
SGLT2 Inhibitor—Dapagliflozin Attenuates Diabetes-Induced Renal Injury by Regulating Inflammation through a CYP4A/20-HETE Signaling Mechanism
 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Models
2.2. Immunohistochemical Analysis
2.3. Detection of Intracellular Superoxide
2.4. NADPH Oxidase Activity
2.5. Inflammatory Markers
2.6. 20-HETE Formation
2.7. mRNA Analysis
2.8. Western Blot Analysis
2.9. Statistical Analysis
3. Results
3.1. Dapagliflozin Treatment Attenuates Functional and Structural Renal Damage in T2DM Mice
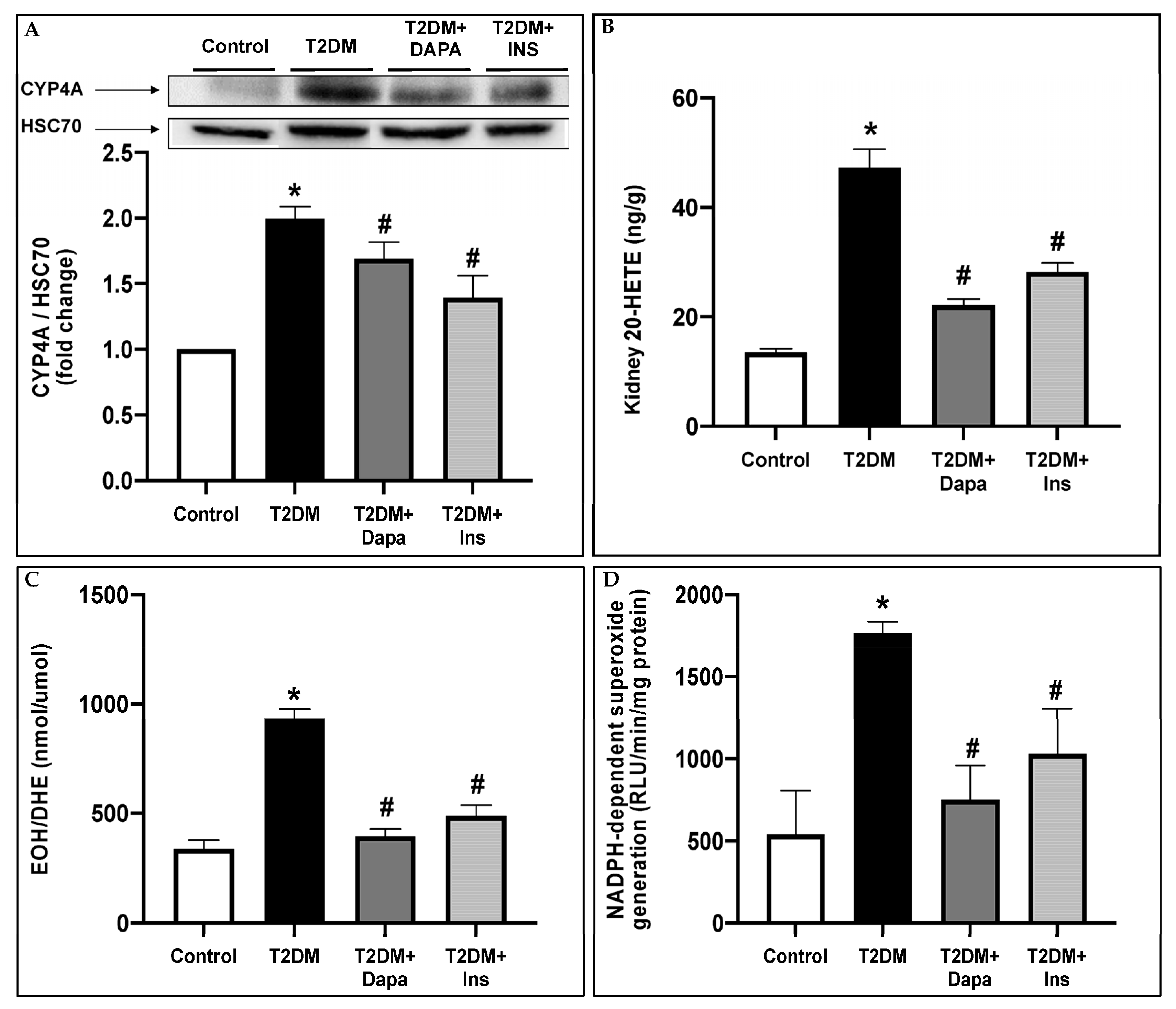
3.2. Dapagliflozin Inhibits CYP4A-Induced 20-HETE Production and Attenuates Oxidative Stress in the Kidneys of T2DM Mice
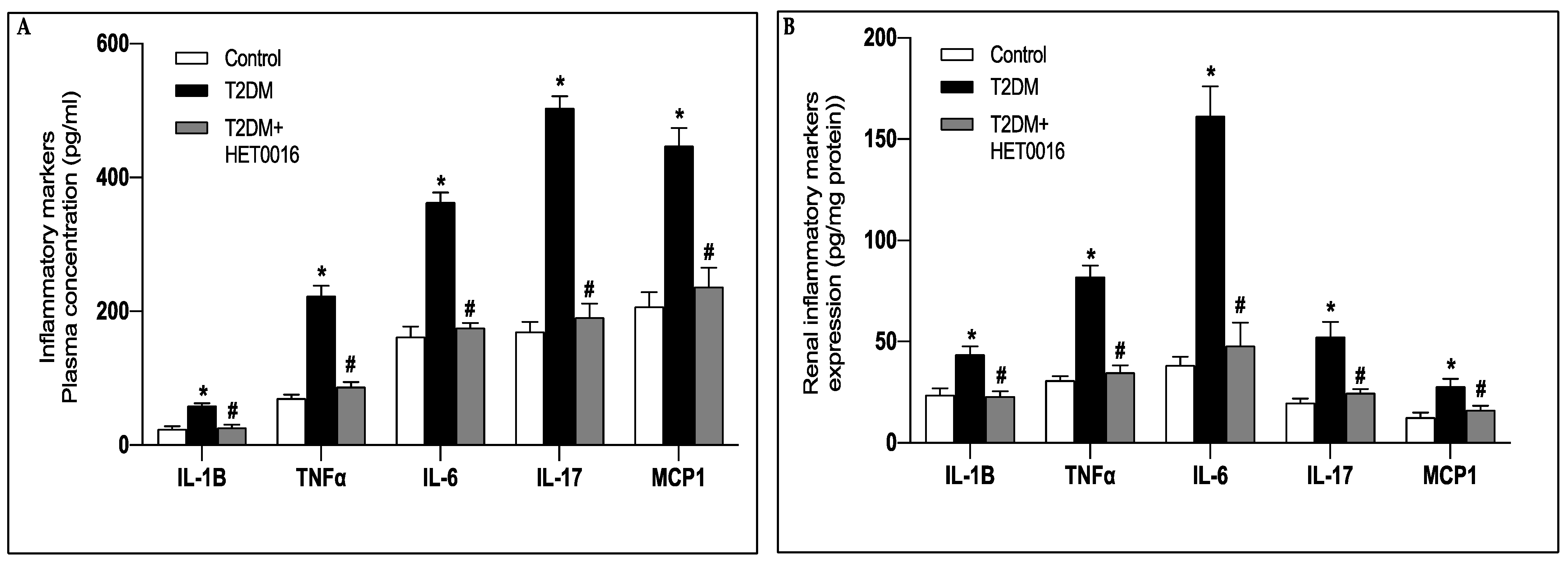
3.3. Treatment with Dapagliflozin Reduces the Systemic and Renal Inflammation Observed in the T2DM Mice
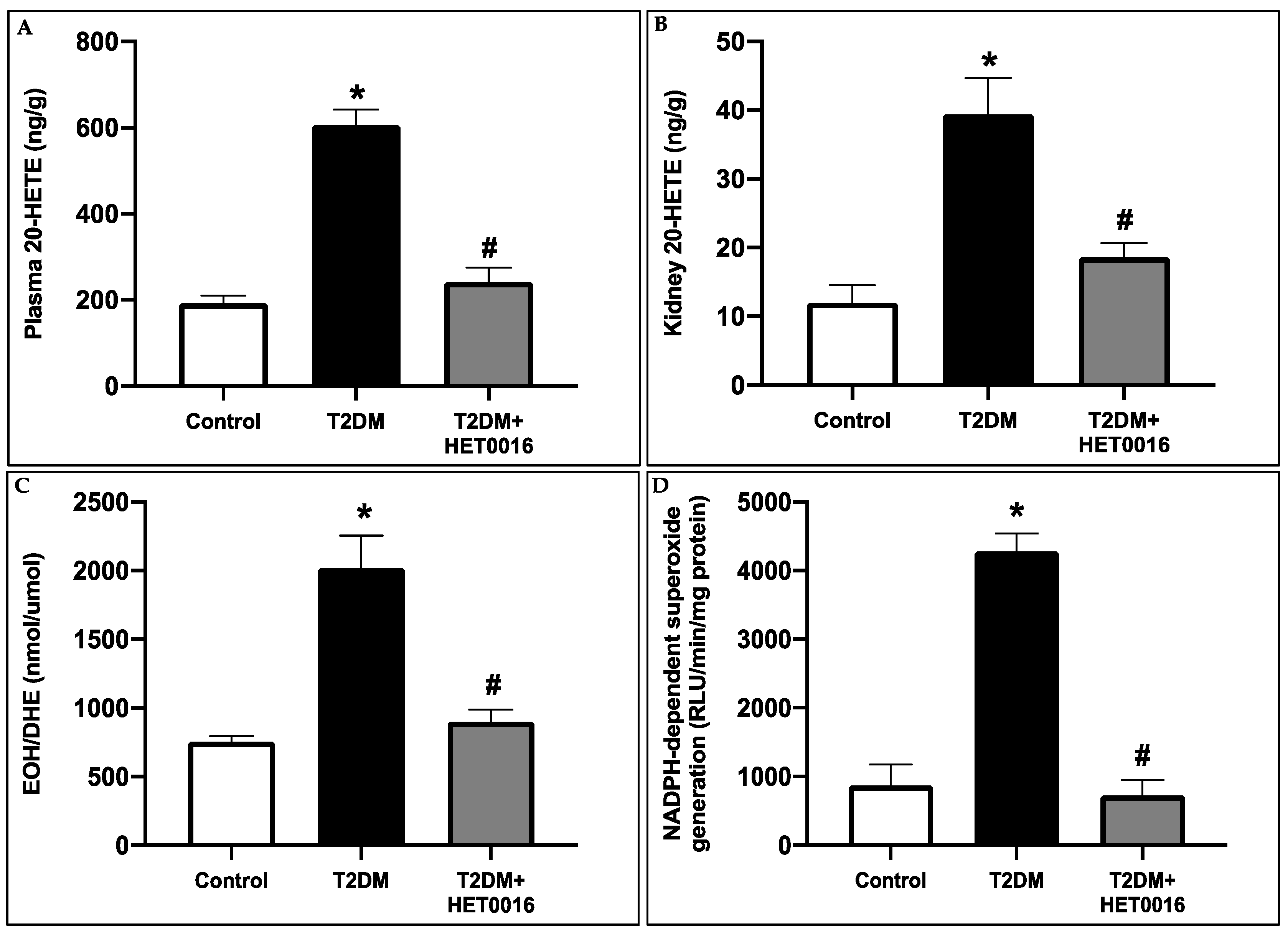
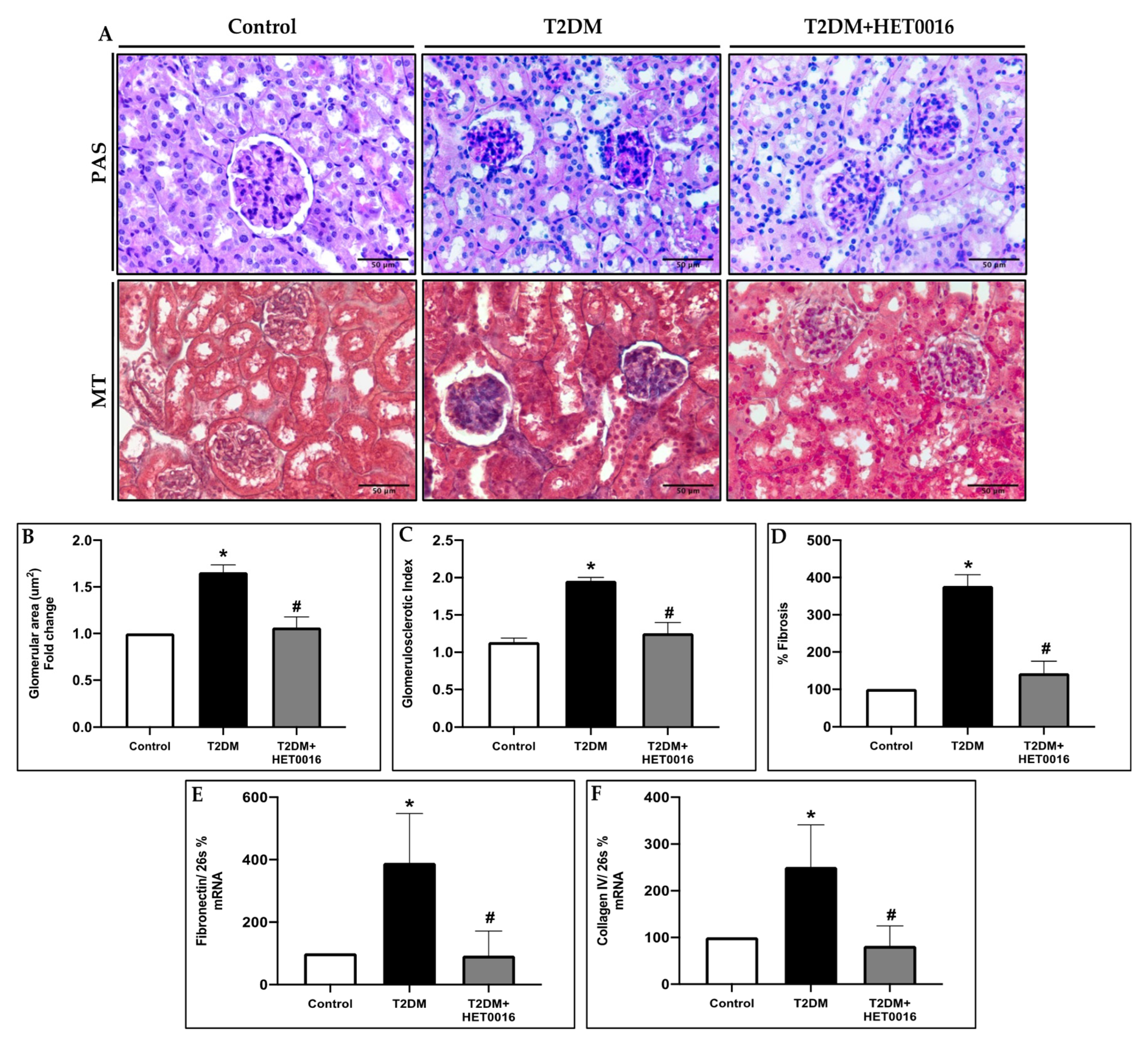
3.4. CYP4A/20-HETE Inhibition by HET0016 Attenuates Renal Injury in T2DM
3.5. CYP4A-Induced 20-HETE Production Prompts the Increase in Reactive Oxygen Species Production and the Rise in Proinflamatory Markers
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marcovecchio, M.L.; Chiarelli, F. Microvascular disease in children and adolescents with type 1 diabetes and obesity. Pediatr. Nephrol. 2011, 26, 365–375. [Google Scholar] [CrossRef]
- Nathan, D.M.; Genuth, S.; Lachin, J.; Cleary, P.; Crofford, O.; Davis, M.; Rand, L.; Siebert, C. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar] [CrossRef]
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998, 352, 837–853. [Google Scholar] [CrossRef]
- Patel, A.; MacMahon, S.; Chalmers, J.; Neal, B.; Billot, L.; Woodward, M.; Marre, M.; Cooper, M.; Glasziou, P.; Grobbee, D.; et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2008, 358, 2560–2572. [Google Scholar] [CrossRef] [Green Version]
- Zoungas, S.; Chalmers, J.; Neal, B.; Billot, L.; Li, Q.; Hirakawa, Y.; Arima, H.; Monaghan, H.; Joshi, R.; Colagiuri, S.; et al. Follow-up of blood-pressure lowering and glucose control in type 2 diabetes. N. Engl. J. Med. 2014, 371, 1392–1406. [Google Scholar] [CrossRef] [Green Version]
- Lewis, E.J.; Hunsicker, L.G.; Bain, R.P.; Rohde, R.D. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N. Engl. J. Med. 1993, 329, 1456–1462. [Google Scholar] [CrossRef]
- Lewis, E.J.; Hunsicker, L.G.; Clarke, W.R.; Berl, T.; Pohl, M.A.; Lewis, J.B.; Ritz, E.; Atkins, R.C.; Rohde, R.; Raz, I. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N. Engl. J. Med. 2001, 345, 851–860. [Google Scholar] [CrossRef] [Green Version]
- Brenner, B.M.; Cooper, M.E.; de Zeeuw, D.; Keane, W.F.; Mitch, W.E.; Parving, H.H.; Remuzzi, G.; Snapinn, S.M.; Zhang, Z.; Shahinfar, S. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N. Engl. J. Med. 2001, 345, 861–869. [Google Scholar] [CrossRef] [Green Version]
- Greco, M.; Chiefari, E.; Mirabelli, M.; Salatino, A.; Tocci, V.; Cianfrone, P.; Foti, D.P.; Brunetti, A. Plasma or Urine Neutrophil Gelatinase-Associated Lipocalin (NGAL): Which Is Better at Detecting Chronic Kidney Damage in Type 2 Diabetes? Endocrines 2022, 3, 175–186. [Google Scholar] [CrossRef]
- Njeim, R.; Azar, W.S.; Fares, A.H.; Azar, S.T.; Kfoury Kassouf, H.; Eid, A.A. NETosis contributes to the pathogenesis of diabetes and its complications. J. Mol. Endocrinol. 2020, 65, R65–R76. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.S.; Han, J.H.; Park, J.H.; Choi, J.S.; Kim, S.H.; Kim, H.S. Pyruvate Kinase M2: A New Biomarker for the Early Detection of Diabetes-Induced Nephropathy. Int. J. Mol. Sci. 2023, 24, 2683. [Google Scholar] [CrossRef]
