
Micro-Injection Moulding of PEO/PCL Blend–Based Matrices for Extended Oral Delivery of Fenbendazole
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Hot-Melt Extrusion
2.3. Melt Flow Index
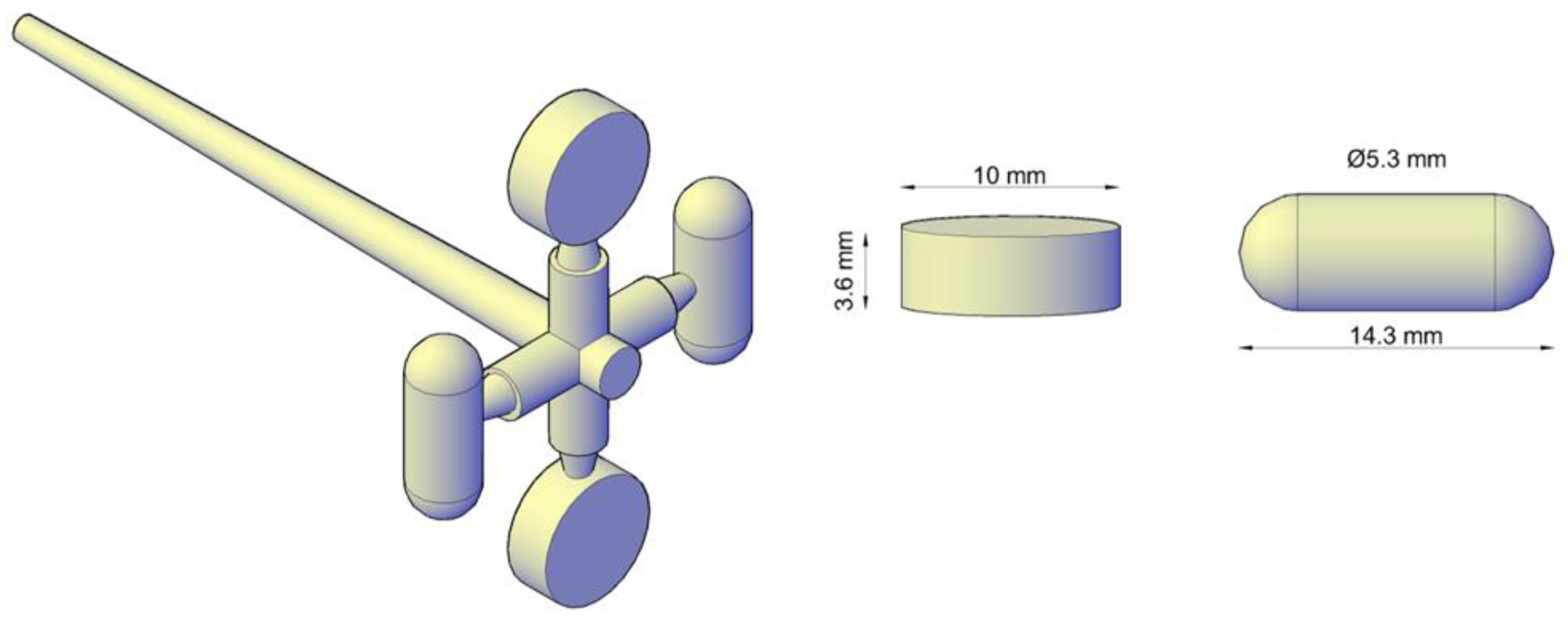
2.4. Micro-Injection Moulding
2.5. Shore D Hardness Evaluation
2.6. Thermal Characterisation by DSC and TGA/DTG
2.7. Attenuated Total Reflectance-Fourier Transform Infrared Spectroscopy
2.8. Powder X-ray Diffraction Spectroscopy
2.9. Scanning Electron Microscopy with Energy-Dispersive X-ray Spectroscopy
2.10. Determination of Drug Content
2.11. Release Studies
2.12. Percentage Mass Loss of Moulded Tablets
2.13. Statistical Analyses
3. Results
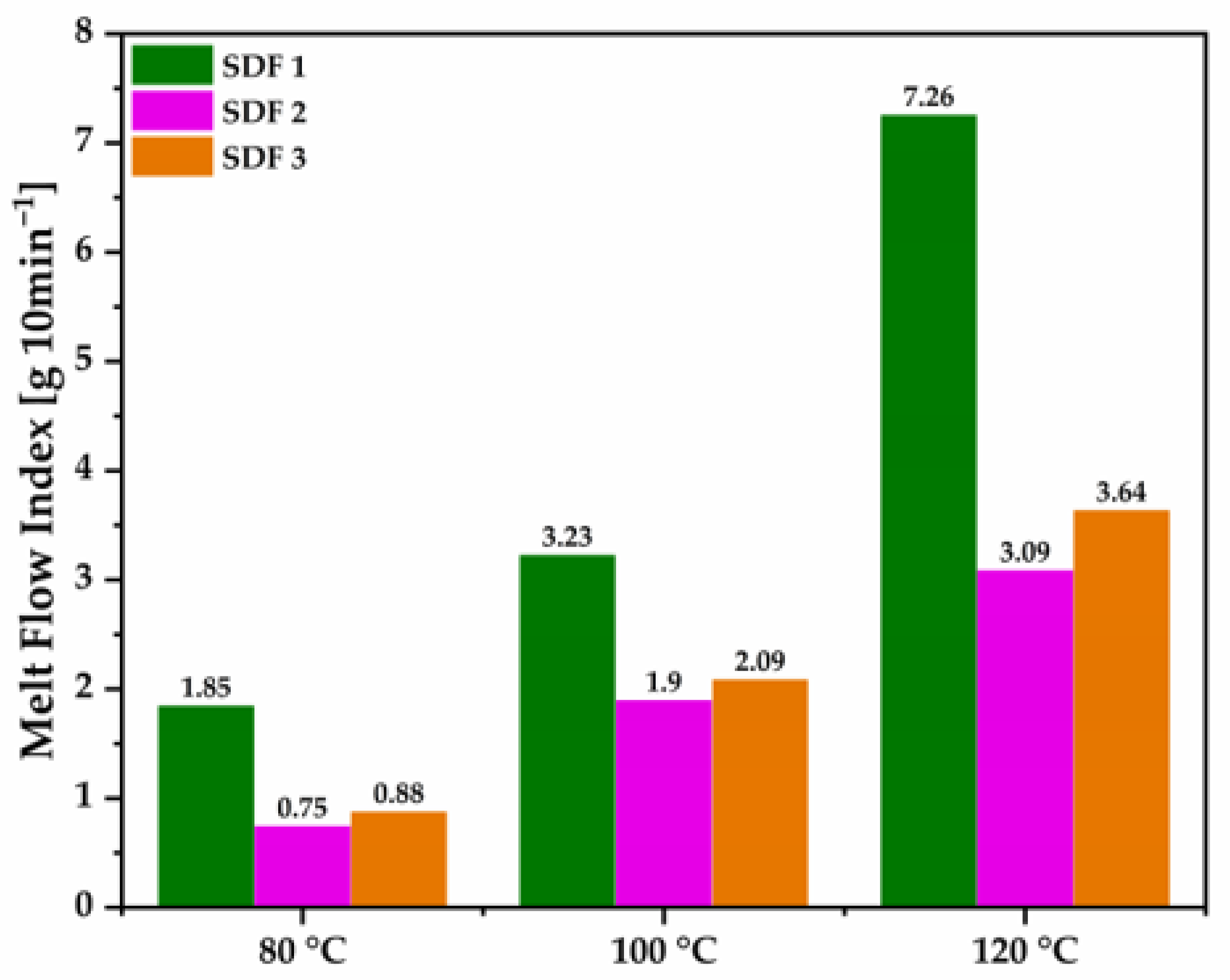
3.1. Material Melt Viscosity by MFI
3.2. Micro-Injection Moulding Tablets
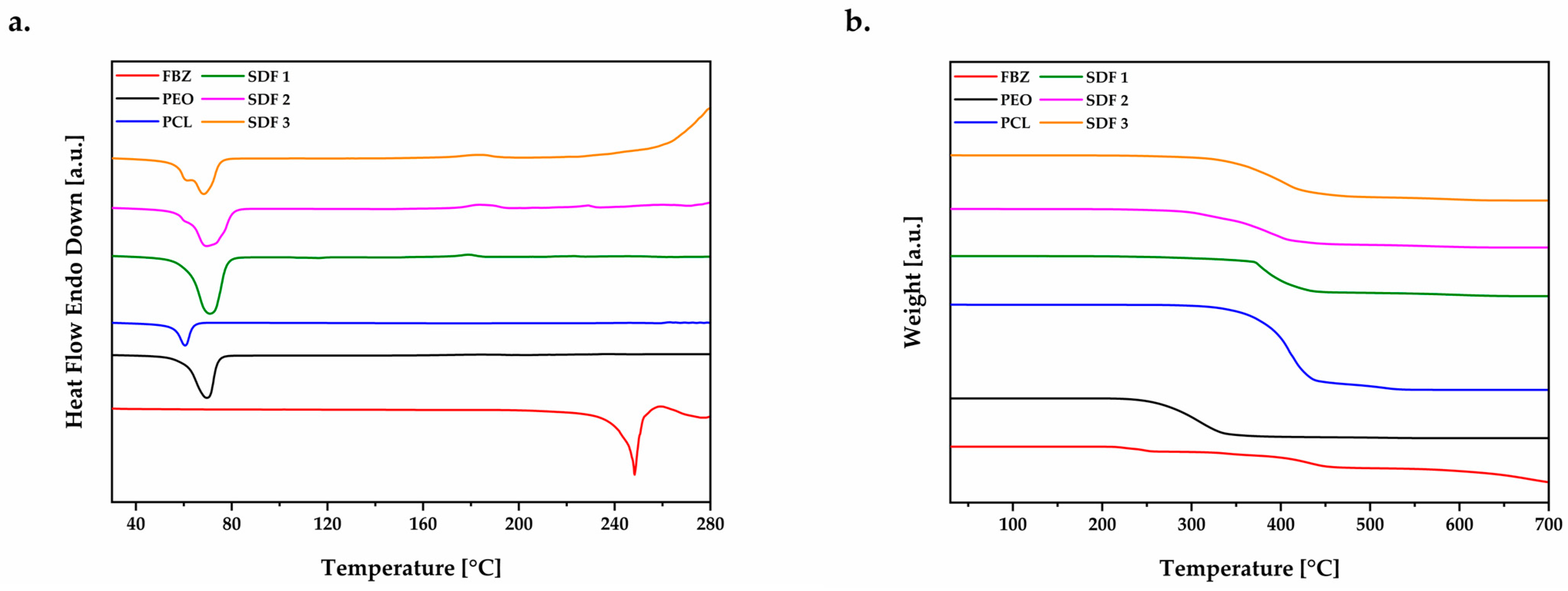
3.3. Thermal Study of the Moulded Tablets by DSC and TGA
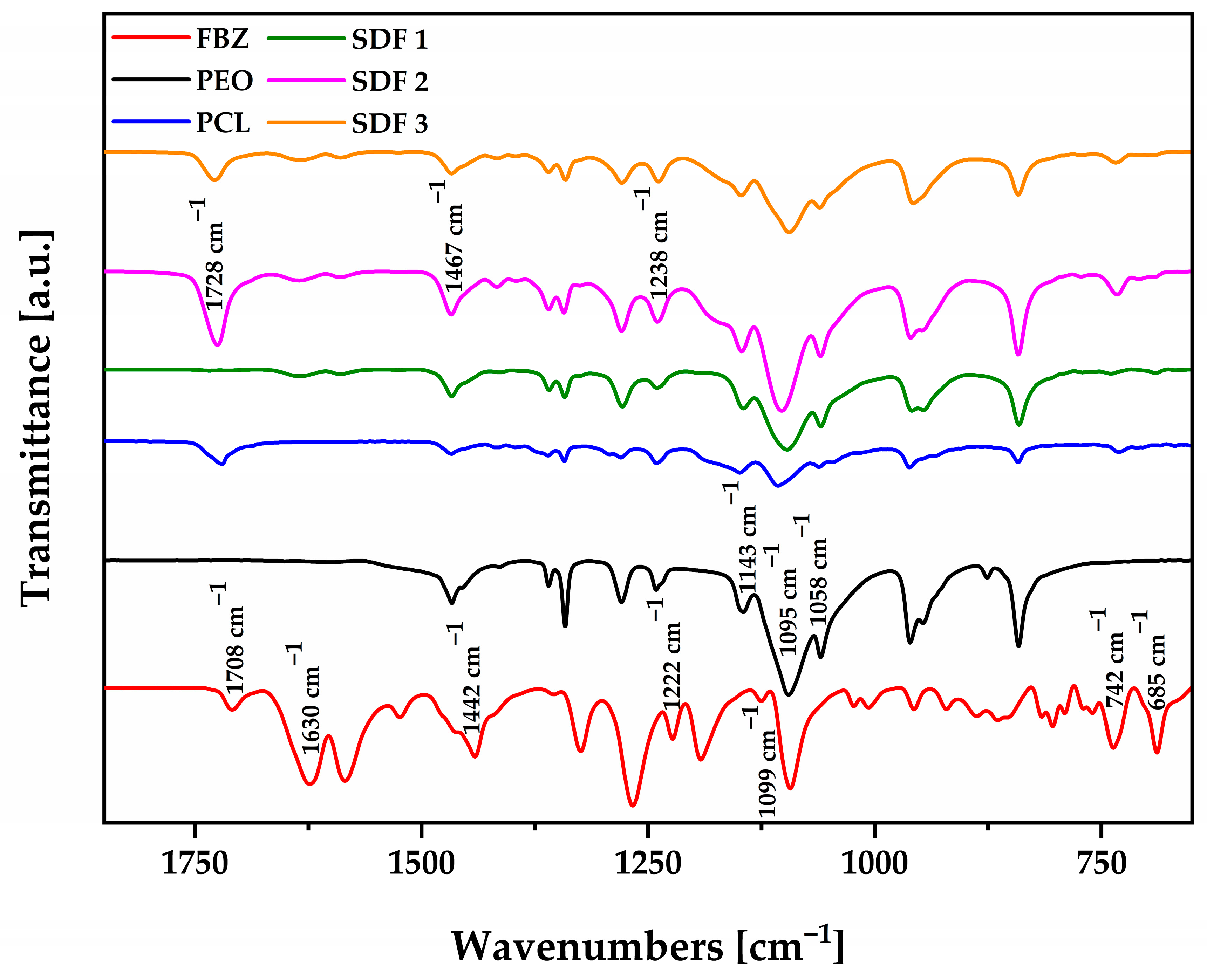
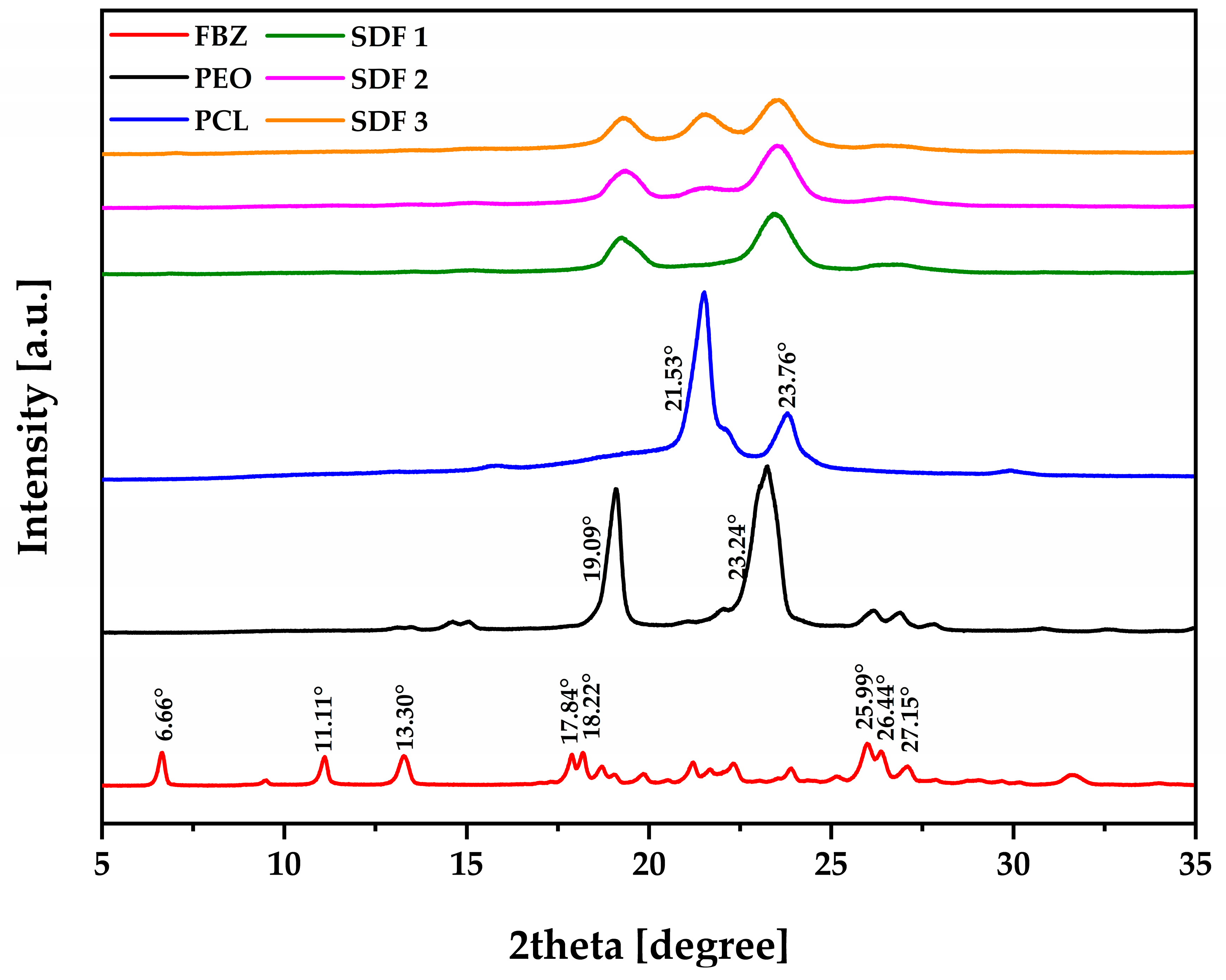
3.4. Physical and Chemical Evaluation of the Moulded Tablets by FTIR and pXRD
3.5. Drug Dispersion Assessment by SEM and EDX
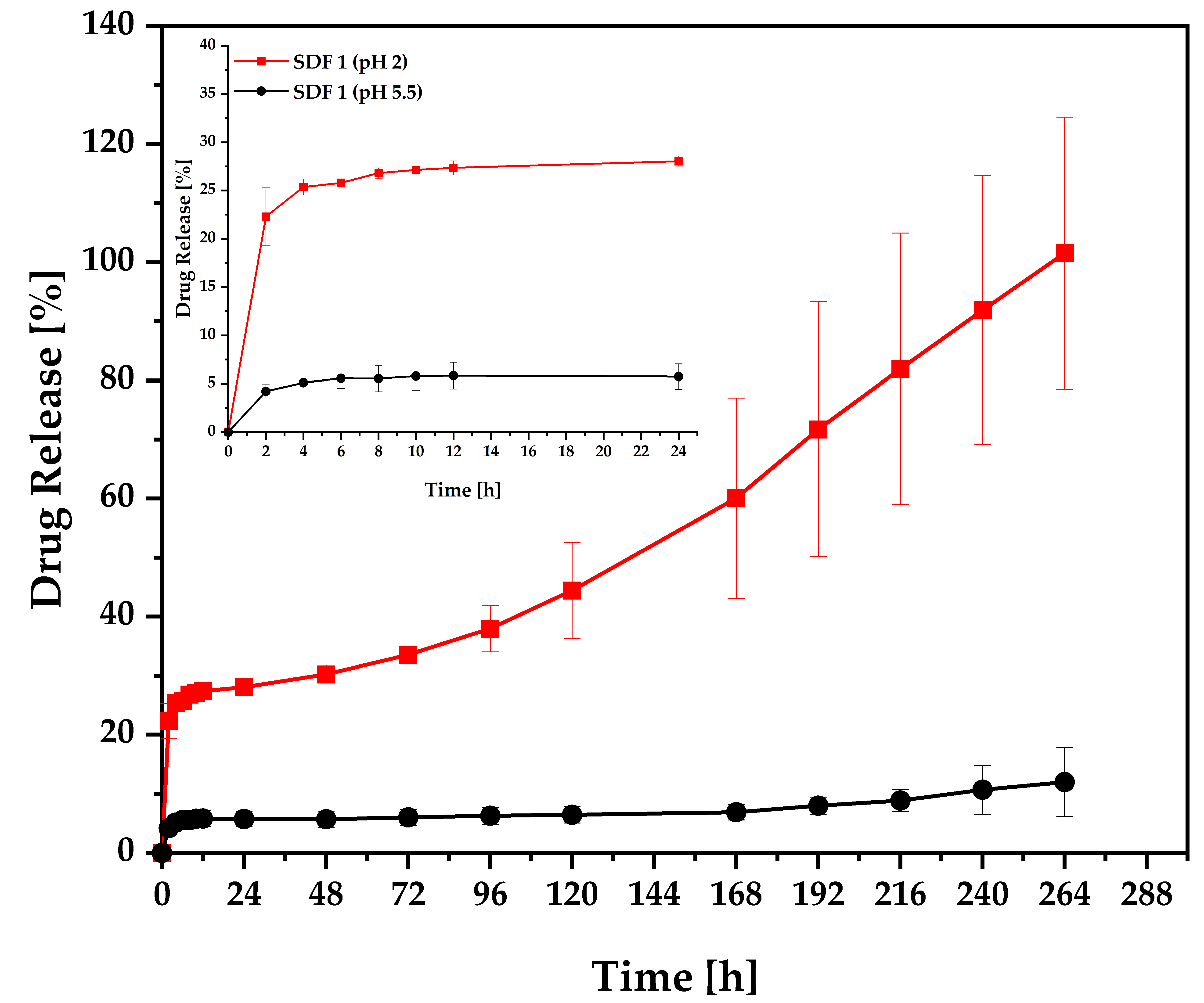
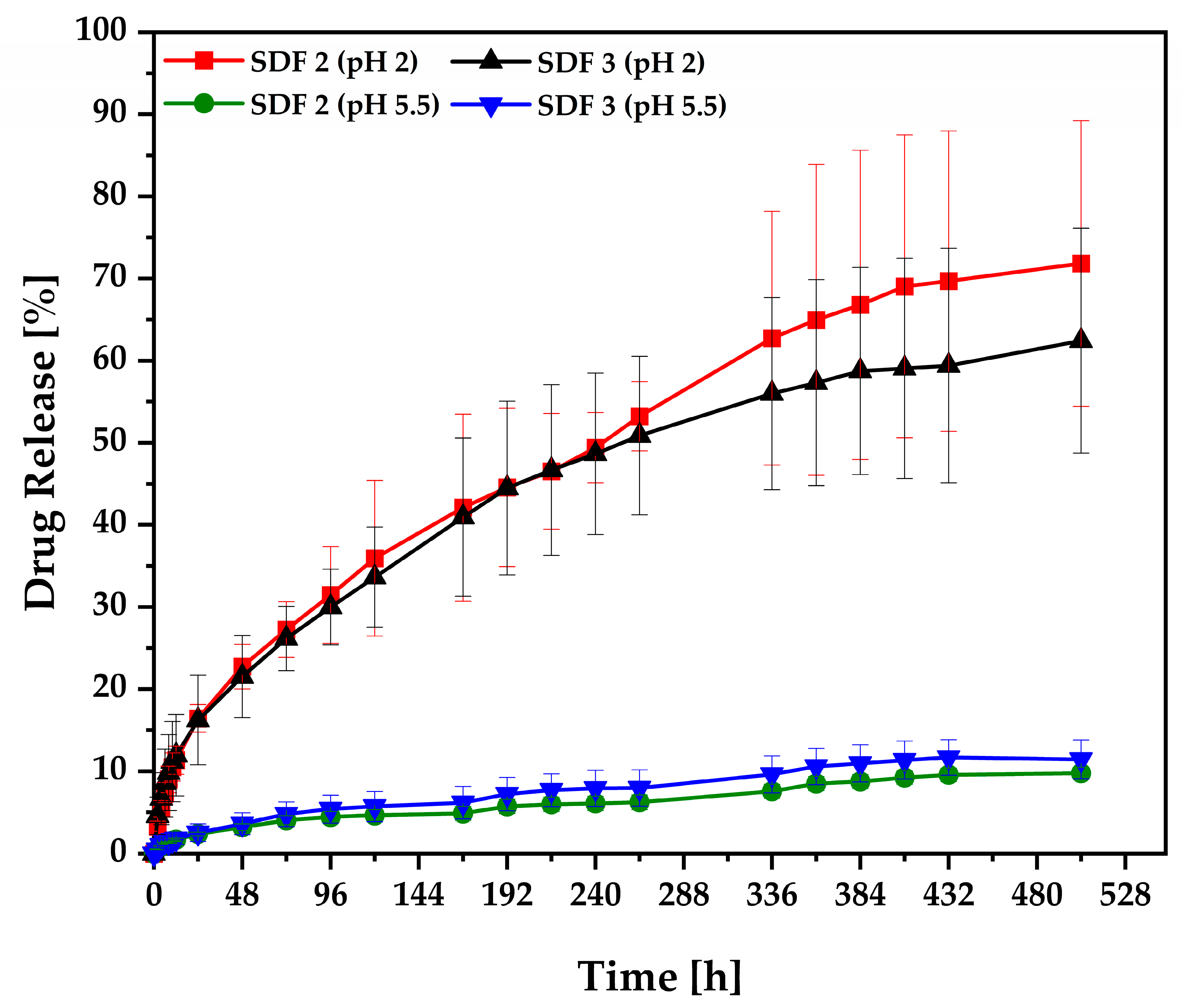
3.6. Release Studies
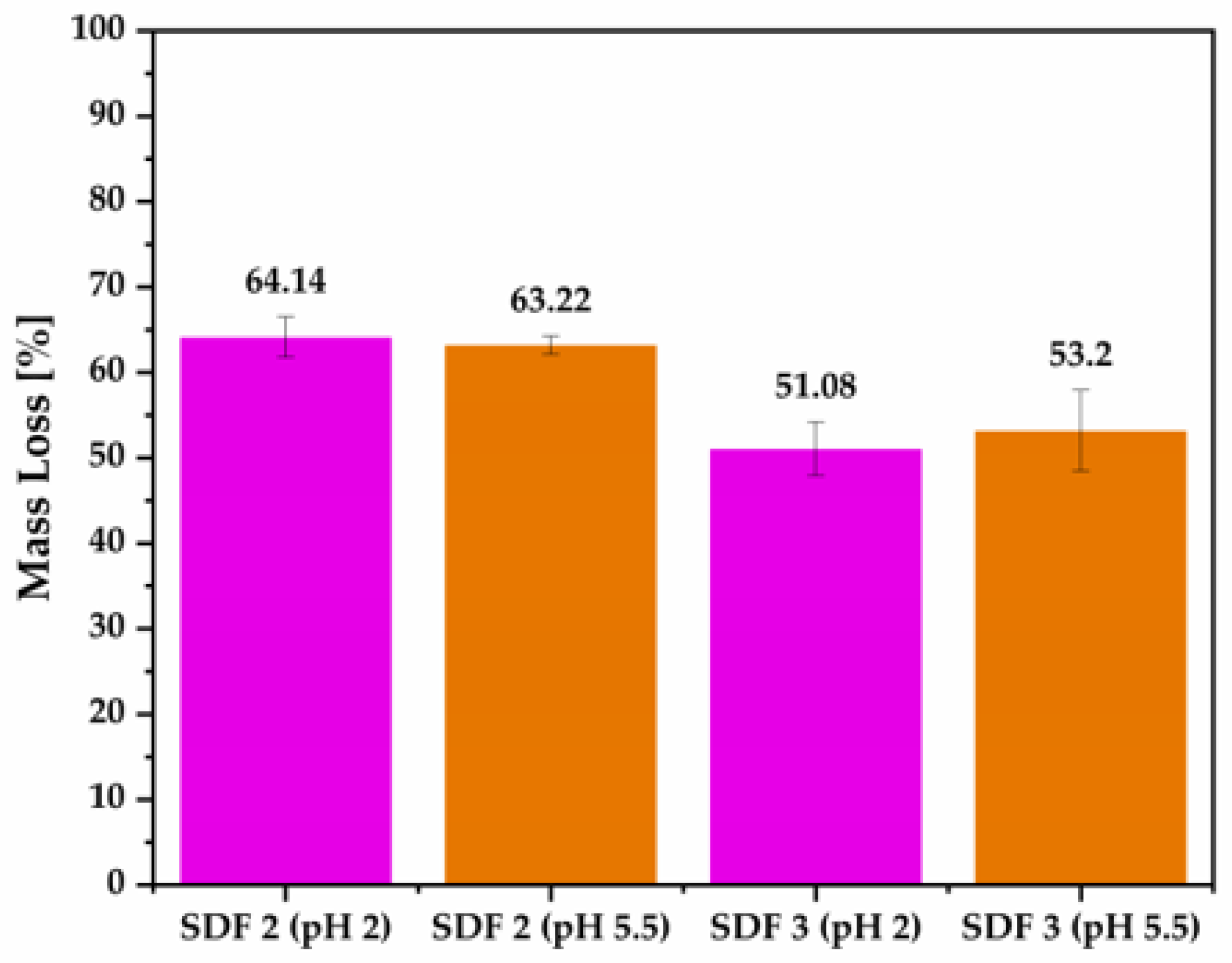
3.7. Weight Loss of Moulded Tablets
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sabnis, S.; Rathbone, M.J. Animal Health Markets and Opportunities: Farmed Animal Landscape. In Long Acting Animal Health Drug Products. Advances in Delivery Science and Technology; Springer: New York, NY, USA, 2013; pp. 1–14. ISBN 9781461444398. [Google Scholar]
