; Beeraka, N.M.; Basavaraju, C.G.; Tulimilli, S.V.; Sadhu, S.P.; Rajesh, K.; Aliev, G.; Madhunapantula, S.V. The Role of Tumor Associated Macrophages (TAMs) in Cancer Progression, Chemoresistance, Angiogenesis and Metastasis—Current Status. Curr. Med. Chem. 2021, 28, 8203–8236. [Google Scholar] [CrossRef]
- Demaria, O.; Cornen, S.; Daëron, M.; Morel, Y.; Medzhitov, R.; Vivier, E. Harnessing innate immunity in cancer therapy. Nature 2019, 574, 45–56. [Google Scholar] [CrossRef]
- Li, H.; Han, Y.; Guo, Q.; Zhang, M.; Cao, X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J. Immunol. 2009, 182, 240–249. [Google Scholar] [CrossRef] [Green Version]
- Bruger, A.M.; Dorhoi, A.; Esendagli, G.; Barczyk-Kahlert, K.; van der Bruggen, P.; Lipoldova, M.; Perecko, T.; Santibanez, J.; Saraiva, M.; Van Ginderachter, J.A.; et al. How to measure the immunosuppressive activity of MDSC: Assays, problems and potential solutions. Cancer Immunol. Immunother. 2019, 68, 631–644. [Google Scholar] [CrossRef]
- Bai, R.; Chen, N.; Li, L.; Du, N.; Bai, L.; Lv, Z.; Tian, H.; Cui, J. Mechanisms of Cancer Resistance to Immunotherapy. Front. Oncol. 2020, 10, 1290. [Google Scholar] [CrossRef]
- Staudt, R.E.; Carlson, R.D.; Snook, A.E. Targeting gastrointestinal cancers with chimeric antigen receptor (CAR)-T cell therapy. Cancer Biol. Ther. 2022, 23, 127–133. [Google Scholar] [CrossRef]
- Sprent, J. Antigen-presenting cells. Professionals and amateurs. Curr. Biol. 1995, 5, 1095–1097. [Google Scholar] [CrossRef] [Green Version]
- Harryvan, T.J.; de Lange, S.; Hawinkels, L.; Verdegaal, E.M.E. The ABCs of Antigen Presentation by Stromal Non-Professional Antigen-Presenting Cells. Int. J. Mol. Sci. 2021, 23, 137. [Google Scholar] [CrossRef]
- Sholl, L.M.; Hirsch, F.R.; Hwang, D.; Botling, J.; Lopez-Rios, F.; Bubendorf, L.; Mino-Kenudson, M.; Roden, A.C.; Beasley, M.B.; Borczuk, A.; et al. The Promises and Challenges of Tumor Mutation Burden as an Immunotherapy Biomarker: A Perspective from the International Association for the Study of Lung Cancer Pathology Committee. J. Thorac. Oncol. 2020, 15, 1409–1424. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, Y.; Mittra, A.; Naqash, A.R.; Takebe, N. A review of mechanisms of resistance to immune checkpoint inhibitors and potential strategies for therapy. Cancer Drug Resist. 2020, 3, 252–275. [Google Scholar] [CrossRef]
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov. 2017, 7, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, A.; Kumar, Y. Cellular and molecular mechanisms in cancer immune escape: A comprehensive review. Expert Rev. Clin. Immunol. 2014, 10, 41–62. [Google Scholar] [CrossRef]
- Sokol, L.; Koelzer, V.H.; Rau, T.T.; Karamitopoulou, E.; Zlobec, I.; Lugli, A. Loss of tapasin correlates with diminished CD8(+) T-cell immunity and prognosis in colorectal cancer. J. Transl. Med. 2015, 13, 279. [Google Scholar] [CrossRef] [Green Version]
- Horvath, L.; Thienpont, B.; Zhao, L.; Wolf, D.; Pircher, A. Overcoming immunotherapy resistance in non-small cell lung cancer (NSCLC)—Novel approaches and future outlook. Mol. Cancer 2020, 19, 141. [Google Scholar] [CrossRef]
- da Silva, I.L.; Montero-Montero, L.; Ferreira, E.; Quintanilla, M. New Insights into the Role of Qa-2 and HLA-G Non-classical MHC-I Complexes in Malignancy. Front. Immunol. 2018, 9, 2894. [Google Scholar] [CrossRef]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404.e9. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]
- Jerby-Arnon, L.; Shah, P.; Cuoco, M.S.; Rodman, C.; Su, M.J.; Melms, J.C.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.R.; et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 2018, 175, 984–997.e24. [Google Scholar] [CrossRef] [Green Version]
- Pereira, C.; Gimenez-Xavier, P.; Pros, E.; Pajares, M.J.; Moro, M.; Gomez, A.; Navarro, A.; Condom, E.; Moran, S.; Gomez-Lopez, G.; et al. Genomic Profiling of Patient-Derived Xenografts for Lung Cancer Identifies B2M Inactivation Impairing Immunorecognition. Clin. Cancer Res. 2017, 23, 3203–3213. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [Green Version]