- Fioretto, P.; Zambon, A.; Rossato, M.; Busetto, L.; Vettor, R. SGLT2 Inhibitors and the Diabetic Kidney. Diabetes Care 2016, 39 (Suppl. S2), S165–S171. [Google Scholar] [CrossRef] [Green Version]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [Green Version]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2018, 380, 347–357. [Google Scholar] [CrossRef]
- Perkovic, V.; de Zeeuw, D.; Mahaffey, K.W.; Fulcher, G.; Erondu, N.; Shaw, W.; Barrett, T.D.; Weidner-Wells, M.; Deng, H.; Matthews, D.R.; et al. Canagliflozin and renal outcomes in type 2 diabetes: Results from the CANVAS Program randomised clinical trials. Lancet Diabetes Endocrinol. 2018, 6, 691–704. [Google Scholar] [CrossRef]
- Mirabelli, M.; Chiefari, E.; Caroleo, P.; Vero, R.; Brunetti, F.S.; Corigliano, D.M.; Arcidiacono, B.; Foti, D.P.; Puccio, L.; Brunetti, A. Long-Term Effectiveness and Safety of SGLT-2 Inhibitors in an Italian Cohort of Patients with Type 2 Diabetes Mellitus. J. Diabetes Res. 2019, 2019, 3971060. [Google Scholar] [CrossRef] [Green Version]
- Nauck, M.A.; Del Prato, S.; Durán-García, S.; Rohwedder, K.; Langkilde, A.M.; Sugg, J.; Parikh, S.J. Durability of glycaemic efficacy over 2 years with dapagliflozin versus glipizide as add-on therapies in patients whose type 2 diabetes mellitus is inadequately controlled with metformin. Diabetes Obes. Metab. 2014, 16, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Vasilakou, D.; Karagiannis, T.; Athanasiadou, E.; Mainou, M.; Liakos, A.; Bekiari, E.; Sarigianni, M.; Matthews, D.R.; Tsapas, A. Sodium-glucose cotransporter 2 inhibitors for type 2 diabetes: A systematic review and meta-analysis. Ann. Intern. Med. 2013, 159, 262–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komala, M.G.; Panchapakesan, U.; Pollock, C.; Mather, A. Sodium glucose cotransporter 2 and the diabetic kidney. Curr. Opin. Nephrol. Hypertens. 2013, 22, 113–119. [Google Scholar] [CrossRef]
- Ojima, A.; Matsui, T.; Nishino, Y.; Nakamura, N.; Yamagishi, S. Empagliflozin, an Inhibitor of Sodium-Glucose Cotransporter 2 Exerts Anti-Inflammatory and Antifibrotic Effects on Experimental Diabetic Nephropathy Partly by Suppressing AGEs-Receptor Axis. Horm. Metab. Res. 2015, 47, 686–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terami, N.; Ogawa, D.; Tachibana, H.; Hatanaka, T.; Wada, J.; Nakatsuka, A.; Eguchi, J.; Horiguchi, C.S.; Nishii, N.; Yamada, H.; et al. Long-term treatment with the sodium glucose cotransporter 2 inhibitor, dapagliflozin, ameliorates glucose homeostasis and diabetic nephropathy in db/db mice. PLoS ONE 2014, 9, e100777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaikumkao, K.; Pongchaidecha, A.; Chueakula, N.; Thongnak, L.; Wanchai, K.; Chatsudthipong, V.; Chattipakorn, N.; Lungkaphin, A. Renal outcomes with sodium glucose cotransporter 2 (SGLT2) inhibitor, dapagliflozin, in obese insulin-resistant model. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2021–2033. [Google Scholar] [CrossRef]
- Swe, M.T.; Thongnak, L.; Jaikumkao, K.; Pongchaidecha, A.; Chatsudthipong, V.; Lungkaphin, A. Dapagliflozin not only improves hepatic injury and pancreatic endoplasmic reticulum stress, but also induces hepatic gluconeogenic enzymes expression in obese rats. Clin. Sci. 2019, 133, 2415–2430. [Google Scholar] [CrossRef] [PubMed]
- Simpson, A.E. The cytochrome P450 4 (CYP4) family. Gen. Pharmacol. 1997, 28, 351–359. [Google Scholar] [CrossRef]
- Capdevila, J.H.; Wang, W.; Falck, J.R. Arachidonic acid monooxygenase: Genetic and biochemical approaches to physiological/pathophysiological relevance. Prostaglandins Other Lipid Mediat. 2015, 120, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Muller, D.N.; Schmidt, C.; Barbosa-Sicard, E.; Wellner, M.; Gross, V.; Hercule, H.; Markovic, M.; Honeck, H.; Luft, F.C.; Schunck, W.H. Mouse Cyp4a isoforms: Enzymatic properties, gender- and strain-specific expression, and role in renal 20-hydroxyeicosatetraenoic acid formation. Biochem. J. 2007, 403, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Roman, R.J. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol. Rev. 2002, 82, 131–185. [Google Scholar] [CrossRef] [Green Version]