- Dziduch, K.; Greniuk, D.; Wujec, M. The Current Directions of Searching for Antiparasitic Drugs. Molecules 2022, 27, 1534. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, L.I.; Mottier, M.L.; Lanusse, C.E. Drug Transfer into Target Helminth Parasites. Trends Parasitol. 2007, 23, 97–104. [Google Scholar] [CrossRef]
- Ellis, K.J. Anatomy and Physiology of the Farmed Animal. In Long Acting Animal Health Drug Products. Advances in Delivery Science and Technology; Springer: New York, NY, USA, 2013; pp. 47–58. ISBN 9781461444398. [Google Scholar]
- Lanusse, C.E.; Prichard, R.K. Clinical Pharmacokinetics and Metabolism of Benzimidazole Anthelmintics in Ruminants. Drug Metab. Rev. 1993, 25, 235–279. [Google Scholar] [CrossRef]
- Köhler, P. The Biochemical Basis of Anthelmintic Action and Resistance. Int. J. Parasitol. 2001, 31, 336–345. [Google Scholar] [CrossRef]
- Martinez, M. Applying the Biopharmaceutics Classification System to Veterinary Pharmaceutical Products Part I: Biopharmaceutics and Formulation Considerations. Adv. Drug Deliv. Rev. 2002, 54, 805–824. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, S.C.; Tichý, T.; Vávra, J.; Dash, R.P.; Slusher, C.E.; Gadiano, A.J.; Wu, Y.; Jančařík, A.; Tenora, L.; Monincová, L.; et al. N-Substituted Prodrugs of Mebendazole Provide Improved Aqueous Solubility and Oral Bioavailability in Mice and Dogs. J. Med. Chem. 2018, 61, 3918–3929. [Google Scholar] [CrossRef] [PubMed]
