
The Hedgehog Pathway as a Therapeutic Target in Chronic Myeloid Leukemia

{kind=link}
{kind=link}
Abstract
:1. Introduction of the Hedgehog Signaling Pathway
2. Role of HH Signaling in Development
3. HH Signaling in Hematopoiesis
4. HH Signaling in Cancer and Development of Small Molecule SMO Inhibitors
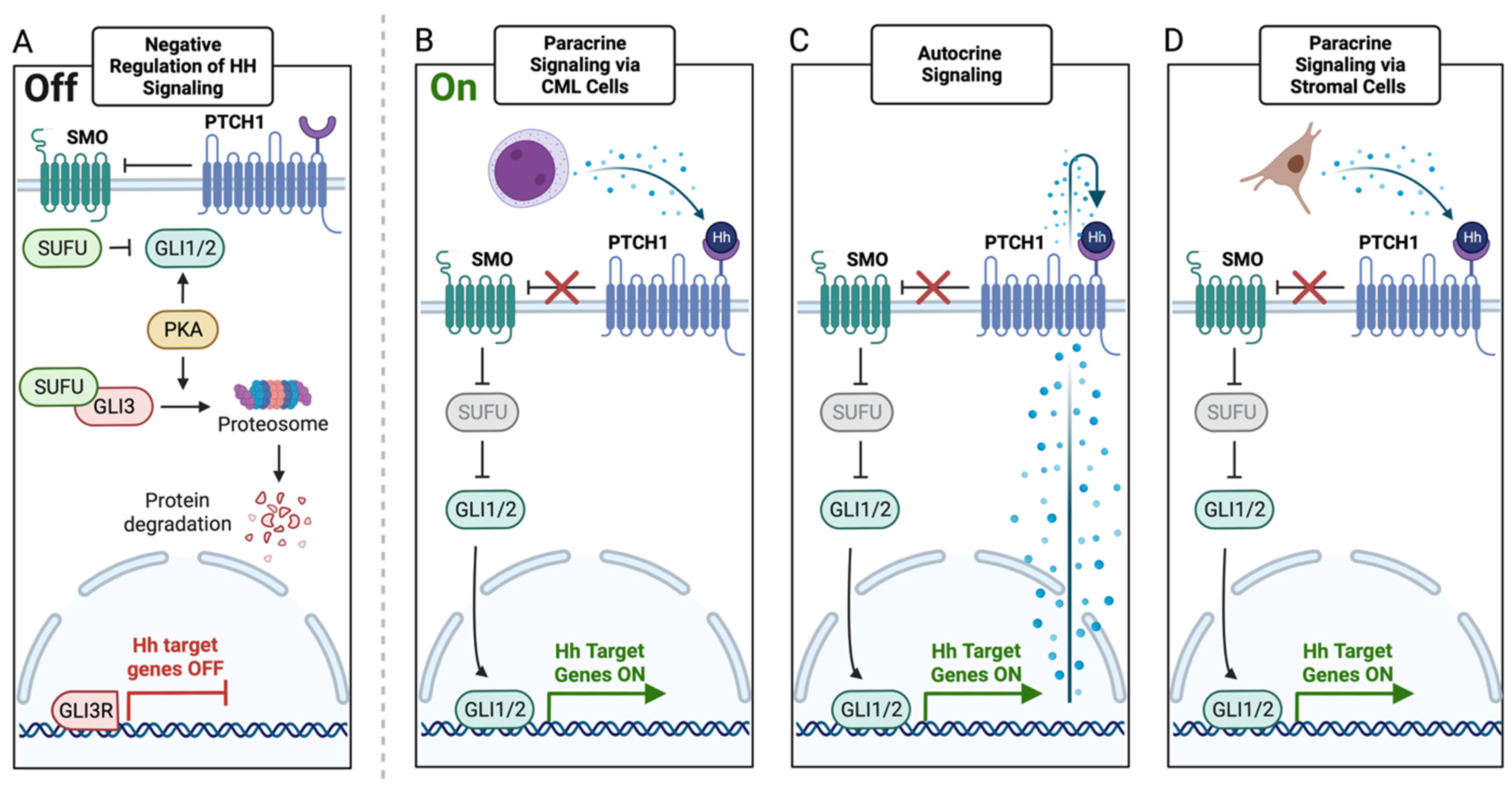
5. HH Signaling in CML
6. Clinical Implications
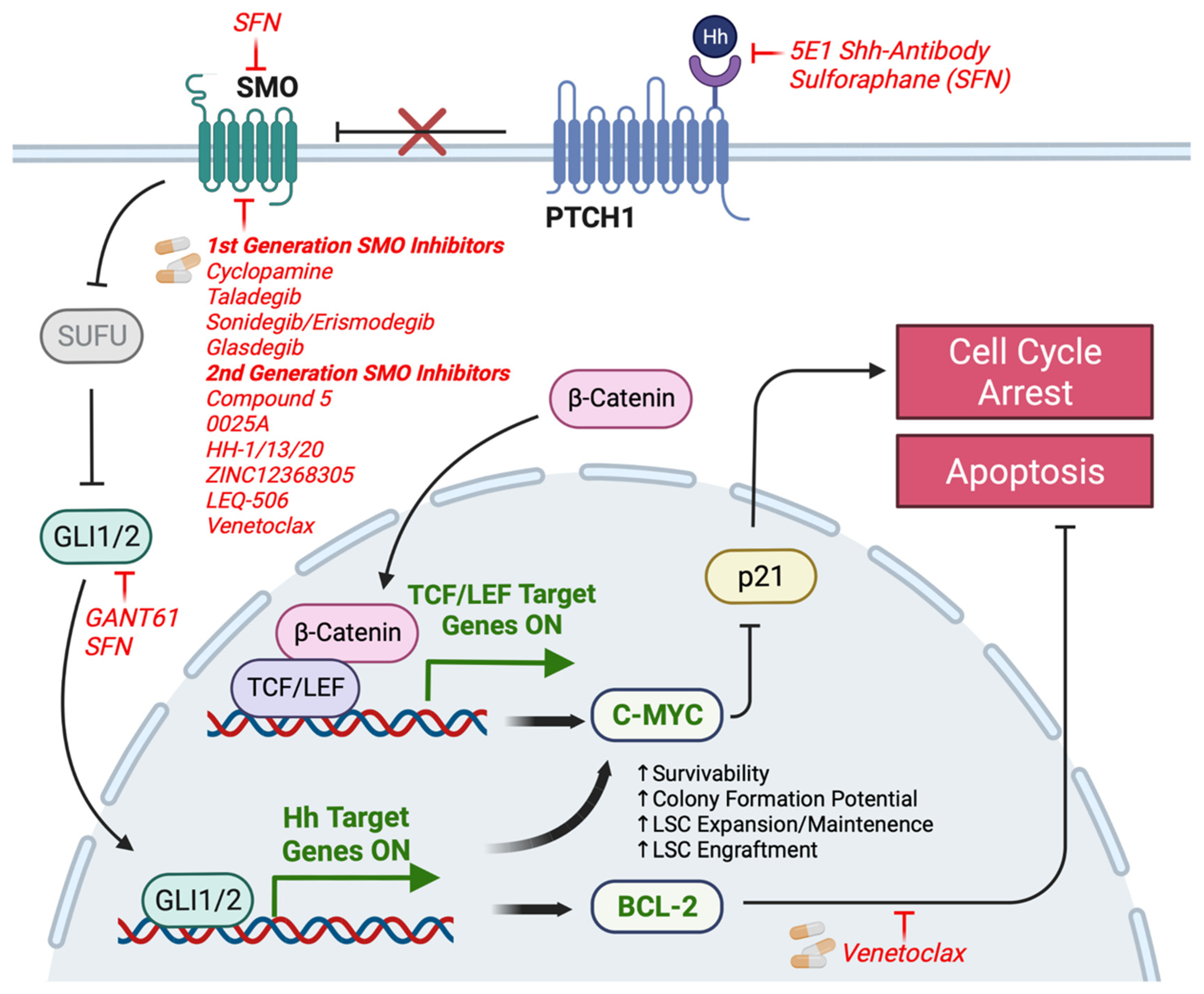
7. Mechanisms of Resistance to Small Molecule Inhibitors of HH Signaling
8. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nüsslein-Volhard, C.; Wieschaus, E. Mutations Affecting Segment Number and Polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef]
- Petrova, R.; Joyner, A.L. Roles for Hedgehog Signaling in Adult Organ Homeostasis and Repair. Development 2014, 141, 3445–3457. [Google Scholar] [CrossRef] [Green Version]
- Bitgood, M.J.; McMahon, A.P. HedgehogandBmpGenes Are Coexpressed at Many Diverse Sites of Cell–Cell Interaction in the Mouse Embryo. Dev. Biol. 1995, 172, 126–138. [Google Scholar] [CrossRef] [Green Version]
- Hammerschmidt, M.; Brook, A.; McMahon, A.P. The world according to bedgebog. Trends Genet. 1997, 13, 14–21. [Google Scholar] [CrossRef]
- Dyer, M.; Farrington, S.; Mohn, D.; Munday, J.; Baron, M. Indian hedgehog activates hematopoiesis and vasculogenesis and can respecify prospective neurectodermal cell fate in the mouse embryo. Development 2001, 128, 1717–1730. [Google Scholar] [CrossRef]
- Lim, Y.; Matsui, W. Hedgehog Signaling in Hematopoiesis. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 129. [Google Scholar] [CrossRef] [Green Version]
- St-Jacques, B.; Hammerschmidt, M.; McMahon, A.P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999, 13, 2072–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitgood, M.J.; Shen, L.; McMahon, A.P. Sertoli cell signaling by Desert hedgehog regulates the male germline. Curr. Biol. 1996, 6, 298–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukui, T.; Capdevila, J.; Tamura, K.; Ruiz-Lozano, P.; Rodriguez-Esteban, C.; Yonei-Tamura, S.; Magallón, J.; Chandraratna, R.A.; Chien, K.; Blumberg, B.; et al. Multiple Left-Right Asymmetry Defects in Shh(−/−) Mutant Mice Unveil a Convergence of the Shh and Retinoic Acid Pathways in the Control of Lefty-1. Proc. Natl. Acad. Sci. USA 1999, 96, 11376–11381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ericson, J.; Rashbass, P.; Schedl, A.; Brenner-Morton, S.; Kawakami, A.; van Heyningen, V.; Jessell, T.; Briscoe, J. Pax6 Controls Progenitor Cell Identity and Neuronal Fate in Response to Graded Shh Signaling. Cell 1997, 90, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Riddle, R.D.; Johnson, R.L.; Laufer, E.; Tabin, C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell 1993, 75, 1401–1416. [Google Scholar] [CrossRef] [PubMed]