- Hu-Lieskovan, S.; Mok, S.; Homet Moreno, B.; Tsoi, J.; Robert, L.; Goedert, L.; Pinheiro, E.M.; Koya, R.C.; Graeber, T.G.; Comin-Anduix, B.; et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci. Transl. Med. 2015, 7, 279ra241. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Mayes, P.A.; Eastman, S.; Shi, H.; Yadavilli, S.; Zhang, T.; Yang, J.; Seestaller-Wehr, L.; Zhang, S.-Y.; Hopson, C.; et al. The BRAF and MEK Inhibitors Dabrafenib and Trametinib: Effects on Immune Function and in Combination with Immunomodulatory Antibodies Targeting PD-1, PD-L1, and CTLA-4. Clin. Cancer Res. 2015, 21, 1639–1651. [Google Scholar]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Gwangwa, M.V.; Joubert, A.M.; Visagie, M.H. Crosstalk between the Warburg effect, redox regulation and autophagy induction in tumourigenesis. Cell. Mol. Biol. Lett. 2018, 23, 20. [Google Scholar] [CrossRef] [Green Version]
- Abou Khouzam, R.; Zaarour, R.F.; Brodaczewska, K.; Azakir, B.; Venkatesh, G.H.; Thiery, J.; Terry, S.; Chouaib, S. The Effect of Hypoxia and Hypoxia-Associated Pathways in the Regulation of Antitumor Response: Friends or Foes? Front. Immunol. 2022, 13, 828875. [Google Scholar] [CrossRef]
- Baek, A.E.; Yu, Y.A.; He, S.; Wardell, S.E.; Chang, C.Y.; Kwon, S.; Pillai, R.V.; McDowell, H.B.; Thompson, J.W.; Dubois, L.G.; et al. The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat. Commun. 2017, 8, 864. [Google Scholar] [CrossRef] [Green Version]
- Leone, R.D.; Emens, L.A. Targeting adenosine for cancer immunotherapy. J. Immunother. Cancer 2018, 6, 57. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Cho, H.; Hong, S.O.; Oh, S.J.; Lee, H.J.; Cho, E.; Woo, S.R.; Song, J.S.; Chung, J.Y.; Son, S.W.; et al. LC3B upregulation by NANOG promotes immune resistance and stem-like property through hyperactivation of EGFR signaling in immune-refractory tumor cells. Autophagy 2021, 17, 1978–1997. [Google Scholar] [CrossRef]
- Fucikova, J.; Becht, E.; Iribarren, K.; Goc, J.; Remark, R.; Damotte, D.; Alifano, M.; Devi, P.; Biton, J.; Germain, C.; et al. Calreticulin Expression in Human Non-Small Cell Lung Cancers Correlates with Increased Accumulation of Antitumor Immune Cells and Favorable Prognosis. Cancer Res. 2016, 76, 1746–1756. [Google Scholar] [CrossRef] [Green Version]
- van der Merwe, M.; van Niekerk, G.; Fourie, C.; du Plessis, M.; Engelbrecht, A.M. The impact of mitochondria on cancer treatment resistance. Cell. Oncol. 2021, 44, 983–995. [Google Scholar] [CrossRef]
- Neophytou, C.M.; Trougakos, I.P.; Erin, N.; Papageorgis, P. Apoptosis Deregulation and the Development of Cancer Multi-Drug Resistance. Cancers 2021, 13, 4363. [Google Scholar] [CrossRef]
- Smith, H.A.; Cronk, R.J.; Lang, J.M.; McNeel, D.G. Expression and immunotherapeutic targeting of the SSX family of cancer-testis antigens in prostate cancer. Cancer Res. 2011, 71, 6785–6795. [Google Scholar] [CrossRef] [Green Version]
- Toor, A.A.; Payne, K.K.; Chung, H.M.; Sabo, R.T.; Hazlett, A.F.; Kmieciak, M.; Sanford, K.; Williams, D.C.; Clark, W.B.; Roberts, C.H.; et al. Epigenetic induction of adaptive immune response in multiple myeloma: Sequential azacitidine and lenalidomide generate cancer testis antigen-specific cellular immunity. Br. J. Haematol. 2012, 158, 700–711. [Google Scholar] [CrossRef]
- Li, H.; Chiappinelli, K.B.; Guzzetta, A.A.; Easwaran, H.; Yen, R.W.; Vatapalli, R.; Topper, M.J.; Luo, J.; Connolly, R.M.; Azad, N.S.; et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget 2014, 5, 587–598. [Google Scholar] [CrossRef]
- Hervouet, E.; Cheray, M.; Vallette, F.M.; Cartron, P.F. DNA methylation and apoptosis resistance in cancer cells. Cells 2013, 2, 545–573. [Google Scholar] [CrossRef]
- Lucas, D.M.; Davis, M.E.; Parthun, M.R.; Mone, A.P.; Kitada, S.; Cunningham, K.D.; Flax, E.L.; Wickham, J.; Reed, J.C.; Byrd, J.C.; et al. The histone deacetylase inhibitor MS-275 induces caspase-dependent apoptosis in B-cell chronic lymphocytic leukemia cells. Leukemia 2004, 18, 1207–1214. [Google Scholar] [CrossRef]
- Ohkura, N.; Hamaguchi, M.; Morikawa, H.; Sugimura, K.; Tanaka, A.; Ito, Y.; Osaki, M.; Tanaka, Y.; Yamashita, R.; Nakano, N.; et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity 2012, 37, 785–799. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Du, F.; Huang, W.; Ding, X.; Wang, Z.; Yan, F.; Wu, Z. Epigenetic control of Foxp3 in intratumoral T-cells regulates growth of hepatocellular carcinoma. Aging 2019, 11, 2343–2351. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Ye, X.; Weinberg, R.A. Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef] [Green Version]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Dongre, A.; Rashidian, M.; Reinhardt, F.; Bagnato, A.; Keckesova, Z.; Ploegh, H.L.; Weinberg, R.A. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res. 2017, 77, 3982–3989. [Google Scholar] [CrossRef] [Green Version]