- Lanusse, C.E.; Prichard, R.K. Relationship between Pharmacological Properties and Clinical Efficacy of Ruminant Anthelmintics. Vet. Parasitol. 1993, 49, 123–158. [Google Scholar] [CrossRef]
- Prichard, R.K. Interaction of Host Physiology and Efficacy of Antiparasitic Drugs. Vet. Parasitol. 1985, 18, 103–110. [Google Scholar] [CrossRef]
- Campbell, W.C. Benzimidazoles: Veterinary Uses. Parasitol. Today 1990, 6, 130–133. [Google Scholar] [CrossRef]
- Mitra, A.; Li, L.; Marsac, P.; Marks, B.; Liu, Z.; Brown, C. Impact of Polymer Type on Bioperformance and Physical Stability of Hot Melt Extruded Formulations of a Poorly Water Soluble Drug. Int. J. Pharm. 2016, 505, 107–114. [Google Scholar] [CrossRef]
- Zhang, F.; Mcginity, J.W. Properties of Sustained-Release Tablets Prepared by Hot-Melt Extrusion. Pharm. Dev. Technol. 1999, 4, 241–250. [Google Scholar] [CrossRef]
- Li, Y.; Pang, H.; Guo, Z.; Lin, L.; Dong, Y.; Li, G.; Lu, M.; Wu, C. Interactions between Drugs and Polymers Influencing Hot Melt Extrusion. J. Pharm. Pharmacol. 2014, 66, 148–166. [Google Scholar] [CrossRef]
- Arkhipov, I.A.; Khalikov, S.S.; Sadov, K.M.; Dushkin, A.V.; Meteleva, E.S.; Varlamova, A.I.; Odoevskaya, I.M.; Danilevskaya, N.V. Influence of Mechanochemical Technology on Anthelmintic Efficacy of the Supramolecular Complex of Fenbendazole with Polyvinylpyrrolidone. J. Adv. Vet. Anim. Res. 2019, 6, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.B.; Rajabi-Siahboomi, A.R. Extended-Release Oral Drug Delivery Technologies: Monolithic Matrix Systems. In Drug Delivery Systems. Methods in Molecular Biology™; Humana: Totowa, NJ, USA, 2008; pp. 217–243. ISBN 978-1-59745-210-6. [Google Scholar]