- Tukachinsky, H.; Petrov, K.; Watanabe, M.; Salic, A. Mechanism of Inhibition of the Tumor Suppressor Patched by Sonic Hedgehog. Proc. Natl. Acad. Sci. USA 2016, 113, E5866–E5875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, T.T.; Gratwick, K.S.; Kollman, J.; Park, D.; Nies, D.H.; Goffeau, A.; Saier, M.H. The RND Permease Superfamily: An Ancient, Ubiquitous and Diverse Family That Includes Human Disease and Development Proteins. J. Mol. Microbiol. Biotechnol. 1999, 1, 107–125. [Google Scholar]
- Taipale, J.; Cooper, M.K.; Maiti, T.; Beachy, P.A. Patched acts catalytically to suppress the activity of Smoothened. Nature 2002, 418, 892–896. [Google Scholar] [CrossRef]
- Ruat, M.; Hoch, L.; Faure, H.; Rognan, D. Targeting of Smoothened for therapeutic gain. Trends Pharmacol. Sci. 2014, 35, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetz, S.C.; Ocbina, P.J.R.; Anderson, K.V. The Primary Cilium as a Hedgehog Signal Transduction Machine. Methods Cell Biol. 2009, 94, 199. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Ren, X.-R.; Nelson, C.D.; Barak, L.S.; Chen, J.K.; Beachy, P.A.; de Sauvage, F.; Lefkowitz, R.J. Activity-Dependent Internalization of Smoothened Mediated by ß-Arrestin 2 and GRK2. Science 2004, 306, 2257–2260. [Google Scholar] [CrossRef]
- Kovacs, J.J.; Whalen, E.J.; Liu, R.; Xiao, K.; Kim, J.; Chen, M.; Wang, J.; Chen, W.; Lefkowitz, R.J. Beta-Arrestin-Mediated Localization of Smoothened to the Primary Cilium. Science 2008, 320, 1777–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.-H.; Wilson, C.W.; Li, Y.-J.; Law, K.K.L.; Lu, C.-S.; Gacayan, R.; Zhang, X.; Hui, C.; Chuang, P.-T. Cilium-Independent Regulation of Gli Protein Function by Sufu in Hedgehog Signaling Is Evolutionarily Conserved. Genes Dev. 2009, 23, 1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, X.; Lai, C.K.; Evangelista, M.; Hongo, J.-A.; de Sauvage, F.J.; Scales, S.J. Kinetics of Hedgehog-Dependent Full-Length Gli3 Accumulation in Primary Cilia and Subsequent Degradation. Mol. Cell. Biol. 2010, 30, 1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenney, A.M.; Widlund, H.; Rowitch, D. Hedgehog and PI-3 kinase signaling converge on Nmyc1 to promote cell cycle progression in cerebellar neuronal precursors. Development 2004, 131, 217–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenney, A.M.; Rowitch, D.H. Sonic Hedgehog Promotes G1 Cyclin Expression and Sustained Cell Cycle Progression in Mammalian Neuronal Precursors. Mol. Cell. Biol. 2000, 20, 9055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niewiadomski, P.; Kong, J.H.; Ahrends, R.; Ma, Y.; Humke, E.W.; Khan, S.; Teruel, M.N.; Novitch, B.G.; Rohatgi, R. Gli Protein Activity Is Controlled by Multisite Phosphorylation in Vertebrate Hedgehog Signaling. Cell Rep. 2014, 6, 168–181. [Google Scholar] [CrossRef] [Green Version]
