
Gene Therapy for Regenerative Medicine

Abstract
:1. Introduction
2. Gene Therapy Systems
2.1. Viral Gene Therapy-Based Systems
2.1.1. Adenoviral Vectors
Barriers to Adenovirus-Mediated Gene Therapy
2.1.2. Adeno-Associated Viral Vectors
Barriers to Adeno-Associated Virus
2.1.3. Retroviral Vectors
2.2. Non-Viral Gene Therapy-Based Systems
2.2.1. Challenges of Non-Viral Gene Therapy
3. Regenerative Medicine Based on Gene Therapy
3.1. Tissue Engineering Approach
3.1.1. Angiogenesis
3.1.2. Bone Regeneration
Viral Gene Delivery
Non-Viral Gene Delivery
3.1.3. Gene Therapy for Cartilage Regeneration
3.1.4. Cardiac Regeneration
3.1.5. Nerve Regeneration
3.1.6. Tooth Regeneration
3.1.7. Hair Cell Regeneration for Hearing Loss Treatment
3.1.8. Skin Regeneration
3.1.9. Disc Regeneration
3.2. Biomaterials-Mediated Regeneration
3.2.1. Hydrogels-Mediated Regeneration
3.2.2. Scaffolds-Mediated Regeneration
3.3. Nanomaterials-Mediated Regeneration
4. Recent Progress in Non-Viral Gene Therapy
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bessis, N.; GarciaCozar, F.J.; Boissier, M.C. Immune responses to gene therapy vectors: Influence on vector function and effector mechanisms. Gene Ther. 2004, 11 (Suppl. S1), S10–S17. [Google Scholar] [CrossRef] [Green Version]
- Bouard, D.; Alazard-Dany, D.; Cosset, F.L. Viral vectors: From virology to transgene expression. Br. J. Pharmacol. 2009, 157, 153–165. [Google Scholar] [CrossRef] [Green Version]
- Baum, C.; Kustikova, O.; Modlich, U.; Li, Z.; Fehse, B. Mutagenesis and oncogenesis by chromosomal insertion of gene transfer vectors. Hum. Gene Ther. 2006, 17, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Waehler, R.; Russell, S.J.; Curiel, D.T. Engineering targeted viral vectors for gene therapy. Nat. Rev. Genet. 2007, 8, 573–587. [Google Scholar] [CrossRef]
- Räty, J.K.; Pikkarainen, J.T.; Wirth, V.; Ylä-Herttuala, S. Gene Therapy: The First Approved Gene-Based Medicines, Molecular Mechanisms and Clinical Indications. Curr. Mol. Pharmacol. 2008, 1, 13−23. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Kirn, D. Gene Therapy Progress and Prospects Cancer: Oncolytic Viruses. Gene Ther. 2008, 15, 877−884. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, H.L.; Kim, D.W.; Deraffele, G.; Mitcham, K.; Coffin, R.S.; Kim-schulze, S. Local and Distant Immunity Induced by Intralesional Vaccination with an Oncolytic Herpes Virus Encoding GM-CSF in Patients with Stage IIIc and IV Melanoma. Ann Surg. Oncol. 2010, 17, 718–730. [Google Scholar] [CrossRef]
- Scott, L.J. Alipogene Tiparvovec: A Review of Its Use in Adults with Familial Lipoprotein Lipase Deficiency. Drugs 2015, 75, 175−182. [Google Scholar] [CrossRef]
- Crystal, R.G. Adenovirus: The first effective In vivo gene delivery vector. Hum Gene Ther. 2014, 25, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Breyer, B.; Jiang, W.; Cheng, H.; Zhou, L.; Paul, R.; Feng, T.; He, T.C. Adenoviral vector-mediated gene transfer for human gene therapy. Curr Gene Ther. 2001, 1, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Seymour, L.W.; Fisher, K.D. Adenovirus: Teaching an old dog new tricks. Hum Gene Ther. 2011, 22, 1041–1042. [Google Scholar] [CrossRef]
- Tatsis, N.; Ertl, H.C.J. Adenoviruses as vaccine vectors. Mol. Ther. 2004, 10, 616–629. [Google Scholar]
- Vannucci, L.; Lai, M.; Chiuppesi, F.; Ceccherini-Nelli, L.; Pistello, M. Viral vectors: A look back and ahead on gene transfer technology. New Microbiol. 2013, 36, 1–22. [Google Scholar]