- Lin, B.; Du, L.; Li, H.; Zhu, X.; Cui, L.; Li, X. Tumor-infiltrating lymphocytes: Warriors fight against tumors powerfully. Biomed. Pharmacother. 2020, 132, 110873. [Google Scholar] [CrossRef]
- Ibrahim, W.N.; Doolaanea, A.A.; Bin Abdull Rasad, M.S.B. Effect of shRNA Mediated Silencing of YB-1 Protein on the Expression of Matrix Collagenases in Malignant Melanoma Cell In Vitro. Cells 2018, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- Saleh, R.; Elkord, E. Treg-mediated acquired resistance to immune checkpoint inhibitors. Cancer Lett. 2019, 457, 168–179. [Google Scholar] [CrossRef]
- De Palma, M.; Lewis, C.E. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 2013, 23, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Kieffer, Y.; Hocine, H.R.; Gentric, G.; Pelon, F.; Bernard, C.; Bourachot, B.; Lameiras, S.; Albergante, L.; Bonneau, C.; Guyard, A.; et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020, 10, 1330–1351. [Google Scholar] [CrossRef]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limón, P. The polarization of immune cells in the tumour environment by TGFbeta. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Cong, M.; Li, J.; He, D.; Wu, Q.; Tian, P.; Wang, Y.; Yang, S.; Liang, C.; Liang, Y.; et al. Cathepsin C promotes breast cancer lung metastasis by modulating neutrophil infiltration and neutrophil extracellular trap formation. Cancer Cell 2021, 39, 423–437.e7. [Google Scholar] [CrossRef]
- OuYang, L.Y.; Wu, X.J.; Ye, S.B.; Zhang, R.X.; Li, Z.L.; Liao, W.; Pan, Z.Z.; Zheng, L.M.; Zhang, X.S.; Wang, Z.; et al. Tumor-induced myeloid-derived suppressor cells promote tumor progression through oxidative metabolism in human colorectal cancer. J. Transl. Med. 2015, 13, 47. [Google Scholar] [CrossRef] [Green Version]
- Byrne, S.N.; Knox, M.C.; Halliday, G.M. TGFbeta is responsible for skin tumour infiltration by macrophages enabling the tumours to escape immune destruction. Immunol. Cell Biol. 2008, 86, 92–97. [Google Scholar] [CrossRef]
- Harryvan, T.J.; Verdegaal, E.M.E.; Hardwick, J.C.H.; Hawinkels, L.; van der Burg, S.H. Targeting of the Cancer-Associated Fibroblast-T-Cell Axis in Solid Malignancies. J. Clin. Med. 2019, 8, 1989. [Google Scholar] [CrossRef] [Green Version]
- Chakravarthy, A.; Khan, L.; Bensler, N.P.; Bose, P.; De Carvalho, D.D. TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat. Commun. 2018, 9, 4692. [Google Scholar] [CrossRef] [Green Version]
- Lakins, M.A.; Ghorani, E.; Munir, H.; Martins, C.P.; Shields, J.D. Cancer-associated fibroblasts induce antigen-specific deletion of CD8 (+) T Cells to protect tumour cells. Nat. Commun. 2018, 9, 948. [Google Scholar] [CrossRef]
- Wang, Y.; Hays, E.; Rama, M.; Bonavida, B. Cell-mediated immune resistance in cancer. Cancer Drug Resist. 2020, 3, 232–251. [Google Scholar] [CrossRef] [Green Version]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [Green Version]
- Bule, P.; Aguiar, S.I.; Aires-Da-Silva, F.; Dias, J.N.R. Chemokine-Directed Tumor Microenvironment Modulation in Cancer Immunotherapy. Int. J. Mol. Sci. 2021, 22, 9804. [Google Scholar] [CrossRef]
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; McSkane, M.; Baba, H.; Lenz, H.J. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation—A target for novel cancer therapy. Cancer Treat. Rev. 2018, 63, 40–47. [Google Scholar] [CrossRef]
- Goulart, M.R.; Stasinos, K.; Fincham, R.E.A.; Delvecchio, F.R.; Kocher, H.M. T cells in pancreatic cancer stroma. World J. Gastroenterol. 2021, 27, 7956–7968. [Google Scholar] [CrossRef]
- Strazza, M.; Mor, A. The Complexity of Targeting Chemokines to Promote a Tumor Immune Response. Inflammation 2020, 43, 1201–1208. [Google Scholar] [CrossRef]
- Marcuzzi, E.; Angioni, R.; Molon, B.; Calì, B. Chemokines and Chemokine Receptors: Orchestrating Tumor Metastasization. Int. J. Mol. Sci. 2018, 20, 96. [Google Scholar] [CrossRef] [Green Version]