- Benzine, Y.; Siepmann, F.; Neut, C.; Danede, F.; Francois Willart, J.; Siepmann, J.; Karrout, Y. Injection-Molded Capsule Bodies and Caps Based on Polymer Blends for Controlled Drug Delivery. Eur. J. Pharm. Biopharm. 2021, 168, 1–14. [Google Scholar] [CrossRef]
- Grehan, L.; Killion, J.A.; Devine, D.M.; Kenny, E.K.; Devery, S.; Higginbotham, C.L.; Geever, L.M. The Development of Hot Melt Extruded Biocompatible Controlled Release Drug Delivery Devices. Int. J. Polym. Mater. Polym. Biomater. 2014, 63, 476–485. [Google Scholar] [CrossRef]
- Lyons, J.G.; Blackie, P.; Higginbotham, C.L. The Significance of Variation in Extrusion Speeds and Temperatures on a PEO/PCL Blend Based Matrix for Oral Drug Delivery. Int. J. Pharm. 2008, 351, 201–208. [Google Scholar] [CrossRef]
- Bezerra, G.S.N.; de Lima, T.A.D.M.; Colbert, D.M.; Geever, J.; Geever, L. Formulation and Evaluation of Fenbendazole Extended-Release Extrudes Processed by Hot-Melt Extrusion. Polymers 2022, 14, 4188. [Google Scholar] [CrossRef]
- Giboz, J.; Copponnex, T.; Mélé, P. Microinjection Molding of Thermoplastic Polymers: A Review. J. Micromechanics Microengineering 2007, 17, R96–R109. [Google Scholar] [CrossRef]