- Lehrman, S. Virus treatment questioned after gene therapy death. Nature 1999, 401, 517–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bett, A.J.; Prevec, L.; Graham, F.L. Packaging capacity and stability of human adenovirus type 5 vectors. J. Virol. 1993, 67, 5911–5921. [Google Scholar] [PubMed]
- Reid, T.; Warren, R.; Kirn, D. Intravascular adenoviral agents in cancer patients: Lessons from clinical trials. Cancer Gene Ther. 2002, 9, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Raper, S.E.; Chirmule, N.; Lee, F.S. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol. Genet. Metab. 2003, 80, 148–158. [Google Scholar] [CrossRef]
- Marshall, E. Gene therapy death prompts review of adenovirus vector. Science 1999, 286, 2244–2245. [Google Scholar] [PubMed]
- Choi, I.K.; Yun, C.O. Recent developments in oncolytic adenovirus-based immunotherapeutic agents for use against metastatic cancers. Cancer Gene Ther. 2013, 20, 70–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Gao, G. State-of-the-art human gene therapy: Part I. Gene Delivery Technology. Discov. Med. 2014, 18, 67–77. [Google Scholar]
- Lasaro, M.O.; Ertl, H.C.J. New insights on adenovirus as vaccine vectors. Mol. Ther. 2009, 17, 1333–1339. [Google Scholar] [CrossRef]
- Kaufmann, J.K.; Nettelbeck, D.M. Virus chimeras for gene therapy, vaccination, and oncolysis: Adenoviruses and beyond. Trends Mol. Med. 2012, 18, 365–376. [Google Scholar] [CrossRef]
- Cerullo, V.; Pesonen, S.; Diaconu, I. Oncolytic adenovirus coding for granulocyte macrophage colony-stimulating factor induces antitumoral immunity in cancer patients. Cancer Res. 2010, 70, 4297–4309. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Kuri, M.; Rapti, K.; Mehel, H. Dominant negative Ras attenuates pathological ventricular remodeling in pressure overload cardiac hypertrophy. Biochim. Biophys. Acta 2015, 1853, 2870–2884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vorburger, S.A.; Hunt, K.K. Adenoviral gene therapy. Oncologist 2002, 7, 46–59. [Google Scholar] [CrossRef]
- Lee, C.S.; Bishop, E.S.; Zhang, R.; Yu, X.; Farina, E.M.; Yan, S.; Zhao, C.; Zeng, Z.; Shu, Y.; Wu, X.; et al. Adenovirus-mediated gene delivery: Potential applications for gene and cell-based therapies in the new era of personalized medicine. Genes Dis. 2017, 4, 43–63. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Goverdhana, S.; Sciascia, S.A.; Candolfi, M.; Zirger, J.M.; Barcia, C. Regulatable gutless adenovirus vectors sustain inducible transgene expression in the brain in the presence of an immune response against adenoviruses. J. Virol. 2006, 80, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Xiong, W.; Candolfi, M.; Kroeger, K.M.; Puntel, M.; Mondkar, S.; Larocque, D.; Liu, C.; Curtin, J.F.; Palmer, D.; Ng, P.; et al. Immunization against the transgene but not the TetReduces expression from gutless adenoviral vectors in the brain. Mol. Ther. 2008, 16, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.X.; Graham, F.L.; Hitt, M.M. A convenient plasmid system for construction of helper-dependent adenoviral vectors and its application for analysis of the breast-cancer-specific mammaglobin promoter. J. Gene Med. 2006, 8, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Aparicio, M.; Mauleon, I.; Alzuguren, P.; Bunuales, M.; Gonzalez-Aseguinolaza, G.; San Martín, C.; Prieto, J.; Hernandez-Alcoceba, R. Self-inactivating helper virus for the production of high-capacity adenoviral vectors. Gene Ther. 2011, 18, 1025–1033. [Google Scholar] [CrossRef] [Green Version]
