
Prospects of Using Protein Engineering for Selective Drug Delivery into a Specific Compartment of Target Cells

Abstract
:1. Introduction
2. Specificity
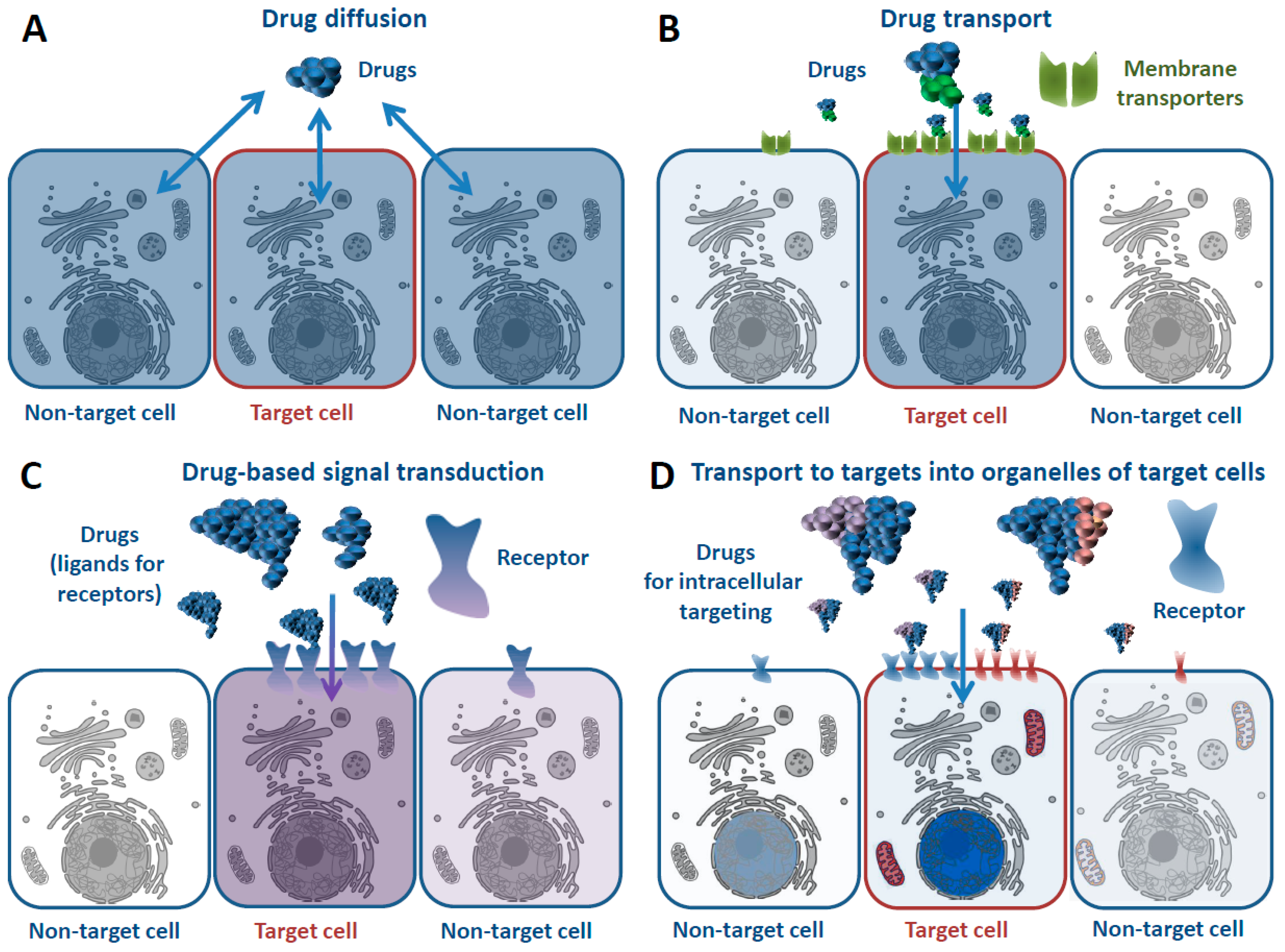
2.1. Cell Specificity
2.2. Subcellular Specificity
3. Extracellular Obstacles to Intracellular Targets during Systemic Drug Administration
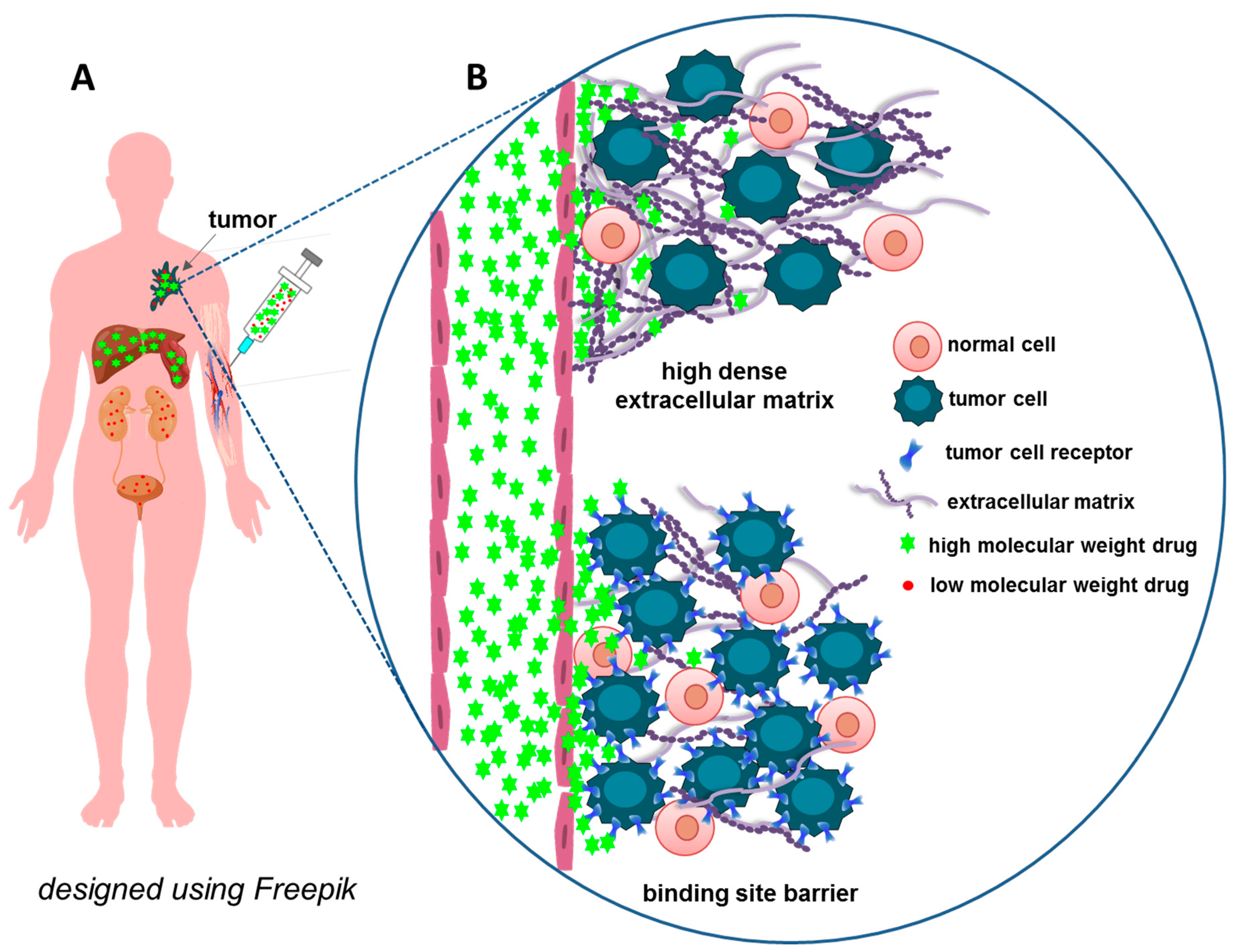
3.1. Extracellular Barriers: Interaction with Blood Proteins and Cells
3.2. Penetration through the Walls of Blood Vessels and Extracellular Matrix
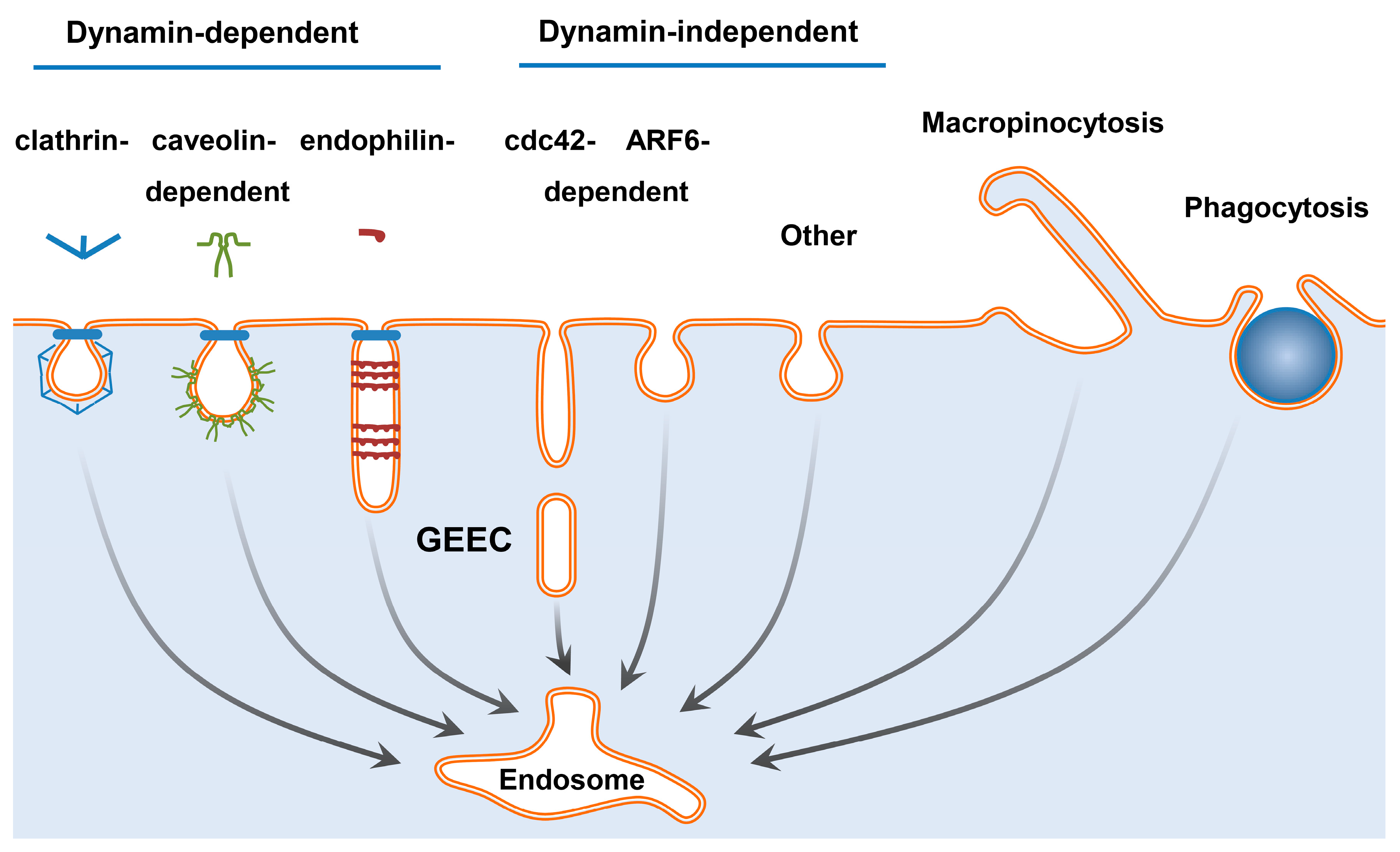
4. Intracellular Targets and Barriers on the Way to Them
5. Macromolecular Delivery Systems
5.1. Properties of Macromolecular Delivery Systems Predetermined by Extracellular Barriers
5.2. Properties of Macromolecular Delivery Systems Predetermined by Active Principles
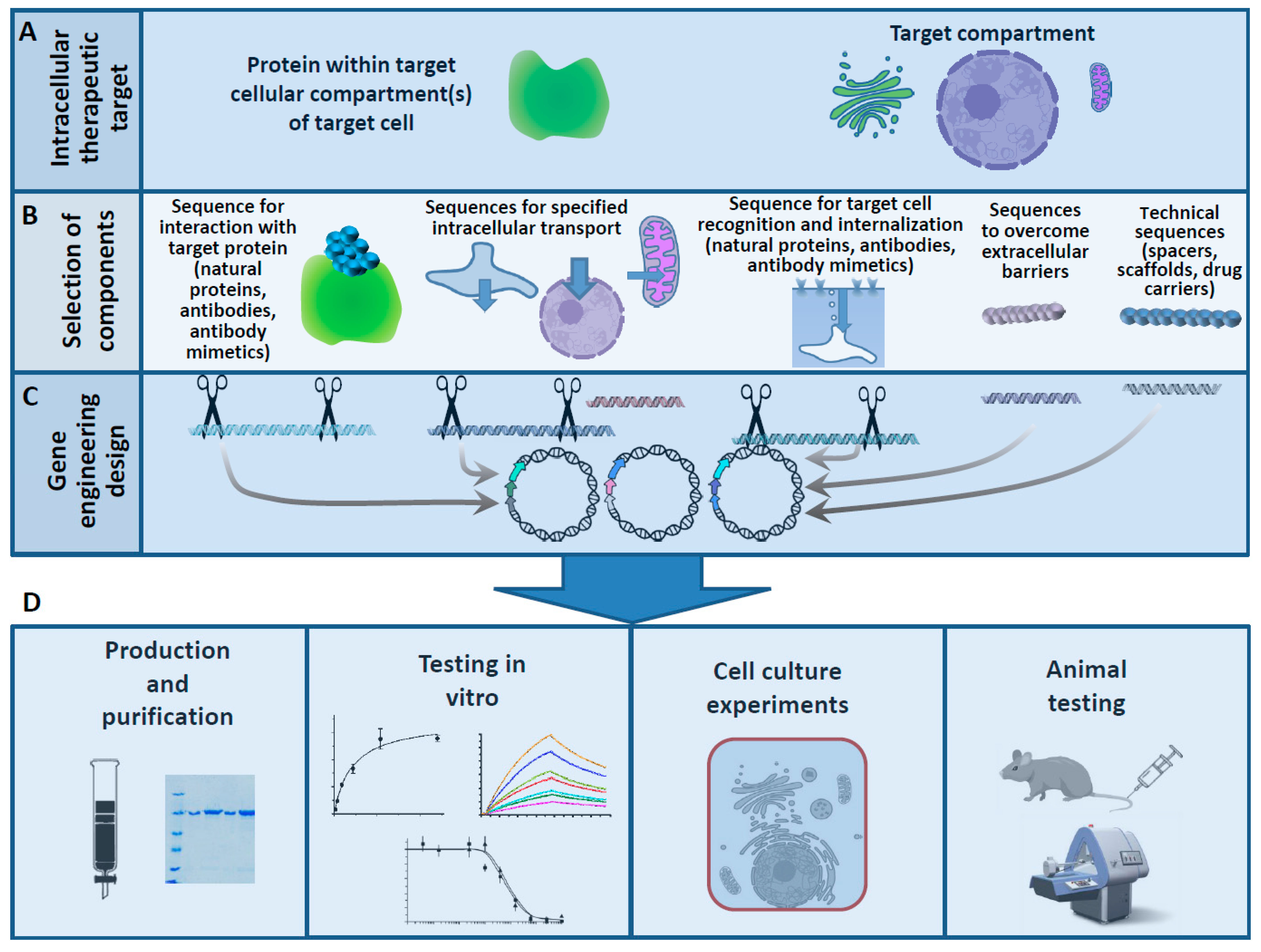
5.3. Combining Transport Functions in a Single Polypeptide Chain
6. Protein-Based Ligands for Intracellular Delivery
7. Use of Receptor-Mediated Endocytosis for Cell-Specific Drug Delivery
8. Branching Delivery Pathways for Endocytosed Drugs
8.1. Endosomes Acidification and Protein Drug Delivery
8.2. Drug Delivery into the Nucleus
8.3. Mitochondrial Drug Delivery
8.4. Peroxisomal Drug Delivery
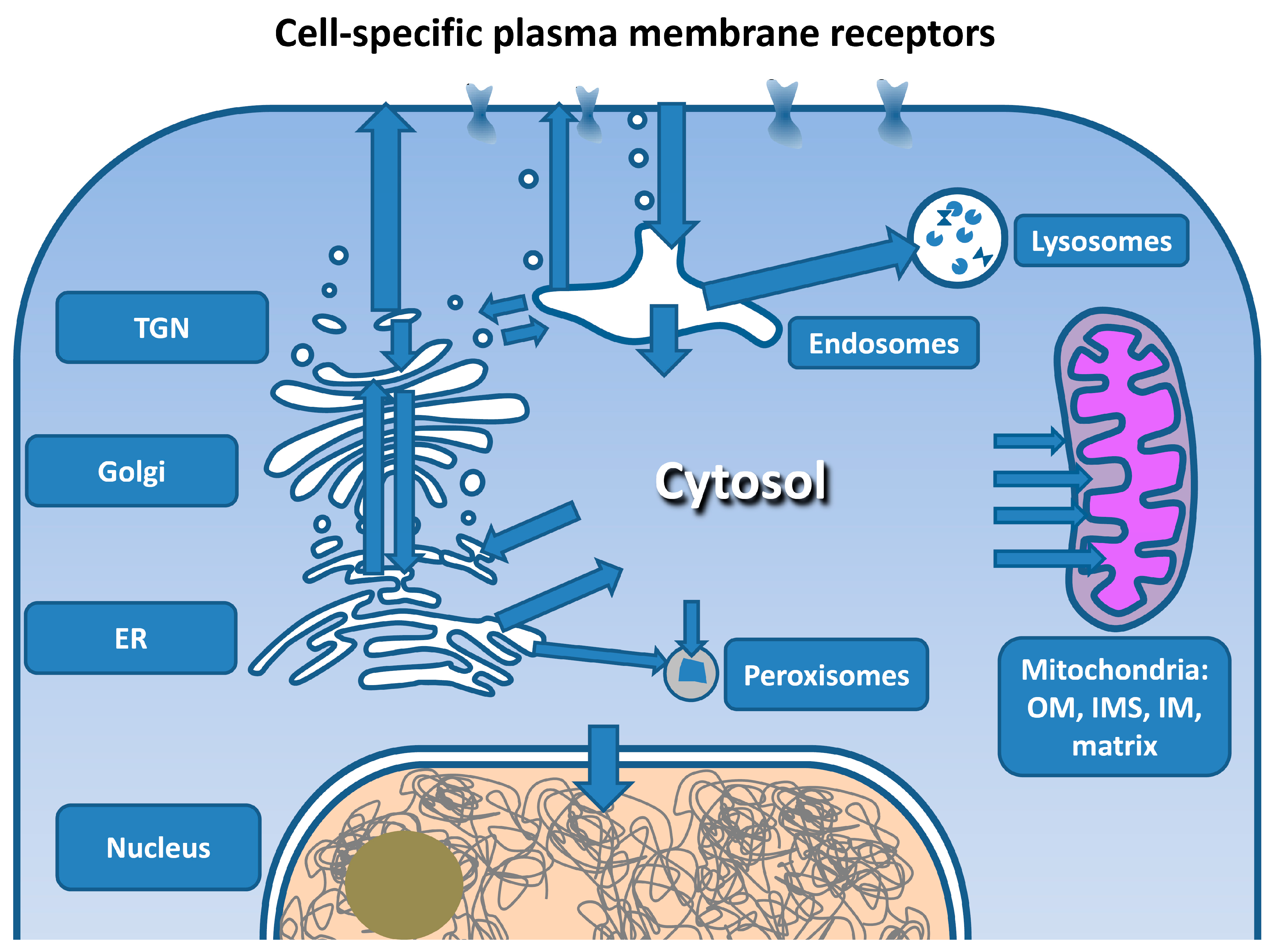
8.5. Vesicular Transport to Intracellular Targets
9. Future Prospects
10. Conclusions
- Generation of datasets on protein expression in individual cell types;
- Many-fold differing expression of a significant part of the protein targets, both by cell types and by subcellular localization;
- A significant change of protein expression in various pathologies, primarily in cancer;
- Elucidation of the plethora of barriers on the way of a macromolecule to its target site after administration at the whole-body-, tissue- and cellular-level and the ways to overcome them.
- Accumulation of data on the subcellular distribution of proteins in cells of various types and their individual differences;
- Significant development of interactome maps with its transition from ascertaining the presence of interaction between proteins to its concentration and kinetic dependences;
- Progress in predicting the spatial structure of the created artificial protein constructs and their interaction with the proteome.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Xie, J.; Bi, Y.; Zhang, H.; Dong, S.; Teng, L.; Lee, R.J.; Yang, Z. Cell-Penetrating Peptides in Diagnosis and Treatment of Human Diseases: From Preclinical Research to Clinical Application. Front. Pharmacol. 2020, 11, 697. [Google Scholar] [CrossRef]
- Raman, V.; Van, D.N.; Hall, C.L.; Wetherby, V.E.; Whitney, S.A.; Kolewe, E.L.; Bloom, S.M.K.; Sharma, A.; Hardy, J.A.; Bollen, M.; et al. Intracellular delivery of protein drugs with an autonomously lysing bacterial system reduces tumor growth and metastases. Nat. Commun. 2021, 12, 6116. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Yu, C.; Li, L.; Yao, S.Q. Intracellular Delivery of Functional Proteins and Native Drugs by Cell-Penetrating Poly(disulfide)s. J. Am. Chem. Soc. 2015, 137, 12153–12160. [Google Scholar] [CrossRef]
- Slowing, I.I.; Trewyn, B.G.; Lin, V.S. Mesoporous silica nanoparticles for intracellular delivery of membrane-impermeable proteins. J. Am. Chem. Soc. 2007, 129, 8845–8849. [Google Scholar] [CrossRef] [Green Version]
- Bae, S.E.; Lyu, S.K.; Kim, K.J.; Shin, H.J.; Kwon, H.; Huh, S. Intracellular delivery of a native functional protein using cell-penetrating peptide functionalized cubic MSNs with ultra-large mesopores. J. Mater. Chem. B 2018, 6, 3456–3465. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Ding, J.; Chen, X. Reduction-Responsive Polypeptide Micelles for Intracellular Delivery of Antineoplastic Agent. Biomacromolecules 2017, 18, 3291–3301. [Google Scholar] [CrossRef]
- Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug. Discov. 2010, 9, 215–236. [Google Scholar]
- Hafliger, P.; Charles, R.P. The L-Type Amino Acid Transporter LAT1-An Emerging Target in Cancer. Int. J. Mol. Sci. 2019, 20, 2428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puris, E.; Gynther, M.; Auriola, S.; Huttunen, K.M. L-Type amino acid transporter 1 as a target for drug delivery. Pharm. Res. 2020, 37, 88. [Google Scholar] [CrossRef]
- Scalise, M.; Pochini, L.; Console, L.; Losso, M.A.; Indiveri, C. The Human SLC1A5 (ASCT2) Amino Acid Transporter: From Function to Structure and Role in Cell Biology. Front. Cell Dev. Biol. 2018, 6, 96. [Google Scholar] [CrossRef] [Green Version]
- Rask-Andersen, M.; Almen, M.S.; Schioth, H.B. Trends in the exploitation of novel drug targets. Nat. Rev. Drug Discov. 2011, 10, 579–590. [Google Scholar] [CrossRef]
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef] [PubMed]
- Attwood, M.M.; Jonsson, J.; Rask-Andersen, M.; Schioth, H.B. Soluble ligands as drug targets. Nat. Rev. Drug Discov. 2020, 19, 695–710. [Google Scholar] [CrossRef]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018, 9, 1410. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Wei, X.; Chen, Z.; Zhang, X.; Yang, G.; Zhou, S. Multifunctional nanoplatforms for subcellular delivery of drugs in cancer therapy. Progr. Mater. Sci. 2020, 107, 100599. [Google Scholar] [CrossRef]
- Rosenkranz, A.A.; Slastnikova, T.A.; Georgiev, G.P.; Zalutsky, M.R.; Sobolev, A.S. Delivery systems exploiting natural cell transport processes of macromolecules for intracellular targeting of Auger electron emitters. Nucl. Med. Biol. 2020, 80–81, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Slastnikova, T.A.; Ulasov, A.V.; Rosenkranz, A.A.; Sobolev, A.S. Targeted Intracellular Delivery of Antibodies: The State of the Art. Front. Pharmacol. 2018, 9, 1208. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.; Liu, J.; Shi, J. Cancer cell nucleus-targeting nanocomposites for advanced tumor therapeutics. Chem. Soc. Rev. 2018, 47, 6930–6946. [Google Scholar] [CrossRef]
- Torchilin, V.P. Multifunctional, stimuli-sensitive nanoparticulate systems for drug delivery. Nat. Rev. Drug Discov. 2014, 13, 813–827. [Google Scholar] [CrossRef] [Green Version]
- Nikitin, M.P.; Zdobnova, T.A.; Lukash, S.V.; Stremovskiy, O.A.; Deyev, S.M. Protein-assisted self-assembly of multifunctional nanoparticles. Proc. Natl. Acad. Sci. USA 2010, 107, 5827–5832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsomaia, N. Peptide therapeutics: Targeting the undruggable space. Eur. J. Med. Chem. 2015, 94, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Verdine, G.L.; Walensky, L.D. The challenge of drugging undruggable targets in cancer: Lessons learned from targeting BCL-2 family members. Clin. Cancer Res. 2007, 13, 7264–7270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibraheem, D.; Elaissari, A.; Fessi, H. Gene therapy and DNA delivery systems. Int. J. Pharm. 2014, 459, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, J.C.; Kowalski, P.S.; Anderson, D.G. Advances in the delivery of RNA therapeutics: From concept to clinical reality. Genome Med. 2017, 9, 60. [Google Scholar] [CrossRef] [Green Version]
- Almen, M.S.; Nordstrom, K.J.; Fredriksson, R.; Schioth, H.B. Mapping the human membrane proteome: A majority of the human membrane proteins can be classified according to function and evolutionary origin. BMC. Biol. 2009, 7, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.S.; Jun, S.Y.; Kim, Y.S. Critical Issues in the Development of Immunotoxins for Anticancer Therapy. J. Pharm. Sci. 2020, 109, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Marei, H.E.; Cenciarelli, C.; Hasan, A. Potential of antibody-drug conjugates (ADCs) for cancer therapy. Cancer Cell Int. 2022, 22, 255. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Eraslan, B.; Wieland, T.; Hallstrom, B.; Hopf, T.; Zolg, D.P.; Zecha, J.; Asplund, A.; Li, L.H.; Meng, C.; et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 2019, 15, e8503. [Google Scholar] [CrossRef]
- Panina, Y.; Karagiannis, P.; Kurtz, A.; Stacey, G.N.; Fujibuchi, W. Human Cell Atlas and cell-type authentication for regenerative medicine. Exp. Mol. Med. 2020, 52, 1443–1451. [Google Scholar] [CrossRef]
- Karlsson, M.; Zhang, C.; Mear, L.; Zhong, W.; Digre, A.; Katona, B.; Sjostedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A single-cell type transcriptomics map of human tissues. Sci. Adv. 2021, 7, eabh2169. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.C.; Karkanias, J.; Krasnow, M.A.; Pisco, A.O.; Quake, S.R.; Salzman, J.; Yosef, N.; Bulthaup, B.; Brown, P.; Harper, W.; et al. The Tabula Sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science 2022, 376, eabl4896. [Google Scholar] [PubMed]