- Do, H.T.T.; Lee, C.H.; Cho, J. Chemokines and their Receptors: Multifaceted Roles in Cancer Progression and Potential Value as Cancer Prognostic Markers. Cancers 2020, 12, 287. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Fertig, E.J.; Jin, K.; Sukumar, S.; Pandey, N.B.; Popel, A.S. Breast cancer cells condition lymphatic endothelial cells within pre-metastatic niches to promote metastasis. Nat. Commun. 2014, 5, 4715. [Google Scholar] [CrossRef] [Green Version]
- de Aguiar, R.B.; de Moraes, J.Z. Exploring the Immunological Mechanisms Underlying the Anti-vascular Endothelial Growth Factor Activity in Tumors. Front. Immunol. 2019, 10, 1023. [Google Scholar] [CrossRef] [Green Version]
- Harjunpää, H.; Llort Asens, M.; Guenther, C.; Fagerholm, S.C. Cell Adhesion Molecules and Their Roles and Regulation in the Immune and Tumor Microenvironment. Front. Immunol. 2019, 10, 1078. [Google Scholar] [CrossRef] [Green Version]
- Lamplugh, Z.; Fan, Y. Vascular Microenvironment, Tumor Immunity and Immunotherapy. Front. Immunol. 2021, 12, 811485. [Google Scholar] [CrossRef]
- Dieterich, L.C.; Ikenberg, K.; Cetintas, T.; Kapaklikaya, K.; Hutmacher, C.; Detmar, M. Tumor-Associated Lymphatic Vessels Upregulate PDL1 to Inhibit T-Cell Activation. Front. Immunol. 2017, 8, 66. [Google Scholar] [CrossRef] [Green Version]
- Hu, K.; Babapoor-Farrokhran, S.; Rodrigues, M.; Deshpande, M.; Puchner, B.; Kashiwabuchi, F.; Hassan, S.J.; Asnaghi, L.; Handa, J.T.; Merbs, S.; et al. Hypoxia-inducible factor 1 upregulation of both VEGF and ANGPTL4 is required to promote the angiogenic phenotype in uveal melanoma. Oncotarget 2016, 7, 7816–7828. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Shen, L.; Jiang, J.; Zhang, L.; Zhang, Z.; Pan, J.; Ni, C.; Chen, Z. Antiangiogenic therapy reverses the immunosuppressive breast cancer microenvironment. Biomark. Res. 2021, 9, 59. [Google Scholar] [CrossRef]
- Tang, F.; Zheng, P. Tumor cells versus host immune cells: Whose PD-L1 contributes to PD-1/PD-L1 blockade mediated cancer immunotherapy? Cell Biosci. 2018, 8, 34. [Google Scholar] [CrossRef] [Green Version]
- Oberholtzer, N.; Quinn, K.M.; Chakraborty, P.; Mehrotra, S. New Developments in T Cell Immunometabolism and Implications for Cancer Immunotherapy. Cells 2022, 11, 708. [Google Scholar] [CrossRef]
- Al-Fahdawi, M.Q.; Al-Doghachi, F.A.J.; Abdullah, Q.K.; Hammad, R.T.; Rasedee, A.; Ibrahim, W.N.; Alshwyeh, H.A.; Alosaimi, A.A.; Aldosary, S.K.; Eid, E.E.M.; et al. Oxidative stress cytotoxicity induced by platinum-doped magnesia nanoparticles in cancer cells. Biomed. Pharmacother. 2021, 138, 111483. [Google Scholar] [CrossRef]
- Zhang, H.; Conrad, D.M.; Butler, J.J.; Zhao, C.; Blay, J.; Hoskin, D.W. Adenosine acts through A2 receptors to inhibit IL-2-induced tyrosine phosphorylation of STAT5 in T lymphocytes: Role of cyclic adenosine 3’,5’-monophosphate and phosphatases. J. Immunol. 2004, 173, 932–944. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Hsu, J.L.; Li, Y.; Hortobagyi, G.N.; Hung, M.C. Cancer Cell Metabolism Bolsters Immunotherapy Resistance by Promoting an Immunosuppressive Tumor Microenvironment. Front. Oncol. 2020, 10, 1197. [Google Scholar] [CrossRef]
- Wang, H.C.; Chen, C.W.; Yang, C.L.; Tsai, I.M.; Hou, Y.C.; Chen, C.J.; Shan, Y.S. Tumor-Associated Macrophages Promote Epigenetic Silencing of Gelsolin through DNA Methyltransferase 1 in Gastric Cancer Cells. Cancer Immunol. Res. 2017, 5, 885–897. [Google Scholar] [CrossRef] [Green Version]
- Fulop, T.; Larbi, A.; Kotb, R.; de Angelis, F.; Pawelec, G. Aging, immunity, and cancer. Discov. Med. 2011, 11, 537–550. [Google Scholar]
- Conforti, F.; Pala, L.; Bagnardi, V.; De Pas, T.; Martinetti, M.; Viale, G.; Gelber, R.D.; Goldhirsch, A. Cancer immunotherapy efficacy and patients’ sex: A systematic review and meta-analysis. Lancet Oncol. 2018, 19, 737–746. [Google Scholar] [CrossRef]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Wang, Q.; Tang, X.; Xu, R.; Zhang, L.; Chen, X.; Xue, Q.; Wang, Z.; Shi, R.; Wang, F.; et al. Correlation between patients’ age and cancer immunotherapy efficacy. Oncoimmunology 2019, 8, e1568810. [Google Scholar] [CrossRef]
- Vavalà, T.; Catino, A.; Pizzutilo, P.; Longo, V.; Galetta, D. Gender Differences and Immunotherapy Outcome in Advanced Lung Cancer. Int. J. Mol. Sci. 2021, 22, 11942. [Google Scholar] [CrossRef]