- Candolfi, M.; Curtin, J.; Xiong, W.; Kroeger, K.; Liu, C.; Rentsendorj, A. Effective high-capacity gutless adenoviral vectors mediate transgene expression in human glioma cells. Mol. Ther. 2006, 14, 371–381. [Google Scholar] [CrossRef]
- Balakrishnan, B.; Jayandharan, G.R. Basic biology of adenoassociated virus (AAV) vectors used in gene therapy. Curr. Gene Ther. 2014, 14, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Samulski, R.J.; Muzyczka, N. AAV-mediated gene therapy for research and therapeutic purposes. Annu. Rev. Virol. 2014, 1, 427–451. [Google Scholar] [CrossRef]
- Kay, M.A.; Glorioso, J.C.; Naldini, L. Viral vectors for gene therapy: The art of turning infectious agents into vehicles of therapeutics. Nat. Med. 2001, 7, 33–40. [Google Scholar] [CrossRef]
- Giacca, M.; Zacchigna, S. Virus-mediated gene delivery for human gene therapy. J. Control. Release 2012, 161, 377–388. [Google Scholar] [CrossRef]
- Flotte, T.R. Gene therapy progress and prospects: Recombinant adeno-associated virus (rAAV) vectors. Gene Ther. 2004, 11, 805–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Judd, J.; Ho, M.L.; Tiwari, A.; Gomez, E.J.; Dempsey, C.; Van Vliet, K.; Igoshin, O.A.; Silberg, J.J.; Agbandje-McKenna, M.; Suh, J. Tunable protease-activatable virus nanonodes. ACS Nano 2014, 8, 4740–4746. [Google Scholar] [CrossRef]
- Tonga, J.G.; Evansa, A.C.; Ho, M.L.; Guenther, C.M.; Brun, M.J.; Judd, J.; Wu, E.; Suha, J. Reducing off target viral delivery in ovarian cancer gene therapy using a protease-activated AAV2 vector platform. J. Control. Release 2019, 307, 292–301. [Google Scholar] [CrossRef]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef]
- Flotte, T.R. Birth of a new therapeutic platform: 47 years of adeno-associated virus biology from virus discovery to licensed gene therapy. Mol. Ther. 2013, 21, 1976–1981. [Google Scholar] [CrossRef] [Green Version]
- Vandamme, C.; Adjali, O.; Mingozzi, F. Unraveling the complex story of immune responses to AAV Vectors trial after trial. Hum. Gene Ther. 2017, 28, 1061–1074. [Google Scholar] [CrossRef] [PubMed]
- Mingozzi, F.; High, K.A. Overcoming the host immune response to adeno-associated virus gene delivery vectors: The race between clearance, tolerance, neutralization, and escape. Annu. Rev. Virol. 2017, 4, 511–534. [Google Scholar] [CrossRef]
- Coura, R.D.; Nardi, N.B. The state of the art of adeno-associated virus-based vectors in gene therapy. Virol. J. 2007, 4, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clement, N.; Knop, D.R.; Byrne, B.J. Large-scale adeno-associated viral vector production using a herpesvirus-based system enables manufacturing for clinical studies. Hum. Gene Ther. 2009, 20, 796–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foust, K.D.; Nurre, E.; Montgomery, C.L.; Hernandez, A.; Chan, C.M.; Kaspar, B.K. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2009, 27, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Nagabhushan Kalburgi, S.; Khan, N.N.; Gray, S.J. Recent gene therapy advancements for neurological diseases. Discov. Med. 2013, 15, 111–119. [Google Scholar]
- Yang, B.; Li, S.; Wang, H.; Guo, Y.; Gessler, D.J.; Cao, C.; Su, Q.; Kramer, J.; Zhong, L.; Ahmed, S.S.; et al. Global CNS transduction of adult mice by intravenously delivered rAAVrh.8 and rAAVrh.10 and nonhuman primates by rAAVrh.10. Mol. Ther. 2014, 22, 1299–1309. [Google Scholar] [CrossRef] [Green Version]
- Maier, P.; von Kalle, C.; Laufs, S. Retroviral vectors for gene therapy. Future Microbiol. 2010, 5, 1507–1523. [Google Scholar] [CrossRef]