- Pietrobono, S.; Gagliardi, S.; Stecca, B. Non-Canonical Hedgehog Signaling Pathway in Cancer: Activation of GLI Transcription Factors Beyond Smoothened. Front. Genet. 2019, 10, 556. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.M.; Cho, J. Hedgehog Pathway Inhibitors as Targeted Cancer Therapy and Strategies to Overcome Drug Resistance. Int. J. Mol. Sci. 2022, 23, 1733. [Google Scholar] [CrossRef]
- Kenney, A.M.; Cole, M.D.; Rowitch, D.H. Nmycupregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development 2003, 130, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Dessaud, E.; McMahon, A.P.; Briscoe, J. Pattern formation in the vertebrate neural tube: A sonic hedgehog morphogen-regulated transcriptional network. Development 2008, 135, 2489–2503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saldaña, J.I.; Solanki, A.; Lau, C.-I.; Sahni, H.; Ross, S.; Furmanski, A.L.; Ono, M.; Holländer, G.; Crompton, T. Sonic Hedgehog regulates thymic epithelial cell differentiation. J. Autoimmun. 2016, 68, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Echelard, Y.; Epstein, D.J.; St-Jacques, B.; Shen, L.; Mohler, J.; McMahon, J.A.; McMahon, A.P. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 1993, 75, 1417–1430. [Google Scholar] [CrossRef]
- Lupo, G.; Harris, W.A.; Lewis, K.E. Mechanisms of ventral patterning in the vertebrate nervous system. Nat. Rev. Neurosci. 2006, 7, 103–114. [Google Scholar] [CrossRef]
- Wu, C.; Shimo, T.; Liu, M.; Pacifici, M.; Koyama, E. Sonic Hedgehog Functions as a Mitogen during Bell Stage of Odontogenesis. Connect. Tissue Res. 2003, 44 (Suppl. 1), 92–96. [Google Scholar] [CrossRef]
- Cooper, O.; Hargus, G.; Deleidi, M.; Blak, A.; Osborn, T.; Marlow, E.; Lee, K.; Levy, A.; Perez-Torres, E.; Yow, A.; et al. Differentiation of human ES and Parkinson’s disease iPS cells into ventral midbrain dopaminergic neurons requires a high activity form of SHH, FGF8a and specific regionalization by retinoic acid. Mol. Cell. Neurosci. 2010, 45, 258–266. [Google Scholar] [CrossRef] [Green Version]
- Seita, J.; Weissman, I.L. Hematopoietic Stem Cell: Self-Renewal versus Differentiation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 640–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eaves, C.J. Hematopoietic Stem Cells: Concepts, Definitions, and the New Reality. Blood 2015, 125, 2605–2613. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, G.; Murdoch, B.; Wu, D.; Baker, D.P.; Williams, K.P.; Chadwick, K.; Ling, L.E.; Karanu, F.N.; Bhatia, M. Sonic Hedgehog Induces the Proliferation of Primitive Human Hematopoietic Cells via BMP Regulation. Nat. Immunol. 2001, 2, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Trowbridge, J.J.; Scott, M.P.; Bhatia, M. Hedgehog Modulates Cell Cycle Regulators in Stem Cells to Control Hematopoietic Regeneration. Proc. Natl. Acad. Sci. USA 2006, 103, 14134–14139. [Google Scholar] [CrossRef] [Green Version]