- Carroll, M.A.; McGiff, J.C. A new class of lipid mediators: Cytochrome P450 arachidonate metabolites. Thorax 2000, 55 (Suppl. S2), S13–S16. [Google Scholar] [CrossRef] [Green Version]
- Wink, D.A.; Osawa, Y.; Darbyshire, J.F.; Jones, C.R.; Eshenaur, S.C.; Nims, R.W. Inhibition of cytochromes P450 by nitric oxide and a nitric oxide-releasing agent. Arch Biochem. Biophys. 1993, 300, 115–123. [Google Scholar] [CrossRef]
- Michaelis, U.R.; Fisslthaler, B.; Medhora, M.; Harder, D.; Fleming, I.; Busse, R. Cytochrome P450 2C9-derived epoxyeicosatrienoic acids induce angiogenesis via cross-talk with the epidermal growth factor receptor (EGFR). FASEB J. 2003, 17, 770–772. [Google Scholar] [CrossRef] [PubMed]
- Ward, N.C.; Rivera, J.; Hodgson, J.; Puddey, I.B.; Beilin, L.J.; Falck, J.R.; Croft, K.D. Urinary 20-hydroxyeicosatetraenoic acid is associated with endothelial dysfunction in humans. Circulation 2004, 110, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Hoagland, K.M.; Maier, K.G.; Roman, R.J. Contributions of 20-HETE to the Antihypertensive Effects of Tempol in Dahl Salt-Sensitive Rats. Hypertension 2003, 41, 697–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, N.C.; Puddey, I.B.; Hodgson, J.M.; Beilin, L.J.; Croft, K.D. Urinary 20-hydroxyeicosatetraenoic acid excretion is associated with oxidative stress in hypertensive subjects. Free Radic. Biol. Med. 2005, 38, 1032–1036. [Google Scholar] [CrossRef]
- Eid, S.; Maalouf, R.; Jaffa, A.A.; Nassif, J.; Hamdy, A.; Rashid, A.; Ziyadeh, F.N.; Eid, A.A. 20-HETE and EETs in diabetic nephropathy: A novel mechanistic pathway. PLoS ONE 2013, 8, e70029. [Google Scholar] [CrossRef] [Green Version]
- Eid, A.A.; Gorin, Y.; Fagg, B.M.; Maalouf, R.; Barnes, J.L.; Block, K.; Abboud, H.E. Mechanisms of podocyte injury in diabetes: Role of cytochrome P450 and NADPH oxidases. Diabetes 2009, 58, 1201–1211. [Google Scholar] [CrossRef] [Green Version]
- Eid, S.; Abou-Kheir, W.; Sabra, R.; Daoud, G.; Jaffa, A.; Ziyadeh, F.N.; Roman, L.; Eid, A.A. Involvement of renal cytochromes P450 and arachidonic acid metabolites in diabetic nephropathy. J. Biol. Regul. Homeost. Agents 2013, 27, 693–703. [Google Scholar]
- Nishikawa, T.; Edelstein, D.; Brownlee, M. The missing link: A single unifying mechanism for diabetic complications. Kidney Int. 2000, 58, S26–S30. [Google Scholar] [CrossRef] [Green Version]
- Pichler, R.; Afkarian, M.; Dieter, B.P.; Tuttle, K.R. Immunity and inflammation in diabetic kidney disease: Translating mechanisms to biomarkers and treatment targets. Am. J. Physiol.-Ren. Physiol. 2017, 312, F716–F731. [Google Scholar] [CrossRef] [Green Version]
- Donate-Correa, J.; Luis-Rodríguez, D.; Martín-Núñez, E.; Tagua, V.G.; Hernández-Carballo, C.; Ferri, C.; Rodríguez-Rodríguez, A.E.; Mora-Fernández, C.; Navarro-González, J.F. Inflammatory Targets in Diabetic Nephropathy. J. Clin. Med. 2020, 9, 458. [Google Scholar] [CrossRef] [Green Version]
- Segerer, S.; Nelson, P.J.; Schlöndorff, D. Chemokines, chemokine receptors, and renal disease: From basic science to pathophysiologic and therapeutic studies. J. Am. Soc. Nephrol. 2000, 11, 152–176. [Google Scholar] [CrossRef]
- Satirapoj, B.; Dispan, R.; Radinahamed, P.; Kitiyakara, C. Urinary epidermal growth factor, monocyte chemoattractant protein-1 or their ratio as predictors for rapid loss of renal function in type 2 diabetic patients with diabetic kidney disease. BMC Nephrol. 2018, 19, 246. [Google Scholar] [CrossRef] [Green Version]