- Guntaka, R.V. Transcription termination and polyadenylation in retroviruses. Microbiol. Rev. 1993, 57, 511–521. [Google Scholar] [CrossRef]
- Suzuki, Y.; Craigie, R. The road to chromatin -nuclear entry of retroviruses. Nat. Rev. Microbiol. 2007, 5, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Emerman, M. Retroviral infection of non-dividing cells: Old and new perspective. Virology 2006, 344, 88–93. [Google Scholar] [CrossRef] [Green Version]
- Urban, J.H.; Merten, C.A. Retroviral display in gene therapy, protein engineering, and vaccine development. ACS Chem. Biol. 2011, 6, 61–74. [Google Scholar] [CrossRef]
- Hosseinkhani, H.; Tabata, Y. Self-assembly of DNA nanoparticles with polycations for the delivery of genetic materials into cells. J. Nanosci. Nanotechnol. 2006, 6, 2320–2328. [Google Scholar] [CrossRef]
- Hosseinkhani, H.; Azzam, T.; Tabata, Y.; Domb, A.J. Dextran–spermine polycation: An efficient nonviral vector for in vitro and In vivo gene transfection. Gene Therapy 2004, 11, 194–203. [Google Scholar] [CrossRef] [Green Version]
- Abedini, F.; Hosseinkhani, H.; Ismail, M.; Chen, Y.R.; Omar, A.R.; Chong, P.P.; Domb, A.J. In vitro intracellular trafficking of biodegradable nanoparticles dextran–spermine in cancer cell lines. Int. J. Nanotechnol. 2011, 8, 712–723. [Google Scholar] [CrossRef]
- Hosseinkhani, H.; Abedini, F.; Ou, K.L.; Domb, A.J. Polymers in gene therapy technology. Polym. Adv. Technol. 2015, 26, 198–211. [Google Scholar] [CrossRef]
- Abdullah, S.; Wendy–Yeo, W.Y.; Hosseinkhani, H.; Hosseinkhani, M.; Masrawa, E.; Ramasamy, R.; Rosli, R.; Rahman, S.A.; Domb, A.J. Gene transfer into the lung by nanoparticle dextran–spermine/plasmid DNA complexes. J. Biomed. Biotechnol. 2010, 2010, 284840. [Google Scholar] [CrossRef] [Green Version]
- Abedini, F.; Ismail, M.; Hosseinkhani, H.; Azmi, T.; Omar, A.R.; Chong, P.P.; Ismail, N.; Farber, I.Y.; Domb, A.J. Toxicity evaluation of dextran–spermine polycation as a tool for gene therapy in vitro. J. Cell Anim. Biol. 2010, 4, 170–176. [Google Scholar]
- Abedini, F.; Ismail, M.; Hosseinkhani, H.; Ibrahim, T.A.T.; Omar, A.R.; Chong, P.P.; Bejo, M.H.; Domb, A.J. Effects of CXCR4 siRNA/ dextran–spermine nanoparticles on CXCR4 expression and serum LDH levels in a mouse model of colorectal cancer metastasis to the liver. Cancer Manag. Res. 2011, 3, 301–309. [Google Scholar]
- Yeo, W.W.; Hosseinkhani, H.; Rahman, S.A.; Rosli, R.; Domb, A.J.; Abdullah, S. Safety profile of dextran–spermine gene delivery vector in mouse lungs. J. Nanosci. Nanotechnol. 2014, 14, 3328–3336. [Google Scholar] [CrossRef]
- Ghadiri, M.; Vasheghani–Farahani, E.; Atyabi, F.; Kobarfard, F.; Hosseinkhani, H. In–vitro assessment of magnetic dextran–spermine nanoparticles for capecitabine delivery to cancerous cells. Iran J. Pharm. Res. 2017, 16, 1320–1334. [Google Scholar] [PubMed]
- Jain, A.; Hosseinkhani, H.; Domb, A.J.; Khan, W. Cationic Polymers for the Delivery of Therapeutic Nucleotides. In Polysaccharides; Ramawat, K.G., Mérillon, J.M., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 1969–1990. [Google Scholar]
- He, W.J.; Hosseinkhani, H.; Mohammadinejad, R.; Roveimiab, Z.; Hueng, D.Y.; Ou, K.L.; Domb, A.J. Polymeric nanoparticles for therapy and imaging. Polym. Adv. Technol. 2014, 25, 1216–1225. [Google Scholar] [CrossRef]
- Hosseinkhani, H.; Chen, Y.R.; He, W.; Hong, P.D.; Yu, D.S.; Domb, A.J. Engineering of magnetic DNA nanoparticles for tumor–targeted therapy. J. Nanopart. Res. 2013, 15, 1345–1355. [Google Scholar] [CrossRef]