- Dierks, C.; Beigi, R.; Guo, G.-R.; Zirlik, K.; Stegert, M.R.; Manley, P.; Trussell, C.; Schmitt-Graeff, A.; Landwerlin, K.; Veelken, H.; et al. Expansion of Bcr-Abl-Positive Leukemic Stem Cells Is Dependent on Hedgehog Pathway Activation. Cancer Cell 2008, 14, 238–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, I.; Stover, E.H.; Cullen, D.E.; Mao, J.; Morgan, K.J.; Lee, B.H.; Kharas, M.G.; Miller, P.G.; Cornejo, M.G.; Okabe, R.; et al. Hedgehog Signaling Is Dispensable for Adult Murine Hematopoietic Stem Cell Function and Hematopoiesis. Cell Stem Cell 2009, 4, 559–567. [Google Scholar] [CrossRef] [Green Version]
- Merchant, A.A.; Matsui, W. Targeting Hedgehog—A Cancer Stem Cell Pathway. Clin. Cancer Res. 2010, 16, 3130–3140. [Google Scholar] [CrossRef] [Green Version]
- Merchant, A.; Joseph, G.; Wang, Q.; Brennan, S.; Matsui, W. Gli1 Regulates the Proliferation and Differentiation of HSCs and Myeloid Progenitors. Blood 2010, 115, 2391–2396. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Graves, S.; Koch, U.; Liu, S.; Jankovic, V.; Buonamici, S.; Andaloussi, A.E.; Nimer, S.; Kee, B.L.; Taichman, R.; et al. Hedgehog Signaling Is Dispensable for Adult Hematopoietic Stem Cell Function. Cell Stem Cell 2009, 4, 548–558. [Google Scholar] [CrossRef] [Green Version]
- Niyaz, M.; Khan, M.S.; Mudassar, S. Hedgehog Signaling: An Achilles’ Heel in Cancer. Transl. Oncol. 2019, 12, 1334–1344. [Google Scholar] [CrossRef]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The Role of the Hedgehog Signaling Pathway in Cancer: A Comprehensive Review. Bosn. J. Basic Med. Sci. 2018, 18, 8. [Google Scholar] [CrossRef] [Green Version]
- Oro, A.E.; Higgins, K.M.; Hu, Z.; Bonifas, J.M.; Epstein, E.H.; Scott, M.P. Basal Cell Carcinomas in Mice Overexpressing Sonic Hedgehog. Science 1997, 276, 817–821. [Google Scholar] [CrossRef] [Green Version]
- Gorlin, R.J. Nevoid Basal Cell Carcinoma (Gorlin) Syndrome. Genet. Med. 2004, 6, 530–539. [Google Scholar] [CrossRef] [Green Version]
- Kubo, M.; Nakamura, M.; Tasaki, A.; Yamanaka, N.; Nakashima, H.; Nomura, M.; Kuroki, S.; Katano, M. Hedgehog Signaling Pathway Is a New Therapeutic Target for Patients with Breast Cancer. Cancer Res. 2004, 64, 6071–6074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watkins, D.N.; Berman, D.M.; Burkholder, S.G.; Wang, B.; Beachy, P.A.; Baylin, S.B. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 2003, 422, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Theunissen, J.-W.; de Sauvage, F.J. Paracrine Hedgehog Signaling in Cancer. Cancer Res. 2009, 69, 6007–6010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellbrock, J.; Latuske, E.; Köhler, J.; Wagner, K.; Stamm, H.; Vettorazzi, E.; Vohwinkel, G.; Klokow, M.; Uibeleisen, R.; Ehm, P.; et al. Expression of Hedgehog Pathway Mediator GLI Represents a Negative Prognostic Marker in Human Acute Myeloid Leukemia and Its Inhibition Exerts Antileukemic Effects. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 2388–2398. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Chen, A.; Jamieson, C.H.; Fereshteh, M.; Abrahamsson, A.; Blum, J.; Kwon, H.Y.; Kim, J.; Chute, J.P.; Rizzieri, D.; et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009, 458, 776–779. [Google Scholar] [CrossRef] [Green Version]