- Wada, T.; Furuichi, K.; Sakai, N.; Iwata, Y.; Yoshimoto, K.; Shimizu, M.; Takeda, S.I.; Takasawa, K.; Yoshimura, M.; Kida, H.; et al. Up-regulation of monocyte chemoattractant protein-1 in tubulointerstitial lesions of human diabetic nephropathy. Kidney Int. 2000, 58, 1492–1499. [Google Scholar] [CrossRef] [Green Version]
- Banba, N.; Nakamura, T.; Matsumura, M.; Kuroda, H.; Hattori, Y.; Kasai, K. Possible relationship of monocyte chemoattractant protein-1 with diabetic nephropathy. Kidney Int. 2000, 58, 684–690. [Google Scholar] [CrossRef] [Green Version]
- Ramesh, G.; Reeves, W.B. TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J. Clin. Investig. 2002, 110, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.S.; You, H.; Gao, T.; Cooper, T.K.; Nedospasov, S.A.; Vacher, J.; Wilkinson, P.F.; Farrell, F.X.; Brian Reeves, W. Macrophage-derived tumor necrosis factor-α mediates diabetic renal injury. Kidney Int. 2015, 88, 722–733. [Google Scholar] [CrossRef] [Green Version]
- Moriwaki, Y.; Yamamoto, T.; Shibutani, Y.; Aoki, E.; Tsutsumi, Z.; Takahashi, S.; Okamura, H.; Koga, M.; Fukuchi, M.; Hada, T. Elevated levels of interleukin-18 and tumor necrosis factor-alpha in serum of patients with type 2 diabetes mellitus: Relationship with diabetic nephropathy. Metabolism 2003, 52, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Navarro, J.F.; Mora, C.; Maca, M.; Garca, J. Inflammatory parameters are independently associated with urinary albumin in type 2 diabetes mellitus. Am. J. Kidney Dis. 2003, 42, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Araújo, L.S.; Torquato, B.G.S.; da Silva, C.A.; dos Reis Monteiro, M.L.G.; dos Santos Martins, A.L.M.; da Silva, M.V.; dos Reis, M.A.; Machado, J.R. Renal expression of cytokines and chemokines in diabetic nephropathy. BMC Nephrol. 2020, 21, 308. [Google Scholar] [CrossRef]
- Sanchez Alamo, B.; Shabaka, A.; Cachofeiro Ramos, M.V.; Fernandez Juarez, G.M. FC 085SERUM INTERLEUKIN-6 LEVELS PREDICT RENAL DISEASE PROGRESSION in DIABETIC KIDNEY DISEASE. Nephrol. Dial. Transplant. 2021, 36, gfab143.002. [Google Scholar] [CrossRef]
- Lei, Y.; Devarapu, S.K.; Motrapu, M.; Cohen, C.D.; Lindenmeyer, M.T.; Moll, S.; Kumar, S.V.; Anders, H.J. Interleukin-1β Inhibition for Chronic Kidney Disease in Obese Mice with Type 2 Diabetes. Front. Immunol. 2019, 10, 1223. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Dharmadhikari, G.; Maedler, K.; Meyer-Hermann, M. Possible Role of Interleukin-1β in Type 2 Diabetes Onset and Implications for Anti-inflammatory Therapy Strategies. PLoS Comput. Biol. 2014, 10, e1003798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Li, Y.J.; Chen, X.; Kwan, T.; Chadban, S.J.; Wu, H. Interleukin 17A promotes diabetic kidney injury. Sci. Rep. 2019, 9, 2264. [Google Scholar] [CrossRef] [Green Version]
- Maric, C.; Sandberg, K.; Hinojosa-Laborde, C. Glomerulosclerosis and tubulointerstitial fibrosis are attenuated with 17beta-estradiol in the aging Dahl salt sensitive rat. J. Am. Soc. Nephrol. 2004, 15, 1546–1556. [Google Scholar] [CrossRef] [Green Version]
- Eid, S.; Boutary, S.; Braych, K.; Sabra, R.; Massaad, C.; Hamdy, A.; Rashid, A.; Moodad, S.; Block, K.; Gorin, Y.; et al. mTORC2 Signaling Regulates Nox4-Induced Podocyte Depletion in Diabetes. Antioxid. Redox Signal. 2016, 25, 703–719. [Google Scholar] [CrossRef] [Green Version]
- Eid, A.A.; Ford, B.M.; Block, K.; Kasinath, B.S.; Gorin, Y.; Ghosh-Choudhury, G.; Barnes, J.L.; Abboud, H.E. AMP-activated protein kinase (AMPK) negatively regulates Nox4-dependent activation of p53 and epithelial cell apoptosis in diabetes. J. Biol. Chem. 2010, 285, 37503–37512. [Google Scholar] [CrossRef] [Green Version]