- Martinez-Val, A.; Bekker-Jensen, D.B.; Steigerwald, S.; Koenig, C.; Ostergaard, O.; Mehta, A.; Tran, T.; Sikorski, K.; Torres-Vega, E.; Kwasniewicz, E.; et al. Spatial-proteomics reveals phospho-signaling dynamics at subcellular resolution. Nat. Commun. 2021, 12, 7113. [Google Scholar] [CrossRef]
- A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 2020, 583, 590–595. [CrossRef] [PubMed]
- Schaum, N.; Lehallier, B.; Hahn, O.; Palovics, R.; Hosseinzadeh, S.; Lee, S.E.; Sit, R.; Lee, D.P.; Losada, P.M.; Zardeneta, M.E.; et al. Ageing hallmarks exhibit organ-specific temporal signatures. Nature 2020, 583, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Shokhirev, M.N.; Johnson, A.A. Modeling the human aging transcriptome across tissues, health status, and sex. Aging Cell 2021, 20, e13280. [Google Scholar] [CrossRef] [PubMed]
- Omenn, G.S.; Lane, L.; Overall, C.M.; Paik, Y.K.; Cristea, I.M.; Corrales, F.J.; Lindskog, C.; Weintraub, S.; Roehrl, M.H.A.; Liu, S.; et al. Progress Identifying and Analyzing the Human Proteome: 2021 Metrics from the HUPO Human Proteome Project. J. Proteome. Res. 2021, 20, 5227–5240. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.T. Single-cell Proteomics: Progress and Prospects. Mol. Cell Proteom. 2020, 19, 1739–1748. [Google Scholar] [CrossRef] [PubMed]
- Mund, A.; Brunner, A.D.; Mann, M. Unbiased spatial proteomics with single-cell resolution in tissues. Mol. Cell 2022, 82, 2335–2349. [Google Scholar] [CrossRef]
- Thul, P.J.; Akesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait, B.H.; Alm, T.; Asplund, A.; Bjork, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356, eaal3321. Available online: https://www.proteinatlas.org/ (accessed on 18 March 2023). [CrossRef]
- Louzoun-Zada, S.; Jaber, Q.Z.; Fridman, M. Guiding Drugs to Target-Harboring Organelles: Stretching Drug-Delivery to a Higher Level of Resolution. Angew. Chem. Int. Ed Engl. 2019, 58, 15584–15594. [Google Scholar] [CrossRef] [PubMed]
- Saminathan, A.; Zajac, M.; Anees, P.; Krishnan, Y. Organelle-level precision with next-generation targeting technologies. Nat. Rev. Mater. 2022, 7, 355–371. [Google Scholar] [CrossRef]
- Han, K.; Zhang, W.-Y.; Zhang, J.; Lei, Q.; Wang, S.-B.; Liu, J.W.; Zhang, X.Z.; Han, H.Y. Acidity-triggered tumor-targeted chimeric peptide for enhanced intra-nuclear photodynamic therapy. Adv. Funct. Mater. 2016, 26, 4351–4361. [Google Scholar] [CrossRef]
- Slastnikova, T.A.; Rosenkranz, A.A.; Gulak, P.V.; Schiffelers, R.M.; Lupanova, T.N.; Khramtsov, Y.V.; Zalutsky, M.R.; Sobolev, A.S. Modular nanotransporters: A multipurpose in vivo working platform for targeted drug delivery. Int. J. Nanomed. 2012, 7, 467–482. [Google Scholar]
- Rosenkranz, A.A.; Slastnikova, T.A.; Karmakova, T.A.; Vorontsova, M.S.; Morozova, N.B.; Petriev, V.M.; Abrosimov, A.S.; Khramtsov, Y.V.; Lupanova, T.N.; Ulasov, A.V.; et al. Antitumor Activity of Auger Electron Emitter (111)In Delivered by Modular Nanotransporter for Treatment of Bladder Cancer With EGFR Overexpression. Front. Pharmacol. 2018, 9, 1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bavelaar, B.M.; Lee, B.Q.; Gill, M.R.; Falzone, N.; Vallis, K.A. Subcellular Targeting of Theranostic Radionuclides. Front. Pharmacol. 2018, 9, 996. [Google Scholar] [CrossRef]
- Huang, S.; Zhu, Z.; Jia, B.; Zhang, W.; Song, J. Design of acid-activated cell-penetrating peptides with nuclear localization capacity for anticancer drug delivery. J. Pept. Sci. 2021, 27, e3354. [Google Scholar] [CrossRef]
- Pan, L.; Liu, J.; Shi, J. Nuclear-Targeting Gold Nanorods for Extremely Low NIR Activated Photothermal Therapy. ACS Appl. Mater. Interfaces. 2017, 9, 15952–15961. [Google Scholar] [CrossRef]
- Li, Q.; Yang, J.; Chen, C.; Lin, X.; Zhou, M.; Zhou, Z.; Huang, Y. A novel mitochondrial targeted hybrid peptide modified HPMA copolymers for breast cancer metastasis suppression. J. Control. Release 2020, 325, 38–51. [Google Scholar] [CrossRef]
- D’Souza, G.G.; Rammohan, R.; Cheng, S.M.; Torchilin, V.P.; Weissig, V. DQAsome-mediated delivery of plasmid DNA toward mitochondria in living cells. J. Control. Release 2003, 92, 189–197. [Google Scholar] [CrossRef]
- Shah, B.P.; Pasquale, N.; De, G.; Tan, T.; Ma, J.; Lee, K.B. Core-shell nanoparticle-based peptide therapeutics and combined hyperthermia for enhanced cancer cell apoptosis. ACS Nano 2014, 8, 9379–9387. [Google Scholar] [CrossRef] [Green Version]
- Yaqoob, M.D.; Xu, L.; Li, C.; Leong, M.M.L.; Xu, D.D. Targeting mitochondria for cancer photodynamic therapy. Photodiagnosis Photodyn. Ther. 2022, 38, 102830. [Google Scholar] [CrossRef]
- Jin, X.; Yang, H.; Mao, Z.; Wang, B. Cathepsin B-responsive multifunctional peptide conjugated gold nanorods for mitochondrial targeting and precise photothermal cancer therapy. J. Colloid Interface Sci. 2021, 601, 714–726. [Google Scholar] [CrossRef]
- Jiang, W.; Cheng, C.; Qiu, X.; Chen, L.; Guo, X.; Luo, Y.; Wang, J.; Wang, J.; Xie, Z.; Li, P.; et al. Peptide Supramolecular Assembly-Instructed In Situ Self-Aggregation for Stratified Targeting Sonodynamic Therapy Enhancement of AIE Luminogens. Adv. Sci. 2022, 10, e2204989. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug. Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primers. 2018, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Paulk, J. Lysosome-targeting chimeras evolve. Nat. Chem. Biol. 2021, 17, 931–933. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, B.; Pan, J.; Feng, Y.; Ye, W.; Xu, J.; Lan, M.; Sun, H.; Zhang, X.; Sun, Y.; et al. Construction and evaluation of DNA vaccine encoding Ebola virus glycoprotein fused with lysosome-associated membrane protein. Antivir. Res. 2021, 193, 105141. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Chambers, J.E.; Ron, D. Pharmacological targeting of endoplasmic reticulum stress in disease. Nat. Rev. Drug. Discov. 2022, 21, 115–140. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wang, S.; Wu, J.; Jin, X.; You, J. Pharmaceutical strategies for endoplasmic reticulum-targeting and their prospects of application. J. Control. Release 2021, 329, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Griffin, A.; Qiang, Z.; Ren, J. Organelle-targeted therapies: A comprehensive review on system design for enabling precision oncology. Signal. Transduct. Target Ther. 2022, 7, 379. [Google Scholar] [CrossRef]
- Jin, J.; Wu, Y.; Chen, J.; Shen, Y.; Zhang, L.; Zhang, H.; Chen, L.; Yuan, H.; Chen, H.; Zhang, W.; et al. The peptide PROTAC modality: A novel strategy for targeted protein ubiquitination. Theranostics 2020, 10, 10141–10153. [Google Scholar] [CrossRef]
- Cotton, A.D.; Nguyen, D.P.; Gramespacher, J.A.; Seiple, I.B.; Wells, J.A. Development of Antibody-Based PROTACs for the Degradation of the Cell-Surface Immune Checkpoint Protein PD-L1. J. Am. Chem. Soc. 2021, 143, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Costantini, D.L.; McLarty, K.; Lee, H.; Done, S.J.; Vallis, K.A.; Reilly, R.M. Antitumor effects and normal-tissue toxicity of 111In-nuclear localization sequence-trastuzumab in athymic mice bearing HER-positive human breast cancer xenografts. J. Nucl. Med. 2010, 51, 1084–1091. [Google Scholar] [CrossRef] [Green Version]
- Cornelissen, B.; Waller, A.; Target, C.; Kersemans, V.; Smart, S.; Vallis, K.A. 111In-BnDTPA-F3: An Auger electron-emitting radiotherapeutic agent that targets nucleolin. EJNMMI. Res. 2012, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Sobolev, A.S. The Delivery of Biologically Active Agents into the Nuclei of Target Cells for the Purposes of Translational Medicine. Acta Nat. 2020, 12, 47–56. [Google Scholar] [CrossRef]
- Li, Q.; Hao, X.; Zaidi, S.S.A.; Guo, J.; Ren, X.; Shi, C.; Zhang, W.; Feng, Y. Oligohistidine and targeting peptide functionalized TAT-NLS for enhancing cellular uptake and promoting angiogenesis in vivo. J. Nanobiotechnology 2018, 16, 29. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Sun, A.; Xu, R.; Tao, X.; Dong, Y.; Lv, X.; Wei, D. Cell-penetrating and endoplasmic reticulum-locating TAT-IL-24-KDEL fusion protein induces tumor apoptosis. J. Cell Physiol. 2016, 231, 84–93. [Google Scholar] [CrossRef]
- He, H.; Guo, J.; Lin, X.; Xu, B. Enzyme-Instructed Assemblies Enable Mitochondria Localization of Histone H2B in Cancer Cells. Angew. Chem. Int. Ed. Engl. 2020, 59, 9330–9334. [Google Scholar] [CrossRef]