- Pietrobono, S.; Stecca, B. Targeting the Oncoprotein Smoothened by Small Molecules: Focus on Novel Acylguanidine Derivatives as Potent Smoothened Inhibitors. Cells 2018, 7, 272. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Paradise, B.D.; Ma, W.W.; Fernandez-Zapico, M.E. Recent Advances in the Clinical Targeting of Hedgehog/GLI Signaling in Cancer. Cells 2019, 8, 394. [Google Scholar] [CrossRef] [Green Version]
- Deininger, M.W.; Goldman, J.M.; Melo, J.V. The Molecular Biology of Chronic Myeloid Leukemia. Blood 2000, 96, 3343–3356. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.M.; Jørgensen, H.G.; Allan, E.; Pearson, C.; Alcorn, M.J.; Richmond, L.; Holyoake, T.L. Primitive, Quiescent, Philadelphia-Positive Stem Cells from Patients with Chronic Myeloid Leukemia Are Insensitive to STI571 In Vitro. Blood 2002, 99, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Houshmand, M.; Simonetti, G.; Circosta, P.; Gaidano, V.; Cignetti, A.; Martinelli, G.; Saglio, G.; Gale, R.P. Chronic Myeloid Leukemia Stem Cells. Leukemia 2019, 33, 1543–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holyoake, T.; Jiang, X.; Eaves, C.; Eaves, A. Isolation of a Highly Quiescent Subpopulation of Primitive Leukemic Cells in Chronic Myeloid Leukemia. Blood 1999, 94, 2056–2064. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Meng, F.; Huang, L.; Zheng, M.; Liu, W.; Sun, H. Sonic Hedgehog Maintains Survival and Growth of Chronic Myeloid Leukemia Progenitor Cells through β-Catenin Signaling. Exp. Hematol. 2012, 40, 418–427. [Google Scholar] [CrossRef]
- Irvine, D.A.; Zhang, B.; Kinstrie, R.; Tarafdar, A.; Morrison, H.; Campbell, V.L.; Moka, H.A.; Ho, Y.; Nixon, C.; Manley, P.W.; et al. Deregulated Hedgehog Pathway Signaling Is Inhibited by the Smoothened Antagonist LDE225 (Sonidegib) in Chronic Phase Chronic Myeloid Leukaemia. Sci. Rep. 2016, 6, 25476. [Google Scholar] [CrossRef] [Green Version]
- Turner, K.A. Assessment of A Potential Therapeutic Target in the Hedgehog Pathway for the Eradication of Primitive Chronic Myeloid Leukemia Cells. Available online: https://open.library.ubc.ca/media/stream/pdf/24/1.0354447/4 (accessed on 14 March 2023).
- Turner, K.; Rothe, K.; Woolfson, A.; Jiang, X. SMO and GLI2 Are Key Regulators Mediating Resistance of CML Stem/Progenitor Cells to Tyrosine Kinase Inhibitors. Exp. Hematol. 2017, 53, S62. [Google Scholar] [CrossRef]
- Sadarangani, A.; Pineda, G.; Lennon, K.M.; Chun, H.-J.; Shih, A.; Schairer, A.E.; Court, A.C.; Goff, D.J.; Prashad, S.L.; Geron, I.; et al. GLI2 Inhibition Abrogates Human Leukemia Stem Cell Dormancy. J. Transl. Med. 2015, 13, 98. [Google Scholar] [CrossRef] [Green Version]