- Eid, A.A.; Lee, D.Y.; Roman, L.J.; Khazim, K.; Gorin, Y. Sestrin 2 and AMPK connect hyperglycemia to Nox4-dependent endothelial nitric oxide synthase uncoupling and matrix protein expression. Mol. Cell. Biol. 2013, 33, 3439–3460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakris, G.; Oshima, M.; Mahaffey, K.W.; Agarwal, R.; Cannon, C.P.; Capuano, G.; Charytan, D.M.; de Zeeuw, D.; Edwards, R.; Greene, T.; et al. Effects of Canagliflozin in Patients with Baseline eGFR <30 mL/min per 1.73 m2: Subgroup analysis of the randomized CREDENCE trial. Clin. J. Am. Soc. Nephrol. 2020, 15, 1705–1714. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [Green Version]
- Kusakabe, T.; Tanioka, H.; Ebihara, K.; Hirata, M.; Miyamoto, L.; Miyanaga, F.; Hige, H.; Aotani, D.; Fujisawa, T.; Masuzaki, H.; et al. Beneficial effects of leptin on glycaemic and lipid control in a mouse model of type 2 diabetes with increased adiposity induced by streptozotocin and a high-fat diet. Diabetologia 2009, 52, 675–683. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, K.; Viswanad, B.; Asrat, L.; Kaul, C.L.; Ramarao, P. Combination of high-fat diet-fed and low-dose streptozotocin-treated rat: A model for type 2 diabetes and pharmacological screening. Pharmacol. Res. 2005, 52, 313–320. [Google Scholar] [CrossRef]
- Ban, C.R.; Twigg, S.M. Fibrosis in diabetes complications: Pathogenic mechanisms and circulating and urinary markers. Vasc. Health Risk Manag. 2008, 4, 575–596. [Google Scholar] [CrossRef] [Green Version]
- Miyata, N.; Taniguchi, K.; Seki, T.; Ishimoto, T.; Sato-Watanabe, M.; Yasuda, Y.; Doi, M.; Kametani, S.; Tomishima, Y.; Ueki, T.; et al. HET0016, a potent and selective inhibitor of 20-HETE synthesizing enzyme. Br. J. Pharmacol. 2001, 133, 325–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alaeddine, L.M.; Harb, F.; Hamza, M.; Dia, B.; Mogharbil, N.; Azar, N.S.; Noureldein, M.H.; El Khoury, M.; Sabra, R.; Eid, A.A. Pharmacological regulation of cytochrome P450 metabolites of arachidonic acid attenuates cardiac injury in diabetic rats. Transl. Res. 2021, 235, 85–101. [Google Scholar] [CrossRef]
- Gangadhariah, M.H.; Luther, J.M.; Garcia, V.; Paueksakon, P.; Zhang, M.Z.; Hayward, S.W.; Love, H.D.; Falck, J.R.; Manthati, V.L.; Imig, J.D.; et al. Hypertension is a major contributor to 20-hydroxyeicosatetraenoic acid-mediated kidney injury in diabetic nephropathy. J. Am. Soc. Nephrol. 2015, 26, 597–610. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.M.; Sharma, M.; Anjaiahh, S.; Falck, J.R.; Roman, R.J. Role of endogenous CYP450 metabolites of arachidonic acid in maintaining the glomerular protein permeability barrier. Am. J. Physiol.-Ren. Physiol. 2007, 293, F501–F505. [Google Scholar] [CrossRef] [PubMed]
- Dahly-Vernon, A.J.; Sharma, M.; McCarthy, E.T.; Savin, V.J.; Ledbetter, S.R.; Roman, R.J. Transforming growth factor-beta, 20-HETE interaction, and glomerular injury in Dahl salt-sensitive rats. Hypertension 2005, 45, 643–648. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, E.T.; Sharma, R.; Sharma, M. Protective effect of 20-hydroxyeicosatetraenoic acid (20-HETE) on glomerular protein permeability barrier. Kidney Int. 2005, 67, 152–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Zou, Y.; Liu, F. Transforming Growth Factor-Beta1 in Diabetic Kidney Disease. Front. Cell Dev. Biol. 2020, 8, 187. [Google Scholar] [CrossRef] [Green Version]
- Luo, P.; Zhou, Y.; Chang, H.-H.; Zhang, J.; Seki, T.; Wang, C.-Y.; Inscho, E.W.; Wang, M.-H. Glomerular 20-HETE, EETs, and TGF-beta1 in diabetic nephropathy. Am. J. Physiol.-Ren. Physiol. 2009, 296, F556–F563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Q.; Han, Y.; Bao, Y.; Li, W.; Li, X.; Shen, X.; Wang, X.; Yao, F.; O’Rourke, S.T.; Sun, C. 20-HETE increases NADPH oxidase-derived ROS production and stimulates the L-type Ca2+ channel via a PKC-dependent mechanism in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1109–H1117. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, M.; López-Oliva, E.; Pinilla, E.; Rodríguez, C.; Martínez, M.P.; Contreras, C.; Gómez, A.; Benedito, S.; Sáenz-Medina, J.; Rivera, L.; et al. Differential contribution of renal cytochrome P450 enzymes to kidney endothelial dysfunction and vascular oxidative stress in obesity. Biochem. Pharmacol. 2022, 195, 114850. [Google Scholar] [CrossRef]