- He, H.; Wang, J.; Wang, H.; Zhou, N.; Yang, D.; Green, D.R.; Xu, B. Enzymatic Cleavage of Branched Peptides for Targeting Mitochondria. J. Am. Chem. Soc. 2018, 140, 1215–1218. [Google Scholar] [CrossRef]
- Wagstaff, K.M.; Glover, D.J.; Tremethick, D.J.; Jans, D.A. Histone-mediated transduction as an efficient means for gene delivery. Mol. Ther. 2007, 15, 721–731. [Google Scholar] [CrossRef]
- Khan, I.; Saeed, K.; Khan, I. Nanoparticles: Properties, applications and toxicities. Arab. J. Chem. 2019, 12, 908–931. [Google Scholar] [CrossRef]
- Farokhzad, O.C.; Langer, R. Impact of nanotechnology on drug delivery. ACS Nano 2009, 3, 16–20. [Google Scholar] [CrossRef]
- Erickson, H.P. Size and shape of protein molecules at the nanometer level determined by sedimentation, gel filtration, and electron microscopy. Biol. Proced. Online 2009, 11, 32–51. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, N.; Leroux, J.C. The journey of a drug-carrier in the body: An anatomo-physiological perspective. J. Control. Release 2012, 161, 152–163. [Google Scholar] [CrossRef]
- Gustafson, H.H.; Holt-Casper, D.; Grainger, D.W.; Ghandehari, H. Nanoparticle Uptake: The Phagocyte Problem. Nano. Today 2015, 10, 487–510. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Yang, M.; Morimoto, T.; Tajima, N.; Ichiraku, K.; Fujita, K.; Iijima, S.; Yudasaka, M.; Okazaki, T. Size-dependent cell uptake of carbon nanotubes by macrophages: A comparative and quantitative study. Carbon 2018, 127, 93–101. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, X.; Huang, J.; Wang, J.; Wang, Z. Harnessing Protein Corona for Biomimetic Nanomedicine Design. Biomimetics 2022, 7, 126. [Google Scholar] [CrossRef] [PubMed]
- Sleep, D. Albumin and its application in drug delivery. Expert. Opin. Drug Deliv. 2015, 12, 793–812. [Google Scholar] [CrossRef]
- Challa, D.K.; Velmurugan, R.; Ober, R.J.; Sally, W.E. FcRn: From molecular interactions to regulation of IgG pharmacokinetics and functions. Curr. Top. Microbiol. Immunol. 2014, 382, 249–272. [Google Scholar] [PubMed]
- Chen, F.; Wang, G.; Griffin, J.I.; Brenneman, B.; Banda, N.K.; Holers, V.M.; Backos, D.S.; Wu, L.; Moghimi, S.M.; Simberg, D. Complement proteins bind to nanoparticle protein corona and undergo dynamic exchange in vivo. Nat. Nanotechnol. 2017, 12, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Owens, D.E., III; Peppas, N.A. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006, 307, 93–102. [Google Scholar] [CrossRef]
- Vu, V.P.; Gifford, G.B.; Chen, F.; Benasutti, H.; Wang, G.; Groman, E.V.; Scheinman, R.; Saba, L.; Moghimi, S.M.; Simberg, D. Immunoglobulin deposition on biomolecule corona determines complement opsonization efficiency of preclinical and clinical nanoparticles. Nat. Nanotechnol. 2019, 14, 260–268. [Google Scholar] [CrossRef]
- Sarin, H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J. Angiogenes Res. 2010, 2, 14. [Google Scholar] [CrossRef] [Green Version]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Maeda, H.; Nakamura, H.; Fang, J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv. Drug Deliv. Rev. 2013, 65, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv. Drug Deliv. Rev. 2011, 63, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv. Drug Deliv. Rev. 2015, 91, 3–6. [Google Scholar] [CrossRef]
- Nichols, J.W.; Bae, Y.H. EPR: Evidence and fallacy. J. Control. Release 2014, 190, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.; Wilhelm, S.; Ding, D.; Syed, A.M.; Sindhwani, S.; Zhang, Y.; Chen, Y.Y.; MacMillan, P.; Chan, W.C.W. Quantifying the Ligand-Coated Nanoparticle Delivery to Cancer Cells in Solid Tumors. ACS Nano. 2018, 12, 8423–8435. [Google Scholar] [CrossRef]
- Sindhwani, S.; Syed, A.M.; Ngai, J.; Kingston, B.R.; Maiorino, L.; Rothschild, J.; MacMillan, P.; Zhang, Y.; Rajesh, N.U.; Hoang, T.; et al. The entry of nanoparticles into solid tumours. Nat. Mater. 2020, 19, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Malik, A.B. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef]
- Fung, K.Y.Y.; Fairn, G.D.; Lee, W.L. Transcellular vesicular transport in epithelial and endothelial cells: Challenges and opportunities. Traffic 2018, 19, 5–18. [Google Scholar] [CrossRef] [Green Version]
- Yazdani, S.; Jaldin-Fincati, J.R.; Pereira, R.V.S.; Klip, A. Endothelial cell barriers: Transport of molecules between blood and tissues. Traffic 2019, 20, 390–403. [Google Scholar] [CrossRef] [Green Version]
- Komarova, Y.A.; Kruse, K.; Mehta, D.; Malik, A.B. Protein Interactions at Endothelial Junctions and Signaling Mechanisms Regulating Endothelial Permeability. Circ. Res. 2017, 120, 179–206. [Google Scholar] [CrossRef] [Green Version]
- Wettschureck, N.; Strilic, B.; Offermanns, S. Passing the Vascular Barrier: Endothelial Signaling Processes Controlling Extravasation. Physiol. Rev. 2019, 99, 1467–1525. [Google Scholar] [CrossRef] [PubMed]
- Pulgar, V.M. Transcytosis to Cross the Blood Brain Barrier, New Advancements and Challenges. Front Neurosci. 2018, 12, 1019. [Google Scholar] [CrossRef]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Lieleg, O.; Baumgartel, R.M.; Bausch, A.R. Selective filtering of particles by the extracellular matrix: An electrostatic bandpass. Biophys. J. 2009, 97, 1569–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stylianopoulos, T.; Poh, M.Z.; Insin, N.; Bawendi, M.G.; Fukumura, D.; Munn, L.L.; Jain, R.K. Diffusion of particles in the extracellular matrix: The effect of repulsive electrostatic interactions. Biophys. J. 2010, 99, 1342–1349. [Google Scholar] [CrossRef] [Green Version]
- Uhl, B.; Hirn, S.; Immler, R.; Mildner, K.; Mockl, L.; Sperandio, M.; Brauchle, C.; Reichel, C.A.; Zeuschner, D.; Krombach, F. The Endothelial Glycocalyx Controls Interactions of Quantum Dots with the Endothelium and Their Translocation across the Blood-Tissue Border. ACS Nano 2017, 11, 1498–1508. [Google Scholar] [CrossRef]
- Dolor, A.; Szoka, F.C., Jr. Digesting a Path Forward: The Utility of Collagenase Tumor Treatment for Improved Drug Delivery. Mol. Pharm. 2018, 15, 2069–2083. [Google Scholar] [CrossRef]
- Yang, H.; Tong, Z.; Sun, S.; Mao, Z. Enhancement of tumour penetration by nanomedicines through strategies based on transport processes and barriers. J. Control. Release 2020, 328, 28–44. [Google Scholar] [CrossRef] [PubMed]
- Christopher, J.A.; Stadler, C.; Martin, C.E.; Morgenstern, M.; Pan, Y.; Betsinger, C.N.; Rattray, D.G.; Mahdessian, D.; Gingras, A.C.; Warscheid, B.; et al. Subcellular proteomics. Nat. Rev. Methods Primers. 2021, 1, 32. [Google Scholar] [CrossRef]
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rennick, J.J.; Johnston, A.P.R.; Parton, R.G. Key principles and methods for studying the endocytosis of biological and nanoparticle therapeutics. Nat. Nanotechnol. 2021, 16, 266–276. [Google Scholar] [CrossRef]
- Mayor, S.; Parton, R.G.; Donaldson, J.G. Clathrin-independent pathways of endocytosis. Cold Spring Harb. Perspect. Biol. 2014, 6, a016758. [Google Scholar] [CrossRef] [Green Version]
- Casamento, A.; Boucrot, E. Molecular mechanism of Fast Endophilin-Mediated Endocytosis. Biochem. J. 2020, 477, 2327–2345. [Google Scholar] [CrossRef]
- Watanabe, S.; Boucrot, E. Fast and ultrafast endocytosis. Curr. Opin. Cell Biol. 2017, 47, 64–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renard, H.F.; Boucrot, E. Unconventional endocytic mechanisms. Curr. Opin. Cell Biol. 2021, 71, 120–129. [Google Scholar] [CrossRef]
- Grant, B.D.; Donaldson, J.G. Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2009, 10, 597–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef]
- Scott, C.C.; Vacca, F.; Gruenberg, J. Endosome maturation, transport and functions. Semin. Cell Dev. Biol. 2014, 31, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Bhuin, T.; Roy, J.K. Rab proteins: The key regulators of intracellular vesicle transport. Exp. Cell Res. 2014, 328, 1–19. [Google Scholar] [CrossRef]
- Kumar, A.; Ahmad, A.; Vyawahare, A.; Khan, R. Membrane Trafficking and Subcellular Drug Targeting Pathways. Front. Pharmacol. 2020, 11, 629. [Google Scholar] [CrossRef]
- Wente, S.R.; Rout, M.P. The nuclear pore complex and nuclear transport. Cold Spring Harb. Perspect. Biol. 2010, 2, a000562. [Google Scholar] [CrossRef]
- Hansen, K.G.; Herrmann, J.M. Transport of Proteins into Mitochondria. Protein J. 2019, 38, 330–342. [Google Scholar] [CrossRef]