- Schairer, A.; Shih, A.; Geron, I.; Reya, T.; Levin, W.J.; Van Arsdale, T.; Jamieson, C. Human Blast Crisis Leukemia Stem Cell Inhibition with a Novel Smoothened Antagonist. Blood 2010, 116, 1223. [Google Scholar] [CrossRef]
- Anusha; Dalal, H.; Subramanian, S.; VP, S.; Gowda, D.A.; Damodar, S.; Vyas, N. Exovesicular-Shh Confers Imatinib Resistance by Upregulating Bcl2 Expression in Chronic Myeloid Leukemia with Variant Chromosomes. Cell Death Dis. 2021, 12, 1–17. [Google Scholar] [CrossRef]
- Jamieson, C.; Martinelli, G.; Papayannidis, C.; Cortes, J.E. Hedgehog Pathway Inhibitors: A New Therapeutic Class for the Treatment of Acute Myeloid Leukemia. Blood Cancer Discov. 2020, 1, 134–145. [Google Scholar] [CrossRef]
- Lachowiez, C.; DiNardo, C.D.; Konopleva, M. Venetoclax in Acute Myeloid Leukemia—Current and Future Directions. Leuk. Lymphoma 2020, 61, 1313–1322. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Y.; Huang, W.-J.; Yang, J.; Tang, W.-G.; Huang, T.-M.; Tan, W.-F. ABT-199 Inhibits Hedgehog Pathway by Acting as a Competitive Inhibitor of Oxysterol, Rather as a BH3 Mimetic. Acta Pharm. Sin. 2021, 42, 1005–1013. [Google Scholar] [CrossRef]
- Pelullo, M.; Zema, S.; Nardozza, F.; Checquolo, S.; Screpanti, I.; Bellavia, D. Wnt, Notch, and TGF-β Pathways Impinge on Hedgehog Signaling Complexity: An Open Window on Cancer. Front. Genet. 2019, 10, 711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riobo, N.A.; Haines, G.M.; Emerson, C.P., Jr. Protein Kinase C-δ and Mitogen-Activated Protein/Extracellular Signal–Regulated Kinase-1 Control GLI Activation in Hedgehog Signaling. Cancer Res. 2006, 66, 839–845. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, G.; Oehler, V.G.; Papayannidis, C.; Courtney, R.; Shaik, M.N.; Zhang, X.; O’Connell, A.; McLachlan, K.R.; Zheng, X.; Radich, J.; et al. Treatment with PF-04449913, an Oral Smoothened Antagonist, in Patients with Myeloid Malignancies: A Phase 1 Safety and Pharmacokinetics Study. Lancet Haematol. 2015, 2, e339–e346. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Gotlib, J.R.; Mesa, R.A.; Newberry, K.J.; Ravandi, F.; Cortes, J.E.; Kelly, P.; Kutok, J.L.; Kantarjian, H.M.; Verstovsek, S. Phase II Evaluation of IPI-926, an Oral Hedgehog Inhibitor, in Patients with Myelofibrosis. Leuk. Lymphoma 2015, 56, 2092–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lainez-González, D.; Serrano-López, J.; Alonso-Domínguez, J.M. Understanding the Hedgehog Signaling Pathway in Acute Myeloid Leukemia Stem Cells: A Necessary Step toward a Cure. Biology 2021, 10, 255. [Google Scholar] [CrossRef]