- Zema, L.; Loreti, G.; Melocchi, A.; Maroni, A.; Gazzaniga, A. Injection Molding and Its Application to Drug Delivery. J. Control. Release 2012, 159, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Puri, V.; Brancazio, D.; Desai, P.M.; Jensen, K.D.; Chun, J.-H.; Myerson, A.S.; Trout, B.L. Development of Maltodextrin-Based Immediate-Release Tablets Using an Integrated Twin-Screw Hot-Melt Extrusion and Injection-Molding Continuous Manufacturing Process. J. Pharm. Sci. 2017, 106, 3328–3336. [Google Scholar] [CrossRef]
- Chatterjee, S. FDA Perspective on Continuous Manufacturing. In Proceedings of the IFPAC Annual Meeting, Baltimore, MD, USA, 22–25 January 2012; p. 22. [Google Scholar]
- Bezerra, G.S.N.; Colbert, D.M.; O’Donnell, C.; Cao, Z.; Geever, J.; Geever, L. Compatibility Study Between Fenbendazole and Poly(Ethylene Oxide) with Application in Solid Dispersion Formulations Using Hot-Melt Extrusion. J. Pharm. Innov. 2022. [Google Scholar] [CrossRef]
- Allaf, R.M.; Albarahmieh, E.; AlHamarneh, B.M. Solid-State Compounding of Immiscible PCL-PEO Blend Powders for Molding Processes. J. Mech. Behav. Biomed. Mater. 2019, 97, 198–211. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information PubChem Compound Summary for CID 3334, Fenbendazole. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Fenbendazole (accessed on 7 November 2021).
- Danaher, M.; De Ruyck, H.; Crooks, S.R.H.; Dowling, G.; O’Keeffe, M. Review of Methodology for the Determination of Benzimidazole Residues in Biological Matrices. J. Chromatogr. B 2007, 845, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Plastics Technology Magazine Micro Molding with an Easy, Affordable, and Established Solution. Available online: http://albaent.com/media-publications/micro-molding-easy-affordable-established-solution/ (accessed on 23 January 2023).