- Kosugi, S.; Hasebe, M.; Matsumura, N.; Takashima, H.; Miyamoto-Sato, E.; Tomita, M.; Yanagawa, H. Six classes of nuclear localization signals specific to different binding grooves of importin alpha. J. Biol. Chem. 2009, 284, 478–485. [Google Scholar] [CrossRef] [Green Version]
- Soniat, M.; Chook, Y.M. Nuclear localization signals for four distinct karyopherin-beta nuclear import systems. Biochem. J. 2015, 468, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Wu, T.; Zhang, B.; Liu, S.; Song, W.; Qiao, J.; Ruan, H. Types of nuclear localization signals and mechanisms of protein import into the nucleus. Cell Commun. Signal. 2021, 19, 60. [Google Scholar] [CrossRef] [PubMed]
- Jans, D.A.; Xiao, C.Y.; Lam, M.H. Nuclear targeting signal recognition: A key control point in nuclear transport? Bioessays 2000, 22, 532–544. [Google Scholar] [CrossRef]
- Hampoelz, B.; Andres-Pons, A.; Kastritis, P.; Beck, M. Structure and Assembly of the Nuclear Pore Complex. Annu. Rev. Biophys. 2019, 48, 515–536. [Google Scholar] [CrossRef] [PubMed]
- Chacinska, A.; Koehler, C.M.; Milenkovic, D.; Lithgow, T.; Pfanner, N. Importing mitochondrial proteins: Machineries and mechanisms. Cell 2009, 138, 628–644. [Google Scholar] [CrossRef] [Green Version]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, O.; Pfanner, N.; Meisinger, C. Mitochondrial protein import: From proteomics to functional mechanisms. Nat. Rev. Mol. Cell Biol. 2010, 11, 655–667. [Google Scholar] [CrossRef]
- Schulz, C.; Schendzielorz, A.; Rehling, P. Unlocking the presequence import pathway. Trends Cell Biol. 2015, 25, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Callegari, S.; Cruz-Zaragoza, L.D.; Rehling, P. From TOM to the TIM23 complex-handing over of a precursor. Biol. Chem. 2020, 401, 709–721. [Google Scholar] [CrossRef]
- Guan, Z.; Yan, L.; Wang, Q.; Qi, L.; Hong, S.; Gong, Z.; Yan, C.; Yin, P. Structural insights into assembly of human mitochondrial translocase TOM complex. Cell Discov. 2021, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J. Macromolecular crowding: Obvious but underappreciated. Trends Biochem. Sci. 2001, 26, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, I.M.; Turoverov, K.K.; Uversky, V.N. What macromolecular crowding can do to a protein. Int. J. Mol. Sci. 2014, 15, 23090–23140. [Google Scholar] [CrossRef] [Green Version]
- Skora, T.; Vaghefikia, F.; Fitter, J.; Kondrat, S. Macromolecular Crowding: How Shape and Interactions Affect Diffusion. J. Phys. Chem. B 2020, 124, 7537–7543. [Google Scholar] [CrossRef] [PubMed]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Hyman, A.A.; Weber, C.A.; Julicher, F. Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 2014, 30, 39–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khramtsov, Y.V.; Ulasov, A.V.; Rosenkranz, A.A.; Slastnikova, T.A.; Lupanova, T.N.; Georgiev, G.P.; Sobolev, A.S. An Approach to Evaluate the Effective Cytoplasmic Concentration of Bioactive Agents Interacting with a Selected Intracellular Target Protein. Pharmaceutics 2023, 15, 324. [Google Scholar] [CrossRef] [PubMed]
- Jevsevar, S.; Kunstelj, M.; Porekar, V.G. PEGylation of therapeutic proteins. Biotechnol. J. 2010, 5, 113–128. [Google Scholar] [CrossRef] [Green Version]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U.S. Poly(ethylene glycol) in drug delivery: Pros and cons as well as potential alternatives. Angew. Chem. Int. Ed Engl. 2010, 49, 6288–6308. [Google Scholar] [CrossRef]
- Qi, Y.; Chilkoti, A. Protein-polymer conjugation-moving beyond PEGylation. Curr. Opin. Chem. Biol. 2015, 28, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Podust, V.N.; Balan, S.; Sim, B.C.; Coyle, M.P.; Ernst, U.; Peters, R.T.; Schellenberger, V. Extension of in vivo half-life of biologically active molecules by XTEN protein polymers. J. Control. Release 2016, 240, 52–66. [Google Scholar] [CrossRef] [Green Version]
- Schellenberger, V.; Wang, C.W.; Geething, N.C.; Spink, B.J.; Campbell, A.; To, W.; Scholle, M.D.; Yin, Y.; Yao, Y.; Bogin, O.; et al. A recombinant polypeptide extends the in vivo half-life of peptides and proteins in a tunable manner. Nat. Biotechnol. 2009, 27, 1186–1190. [Google Scholar] [CrossRef] [PubMed]
- Schlapschy, M.; Binder, U.; Borger, C.; Theobald, I.; Wachinger, K.; Kisling, S.; Haller, D.; Skerra, A. PASylation: A biological alternative to PEGylation for extending the plasma half-life of pharmaceutically active proteins. Protein Eng. Des. Sel. 2013, 26, 489–501. [Google Scholar] [CrossRef] [Green Version]
- Varanko, A.K.; Su, J.C.; Chilkoti, A. Elastin-Like Polypeptides for Biomedical Applications. Annu. Rev. Biomed. Eng. 2020, 22, 343–369. [Google Scholar] [CrossRef] [PubMed]
- Pastan, I.; Hassan, R.; Fitzgerald, D.J.; Kreitman, R.J. Immunotoxin therapy of cancer. Nat. Rev. Cancer 2006, 6, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Kamaletdinova, T.R.; Rosenkranz, A.A.; Ulasov, A.V.; Khramtsov, Y.V.; Tsvetkova, A.D.; Georgiev, G.P.; Sobolev, A.S. Modular Nanotransporter with P21 Fragment Inhibits DNA Repair after Bleomycin Treatment. Dokl. Biochem. Biophys. 2018, 479, 95–97. [Google Scholar] [CrossRef] [PubMed]
- Sobolev, A.S.; Jans, D.A.; Rosenkranz, A.A. Targeted intracellular delivery of photosensitizers. Prog. Biophys. Mol. Biol. 2000, 73, 51–90. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, A.A.; Salomone, S.; Sobolev, A.S. Editorial: Delivery of Locally-Acting Agents to Intracellular Targets. Front. Pharmacol. 2020, 11, 593064. [Google Scholar] [CrossRef]
- Rosenkranz, A.A.; Jans, D.A.; Sobolev, A.S. Targeted intracellular delivery of photosensitizers to enhance photodynamic efficiency. Immunol. Cell Biol. 2000, 78, 452–464. [Google Scholar] [CrossRef] [Green Version]
- Gilyazova, D.G.; Rosenkranz, A.A.; Gulak, P.V.; Lunin, V.G.; Sergienko, O.V.; Khramtsov, Y.V.; Timofeyev, K.N.; Grin, M.A.; Mironov, A.F.; Rubin, A.B.; et al. Targeting cancer cells by novel engineered modular transporters. Cancer Res. 2006, 66, 10534–10540. [Google Scholar] [CrossRef] [Green Version]
- Rosenkranz, A.A.; Lunin, V.G.; Gulak, P.V.; Sergienko, O.V.; Shumiantseva, M.A.; Voronina, O.L.; Gilyazova, D.G.; John, A.P.; Kofner, A.A.; Mironov, A.F.; et al. Recombinant modular transporters for cell-specific nuclear delivery of locally acting drugs enhance photosensitizer activity. FASEB J. 2003, 17, 1121–1123. [Google Scholar] [CrossRef] [Green Version]
- Rosenkranz, A.A.; Vaidyanathan, G.; Pozzi, O.R.; Lunin, V.G.; Zalutsky, M.R.; Sobolev, A.S. Engineered modular recombinant transporters: Application of new platform for targeted radiotherapeutic agents to alpha-particle emitting 211 At. Int. J. Radiat. Oncol. Biol. Phys. 2008, 72, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Koumarianou, E.; Slastnikova, T.A.; Pruszynski, M.; Rosenkranz, A.A.; Vaidyanathan, G.; Sobolev, A.S.; Zalutsky, M.R. Radiolabeling and in vitro evaluation of (67)Ga-NOTA-modular nanotransporter--a potential Auger electron emitting EGFR-targeted radiotherapeutic. Nucl. Med. Biol. 2014, 41, 441–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slastnikova, T.A.; Rosenkranz, A.A.; Morozova, N.B.; Vorontsova, M.S.; Petriev, V.M.; Lupanova, T.N.; Ulasov, A.V.; Zalutsky, M.R.; Yakubovskaya, R.I.; Sobolev, A.S. Preparation, cytotoxicity, and in vivo antitumor efficacy of (111)In-labeled modular nanotransporters. Int. J. Nanomed. 2017, 12, 395–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slastnikova, T.A.; Koumarianou, E.; Rosenkranz, A.A.; Vaidyanathan, G.; Lupanova, T.N.; Sobolev, A.S.; Zalutsky, M.R. Modular nanotransporters: A versatile approach for enhancing nuclear delivery and cytotoxicity of Auger electron-emitting 125I. EJNMMI Res. 2012, 2, 59. [Google Scholar] [CrossRef] [Green Version]
- Sobolev, A.S. Modular nanotransporters of anticancer drugs conferring cell specificity and higher efficiency. Biochemistry 2009, 74, 1567–1574. [Google Scholar] [CrossRef]
- Slastnikova, T.A.; Rosenkranz, A.A.; Lupanova, T.N.; Gulak, P.V.; Gnuchev, N.V.; Sobolev, A.S. Study of efficiency of the modular nanotransporter for targeted delivery of photosensitizers to melanoma cell nuclei in vivo. Dokl. Biochem. Biophys. 2012, 446, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Slastnikova, T.A.; Rosenkranz, A.A.; Khramtsov, Y.V.; Karyagina, T.S.; Ovechko, S.A.; Sobolev, A.S. Development and evaluation of a new modular nanotransporter for drug delivery into nuclei of pathological cells expressing folate receptors. Drug Des. Devel. Ther. 2017, 11, 1315–1334. [Google Scholar] [CrossRef] [Green Version]