- Woodall, M.J.; Neumann, S.; Campbell, K.; Pattison, S.T.; Young, S.L. The Effects of Obesity on Anti-Cancer Immunity and Cancer Immunotherapy. Cancers 2020, 12, 1230. [Google Scholar] [CrossRef]
- Barbosa, A.M.; Gomes-Gonçalves, A.; Castro, A.G.; Torrado, E. Immune System Efficiency in Cancer and the Microbiota Influence. Pathobiology 2021, 88, 170–186. [Google Scholar] [CrossRef]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Jørgensen, N.; Persson, G.; Hviid, T.V.F. The Tolerogenic Function of Regulatory T Cells in Pregnancy and Cancer. Front. Immunol. 2019, 10, 911. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wei, H. Role of decidual natural killer cells in human pregnancy and related pregnancy complications. Front. Immunol. 2021, 12, 728291. [Google Scholar]
- Tie, Y.; Tang, F.; Wei, Y.-Q.; Wei, X.-W. Immunosuppressive cells in cancer: Mechanisms and potential therapeutic targets. J. Hematol. Oncol. 2022, 15, 61. [Google Scholar] [CrossRef]
- Wiens, L.; Schäffeler, N.; Eigentler, T.; Garbe, C.; Forschner, A. Psychological Distress of Metastatic Melanoma Patients during Treatment with Immune Checkpoint Inhibitors: Results of a Prospective Study. Cancers 2021, 13, 2642. [Google Scholar] [CrossRef]
- Gouez, M.; Delrieu, L.; Bouleuc, C.; Girard, N.; Raynard, B.; Marchal, T. Association between Nutritional Status and Treatment Response and Survival in Patients Treated with Immunotherapy for Lung Cancer: A Retrospective French Study. Cancers 2022, 14, 3439. [Google Scholar] [CrossRef]
- Ziętarska, M.; Krawczyk-Lipiec, J.; Kraj, L.; Zaucha, R.; Małgorzewicz, S. Nutritional status assessment in colorectal cancer patients qualified to systemic treatment. Contemp. Oncol. 2017, 21, 157–161. [Google Scholar] [CrossRef]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Dai, Y.; Zhao, W.; Yue, L.; Dai, X.; Rong, D.; Wu, F.; Gu, J.; Qian, X. Perspectives on Immunotherapy of Metastatic Colorectal Cancer. Front. Oncol. 2021, 11, 659964. [Google Scholar] [CrossRef]
- Escribese, M.M.; Barber, D. New insight into cancer immunotherapy. Allergol. Immunopathol. 2017, 45 (Suppl. S1), 50–55. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Helissey, C.; Vicier, C.; Champiat, S. The development of immunotherapy in older adults: New treatments, new toxicities? J. Geriatr. Oncol. 2016, 7, 325–333. [Google Scholar] [CrossRef]
- Yang, J.C.; Hughes, M.; Kammula, U.; Royal, R.; Sherry, R.M.; Topalian, S.L.; Suri, K.B.; Levy, C.; Allen, T.; Mavroukakis, S.; et al. Ipilimumab (anti-CTLA4 antibody) causes regression of metastatic renal cell cancer associated with enteritis and hypophysitis. J. Immunother. 2007, 30, 825–830. [Google Scholar] [CrossRef] [Green Version]
- Duraiswamy, J.; Kaluza, K.M.; Freeman, G.J.; Coukos, G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 2013, 73, 3591–3603. [Google Scholar] [CrossRef] [Green Version]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E.; et al. Nivolumab plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef]
- Morgado, M.; Plácido, A.; Morgado, S.; Roque, F. Management of the Adverse Effects of Immune Checkpoint Inhibitors. Vaccines 2020, 8, 575. [Google Scholar] [CrossRef]
- Wang, Y.; Tong, Z.; Zhang, W.; Zhang, W.; Buzdin, A.; Mu, X.; Yan, Q.; Zhao, X.; Chang, H.H.; Duhon, M.; et al. FDA-Approved and Emerging Next Generation Predictive Biomarkers for Immune Checkpoint Inhibitors in Cancer Patients. Front. Oncol. 2021, 11, 683419. [Google Scholar] [CrossRef]
- Long, L.; Zhang, X.; Chen, F.; Pan, Q.; Phiphatwatchara, P.; Zeng, Y.; Chen, H. The promising immune checkpoint LAG-3: From tumor microenvironment to cancer immunotherapy. Genes Cancer 2018, 9, 176–189. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Cao, J.; Zhao, C.; Li, X.; Zhou, C.; Hirsch, F.R. TIM-3, a promising target for cancer immunotherapy. Onco. Targets Ther. 2018, 11, 7005–7009. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.X.; Wu, J.W.; Cheng, X.Q.; Wang, J.H.; Wen, X.Z.; Li, J.J.; Zhang, Q.; Jiang, H.; Ding, Q.Y.; Zhu, X.F.; et al. HHLA2 predicts improved prognosis of anti-PD-1/PD-L1 immunotherapy in patients with melanoma. Front. Immunol. 2022, 13, 902167. [Google Scholar] [CrossRef]
- ElTanbouly, M.A.; Croteau, W.; Noelle, R.J.; Lines, J.L. VISTA: A novel immunotherapy target for normalizing innate and adaptive immunity. Semin. Immunol. 2019, 42, 101308. [Google Scholar] [CrossRef]