- Bezerra, G.S.N.; Moritz, V.F.; de Lima, T.A.M.; Colbert, D.M.; Geever, J.; Geever, L. Compatibility Study between Fenbendazole and Polymeric Excipients Used in Pharmaceutical Dosage Forms Using Thermal and Non-Thermal Analytical Techniques. Analytica 2022, 3, 448–461. [Google Scholar] [CrossRef]
- Li, S.; Pollock-Dove, C.; Dong, L.C.; Chen, J.; Creasey, A.A.; Dai, W.-G. Enhanced Bioavailability of a Poorly Water-Soluble Weakly Basic Compound Using a Combination Approach of Solubilization Agents and Precipitation Inhibitors: A Case Study. Mol. Pharm. 2012, 9, 1100–1108. [Google Scholar] [CrossRef]
- Vandamme, T.F.; Ellis, K.J. Issues and Challenges in Developing Ruminal Drug Delivery Systems. Adv. Drug Deliv. Rev. 2004, 56, 1415–1436. [Google Scholar] [CrossRef]
- Sanna, V.; Gavini, E.; Giunchedi, P. Bilayer Tablets Based on Poly (ε-Caprolactone) and Polymethylmethacrilates as Controlled-Release Systems for Ruminants. Pharm. Dev. Technol. 2004, 9, 321–328. [Google Scholar] [CrossRef]
- Pell, A.N.; Wu, S.H.; Welch, J.G. Design Parameters for Post-Ruminal Drug Delivery Systems and Rumen-Stable Products. In Controlled Release Veterinary Drug Delivery; Elsevier: Amsterdam, The Netherlands, 2000; pp. 83–113. [Google Scholar]
- The United States Pharmacopeia Online Buffer Solutions. Available online: http://www.uspbpep.com/usp32/pub/data/v32270/usp32nf27s0_buffer-solutions.html (accessed on 13 January 2023).