- Desnoyers, L.R.; Vasiljeva, O.; Richardson, J.H.; Yang, A.; Menendez, E.E.; Liang, T.W.; Wong, C.; Bessette, P.H.; Kamath, K.; Moore, S.J.; et al. Tumor-specific activation of an EGFR-targeting probody enhances therapeutic index. Sci Transl. Med 2013, 5, 207ra144. [Google Scholar] [CrossRef]
- Mansurov, A.; Hosseinchi, P.; Chang, K.; Lauterbach, A.L.; Gray, L.T.; Alpar, A.T.; Budina, E.; Slezak, A.J.; Kang, S.; Cao, S.; et al. Masking the immunotoxicity of interleukin-12 by fusing it with a domain of its receptor via a tumour-protease-cleavable linker. Nat. Biomed. Eng 2022, 6, 819–829. [Google Scholar] [CrossRef]
- Trang, V.H.; Zhang, X.; Yumul, R.C.; Zeng, W.; Stone, I.J.; Wo, S.W.; Dominguez, M.M.; Cochran, J.H.; Simmons, J.K.; Ryan, M.C.; et al. A coiled-coil masking domain for selective activation of therapeutic antibodies. Nat. Biotechnol. 2019, 37, 761–765. [Google Scholar] [CrossRef]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Parray, H.A.; Shukla, S.; Samal, S.; Shrivastava, T.; Ahmed, S.; Sharma, C.; Kumar, R. Hybridoma technology a versatile method for isolation of monoclonal antibodies, its applicability across species, limitations, advancement and future perspectives. Int. Immunopharmacol. 2020, 85, 106639. [Google Scholar] [CrossRef]
- Shipunova, V.O.; Deyev, S.M. Artificial Scaffold Polypeptides As an Efficient Tool for the Targeted Delivery of Nanostructures In Vitro and In Vivo. Acta Nat. 2022, 14, 54–72. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. FDA approves 100th monoclonal antibody product. Nat. Rev. Drug Discov. 2021, 20, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, H.; Chenoweth, A.; Crescioli, S.; Reichert, J.M. Antibodies to watch in 2022. MAbs. 2022, 14, 2014296. [Google Scholar] [CrossRef]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar] [CrossRef]
- Urquhart, L. Top companies and drugs by sales in 2019. Nat. Rev. Drug Discov. 2020, 19, 228. [Google Scholar] [CrossRef]
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the ’undruggable’ cancer targets. Nat. Rev. Cancer 2017, 17, 502–508. [Google Scholar] [CrossRef]
- Ulasov, A.V.; Rosenkranz, A.A.; Sobolev, A.S. Transcription factors: Time to deliver. J. Control. Release 2018, 269, 24–35. [Google Scholar] [CrossRef]
- Stahl, S.; Graslund, T.; Eriksson, K.A.; Frejd, F.Y.; Nygren, P.A.; Lofblom, J. Affibody Molecules in Biotechnological and Medical Applications. Trends Biotechnol. 2017, 35, 691–712. [Google Scholar] [CrossRef] [PubMed]
- Deuschle, F.C.; Ilyukhina, E.; Skerra, A. Anticalin(R) proteins: From bench to bedside. Expert. Opin. Biol. Ther. 2021, 21, 509–518. [Google Scholar] [CrossRef]
- Pluckthun, A. Designed ankyrin repeat proteins (DARPins): Binding proteins for research, diagnostics, and therapy. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 489–511. [Google Scholar] [CrossRef]
- Kintzing, J.R.; Cochran, J.R. Engineered knottin peptides as diagnostics, therapeutics, and drug delivery vehicles. Curr. Opin. Chem. Biol. 2016, 34, 143–150. [Google Scholar] [CrossRef]
- Chandler, P.G.; Buckle, A.M. Development and Differentiation in Monobodies Based on the Fibronectin Type 3 Domain. Cells 2020, 9, 610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Yang, Y.P.; Dikici, E.; Deo, S.K.; Daunert, S. Beyond Antibodies as Binding Partners: The Role of Antibody Mimetics in Bioanalysis. Annu. Rev. Anal. Chem. 2017, 10, 293–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, D.; Humblet, Y.; Siena, S.; Khayat, D.; Bleiberg, H.; Santoro, A.; Bets, D.; Mueser, M.; Harstrick, A.; Verslype, C.; et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N. Engl. J. Med. 2004, 351, 337–345. [Google Scholar] [CrossRef] [Green Version]
- Suter, T.M.; Procter, M.; van Veldhuisen, D.J.; Muscholl, M.; Bergh, J.; Carlomagno, C.; Perren, T.; Passalacqua, R.; Bighin, C.; Klijn, J.G.; et al. Trastuzumab-associated cardiac adverse effects in the herceptin adjuvant trial. J. Clin. Oncol. 2007, 25, 3859–3865. [Google Scholar] [CrossRef]
- Nordberg, E.; Ekerljung, L.; Sahlberg, S.H.; Carlsson, J.; Lennartsson, J.; Glimelius, B. Effects of an EGFR-binding affibody molecule on intracellular signaling pathways. Int. J. Oncol. 2010, 36, 967–972. [Google Scholar]
- Baumdick, M.; Bruggemann, Y.; Schmick, M.; Xouri, G.; Sabet, O.; Davis, L.; Chin, J.W.; Bastiaens, P.I. EGF-dependent re-routing of vesicular recycling switches spontaneous phosphorylation suppression to EGFR signaling. Elife. 2015, 4, e12223. [Google Scholar] [CrossRef] [PubMed]
- Andreone, B.J.; Chow, B.W.; Tata, A.; Lacoste, B.; Ben-Zvi, A.; Bullock, K.; Deik, A.A.; Ginty, D.D.; Clish, C.B.; Gu, C. Blood-Brain Barrier Permeability Is Regulated by Lipid Transport-Dependent Suppression of Caveolae-Mediated Transcytosis. Neuron 2017, 94, 581–594. [Google Scholar] [CrossRef] [Green Version]
- Arredouani, M.S.; Palecanda, A.; Koziel, H.; Huang, Y.C.; Imrich, A.; Sulahian, T.H.; Ning, Y.Y.; Yang, Z.; Pikkarainen, T.; Sankala, M.; et al. MARCO is the major binding receptor for unopsonized particles and bacteria on human alveolar macrophages. J. Immunol. 2005, 175, 6058–6064. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, N.; Gomez, G.A.; Howes, M.T.; Lo, H.P.; McMahon, K.A.; Rae, J.A.; Schieber, N.L.; Hill, M.M.; Gaus, K.; Yap, A.S.; et al. Endocytic crosstalk: Cavins, caveolins, and caveolae regulate clathrin-independent endocytosis. PLoS Biol. 2014, 12, e1001832. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Xing, X.; Wang, X.; Wu, D.; Wu, W.; Guo, J.; Mitragotri, S. Nanocarrier-mediated cytosolic delivery of biopharmaceuticals. Adv. Funct. Mat. 2020, 30, 1910566. [Google Scholar] [CrossRef]
- Fu, A.; Tang, R.; Hardie, J.; Farkas, M.E.; Rotello, V.M. Promises and pitfalls of intracellular delivery of proteins. Bioconjug. Chem. 2014, 25, 1602–1608. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.W.; Luther, D.C.; Kretzmann, J.A.; Burden, A.; Jeon, T.; Zhai, S.; Rotello, V.M. Protein Delivery into the Cell Cytosol using Non-Viral Nanocarriers. Theranostics 2019, 9, 3280–3292. [Google Scholar] [CrossRef]
- Rosenkranz, A.A.; Ulasov, A.V.; Slastnikova, T.A.; Khramtsov, Y.V.; Sobolev, A.S. Use of intracellular transport processes for targeted drug delivery into a specified cellular compartment. Biochemistry 2014, 79, 928–946. [Google Scholar] [CrossRef] [PubMed]
- Brock, D.J.; Kondow-McConaghy, H.M.; Hager, E.C.; Pellois, J.P. Endosomal Escape and Cytosolic Penetration of Macromolecules Mediated by Synthetic Delivery Agents. Bioconjug. Chem. 2019, 30, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Varkouhi, A.K.; Scholte, M.; Storm, G.; Haisma, H.J. Endosomal escape pathways for delivery of biologicals. J. Control. Release 2011, 151, 220–228. [Google Scholar] [CrossRef]
- Volta-Duran, E.; Parlade, E.; Serna, N.; Villaverde, A.; Vazquez, E.; Unzueta, U. Endosomal escape for cell-targeted proteins. Going out after going in. Biotechnol. Adv. 2023, 63, 108103. [Google Scholar] [CrossRef]
- Ahmad, A.; Khan, J.M.; Haque, S. Strategies in the design of endosomolytic agents for facilitating endosomal escape in nanoparticles. Biochimie 2019, 160, 61–75. [Google Scholar] [CrossRef]
- Kim, G.C.; Cheon, D.H.; Lee, Y. Challenge to overcome current limitations of cell-penetrating peptides. Biochim. Biophys. Acta Proteins Proteom. 2021, 1869, 140604. [Google Scholar] [CrossRef] [PubMed]
- Sahni, A.; Qian, Z.; Pei, D. Cell-Penetrating Peptides Escape the Endosome by Inducing Vesicle Budding and Collapse. ACS Chem. Biol. 2020, 15, 2485–2492. [Google Scholar] [CrossRef]
- Sobolev, A.S. Modular Nanotransporters for Nuclear-Targeted Delivery of Auger Electron Emitters. Front. Pharmacol. 2018, 9, 952. [Google Scholar] [CrossRef]
- Xu, E.; Saltzman, W.M.; Piotrowski-Daspit, A.S. Escaping the endosome: Assessing cellular trafficking mechanisms of non-viral vehicles. J. Control. Release 2021, 335, 465–480. [Google Scholar] [CrossRef]
- Ahmad, A.; Khan, J.M. pH-sensitive endosomolytic peptides in gene and drug delivery: Endosomal escape and current challenges. J. Drug Deliv. Sci. Technol. 2022, 76, 103786. [Google Scholar] [CrossRef]
- Pei, D.; Buyanova, M. Overcoming Endosomal Entrapment in Drug Delivery. Bioconjug. Chem. 2019, 30, 273–283. [Google Scholar] [CrossRef]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef]
- Ruseska, I.; Zimmer, A. Internalization mechanisms of cell-penetrating peptides. Beilstein. J. Nanotechnol. 2020, 11, 101–123. [Google Scholar] [CrossRef] [PubMed]