- Märkl, F.; Huynh, D.; Endres, S.; Kobold, S. Utilizing chemokines in cancer immunotherapy. Trends Cancer 2022, 8, 670–682. [Google Scholar] [CrossRef]
- Qian, C.; Liu, X.Y.; Prieto, J. Therapy of cancer by cytokines mediated by gene therapy approach. Cell Res. 2006, 16, 182–188. [Google Scholar] [CrossRef] [Green Version]
- Song, J.S.; Chang, C.C.; Wu, C.H.; Dinh, T.K.; Jan, J.J.; Huang, K.W.; Chou, M.C.; Shiue, T.Y.; Yeh, K.C.; Ke, Y.Y.; et al. A highly selective and potent CXCR4 antagonist for hepatocellular carcinoma treatment. Proc. Natl. Acad. Sci. USA 2021, 118, e2015433118. [Google Scholar] [CrossRef]
- Gilchrist, A.; Echeverria, S.L. Targeting Chemokine Receptor CCR1 as a Potential Therapeutic Approach for Multiple Myeloma. Front. Endocrinol. 2022, 13, 846310. [Google Scholar] [CrossRef]
- Pradhan, A.K.; Bhoopathi, P.; Maji, S.; Kumar, A.; Guo, C.; Mannangatti, P.; Li, J.; Wang, X.Y.; Sarkar, D.; Emdad, L.; et al. Enhanced Cancer Therapy Using an Engineered Designer Cytokine Alone and in Combination with an Immune Checkpoint Inhibitor. Front. Oncol. 2022, 12, 812560. [Google Scholar] [CrossRef]
- Wurm, M.; Schaaf, O.; Reutner, K.; Ganesan, R.; Mostböck, S.; Pelster, C.; Böttcher, J.; de Andrade Pereira, B.; Taubert, C.; Alt, I.; et al. A Novel Antagonistic CD73 Antibody for Inhibition of the Immunosuppressive Adenosine Pathway. Mol. Cancer Ther. 2021, 20, 2250–2261. [Google Scholar] [CrossRef]
- Zhang, J.; Yan, W.; Duan, W.; Wüthrich, K.; Cheng, J. Tumor Immunotherapy Using A(2A) Adenosine Receptor Antagonists. Pharmaceuticals 2020, 13, 237. [Google Scholar] [CrossRef]
- Hong, Y.; Kim, I.-S. The therapeutic potential of immune cell-derived exosomes as an alternative to adoptive cell transfer. BMB Rep. 2022, 55, 39–47. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Quinn, S.; Lenart, N.; Dronzek, V.; Scurti, G.M.; Hossain, N.M.; Nishimura, M.I. Genetic Modification of T Cells for the Immunotherapy of Cancer. Vaccines 2022, 10, 457. [Google Scholar] [CrossRef]
- Miliotou, A.N.; Papadopoulou, L.C. CAR T-cell Therapy: A New Era in Cancer Immunotherapy. Curr. Pharm. Biotechnol. 2018, 19, 5–18. [Google Scholar] [CrossRef]
- Johnson, L.A.; Morgan, R.A.; Dudley, M.E.; Cassard, L.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Royal, R.E.; Sherry, R.M.; Wunderlich, J.R.; et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009, 114, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Scholler, J.; Brady, T.L.; Binder-Scholl, G.; Hwang, W.T.; Plesa, G.; Hege, K.M.; Vogel, A.N.; Kalos, M.; Riley, J.L.; Deeks, S.G.; et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci. Transl. Med. 2012, 4, 132ra153. [Google Scholar] [CrossRef] [Green Version]
- Bashiri Dezfouli, A.; Yazdi, M.; Pockley, A.G.; Khosravi, M.; Kobold, S.; Wagner, E.; Multhoff, G. NK Cells Armed with Chimeric Antigen Receptors (CAR): Roadblocks to Successful Development. Cells 2021, 10, 3390. [Google Scholar] [CrossRef]
- Tang, X.; Yang, L.; Li, Z.; Nalin, A.P.; Dai, H.; Xu, T.; Yin, J.; You, F.; Zhu, M.; Shen, W.; et al. First-in-man clinical trial of CAR NK-92 cells: Safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 2018, 8, 1083–1089. [Google Scholar]
- Tvedt, T.H.A.; Vo, A.K.; Bruserud, Ø.; Reikvam, H. Cytokine Release Syndrome in the Immunotherapy of Hematological Malignancies: The Biology behind and Possible Clinical Consequences. J. Clin. Med. 2021, 10, 5190. [Google Scholar] [CrossRef]
- Almeida-Porada, G.; Atala, A.J.; Porada, C.D. Therapeutic Mesenchymal Stromal Cells for Immunotherapy and for Gene and Drug Delivery. Mol. Ther. Methods Clin. Dev. 2020, 16, 204–224. [Google Scholar] [CrossRef] [Green Version]
- Lan, T.; Luo, M.; Wei, X. Mesenchymal stem/stromal cells in cancer therapy. J. Hematol. Oncol. 2021, 14, 195. [Google Scholar] [CrossRef]
- Apostolopoulos, V. Cancer Vaccines: Research and Applications. Cancers 2019, 11, 1041. [Google Scholar] [CrossRef] [Green Version]