- Ishizuka, T.; Cheng, J.; Singh, H.; Vitto, M.D.; Manthati, V.L.; Falck, J.R.; Laniado-Schwartzman, M. 20-Hydroxyeicosatetraenoic acid stimulates nuclear factor-kappaB activation and the production of inflammatory cytokines in human endothelial cells. J. Pharmacol. Exp. Ther. 2008, 324, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Chow, F.; Ozols, E.; Nikolic-Paterson, D.J.; Atkins, R.C.; Tesch, G.H. Macrophages in mouse type 2 diabetic nephropathy: Correlation with diabetic state and progressive renal injury. Kidney Int. 2004, 65, 116–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehrabian, M.; Sparkes, R.S.; Mohandas, T.; Fogelman, A.M.; Lusis, A.J. Localization of monocyte chemotactic protein-1 gene (SCYA2) to human chromosome 17q11. 2–q21. 1. Genomics 1991, 9, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-Y.; Guijarro, C.; O’Donnell, M.P.; Kasiske, B.L.; Kim, Y.; Keane, W.F. Human mesangial cell production of monocyte chemoattractant protein-1: Modulation by lovastatin. Kidney Int. 1995, 48, 363–371. [Google Scholar] [CrossRef] [Green Version]
- Stephan, M.; Conrad, S.; Eggert, T.; Heuer, R.; Fernandez, S.; Huland, H. Urinary concentration and tissue messenger RNA expression of monocyte chemoattractant protein-1 as an indicator of the degree of hydronephrotic atrophy in partial ureteral obstruction. J. Urol. 2002, 167, 1497–1502. [Google Scholar] [CrossRef]
- Viedt, C.; Orth, S.R. Monocyte chemoattractant protein-1 (MCP-1) in the kidney: Does it more than simply attract monocytes? Nephrol. Dial. Transplant. 2002, 17, 2043–2047. [Google Scholar] [CrossRef] [Green Version]
- Mine, S.; Okada, Y.; Tanikawa, T.; Kawahara, C.; Tabata, T.; Tanaka, Y. Increased expression levels of monocyte CCR2 and monocyte chemoattractant protein-1 in patients with diabetes mellitus. Biochem. Biophys. Res. Commun. 2006, 344, 780–785. [Google Scholar] [CrossRef]
- Wolf, G.; Jocks, T.; Zahner, G.; Panzer, U.; Stahl, R.A.K. Existence of a regulatory loop between MCP-1 and TGF-β in glomerular immune injury. Am. J. Physiol.-Ren. Physiol. 2002, 283, F1075–F1084. [Google Scholar] [CrossRef] [Green Version]
- Diaz Encarnacion, M.M.; Warner, G.M.; Cheng, J.; Gray, C.E.; Nath, K.A.; Grande, J.P. n-3 Fatty acids block TNF-α-stimulated MCP-1 expression in rat mesangial cells. Am. J. Physiol.-Ren. Physiol. 2011, 300, F1142–F1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, J.C.; Gray, S.P.; Barit, D.; Okabe, J.; El-Osta, A.; Namikoshi, T.; Thallas-Bonke, V.; Wingler, K.; Szyndralewiez, C.; Heitz, F. Genetic targeting or pharmacologic inhibition of NADPH oxidase nox4 provides renoprotection in long-term diabetic nephropathy. J. Am. Soc. Nephrol. 2014, 25, 1237–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Wu, G.; Qi, X.; Lin, H.; Qian, H.; Shen, J.; Lin, S. Protein kinase C β inhibitor LY333531 attenuates intercellular adhesion molecule-1 and monocyte chemotactic protein-1 expression in the kidney in diabetic rats. J. Pharmacol. Sci. 2006, 101, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.J.; Liu, Z.-G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, R.M.; Wahab, N.A. Extracellular matrix metabolism in diabetic nephropathy. J. Am. Soc. Nephrol. 2003, 14, 1358–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahab, N.; Schaefer, L.; Weston, B.; Yiannikouris, O.; Wright, A.; Babelova, A.; Schaefer, R.; Mason, R. Glomerular expression of thrombospondin-1, transforming growth factor beta and connective tissue growth factor at different stages of diabetic nephropathy and their interdependent roles in mesangial response to diabetic stimuli. Diabetologia 2005, 48, 2650–2660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, H.; Li, Y.; Qi, J.; Wang, H.; Liu, K. Peroxynitrite plays a key role in glomerular lesions in diabetic rats. J. Nephrol. 2009, 22, 800–808. [Google Scholar]