- Hosseinkhani, H.; He, W.J.; Chiang, C.H.; Hong, P.D.; Yu, D.S.; Domb, A.J.; Ou, K.L. Biodegradable nanoparticles for gene therapy technology. J. Nanopart. Res. 2013, 15, 1794–1809. [Google Scholar] [CrossRef]
- Singh, J.; Michel, D.; Chitanda, J.M.; Verrall, R.E.; Badea, I. Evaluation of cellular uptake and intracellular trafficking as determining factors of gene expression for amino acid-substituted gemini surfactant-based DNA nanoparticles. J. Nanobiotechnology 2012, 10, 7. [Google Scholar] [CrossRef] [Green Version]
- Hosseinkhani, H. On nanomedicine. Int. J. Nanotechnol. 2011, 8, 615–617. [Google Scholar]
- He, W.J.; Hosseinkhani, H.; Hong, P.D.; Chiang, C.H.; Yu, D.S. Magnetic nanoparticles for imaging technology. Int. J. Nanotechnol. 2013, 10, 930–944. [Google Scholar] [CrossRef]
- Khan, W.; Hosseinkhani, H.; Ickowicz, D.; Hong, P.D.; Yu, D.S.; Domb, A.J. Polysaccharide gene transfection agents. Acta Biomater. 2012, 8, 4224–4232. [Google Scholar] [CrossRef]
- Abedini, F.; Hosseinkhani, H.; Ismail, M.; Domb, A.J.; Omar, A.R.; Chong, P.P.; Hong, P.D.; Yu, D.S.; Farber, I.Y. Cationized dextran nano-particle–encapsulated CXCR4–siRNA enhanced correlation between CXCR4 expression and serum alkaline phosphatase in a mouse model of colorectal cancer. Int. J. Nanomed. 2012, 7, 4159–4168. [Google Scholar]
- Taheri, M.M.; Vasheghani-Farahani, E.; Hosseinkhani, H.; Shojaosadati, S.A.; Soleimani, M. Fabrication and characterization of a new MRI contrast agent based on a magnetic dextran–spermine nanoparticle system. Iran Polym. J. 2012, 21, 239–251. [Google Scholar] [CrossRef]
- Amini, R.; Hosseinkhani, H.; Abdulamir, A.; Rosli, R. Azizi Jalilian, F. Engineered smart biomaterials for gene delivery. Gene Ther. Mol. Biol. 2012, 14, 72–86. [Google Scholar]
- Hoseinkhani, H.; Hosseinkhani, M.; Chen, V.R.; Subramani, K.; Domb, A.J. Innovative technology of engineering magnetic DNA nanoparticles for gene therapy. Int. J. Nanotechnol. 2011, 8, 724–735. [Google Scholar] [CrossRef]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef]
- Hosseinkhani, M.; Hosseinkhani, H.; Chen, V.R.; Subramani, K. In vitro physicochemical evaluation of DNA nanoparticles. Int. J. Nanotechnol. 2011, 8, 736–748. [Google Scholar] [CrossRef]
- Hosseinkhani, H. DNA nanoparticles for gene delivery to cells and tissue. Int. J. Nanotechnol. 2006, 3, 416–461. [Google Scholar] [CrossRef]
- Hosseinkhani, H.; Hong, P.D.; Yu, D.S.; Ickowicz, D.; Farber, I.Y.; Domb, A.J. Development of 3D in vitro platform technology to engineer mesenchymal stem cells. Int. J. Nanomed. 2012, 7, 3035–3043. [Google Scholar] [CrossRef] [Green Version]
- Abedini, F.; Ebrahimi, M.; Hosseinkhani, H. Technology of RNA Interference in advanced medicine. MicroRNA 2018, 7, 74–84. [Google Scholar] [CrossRef]
- Abedini, F.; Ebrahimi, M.; Roozbehani, A.H.; Domb, A.J.; Hosseinkhani, H. Overview on Natural Hydrophilic Polysaccharide Polymers in Drug Delivery. Polym. Adv. Technol. 2018, 29, 2564–2573. [Google Scholar] [CrossRef]
- Mottaghitalab, F.; Rastegari, A.; Farokhi, M.; Dinarvand, R.; Hosseinkhani, H.; Ou, K.L.; Pack, D.W.; Mao, C.; Dinarvand, M.; Fatahi, Y.; et al. Prospects of siRNA applications in regenerative medicine. Int. J. Pharm. 2017, 524, 312–329. [Google Scholar] [CrossRef] [PubMed]
- Ghadiri, M.; Vasheghani-Farahani, E.; Atyabi, F.; Kobarfard, F.; Mohamadyar-Toupkanlou, F.; Hosseinkhani, H. Transferrin-Conjugated Magnetic Dextran-Spermine Nanoparticles for Targeted Drug Transport across Blood-Brain Barrier. J. Biomed. Mater. Res. Part A 2017, 105, 2851–2864. [Google Scholar] [CrossRef]