- Zhang, Y.; Huo, M.; Zhou, J.; Zou, A.; Li, W.; Yao, C.; Xie, S. DDSolver: An Add-In Program for Modeling and Comparison of Drug Dissolution Profiles. AAPS J. 2010, 12, 263–271. [Google Scholar] [CrossRef]
- O’Hara, T.; Dunne, A.; Butler, J.; Devane, J. A Review of Methods Used to Compare Dissolution Profile Data. Pharm. Sci. Technol. Today 1998, 1, 214–223. [Google Scholar] [CrossRef]
- Pezzoli, R.; Hopkins, M.; Direur, G.; Gately, N.; Lyons, J.G.; Higginbotham, C.L. Micro-Injection Moulding of Poly(Vinylpyrrolidone-Vinyl Acetate) Binary and Ternary Amorphous Solid Dispersions. Pharmaceutics 2019, 11, 240. [Google Scholar] [CrossRef]
- Aho, J.; Boetker, J.P.; Baldursdottir, S.; Rantanen, J. Rheology as a Tool for Evaluation of Melt Processability of Innovative Dosage Forms. Int. J. Pharm. 2015, 494, 623–642. [Google Scholar] [CrossRef] [PubMed]
- Guarino, V.; Guaccio, A.; Ambrosio, L. Manipulating Co-Continuous Polymer Blends to Create PCL Scaffolds with Fully Interconnected and Anisotropic Pore Architecture. J. Appl. Biomater. Biomech. 2011, 9, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Stationery Office. British Pharmacopoeia Volume V; Stationery Office: London, UK, 2016; ISBN 9780113230006. [Google Scholar]
- The United States Pharmacopeia Online Fenbendazole. Available online: http://www.uspbpep.com/usp32/pub/data/v32270/usp32nf27s0_m32672.html (accessed on 20 January 2023).
- Attia, A.K.; Saad, A.S.; Alaraki, M.S.; Elzanfaly, E.S. Study of Thermal Analysis Behavior of Fenbendazole and Rafoxanide. Adv. Pharm. Bull. 2017, 7, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Jiang, Y.; Napiwocki, B.; Mi, H.; Peng, X.; Turng, L.-S. Fabrication of Poly(ε-Caprolactone) Tissue Engineering Scaffolds with Fibrillated and Interconnected Pores Utilizing Microcellular Injection Molding and Polymer Leaching. RSC Adv. 2017, 7, 43432–43444. [Google Scholar] [CrossRef]
- Rothen-Weinhold, A.; Besseghir, K.; Vuaridel, E.; Sublet, E.; Oudry, N.; Kubel, F.; Gurny, R. Injection-Molding versus Extrusion as Manufacturing Technique for the Preparation of Biodegradable Implants. Eur. J. Pharm. Biopharm. 1999, 48, 113–121. [Google Scholar] [CrossRef]
- Zhu, Q.; Taylor, L.S.; Harris, M.T. Evaluation of the Microstructure of Semicrystalline Solid Dispersions. Mol. Pharm. 2010, 7, 1291–1300. [Google Scholar] [CrossRef]
- Wong, R.S.H.; Dodou, K. Effect of Drug Loading Method and Drug Physicochemical Properties on the Material and Drug Release Properties of Poly (Ethylene Oxide) Hydrogels for Transdermal Delivery. Polymers 2017, 9, 286. [Google Scholar] [CrossRef]
- Meng, F.; Gala, U.; Chauhan, H. Classification of Solid Dispersions: Correlation to (i) Stability and Solubility (II) Preparation and Characterization Techniques. Drug Dev. Ind. Pharm. 2015, 41, 1401–1415. [Google Scholar] [CrossRef] [PubMed]
- Bunker, A. Poly(Ethylene Glycol) in Drug Delivery, Why Does It Work, and Can We Do Better? All Atom Molecular Dynamics Simulation Provides Some Answers. Phys. Procedia 2012, 34, 24–33. [Google Scholar] [CrossRef]
- Chakrapani, V.Y.; Gnanamani, A.; Giridev, V.R.; Madhusoothanan, M.; Sekaran, G. Electrospinning of Type I Collagen and PCL Nanofibers Using Acetic Acid. J. Appl. Polym. Sci. 2012, 125, 3221–3227. [Google Scholar] [CrossRef]
- Melian, M.E.; Munguía, A.B.; Faccio, R.; Palma, S.; Domínguez, L. The Impact of Solid Dispersion on Formulation, Using Confocal Micro Raman Spectroscopy as Tool to Probe Distribution of Components. J. Pharm. Innov. 2018, 13, 58–68. [Google Scholar] [CrossRef]
- Xu, X.; Jiang, L.; Zhou, Z.; Wu, X.; Wang, Y. Preparation and Properties of Electrospun Soy Protein Isolate/Polyethylene Oxide Nanofiber Membranes. ACS Appl. Mater. Interfaces 2012, 4, 4331–4337. [Google Scholar] [CrossRef]
- Balu, R.; Sampath Kumar, T.S.; Ramalingam, M.; Ramakrishna, S. Electrospun Polycaprolactone/Poly(1,4-Butylene Adipate-Co-Polycaprolactam) Blends: Potential Biodegradable Scaffold for Bone Tissue Regeneration. J. Biomater. Tissue Eng. 2011, 1, 30–39. [Google Scholar] [CrossRef]
- Quinten, T.; Beer, T.; Vervaet, C.; Remon, J. Evaluation of Injection Moulding as a Pharmaceutical Technology to Produce Matrix Tablets. Eur. J. Pharm. Biopharm. 2009, 71, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Desai, P.M.; Hogan, R.C.; Brancazio, D.; Puri, V.; Jensen, K.D.; Chun, J.-H.; Myerson, A.S.; Trout, B.L. Integrated Hot-Melt Extrusion—Injection Molding Continuous Tablet Manufacturing Platform: Effects of Critical Process Parameters and Formulation Attributes on Product Robustness and Dimensional Stability. Int. J. Pharm. 2017, 531, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Quinten, T.; De Beer, T.; Almeida, A.; Vlassenbroeck, J.; Van Hoorebeke, L.; Remon, J.P.; Vervaet, C. Development and Evaluation of Injection-Molded Sustained-Release Tablets Containing Ethylcellulose and Polyethylene Oxide. Drug Dev. Ind. Pharm. 2011, 37, 149–159. [Google Scholar] [CrossRef]