- Navarro, J.F.; Milena, F.J.; Mora, C.; León, C.; García, J. Renal pro-inflammatory cytokine gene expression in diabetic nephropathy: Effect of angiotensin-converting enzyme inhibition and pentoxifylline administration. Am. J. Nephrol. 2006, 26, 562–570. [Google Scholar] [CrossRef]
- Kuhad, A.; Chopra, K. Attenuation of diabetic nephropathy by tocotrienol: Involvement of NFkB signaling pathway. Life Sci. 2009, 84, 296–301. [Google Scholar] [CrossRef]
- Pecoits-Filho, R.; Lindholm, B.; Axelsson, J.; Stenvinkel, P. Update on interleukin-6 and its role in chronic renal failure. Nephrol. Dial. Transplant. 2003, 18, 1042–1045. [Google Scholar] [CrossRef] [Green Version]
- Iyoda, M.; Shibata, T.; Kawaguchi, M.; Hizawa, N.; Yamaoka, T.; Kokubu, F.; Akizawa, T. IL-17A and IL-17F stimulate chemokines via MAPK pathways (ERK1/2 and p38 but not JNK) in mouse cultured mesangial cells: Synergy with TNF-α and IL-1β. Am. J. Physiol.-Ren. Physiol. 2010, 298, F779–F787. [Google Scholar] [CrossRef]
- Van Kooten, C.; Boonstra, J.G.; Paape, M.E.; Fossiez, F.; Banchereau, J.; Lebecque, S.; Bruijn, J.A.; De Fijter, J.; Van Es, L.A.; Daha, M.R. Interleukin-17 activates human renal epithelial cells in vitro and is expressed during renal allograft rejection. J. Am. Soc. Nephrol. 1998, 9, 1526–1534. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.L.; Perco, P.; Mulder, S.; Leierer, J.; Hansen, M.K.; Heinzel, A.; Mayer, G. Canagliflozin reduces inflammation and fibrosis biomarkers: A potential mechanism of action for beneficial effects of SGLT2 inhibitors in diabetic kidney disease. Diabetologia 2019, 62, 1154–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeFronzo, R.A.; Reeves, W.B.; Awad, A.S. Pathophysiology of diabetic kidney disease: Impact of SGLT2 inhibitors. Nat. Rev. Nephrol. 2021, 17, 319–334. [Google Scholar] [CrossRef]
- Rutledge, J.C.; Ng, K.F.; Aung, H.H.; Wilson, D.W. Role of triglyceride-rich lipoproteins in diabetic nephropathy. Nat. Rev. Nephrol. 2010, 6, 361–370. [Google Scholar] [CrossRef]
- Thomson, S.C.; Blantz, R.C. Glomerulotubular Balance, Tubuloglomerular Feedback, and Salt Homeostasis. J. Am. Soc. Nephrol. 2008, 19, 2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Booz, G.W.; Yu, Q.; He, X.; Wang, S.; Fan, F. Conflicting roles of 20-HETE in hypertension and renal end organ damage. Eur. J. Pharmacol. 2018, 833, 190–200. [Google Scholar] [CrossRef]
- Yu, M.; Lopez, B.; Santos, E.A.D.; Falck, J.R.; Roman, R.J. Effects of 20-HETE on Na+ transport and Na+-K+-ATPase activity in the thick ascending loop of Henle. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2007, 292, R2400–R2405. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Breaks, J.; Loutzenhiser, K.; Loutzenhiser, R. Effects of inhibition of the Na+/K+/2Cl− cotransporter on myogenic and angiotensin II responses of the rat afferent arteriole. Am. J. Physiol.-Ren. Physiol. 2007, 292, F999–F1006. [Google Scholar] [CrossRef]
- Oppermann, M.; Mizel, D.; Huang, G.; Li, C.; Deng, C.; Theilig, F.; Bachmann, S.; Briggs, J.; Schnermann, J.; Castrop, H. Macula Densa Control of Renin Secretion and Preglomerular Resistance in Mice with Selective Deletion of the B Isoform of the Na, K, 2Cl Co-Transporter. J. Am. Soc. Nephrol. 2006, 17, 2143. [Google Scholar] [CrossRef] [Green Version]