- Cortes, J.E.; Heidel, F.H.; Heuser, M.; Fiedler, W.; Smith, B.D.; Robak, T.; Montesinos Fernandez, P.; Ma, W.W.; Shaik, M.N.; Zeremski, M.; et al. A Phase 2 Randomized Study of Low Dose Ara-C with or without Glasdegib (PF-04449913) in Untreated Patients with Acute Myeloid Leukemia or High-Risk Myelodysplastic Syndrome. Blood 2016, 128, 99. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Abraham, A.; Matsui, W. Hedgehog Signaling in Myeloid Malignancies. Cancers 2021, 13, 4888. [Google Scholar] [CrossRef]
- Shah, N.P.; Cortes, J.E.; Martinelli, G.; Smith, B.D.; Clarke, E.; Copland, M.; Strauss, L.; Talpaz, M. Dasatinib Plus Smoothened (SMO) Inhibitor BMS-833923 in Chronic Myeloid Leukemia (CML) with Resistance or Suboptimal Response to a Prior Tyrosine Kinase Inhibitor (TKI): Phase I Study CA180323. Blood 2014, 124, 4539. [Google Scholar] [CrossRef]
- Lacouture, M.E.; Dréno, B.; Ascierto, P.A.; Dummer, R.; Basset-Seguin, N.; Fife, K.; Ernst, S.; Licitra, L.; Neves, R.I.; Peris, K.; et al. Characterization and Management of Hedgehog Pathway Inhibitor-Related Adverse Events in Patients With Advanced Basal Cell Carcinoma. Oncologist 2016, 21, 1218–1229. [Google Scholar] [CrossRef] [Green Version]
- Guerra, V.A.; DiNardo, C.; Konopleva, M. Venetoclax-Based Therapies for Acute Myeloid Leukemia. Best Pr. Res. Clin. Haematol. 2019, 32, 145–153. [Google Scholar] [CrossRef]
- Wang, F.; Huang, X.; Sun, Y.; Li, Z.; Sun, R.; Zhao, T.; Wang, M.; Yan, C.; Liu, P. Sulforaphane Regulates the Proliferation of Leukemia Stem-like Cells via Sonic Hedgehog Signaling Pathway. Eur. J. Pharmacol. 2022, 919, 174824. [Google Scholar] [CrossRef]
- Yauch, R.L.; Dijkgraaf, G.J.P.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened Mutation Confers Resistance to a Hedgehog Pathway Inhibitor in Medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Wang, C.; Chen, Z.; Zhao, W. Overcoming the Resistance Mechanisms of Smoothened Inhibitors. Drug Discov. Today 2018, 23, 704–710. [Google Scholar] [CrossRef]
- Zhang, J.; Fan, J.; Zeng, X.; Nie, M.; Luan, J.; Wang, Y.; Ju, D.; Yin, K. Hedgehog Signaling in Gastrointestinal Carcinogenesis and the Gastrointestinal Tumor Microenvironment. Acta Pharm. Sin. B 2021, 11, 609–620. [Google Scholar] [CrossRef]
- Katagiri, T.; Kobayashi, M.; Yoshimura, M.; Morinibu, A.; Itasaka, S.; Hiraoka, M.; Harada, H. HIF-1 Maintains a Functional Relationship between Pancreatic Cancer Cells and Stromal Fibroblasts by Upregulating Expression and Secretion of Sonic Hedgehog. Oncotarget 2018, 9, 10525–10535. [Google Scholar] [CrossRef] [Green Version]
- Lei, J.; Ma, J.; Ma, Q.; Li, X.; Liu, H.; Xu, Q.; Duan, W.; Sun, Q.; Xu, J.; Wu, Z.; et al. Hedgehog Signaling Regulates Hypoxia Induced Epithelial to Mesenchymal Transition and Invasion in Pancreatic Cancer Cells via a Ligand-Independent Manner. Mol. Cancer 2013, 12, 66. [Google Scholar] [CrossRef] [Green Version]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz, I.; Altaba, A. Melanomas Require HEDGEHOG-GLI Signaling Regulated by Interactions between GLI1 and the RAS-MEK/AKT Pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar] [CrossRef] [Green Version]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The Tumour Microenvironment in Pancreatic Cancer—Clinical Challenges and Opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef]