- Melocchi, A.; Loreti, G.; Del Curto, M.D.; Maroni, A.; Gazzaniga, A.; Zema, L. Evaluation of Hot-Melt Extrusion and Injection Molding for Continuous Manufacturing of Immediate-Release Tablets. J. Pharm. Sci. 2015, 104, 1971–1980. [Google Scholar] [CrossRef] [PubMed]
- Surov, A.O.; Vasilev, N.A.; Vener, M.V.; Parashchuk, O.D.; Churakov, A.V.; Magdysyuk, O.V.; Perlovich, G.L. Pharmaceutical Salts of Fenbendazole with Organic Counterions: Structural Analysis and Solubility Performance. Cryst. Growth Des. 2021, 21, 4516–4530. [Google Scholar] [CrossRef]
- Sutar, Y.; Fulton, S.R.; Paul, S.; Altamirano, S.; Mhatre, S.; Saeed, H.; Patel, P.; Mallick, S.; Bhat, R.; Patravale, V.B.; et al. Docusate-Based Ionic Liquids of Anthelmintic Benzimidazoles Show Improved Pharmaceutical Processability, Lipid Solubility, and in Vitro Activity against Cryptococcus Neoformans. ACS Infect. Dis. 2021, 7, 2637–2649. [Google Scholar] [CrossRef]
- Zlomke, C.; Barth, M.; Mäder, K. Polymer Degradation Induced Drug Precipitation in PLGA Implants—Why Less Is Sometimes More. Eur. J. Pharm. Biopharm. 2019, 139, 142–152. [Google Scholar] [CrossRef]
- Xie, T.; Gao, W.; Taylor, L.S. Impact of Eudragit EPO and Hydroxypropyl Methylcellulose on Drug Release Rate, Supersaturation, Precipitation Outcome and Redissolution Rate of Indomethacin Amorphous Solid Dispersions. Int. J. Pharm. 2017, 531, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, T.; Plakogiannis, F.M. Development and Oral Bioavailability Assessment of a Supersaturated Self-Microemulsifying Drug Delivery System (SMEDDS) of Albendazole. J. Pharm. Pharmacol. 2010, 62, 1112–1120. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Grewal, J.; Jyoti, K.; Jain, U.K.; Chandra, R.; Madan, J. Oral Controlled and Sustained Drug Delivery Systems: Concepts, Advances, Preclinical, and Clinical Status. In Drug Targeting and Stimuli Sensitive Drug Delivery Systems; Elsevier Inc.: Amsterdam, The Netherlands, 2018; pp. 567–626. ISBN 9780128136898. [Google Scholar]
- Sriamornsak, P.; Thirawong, N.; Korkerd, K. Swelling, Erosion and Release Behavior of Alginate-Based Matrix Tablets. Eur. J. Pharm. Biopharm. 2007, 66, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Siepmann, J.; Siepmann, F. Mathematical Modeling of Drug Delivery. Int. J. Pharm. 2008, 364, 328–343. [Google Scholar] [CrossRef] [PubMed]
- Sriamornsak, P.; Sungthongjeeh, S. Modification of Theophylline Release with Alginate Gel Formed in Hard Capsules. AAPS PharmSciTech 2007, 8, E1–E8. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Chemical Structure | Molecular Weight | Melting Temperature | Properties |
|---|---|---|---|---|
| FBZ |  | 299.35 g mol−1 | 244 °C | Hydrophobic Crystalline pKa (5.12, 12.72) |
| PEO |  | 100,000–200,000 g mol−1 | 65 °C | Hydrophilic Semicrystalline Biodegradable |
| PCL |  | 50,000 g mol−1 | 58–60 °C | Hydrophobic Semicrystalline Biodegradable |
| Granules | SDF 1 | SDF 2 | SDF 3 |
|---|---|---|---|
| PEO + FBZ | 100% | 65% | 55% |
| PCL | 0% | 35% | 45% |
| Formulation | Weight (mg) | Shore D Hardness | FBZ (%) |
|---|---|---|---|
| SDF 1 | 352.84 (±SD = 0.82) | 49.85 (±SD = 1.68) | 90.47 (±SD = 3.50) |
| SDF 2 | 347.78 (±SD = 0.37) | 50.43 (±SD = 1.50) | 90.94 (±SD = 2.79) |
| SDF3 | 345.25 (±SD = 2.71) | 52.23 (±SD = 2.05) | 100.09 (±SD = 5.25) |
| Release Model | Equation | Parameter | SDF 2 | SDF 3 | ||
|---|---|---|---|---|---|---|
| pH 2 | pH 5.5 | pH 2 | pH 5.5 | |||
| Zero-order | R2adjusted | 0.84 | 0.83 | 0.74 | 0.84 | |
| AIC | 177.55 | 85.00 | 182.39 | 93.71 | ||
| MSC | 1.68 | 1.58 | 1.17 | 1.67 | ||
| First-order | R2adjusted | 0.95 | 0.84 | 0.89 | 0.86 | |
| AIC | 148.98 | 83.21 | 161.65 | 91.08 | ||
| MSC | 2.93 | 1.66 | 2.07 | 1.79 | ||
| Higuchi | R2adjusted | 0.99 | 0.98 | 0.99 | 0.99 | |
| AIC | 87.34 | 28.18 | 102.20 | 26.77 | ||
| MSC | 5.61 | 4.05 | 4.65 | 4.58 | ||
| Korsmeyer–Peppas | R2adjusted | 0.99 | 0.98 | 0.99 | 0.99 | |
| AIC | 85.60 | 30.15 | 87.49 | 27.46 | ||
| MSC | 5.68 | 3.97 | 5.29 | 4.55 | ||
| n | 0.51 | 0.50 | 0.45 | 0.51 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bezerra, G.S.N.; De Lima, G.G.; Colbert, D.M.; Halligan, E.; Geever, J.; Geever, L. Micro-Injection Moulding of PEO/PCL Blend–Based Matrices for Extended Oral Delivery of Fenbendazole. Pharmaceutics 2023, 15, 900. https://doi.org/10.3390/pharmaceutics15030900
Bezerra GSN, De Lima GG, Colbert DM, Halligan E, Geever J, Geever L. Micro-Injection Moulding of PEO/PCL Blend–Based Matrices for Extended Oral Delivery of Fenbendazole. Pharmaceutics. 2023; 15(3):900. https://doi.org/10.3390/pharmaceutics15030900
Chicago/Turabian StyleBezerra, Gilberto S. N., Gabriel G. De Lima, Declan M. Colbert, Elaine Halligan, Joseph Geever, and Luke Geever. 2023. "Micro-Injection Moulding of PEO/PCL Blend–Based Matrices for Extended Oral Delivery of Fenbendazole" Pharmaceutics 15, no. 3: 900. https://doi.org/10.3390/pharmaceutics15030900