- Schnermann, J.; Briggs, J.P. Tubuloglomerular feedback: Mechanistic insights from gene-manipulated mice. Kidney Int. 2008, 74, 418–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croft, K.D.; McGiff, J.C.; Sanchez-Mendoza, A.; Carroll, M.A. Angiotensin II releases 20-HETE from rat renal microvessels. Am. J. Physiol.-Ren. Physiol. 2000, 279, F544–F551. [Google Scholar] [CrossRef] [Green Version]
- Hoopes, S.L.; Garcia, V.; Edin, M.L.; Schwartzman, M.L.; Zeldin, D.C. Vascular actions of 20-HETE. Prostaglandins Other Lipid Mediat. 2015, 120, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso-Galicia, M.; Maier, K.G.; Greene, A.S.; Allen, W.; Cowley, J.; Roman, R.J. Role of 20-hydroxyeicosatetraenoic acid in the renal and vasoconstrictor actions of angiotensin II. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2002, 283, R60–R68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGiff, J.C.; Quilley, J. 20-HETE and the kidney: Resolution of old problems and new beginnings. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 1999, 277, R607–R623. [Google Scholar] [CrossRef]
- Miyata, K.N.; Lo, C.-S.; Zhao, S.; Liao, M.-C.; Pang, Y.; Chang, S.-Y.; Peng, J.; Kretzler, M.; Filep, J.G.; Ingelfinger, J.R.; et al. Angiotensin II up-regulates sodium-glucose co-transporter 2 expression and SGLT2 inhibitor attenuates Ang II-induced hypertensive renal injury in mice. Clin. Sci. 2021, 135, 943–961. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.J.; Chung, S.; Kim, S.J.; Lee, E.-M.; Yoo, Y.-H.; Kim, J.-W.; Ahn, Y.-B.; Kim, E.-S.; Moon, S.-D.; Kim, M.-J.; et al. Effect of Sodium-Glucose Co-Transporter 2 Inhibitor, Dapagliflozin, on Renal Renin-Angiotensin System in an Animal Model of Type 2 Diabetes. PLoS ONE 2016, 11, e0165703. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhao, C.; Ye, Y.; Yu, M.; Qu, X. Prospect of Sodium-Glucose Co-transporter 2 Inhibitors Combined With Insulin for the Treatment of Type 2 Diabetes. Front. Endocrinol. 2020, 11, 190. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | T2DM | T2DM + Dapa | T2DM + Ins | |
| Glucose levels (mg/dL) | 143 ± 17 | 302 ± 55 * | 214 ± 16 # | 196 ± 30 # |
| Body weight (g) | 32.93 ± 2.91 | 35.6 ± 1.06 | 33.15 ± 3.55 | 30.55 ± 2 # |
| Kidney Weight/ Body weight (mg/g) | 6.6 ± 0.33 | 8.5 ± 0.19 * | 6.9 ± 0.37 # | 6.5 ± 0.35 # |
| Proteinuria (mg/24 h) | 26 ± 1.71 | 68 ± 17.57 * | 28 ± 5.59 # | 21 ± 3.47 # |
| UACR (μg/mg) | 31 ± 5 | 88 ± 14 * | 13 ± 4 # | 21 ± 6 # |
| Control | T2DM | T2DM + HET0016 | |
| Glucose levels (mg/dL) | 154 ± 19 | 485 ± 74 * | 476 ± 85 * |
| Body weight (g) | 32 ± 0.51 | 34 ± 2.62 | 37 ± 1.96 * |
| Kidney Weight/ Body weight (mg/g) | 7.3 ± 0.27 | 9 ± 0.45 * | 7.5 ± 0.3 # |
| Proteinuria (mg/24 h) | 24 ± 6.76 | 112 ± 63.26 * | 25 ± 11.63 # |
| UACR (ug/mg) | 52 ± 6 | 198 ± 10 * | 70 ± 5 # |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dia, B.; Alkhansa, S.; Njeim, R.; Al Moussawi, S.; Farhat, T.; Haddad, A.; Riachi, M.E.; Nawfal, R.; Azar, W.S.; Eid, A.A. SGLT2 Inhibitor—Dapagliflozin Attenuates Diabetes-Induced Renal Injury by Regulating Inflammation through a CYP4A/20-HETE Signaling Mechanism. Pharmaceutics 2023, 15, 965. https://doi.org/10.3390/pharmaceutics15030965
Dia B, Alkhansa S, Njeim R, Al Moussawi S, Farhat T, Haddad A, Riachi ME, Nawfal R, Azar WS, Eid AA. SGLT2 Inhibitor—Dapagliflozin Attenuates Diabetes-Induced Renal Injury by Regulating Inflammation through a CYP4A/20-HETE Signaling Mechanism. Pharmaceutics. 2023; 15(3):965. https://doi.org/10.3390/pharmaceutics15030965
Chicago/Turabian StyleDia, Batoul, Sahar Alkhansa, Rachel Njeim, Sarah Al Moussawi, Theresa Farhat, Antony Haddad, Mansour E. Riachi, Rashad Nawfal, William S. Azar, and Assaad A. Eid. 2023. "SGLT2 Inhibitor—Dapagliflozin Attenuates Diabetes-Induced Renal Injury by Regulating Inflammation through a CYP4A/20-HETE Signaling Mechanism" Pharmaceutics 15, no. 3: 965. https://doi.org/10.3390/pharmaceutics15030965