- Koren, E.; Torchilin, V.P. Cell-penetrating peptides: Breaking through to the other side. Trends Mol. Med. 2012, 18, 385–393. [Google Scholar] [CrossRef]
- Kauffman, W.B.; Fuselier, T.; He, J.; Wimley, W.C. Mechanism Matters: A Taxonomy of Cell Penetrating Peptides. Trends Biochem. Sci. 2015, 40, 749–764. [Google Scholar] [CrossRef] [Green Version]
- Murphy, J.R. Mechanism of diphtheria toxin catalytic domain delivery to the eukaryotic cell cytosol and the cellular factors that directly participate in the process. Toxins 2011, 3, 294–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moayeri, M.; Leppla, S.H.; Vrentas, C.; Pomerantsev, A.P.; Liu, S. Anthrax Pathogenesis. Annu. Rev. Microbiol. 2015, 69, 185–208. [Google Scholar] [CrossRef] [PubMed]
- Haug, G.; Leemhuis, J.; Tiemann, D.; Meyer, D.K.; Aktories, K.; Barth, H. The host cell chaperone Hsp90 is essential for translocation of the binary Clostridium botulinum C2 toxin into the cytosol. J. Biol. Chem. 2003, 278, 32266–32274. [Google Scholar] [CrossRef] [Green Version]
- Gerding, D.N.; Johnson, S.; Rupnik, M.; Aktories, K. Clostridium difficile binary toxin CDT: Mechanism, epidemiology, and potential clinical importance. Gut Microbes 2014, 5, 15–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papatheodorou, P.; Barth, H.; Minton, N.; Aktories, K. Cellular Uptake and Mode-of-Action of Clostridium difficile Toxins. Adv. Exp. Med. Biol. 2018, 1050, 77–96. [Google Scholar]
- Stiles, B.G.; Wigelsworth, D.J.; Popoff, M.R.; Barth, H. Clostridial binary toxins: Iota and C2 family portraits. Front. Cell Infect. Microbiol. 2011, 1, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haywood, E.E.; Handy, N.B.; Lopez, J.W.; Ho, M.; Wilson, B.A. Insertion-trigger residues differentially modulate endosomal escape by cytotoxic necrotizing factor toxins. J. Biol. Chem. 2021, 297, 101347. [Google Scholar] [CrossRef]
- Gatsogiannis, C.; Merino, F.; Prumbaum, D.; Roderer, D.; Leidreiter, F.; Meusch, D.; Raunser, S. Membrane insertion of a Tc toxin in near-atomic detail. Nat. Struct. Mol. Biol. 2016, 23, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Pentelute, B.L.; Collier, R.J.; Zhou, Z.H. Atomic structure of anthrax protective antigen pore elucidates toxin translocation. Nature 2015, 521, 545–549. [Google Scholar] [CrossRef]
- Kagan, B.L.; Finkelstein, A.; Colombini, M. Diphtheria toxin fragment forms large pores in phospholipid bilayer membranes. Proc. Natl. Acad. Sci. USA 1981, 78, 4950–4954. [Google Scholar] [CrossRef] [Green Version]
- Khramtsov, Y.V.; Rokitskaya, T.I.; Rosenkranz, A.A.; Trusov, G.A.; Gnuchev, N.V.; Antonenko, Y.N.; Sobolev, A.S. Modular drug transporters with diphtheria toxin translocation domain form edged holes in lipid membranes. J. Control. Release 2008, 128, 241–247. [Google Scholar] [CrossRef]
- Mondal, A.K.; Lata, K.; Singh, M.; Chatterjee, S.; Chauhan, A.; Puravankara, S.; Chattopadhyay, K. Cryo-EM elucidates mechanism of action of bacterial pore-forming toxins. Biochim. Biophys. Acta Biomembr. 2022, 1864, 184013. [Google Scholar] [CrossRef]
- Sharpe, J.C.; London, E. Diphtheria toxin forms pores of different sizes depending on its concentration in membranes: Probable relationship to oligomerization. J. Membr. Biol. 1999, 171, 209–221. [Google Scholar] [CrossRef]
- Barth, H. Exploring the role of host cell chaperones/PPIases during cellular up-take of bacterial ADP-ribosylating toxins as basis for novel pharmacological strategies to protect mammalian cells against these virulence factors. Naunyn Schmiedebergs Arch. Pharmacol. 2011, 383, 237–245. [Google Scholar] [CrossRef]
- Sly, W.S.; Fischer, H.D. The phosphomannosyl recognition system for intracellular and intercellular transport of lysosomal enzymes. J. Cell Biochem. 1982, 18, 67–85. [Google Scholar] [CrossRef]
- Auger, A.; Park, M.; Nitschke, F.; Minassian, L.M.; Beilhartz, G.L.; Minassian, B.A.; Melnyk, R.A. Efficient Delivery of Structurally Diverse Protein Cargo into Mammalian Cells by a Bacterial Toxin. Mol. Pharm. 2015, 12, 2962–2971. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Nicol, F.; Szoka, F.C., Jr. GALA: A designed synthetic pH-responsive amphipathic peptide with applications in drug and gene delivery. Adv. Drug Deliv. Rev. 2004, 56, 967–985. [Google Scholar] [CrossRef]
- Lin, D.H.; Hoelz, A. The Structure of the Nuclear Pore Complex (An Update). Annu. Rev. Biochem. 2019, 88, 725–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timney, B.L.; Raveh, B.; Mironska, R.; Trivedi, J.M.; Kim, S.J.; Russel, D.; Wente, S.R.; Sali, A.; Rout, M.P. Simple rules for passive diffusion through the nuclear pore complex. J. Cell Biol. 2016, 215, 57–76. [Google Scholar] [CrossRef] [Green Version]
- Rosenkranz, A.A.; Slastnikova, T.A.; Durymanov, M.O.; Georgiev, G.P.; Sobolev, A.S. Exploiting active nuclear import for efficient delivery of Auger electron emitters into the cell nucleus. Int. J. Radiat. Biol. 2020, 99, 28–38. [Google Scholar] [CrossRef]
- Keiko, L.H.; Hasegawa, S. Favorable tumor uptake and nuclear transport of Auger electrons by nuclear targeting with 111In-trastuzumab in an intraperitoneal tumor mouse model. Nucl. Med. Commun. 2022, 43, 763–769. [Google Scholar] [CrossRef]
- Hoang, B.; Reilly, R.M.; Allen, C. Block copolymer micelles target Auger electron radiotherapy to the nucleus of HER2-positive breast cancer cells. Biomacromolecules 2012, 13, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, B.; Vallis, K.A. Targeting the nucleus: An overview of Auger-electron radionuclide therapy. Curr. Drug Discov. Technol. 2010, 7, 263–279. [Google Scholar] [CrossRef] [PubMed]
- Karyagina, T.S.; Ulasov, A.V.; Slastnikova, T.A.; Rosenkranz, A.A.; Lupanova, T.N.; Khramtsov, Y.V.; Georgiev, G.P.; Sobolev, A.S. Targeted Delivery of (111)In Into the Nuclei of EGFR Overexpressing Cells via Modular Nanotransporters With Anti-EGFR Affibody. Front. Pharmacol. 2020, 11, 176. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.Y.; Shen, J.M.; Lang, H.; Yue, T.; Sun, C. pH/enzyme dual sensitive and nucleus-targeting dendrimer nanoparticles to enhance the antitumour activity of doxorubicin. Pharm. Dev. Technol. 2022, 27, 357–371. [Google Scholar] [CrossRef]
- Pouton, C.W.; Wagstaff, K.M.; Roth, D.M.; Moseley, G.W.; Jans, D.A. Targeted delivery to the nucleus. Adv. Drug Deliv. Rev. 2007, 59, 698–717. [Google Scholar] [CrossRef]
- Luby-Phelps, K. The physical chemistry of cytoplasm and its influence on cell function: An update. Mol. Biol. Cell 2013, 24, 2593–2596. [Google Scholar] [CrossRef] [PubMed]
- Mogre, S.; Brown, A.I.; Koslover, E.F. Getting around the cell: Physical transport in the intracellular world. Phys. Biol. 2020, 17, 061003. [Google Scholar] [CrossRef]
- Dalmau-Mena, I.; Del, P.P.; Pelaz, B.; Cuesta-Geijo, M.A.; Galindo, I.; Moros, M.; de la Fuente, J.M.; Alonso, C. Nanoparticles engineered to bind cellular motors for efficient delivery. J. Nanobiotechnology 2018, 16, 33. [Google Scholar] [CrossRef]
- Moseley, G.W.; Roth, D.M.; DeJesus, M.A.; Leyton, D.L.; Filmer, R.P.; Pouton, C.W.; Jans, D.A. Dynein light chain association sequences can facilitate nuclear protein import. Mol. Biol. Cell 2007, 18, 3204–3213. [Google Scholar] [CrossRef] [Green Version]
- Fox, T.D. Mitochondrial protein synthesis, import, and assembly. Genetics 2012, 192, 1203–1234. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Bruno, B.J.; Rabenau, M.; Lim, C.S. Delivery of drugs and macromolecules to the mitochondria for cancer therapy. J. Control. Release 2016, 240, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, E.; Tricarico, R.; Savage, M.; Golemis, E.A.; Hall, M.J. Disease-Associated Genetic Variation in Human Mitochondrial Protein Import. Am. J. Hum. Genet. 2019, 104, 784–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moulin, C.; Caumont-Sarcos, A.; Ieva, R. Mitochondrial presequence import: Multiple regulatory knobs fine-tune mitochondrial biogenesis and homeostasis. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 930–944. [Google Scholar] [CrossRef]
- Edwards, R.; Gerlich, S.; Tokatlidis, K. The biogenesis of mitochondrial intermembrane space proteins. Biol. Chem. 2020, 401, 737–747. [Google Scholar]
- Edwards, R.; Eaglesfield, R.; Tokatlidis, K. The mitochondrial intermembrane space: The most constricted mitochondrial sub-compartment with the largest variety of protein import pathways. Open. Biol. 2021, 11, 210002. [Google Scholar] [CrossRef]