- Queiroz, K.C.S.; Ruela-de-Sousa, R.R.; Fuhler, G.M.; Aberson, H.L.; Ferreira, C.V.; Peppelenbosch, M.P.; Spek, C.A. Hedgehog Signaling Maintains Chemoresistance in Myeloid Leukemic Cells. Oncogene 2010, 29, 6314–6322. [Google Scholar] [CrossRef] [Green Version]
- De Araújo, T.B.S.; Rocha, L.D.O.S.D.; Vidal, M.T.A.; Coelho, P.C.; Dos Reis, M.G.; Souza, B.S.D.F.; Soares, M.B.P.; Pereira, T.A.; Della Coletta, R.; Bezerra, D.P.; et al. GANT61 Reduces Hedgehog Molecule (GLI1) Expression and Promotes Apoptosis in Metastatic Oral Squamous Cell Carcinoma Cells. Int. J. Mol. Sci. 2020, 21, 6076. [Google Scholar] [CrossRef]
- Benvenuto, M.; Masuelli, L.; De Smaele, E.; Fantini, M.; Mattera, R.; Cucchi, D.; Bonanno, E.; Di Stefano, E.; Frajese, G.V.; Orlandi, A.; et al. In Vitro and in Vivo Inhibition of Breast Cancer Cell Growth by Targeting the Hedgehog/GLI Pathway with SMO (GDC-0449) or GLI (GANT-61) Inhibitors. Oncotarget 2016, 7, 9250–9270. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Rodova, M.; Roy, S.K.; Sharma, J.; Singh, K.P.; Srivastava, R.K.; Shankar, S. GANT-61 Inhibits Pancreatic Cancer Stem Cell Growth in Vitro and in NOD/SCID/IL2R Gamma Null Mice Xenograft. Cancer Lett. 2013, 330, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, R.K.; Kaylani, S.Z.; Edrees, N.; Li, C.; Talwelkar, S.S.; Xu, J.; Palle, K.; Pressey, J.G.; Athar, M. GLI Inhibitor GANT-61 Diminishes Embryonal and Alveolar Rhabdomyosarcoma Growth by Inhibiting Shh/AKT-MTOR Axis. Oncotarget 2014, 5, 12151–12165. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Gu, S.; Huang, J.; Chen, S.; Zhang, Z.; Xu, M. Inhibition of Autophagy Potentiates the Efficacy of Gli Inhibitor GANT-61 in MYCN-Amplified Neuroblastoma Cells. BMC Cancer 2014, 14, 768. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Lee, J.J.; Kim, J.; Gardner, D.; Beachy, P.A. Arsenic Antagonizes the Hedgehog Pathway by Preventing Ciliary Accumulation and Reducing Stability of the Gli2 Transcriptional Effector. Proc. Natl. Acad. Sci. USA 2010, 107, 13432–13437. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, A.; Turner, K.A.; Woolfson, A.; Jiang, X. The Hedgehog Pathway as a Therapeutic Target in Chronic Myeloid Leukemia. Pharmaceutics 2023, 15, 958. https://doi.org/10.3390/pharmaceutics15030958
Wu A, Turner KA, Woolfson A, Jiang X. The Hedgehog Pathway as a Therapeutic Target in Chronic Myeloid Leukemia. Pharmaceutics. 2023; 15(3):958. https://doi.org/10.3390/pharmaceutics15030958
Chicago/Turabian StyleWu, Andrew, Kelly A. Turner, Adrian Woolfson, and Xiaoyan Jiang. 2023. "The Hedgehog Pathway as a Therapeutic Target in Chronic Myeloid Leukemia" Pharmaceutics 15, no. 3: 958. https://doi.org/10.3390/pharmaceutics15030958