- Apostolopoulos, V. Vaccine Delivery Methods into the Future. Vaccines 2016, 4, 9. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer vaccines as promising immuno-therapeutics: Platforms and current progress. J. Hematol. Oncol. 2022, 15, 28. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Morse, M.A.; Garst, J.; Osada, T.; Khan, S.; Hobeika, A.; Clay, T.M.; Valente, N.; Shreeniwas, R.; Sutton, M.A.; Delcayre, A.; et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J. Transl. Med. 2005, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Wang, H.; Huang, Q.; Peng, C.; Yao, L.; Chen, H.; Qiu, Z.; Wu, Y.; Wang, L.; Chen, W. Exosomes from M1-Polarized Macrophages Enhance Paclitaxel Antitumor Activity by Activating Macrophages-Mediated Inflammation. Theranostics 2019, 9, 1714–1727. [Google Scholar] [CrossRef]
- Pitt, J.M.; André, F.; Amigorena, S.; Soria, J.C.; Eggermont, A.; Kroemer, G.; Zitvogel, L. Dendritic cell-derived exosomes for cancer therapy. J. Clin. Investig. 2016, 126, 1224–1232. [Google Scholar] [CrossRef]
- Bouyahya, A.; Mechchate, H.; Oumeslakht, L.; Zeouk, I.; Aboulaghras, S.; Balahbib, A.; Zengin, G.; Kamal, M.A.; Gallo, M.; Montesano, D.; et al. The Role of Epigenetic Modifications in Human Cancers and the Use of Natural Compounds as Epidrugs: Mechanistic Pathways and Pharmacodynamic Actions. Biomolecules 2022, 12, 367. [Google Scholar] [CrossRef]
- Qiu, Q.; Lin, Y.; Ma, Y.; Li, X.; Liang, J.; Chen, Z.; Liu, K.; Huang, Y.; Luo, H.; Huang, R.; et al. Exploring the Emerging Role of the Gut Microbiota and Tumor Microenvironment in Cancer Immunotherapy. Front. Immunol. 2020, 11, 612202. [Google Scholar] [CrossRef]
- Lu, Y.; Yuan, X.; Wang, M.; He, Z.; Li, H.; Wang, J.; Li, Q. Gut microbiota influence immunotherapy responses: Mechanisms and therapeutic strategies. J. Hematol. Oncol. 2022, 15, 47. [Google Scholar] [CrossRef]
- Davar, D.; Dzutsev, A.K.; McCulloch, J.A.; Rodrigues, R.R.; Chauvin, J.M.; Morrison, R.M.; Deblasio, R.N.; Menna, C.; Ding, Q.; Pagliano, O.; et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 2021, 371, 595–602. [Google Scholar] [CrossRef]
- Lin, C.C.; Doi, T.; Muro, K.; Hou, M.M.; Esaki, T.; Hara, H.; Chung, H.C.; Helwig, C.; Dussault, I.; Osada, M.; et al. Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGFβ and PD-L1, in Patients with Esophageal Squamous Cell Carcinoma: Results from a Phase 1 Cohort in Asia. Target. Oncol. 2021, 16, 447–459. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Chavez, J.C.; Bachmeier, C.; Kharfan-Dabaja, M.A. CAR T-cell therapy for B-cell lymphomas: Clinical trial results of available products. Ther. Adv. Hematol. 2019, 10, 2040620719841581. [Google Scholar] [CrossRef] [Green Version]
- SRIVASTAVA, S.; O’Brien, M.; Cheema, P.; Grohe, C.; Carcereny, E.; Girard, N.; Chiappori, A.; Ross, S.; Rossetti, M.; Dubois, F. EP08. 01-021 Phase 2 Study Evaluating Inupadenant in Combination with Chemotherapy in Adults with NSCLC who Progressed on Immunotherapy. J. Thorac. Oncol. 2022, 17, S346–S347. [Google Scholar]
- Harshman, L.C.; Chu, M.; George, S.; Hughes, B.G.M.; Carthon, B.C.; Fong, L.; Merchan, J.R.; Kwei, L.; Hotson, A.N.; Mobasher, M. Adenosine Receptor Blockade with Ciforadenant+/-Atezolizumab in Advanced Metastatic Castration-Resistant Prostate Cancer (mCRPC); American Society of Clinical Oncology: Alexandria, VA, USA, 2020. [Google Scholar]
- Wei, G.; Zhang, H.; Zhao, H.; Wang, J.; Wu, N.; Li, L.; Wu, J.; Zhang, D. Emerging immune checkpoints in the tumor microenvironment: Implications for cancer immunotherapy. Cancer Lett. 2021, 511, 68–76. [Google Scholar] [CrossRef]
- Wei, Q.; Taskén, K. Immunoregulatory signal networks and tumor immune evasion mechanisms: Insights into therapeutic targets and agents in clinical development. Biochem. J. 2022, 479, 2219–2260. [Google Scholar] [CrossRef]
- Capici, S.; Ammoni, L.C.; Meli, N.; Cogliati, V.; Pepe, F.F.; Piazza, F.; Cazzaniga, M.E. Personalised Therapies for Metastatic Triple-Negative Breast Cancer: When Target Is Not Everything. Cancers 2022, 14, 3729. [Google Scholar] [CrossRef]
- Ernst, M.; Oeser, A.; Besiroglu, B.; Caro-Valenzuela, J.; Abd El Aziz, M.; Monsef, I.; Borchmann, P.; Estcourt, L.J.; Skoetz, N.; Goldkuhle, M. Chimeric antigen receptor (CAR) T-cell therapy for people with relapsed or refractory diffuse large B-cell lymphoma. Cochrane Database Syst. Rev. 2021, 9, Cd013365. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Jänne, P.A.; Opyrchal, M.; Hafez, N.; Raez, L.E.; Gabrilovich, D.I.; Wang, F.; Trepel, J.B.; Lee, M.J.; Yuno, A.; et al. Entinostat plus Pembrolizumab in Patients with Metastatic NSCLC Previously Treated with Anti-PD-(L)1 Therapy. Clin. Cancer Res. 2021, 27, 1019–1028. [Google Scholar] [CrossRef]