- Song, J.; Herrmann, J.M.; Becker, T. Quality control of the mitochondrial proteome. Nat. Rev. Mol. Cell Biol. 2021, 22, 54–70. [Google Scholar] [CrossRef] [PubMed]
- Bykov, Y.S.; Rapaport, D.; Herrmann, J.M.; Schuldiner, M. Cytosolic Events in the Biogenesis of Mitochondrial Proteins. Trends Biochem. Sci. 2020, 45, 650–667. [Google Scholar] [CrossRef]
- Yamada, Y.; Hibino, M.; Sasaki, D.; Abe, J.; Harashima, H. Power of mitochondrial drug delivery systems to produce innovative nanomedicines. Adv. Drug Deliv. Rev. 2020, 154–155, 187–209. [Google Scholar] [CrossRef]
- Cerrato, C.P.; Langel, U. An update on cell-penetrating peptides with intracellular organelle targeting. Expert Opin. Drug Deliv. 2022, 19, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Jean, S.R.; Ahmed, M.; Lei, E.K.; Wisnovsky, S.P.; Kelley, S.O. Peptide-Mediated Delivery of Chemical Probes and Therapeutics to Mitochondria. Acc. Chem. Res. 2016, 49, 1893–1902. [Google Scholar] [CrossRef]
- Kim, S.; Nam, H.Y.; Lee, J.; Seo, J. Mitochondrion-Targeting Peptides and Peptidomimetics: Recent Progress and Design Principles. Biochemistry 2020, 59, 270–284. [Google Scholar] [CrossRef]
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 447–464. [Google Scholar] [CrossRef]
- Kim, P.K.; Hettema, E.H. Multiple pathways for protein transport to peroxisomes. J. Mol. Biol. 2015, 427 6 Pt A, 1176–1190. [Google Scholar] [CrossRef] [Green Version]
- Islinger, M.; Voelkl, A.; Fahimi, H.D.; Schrader, M. The peroxisome: An update on mysteries 2.0. Histochem. Cell Biol. 2018, 150, 443–471. [Google Scholar] [CrossRef] [Green Version]
- Terlecky, S.R.; Koepke, J.I. Drug delivery to peroxisomes: Employing unique trafficking mechanisms to target protein therapeutics. Adv. Drug Deliv. Rev. 2007, 59, 739–747. [Google Scholar] [CrossRef]
- Roncador, A.; Oppici, E.; Talelli, M.; Pariente, A.N.; Donini, M.; Dusi, S.; Voltattorni, C.B.; Vicent, M.J.; Cellini, B. Use of polymer conjugates for the intraperoxisomal delivery of engineered human alanine:glyoxylate aminotransferase as a protein therapy for primary hyperoxaluria type I. Nanomedicine 2017, 13, 897–907. [Google Scholar] [CrossRef]
- Sugiman-Marangos, S.N.; Beilhartz, G.L.; Zhao, X.; Zhou, D.; Hua, R.; Kim, P.K.; Rini, J.M.; Minassian, B.A.; Melnyk, R.A. Exploiting the diphtheria toxin internalization receptor enhances delivery of proteins to lysosomes for enzyme replacement therapy. Sci. Adv. 2020, 6, eabb0385. [Google Scholar] [CrossRef]
- Gary-Bobo, M.; Nirde, P.; Jeanjean, A.; Morere, A.; Garcia, M. Mannose 6-phosphate receptor targeting and its applications in human diseases. Curr. Med. Chem. 2007, 14, 2945–2953. [Google Scholar] [CrossRef] [Green Version]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef]
- Progida, C.; Bakke, O. Bidirectional traffic between the Golgi and the endosomes-machineries and regulation. J. Cell Sci. 2016, 129, 3971–3982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, Y.; Zhao, L.; Billadeau, D.D.; Jia, D. Endosome-to-TGN Trafficking: Organelle-Vesicle and Organelle-Organelle Interactions. Front. Cell Dev. Biol. 2020, 8, 163. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, P.; Dahms, N.M.; Kornfeld, S. Mannose 6-phosphate receptors: New twists in the tale. Nat. Rev. Mol. Cell Biol. 2003, 4, 202–212. [Google Scholar] [CrossRef]
- Lane, R.F.; St George-Hyslop, P.; Hempstead, B.L.; Small, S.A.; Strittmatter, S.M.; Gandy, S. Vps10 family proteins and the retromer complex in aging-related neurodegeneration and diabetes. J. Neurosci. 2012, 32, 14080–14086. [Google Scholar] [CrossRef] [Green Version]
- Braun, E.; Sauter, D. Furin-mediated protein processing in infectious diseases and cancer. Clin. Transl. Immunol. 2019, 8, e1073. [Google Scholar] [CrossRef] [Green Version]
- Sowa-Rogozinska, N.; Sominka, H.; Nowakowska-Golacka, J.; Sandvig, K.; Slominska-Wojewodzka, M. Intracellular Transport and Cytotoxicity of the Protein Toxin Ricin. Toxins 2019, 11, 350. [Google Scholar] [CrossRef] [Green Version]
- Sandvig, K.; Bergan, J.; Dyve, A.B.; Skotland, T.; Torgersen, M.L. Endocytosis and retrograde transport of Shiga toxin. Toxicon 2010, 56, 1181–1185. [Google Scholar] [CrossRef]
- Wernick, N.L.; Chinnapen, D.J.; Cho, J.A.; Lencer, W.I. Cholera toxin: An intracellular journey into the cytosol by way of the endoplasmic reticulum. Toxins 2010, 2, 310–325. [Google Scholar] [CrossRef] [Green Version]
- Beddoe, T.; Paton, A.W.; Le, N.J.; Rossjohn, J.; Paton, J.C. Structure, biological functions and applications of the AB5 toxins. Trends Biochem. Sci. 2010, 35, 411–418. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Xu, N.; Xu, W.; Ling, G.; Zhang, P. Potential therapies and diagnosis based on Golgi-targeted nano drug delivery systems. Pharmacol. Res. 2022, 175, 105861. [Google Scholar] [CrossRef]
- Thomas, L.L.; Fromme, J.C. Extensive GTPase crosstalk regulates Golgi trafficking and maturation. Curr. Opin. Cell Biol. 2020, 65, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lujan, P.; Campelo, F. Should I stay or should I go? Golgi membrane spatial organization for protein sorting and retention. Arch. Biochem. Biophys. 2021, 707, 108921. [Google Scholar] [CrossRef] [PubMed]
- Papanikou, E.; Glick, B.S. Golgi compartmentation and identity. Curr. Opin. Cell Biol. 2014, 29, 74–81. [Google Scholar] [CrossRef] [Green Version]
- Tu, L.; Tai, W.C.; Chen, L.; Banfield, D.K. Signal-mediated dynamic retention of glycosyltransferases in the Golgi. Science 2008, 321, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Sirkis, D.W.; Schekman, R. Protein sorting at the trans-Golgi network. Annu. Rev. Cell Dev. Biol. 2014, 30, 169–206. [Google Scholar] [CrossRef] [PubMed]
- Ford, C.; Parchure, A.; Von, B.J.; Burd, C.G. Cargo sorting at the trans-Golgi network at a glance. J. Cell Sci. 2021, 134, jcs259110. [Google Scholar] [CrossRef]
- Rizzo, R.; Parashuraman, S.; Mirabelli, P.; Puri, C.; Lucocq, J.; Luini, A. The dynamics of engineered resident proteins in the mammalian Golgi complex relies on cisternal maturation. J. Cell Biol. 2013, 201, 1027–1036. [Google Scholar] [CrossRef] [Green Version]
- Brauer, P.; Parker, J.L.; Gerondopoulos, A.; Zimmermann, I.; Seeger, M.A.; Barr, F.A.; Newstead, S. Structural basis for pH-dependent retrieval of ER proteins from the Golgi by the KDEL receptor. Science 2019, 363, 1103–1107. [Google Scholar] [CrossRef]
- Valm, A.M.; Cohen, S.; Legant, W.R.; Melunis, J.; Hershberg, U.; Wait, E.; Cohen, A.R.; Davidson, M.W.; Betzig, E.; Lippincott-Schwartz, J. Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature 2017, 546, 162–167. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Chen, J.; Liu, H.; Chen, S.; Zhang, Y.; Li, P.; Thierry-Mieg, D.; Thierry-Mieg, J.; Mattes, W.; Ning, B.; et al. Comprehensive Identification and Characterization of Human Secretome Based on Integrative Proteomic and Transcriptomic Data. Front. Cell Dev. Biol. 2019, 7, 299. [Google Scholar] [CrossRef] [Green Version]
- Dobson, L.; Remenyi, I.; Tusnady, G.E. The human transmembrane proteome. Biol. Direct 2015, 10, 31. [Google Scholar] [CrossRef] [Green Version]
- Pereira, F.; Rettel, M.; Stein, F.; Savitski, M.M.; Collinson, I.; Romisch, K. Effect of Sec61 interaction with Mpd1 on endoplasmic reticulum-associated degradation. PLoS ONE 2019, 14, e0211180. [Google Scholar] [CrossRef] [Green Version]
- Romisch, K. A case for Sec61 channel involvement in ERAD. Trends Biochem. Sci. 2017, 42, 171–179. [Google Scholar] [CrossRef]
- Schoebel, S.; Mi, W.; Stein, A.; Ovchinnikov, S.; Pavlovicz, R.; DiMaio, F.; Baker, D.; Chambers, M.G.; Su, H.; Li, D.; et al. Cryo-EM structure of the protein-conducting ERAD channel Hrd1 in complex with Hrd3. Nature 2017, 548, 352–355. [Google Scholar] [CrossRef] [Green Version]
- Rao, B.; Li, S.; Yao, D.; Wang, Q.; Xia, Y.; Jia, Y.; Shen, Y.; Cao, Y. The cryo-EM structure of an ERAD protein channel formed by tetrameric human Derlin-1. Sci. Adv. 2021, 7, eabe8591. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Siggel, M.; Ovchinnikov, S.; Mi, W.; Svetlov, V.; Nudler, E.; Liao, M.; Hummer, G.; Rapoport, T.A. Structural basis of ER-associated protein degradation mediated by the Hrd1 ubiquitin ligase complex. Science 2020, 368, eaaz2449. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef]