- Kim, C.G.; Sang, Y.B.; Lee, J.H.; Chon, H.J. Combining Cancer Vaccines with Immunotherapy: Establishing a New Immunological Approach. Int. J. Mol. Sci. 2021, 22, 8035. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Boss, I.; Beach, C.L.; Copeland, W.B.; Thompson, E.; Fox, B.A.; Hasle, V.E.; Hellmann, A.; Taussig, D.C.; Tormo, M.; et al. A randomized phase 2 trial of azacitidine with or without durvalumab as first-line therapy for older patients with AML. Blood Adv. 2022, 6, 2219–2229. [Google Scholar] [CrossRef]
- Valsecchi, M.E. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 1270. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Demaria, S.; Ng, B.; Devitt, M.L.; Babb, J.S.; Kawashima, N.; Liebes, L.; Formenti, S.C. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 862–870. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [Green Version]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- Maus, M.V.; June, C.H. Making Better Chimeric Antigen Receptors for Adoptive T-cell Therapy. Clin. Cancer Res. 2016, 22, 1875–1884. [Google Scholar] [CrossRef] [Green Version]
- Beyar-Katz, O.; Gill, S. Novel Approaches to Acute Myeloid Leukemia Immunotherapy. Clin. Cancer Res. 2018, 24, 5502–5515. [Google Scholar] [CrossRef] [Green Version]
- Dangaj, D.; Bruand, M.; Grimm, A.J.; Ronet, C.; Barras, D.; Duttagupta, P.A.; Lanitis, E.; Duraiswamy, J.; Tanyi, J.L.; Benencia, F.; et al. Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell 2019, 35, 885–900.e10. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Modalities of Combined Immunotherapy | Tumor Type | Phase | Clinical Trial | Ref. |
|---|---|---|---|---|---|
| Durvalumab + autologous anti-CD19CAR-4-1BB-CD3z EGFRt-expressing CD4+/CD8+ central memory T lymphocytes JCAR014 | Anti- PDL-1 + CAR T cells | Diffuse large B-cell lymphoma | 1 | NCT02706405 | [154] |
| Pembrolizumab + CART-EGFRvIII T cells | Anti- PD-1 + CAR T cells | Glioblastoma | 1 | NCT03726515 | [11] |
| Inupadenant + EOS-448 | Adenosine A receptor blocker + anti-TIGIT mAB | Advanced solid tumors | 2 | NCT05060432 | [155] |
| Ciforadenant ± atezolizumab | Adenosine A receptor blocker + anti-PD-1 | Advanced prostate cancer | 1,1b | NCT02655822 | [156] |
| Ciforadenant + daratumumab | Adenosine A receptor blocker + mAb against CD38 | Multiple myeloma | 1 | NCT04280328 | [157] |
| AB928/etrumadenant + zimberelimab + enzalutamide | Adenosine A and B receptor blocker + anti-PD-1 + anti-androgen or chemotherapy | Advanced prostate cancer | 2 | NCT04381832 | [158] |
| AB928/etrumadenant zimberelimab + AB680 | Adenosine A and B receptor blocker + anti-PD-1 + CD73 inhibitor | Colonic cancer | 2 | NCT04660812 | [158] |
| Etrumadenant +PLD + IPI-549 | Adenosine A and B receptor blocker + chemotherapy + PI3K-gamma inhibitor | Breast cancer | 1 | NCT03719326 | [159] |
| Bintrafusp alfa | PD-L1 and TGF β inhibitors | Squamous cell carcinoma | 1 | NCT02699515. | [11,152] |
| CD19 CAR T-expressing IL7 and CCL19 combined with tislelizumab | IL17, CCL9 chemokine, and PD-1 mAb | Relapsed or refractory B-cell lymphoma | 1 | NCT04381741 | [160] |
| HuMax-IL8 + nivolumab | CXCL8-chemokine and PD-1- IC | Head and neck squamous | 2 | NCT04848116 | [123] |
| Entinostat plus pembrolizumab | Histone deacetylase inhibitors plus PD-1 blockade | Non-small-cell lung cancer (NSCLC) | 2 | NCT02437136 | [161] |
| Autogene cevumeran and atezolizumab | mRNA-based cancer vaccine and anti-PD-L1 | Metastatic tumors | 1 | NCT03289962 | [162] |
| Azacitidine and PD-L1 inhibitors | Demethylating agent plus PD-L1 inhibitor | Acute myeloid leukemia | 2 | NCT02775903 | [163] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Said, S.S.; Ibrahim, W.N. Cancer Resistance to Immunotherapy: Comprehensive Insights with Future Perspectives. Pharmaceutics 2023, 15, 1143. https://doi.org/10.3390/pharmaceutics15041143
Said SS, Ibrahim WN. Cancer Resistance to Immunotherapy: Comprehensive Insights with Future Perspectives. Pharmaceutics. 2023; 15(4):1143. https://doi.org/10.3390/pharmaceutics15041143
Chicago/Turabian StyleSaid, Sawsan Sudqi, and Wisam Nabeel Ibrahim. 2023. "Cancer Resistance to Immunotherapy: Comprehensive Insights with Future Perspectives" Pharmaceutics 15, no. 4: 1143. https://doi.org/10.3390/pharmaceutics15041143