- Guerriero, C.J.; Brodsky, J.L. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol. Rev. 2012, 92, 537–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenkranz, A.A.; Slastnikova, T.A. Epidermal Growth Factor Receptor: Key to Selective Intracellular Delivery. Biochemistry 2020, 85, 967–1092. [Google Scholar] [CrossRef]
- Maisel, S.A.; Schroeder, J. Wrong place at the wrong time: How retrograde trafficking drives cancer metastasis through receptor mislocalization. J. Cancer Metastasis Treat. 2019, 5, 7. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.A.; Chinnapen, D.J.; Aamar, E.; te Welscher, Y.M.; Lencer, W.I.; Massol, R. Insights on the trafficking and retro-translocation of glycosphingolipid-binding bacterial toxins. Front. Cell Infect. Microbiol. 2012, 2, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowakowska-Golacka, J.; Sominka, H.; Sowa-Rogozinska, N.; Slominska-Wojewodzka, M. Toxins Utilize the Endoplasmic Reticulum-Associated Protein Degradation Pathway in Their Intoxication Process. Int. J. Mol. Sci. 2019, 20, 1307. [Google Scholar] [CrossRef] [Green Version]
- Teter, K. Intracellular Trafficking and Translocation of Pertussis Toxin. Toxins 2019, 11, 437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, N.; Powis, K.; High, S. Post-translational translocation into the endoplasmic reticulum. Biochim. Biophys. Acta 2013, 1833, 2403–2409. [Google Scholar] [CrossRef] [PubMed]
- Luginbuehl, V.; Meier, N.; Kovar, K.; Rohrer, J. Intracellular drug delivery: Potential usefulness of engineered Shiga toxin subunit B for targeted cancer therapy. Biotechnol. Adv. 2018, 36, 613–623. [Google Scholar] [CrossRef]
- Kakimoto, S.; Hamada, T.; Komatsu, Y.; Takagi, M.; Tanabe, T.; Azuma, H.; Shinkai, S.; Nagasaki, T. The conjugation of diphtheria toxin T domain to poly(ethylenimine) based vectors for enhanced endosomal escape during gene transfection. Biomaterials 2009, 30, 402–408. [Google Scholar] [CrossRef]
- Vargason, A.M.; Anselmo, A.C.; Mitragotri, S. The evolution of commercial drug delivery technologies. Nat. Biomed. Eng. 2021, 5, 951–967. [Google Scholar] [CrossRef]
- Shilova, O.; Kotelnikova, P.; Proshkina, G.; Shramova, E.; Deyev, S. Barnase-Barstar Pair: Contemporary Application in Cancer Research and Nanotechnology. Molecules 2021, 26, 6785. [Google Scholar] [CrossRef]
- Shim, H. Bispecific Antibodies and Antibody-Drug Conjugates for Cancer Therapy: Technological Considerations. Biomolecules 2020, 10, 360. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.J.; Geeson, M.B.; Journeaux, T.; Bernardes, G.J.L. Chemical and Enzymatic Methods for Post-Translational Protein-Protein Conjugation. J. Am. Chem. Soc. 2022, 144, 14404–14419. [Google Scholar] [CrossRef] [PubMed]
- Ta, D.T.; Redeker, E.S.; Billen, B.; Reekmans, G.; Sikulu, J.; Noben, J.P.; Guedens, W.; Adriaensens, P. An efficient protocol towards site-specifically clickable nanobodies in high yield: Cytoplasmic expression in Escherichia coli combined with intein-mediated protein ligation. Protein Eng. Des. Sel. 2015, 28, 351–363. [Google Scholar] [CrossRef] [Green Version]
- Grahn, A.K.; Miller, C.J.; Kay, B.K. Plug and play with recombinant antibody fragments. Cell Chem. Biol. 2021, 28, 745–747. [Google Scholar] [CrossRef] [PubMed]
- Pearce, R.; Zhang, Y. Toward the solution of the protein structure prediction problem. J. Biol. Chem. 2021, 297, 100870. [Google Scholar] [CrossRef] [PubMed]
- Porta-Pardo, E.; Ruiz-Serra, V.; Valentini, S.; Valencia, A. The structural coverage of the human proteome before and after AlphaFold. PLoS Comput. Biol. 2022, 18, e1009818. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Zhou, X.; Zheng, W.; Li, Y.; Pearce, R.; Zhang, C.; Bell, E.W.; Zhang, G.; Zhang, Y. I-TASSER-MTD: A deep-learning-based platform for multi-domain protein structure and function prediction. Nat. Protoc. 2022, 17, 2326–2353. [Google Scholar] [CrossRef]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Zidek, A.; Bates, R.; Blackwell, S.; Yim, J. Protein complex prediction with AlphaFold-Multimer. BioRxiv 2022, 2021-10. [Google Scholar]
- Ruff, K.M.; Pappu, R.V. AlphaFold and Implications for Intrinsically Disordered Proteins. J. Mol. Biol. 2021, 433, 167208. [Google Scholar] [PubMed]
- Khramtsov, Y.V.; Vlasova, A.D.; Vlasov, A.V.; Rosenkranz, A.A.; Ulasov, A.V.; Ryzhykau, Y.L.; Kuklin, A.I.; Orekhov, A.S.; Eydlin, I.B.; Georgiev, G.P.; et al. Low-resolution structures of modular nanotransporters shed light on their functional activity. Acta Crystallogr. D. Struct. Biol. 2020, 76 Pt 12, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Luck, K.; Kim, D.K.; Lambourne, L.; Spirohn, K.; Begg, B.E.; Bian, W.; Brignall, R.; Cafarelli, T.; Campos-Laborie, F.J.; Charloteaux, B.; et al. A reference map of the human binary protein interactome. Nature 2020, 580, 402–408. [Google Scholar] [PubMed]
- Huttlin, E.L.; Bruckner, R.J.; Navarrete-Perea, J.; Cannon, J.R.; Baltier, K.; Gebreab, F.; Gygi, M.P.; Thornock, A.; Zarraga, G.; Tam, S.; et al. Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell 2021, 184, 3022–3040. [Google Scholar] [CrossRef] [PubMed]
- Kondratyeva, L.; Alekseenko, I.; Chernov, I.; Sverdlov, E. Data Incompleteness May form a Hard-to-Overcome Barrier to Decoding Life’s Mechanism. Biology 2022, 11, 1208. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compartment | All | Cell Type Enriched * | Group Enriched ** | Low Cell Type Specificity |
|---|---|---|---|---|
| Plasma membrane | 1999 | 152 | 286 | 258 |
| Cytosol *** | 5550 | 330 | 576 | 1203 |
| Nucleus **** | 7285 | 385 | 725 | 1926 |
| Mitochondria | 1109 | 46 | 100 | 263 |
| Endoplasmic reticulum | 527 | 43 | 51 | 145 |
| Golgi apparatus | 1134 | 87 | 139 | 218 |
| Delivery System | Cargo | Targeted Subcellular Compartment/Complex | Clinical Use | Refs. | |
|---|---|---|---|---|---|
| Delivered | Recruited | ||||
| Antibody drug conjugates | Wide panel of cytotoxic agents (e.g., methotrexate, doxorubicin, radionuclides, calicheamicin) | Usually lysosome, where the drug is cleaved from the conjugate | More than 10 are already used in clinical settings, e.g., trastuzumab emtansine (Kadcyla), inotuzumab ozogamicin (Besponsa), brentuximab vedotin (Adcetris) and others | [28] | |
| Immunotoxins | Catalytic subunits of natural toxins (pseudomonas exotoxin, diphtheria toxin) | Cytosol | Three immunotoxins have been approved for clinical use, with one of them—IL-3 fused to a fragment of diphtheria toxin (Tagraxofusp)—currently used in clinical settings | [27] | |
| p-PROTACs (peptide-based proteolysis-targeting chimaeras) | Intracellular proteins of interest (e.g., protein kinases, estrogen receptor, Tau-protein) | Proteasome | [64] | ||
| LYTACs (lysosome-targeting chimaeras) | Extracellular and membrane-associated proteins of interest (e.g., EGFR, PD-L1, transferrin receptor-1) | Lysosome | [58] | ||
| AbTACs (antibody-based targeting chimaeras) | Cell surface protein of interest (e.g., PD-L1) | Lysosome | [65] | ||
| NLS-modified monoclonal antibody-DTPA | Auger electron emitter (111In) | Nucleus | [66] | ||
| Nucleolin-binding F3-peptide-DTPA | Auger electron emitter (111In) | Nucleus | [67] | ||
| Acid-activated cell-penetrating peptide (CPP) with NLS | Chemotherapeutics (camptothecin) | Nucleus | [48] | ||
| MNTs (modular nanotransporters) | Auger electron emitters (111In, 125I, 67Ga), α-emitter 211At, photosensitizers | Nucleus | [68] | ||
| Cell-targeting peptide functionalized CPP with NLS and oligohistidine | Plasmid DNA (pDNA) | Nucleus | [69] | ||
| CPP with ER retention sequence | Interleukin-24 | ER | [70] | ||
| Self-assembling peptides | Histone protein H2B, chemotherapeutics (doxorubicin) | Mitochondria | [71,72] | ||
| Histone-mediated transduction | pDNA | Nucleus | [73] | ||
| Peptide/pDNA self-assembling complexes | pDNA | Mitochondria | [69] | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosenkranz, A.A.; Slastnikova, T.A. Prospects of Using Protein Engineering for Selective Drug Delivery into a Specific Compartment of Target Cells. Pharmaceutics 2023, 15, 987. https://doi.org/10.3390/pharmaceutics15030987
Rosenkranz AA, Slastnikova TA. Prospects of Using Protein Engineering for Selective Drug Delivery into a Specific Compartment of Target Cells. Pharmaceutics. 2023; 15(3):987. https://doi.org/10.3390/pharmaceutics15030987
Chicago/Turabian StyleRosenkranz, Andrey A., and Tatiana A. Slastnikova. 2023. "Prospects of Using Protein Engineering for Selective Drug Delivery into a Specific Compartment of Target Cells" Pharmaceutics 15, no. 3: 987. https://doi.org/10.3390/pharmaceutics15030987