- Alibolandi, M.; Abnous, K.; Sadeghi, F.; Ramezani, M.; Hosseinkhani, H.; Hadizadeh, F. Folate Receptor-Targeted Multimodal Polymersomes for delivery of quantum dots and doxorubicin to breast adenocarcinoma: In vitro and In vivo Evaluation. Int. J. Pharm. 2016, 500, 162–178. [Google Scholar] [CrossRef] [PubMed]
- Mottaghitalab, F.; Shokrghozar, M.A.; Farokhi, M.; Hosseinkhani, H. Silk fibrin nanoparticles as novel drug delivery systems. J. Control. Release 2015, 206, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Alibolandi, M.; Abnous, K.; Ramezani, M.; Hosseinkhani, H.; Hadizadeh, F. Synthesis of AS1411-Aptamer-Conjugated CdTe Quantum Dots with High Fluorescence Strength for Probe Labeling Tumor Cells. J. Fluoresc. 2014, 24, 1519–1529. [Google Scholar] [CrossRef]
- Chiang, C.H.; Hosseinkhani, H.; Cheng, W.S.; Chen, C.W.; Wang, C.H.; Lo, Y.L. Improving drug loading efficiency and delivery performance of micro- and nanoparticle preparations through optimizing formulation variables. Int. J. Nanotechnol. 2013, 10, 996–1006. [Google Scholar] [CrossRef]
- Mahmoudi, M.; Hosseinkhani, H.; Hosseinkhani, M.; Boutry, S.; Simchi, A.; Journeay, W.S.; Subramani, K.; Laurent, S. Magnetic Resonance Imaging Tracking of Stem Cells In vivo Using Iron Oxide Nanoparticles as a Tool for the Advancement of Clinical Regenerative Medicine. Chem. Rev. 2011, 111, 253–280. [Google Scholar] [CrossRef]
- Sarabi, R.S.; Sadeghi, E.; Hosseinkhani, H.; Mahmoudi, M.; Kalantari, M.; Adeli, M. Polyrotaxane Capped Quantum Dots as New Candidates for Cancer Diagnosis and Therapy. J. Nanostructured Polym. Nanocomposites 2011, 7, 18–31. [Google Scholar]
- Hosseinkhani, H. Nanomaterials in Advanced Medicine, 1st ed.; John Wiley & Sons: Hoboken, NJ, USA, 2019; pp. 150–196. [Google Scholar]
- Li, W.; Szoka, F.C., Jr. Lipid-based nanoparticles for nucleic acid delivery. Pharm. Res. 2007, 24, 438–449. [Google Scholar] [CrossRef]
- Koltover, I.; Salditt, T.; Radler, J.O.; Safinya, C.R. An inverted hexagonal phase of cationic liposome-DNA complexes related to DNA release and delivery. Science 1998, 281, 78–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. 2010, 18, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Wolfrum, C.; Shi, S.; Jayaprakash, K.N.; Jayaraman, M.; Wang, G.; Pandey, R.K.; Rajeev, K.G.; Nakayama, T.; Charrise, K.; Ndungo, E.M.; et al. Mechanisms and optimization of In vivo delivery of lipophilic siRNAs. Nat. Biotech. 2007, 25, 1149–1157. [Google Scholar] [CrossRef]
- Hosseinkhani, H. Biomedical Engineering: Materials, Technology, and Application, 1st ed.; John Wiley & Sons: Hoboken, NJ, USA, 2022; pp. 120–180. [Google Scholar]
- Domb, A.J.; Sharifzadeh, G.; Nahum, V.; Hosseinkhani, H. Safety Evaluation of Nanotechnology Products. Pharmaceutics 2021, 13, 1615. [Google Scholar] [CrossRef]
- Steinman, N.Y.; Campos, L.M.; Feng, Y.; Domb, A.J.; Hosseinkhani, H. Cyclopropenium Nanoparticles and Gene Transfection in Cells. Pharmaceutics 2020, 12, 768. [Google Scholar] [CrossRef] [PubMed]
- Hosseinkhani, H.; Domb, A.J. Biodegradable polymers in gene-silencing technology. Polym. Adv. Technol. 2019, 30, 2647–2655. [Google Scholar] [CrossRef]
- Hu, C.S.; Tang, S.L.; Chiang, C.H.; Hosseinkhani, H.; Hong, P.D.; Yeh, M.K. Characterization and antitumor effects of chondroitin sulfate-chitosan nanoparticles delivery system. J. Nanoparticle Res. 2014, 16, 1–15. [Google Scholar] [CrossRef]
- Amini, R.; Azizi Jalilian, F.; Abdullah, S.; Veerakumarasivam, A.; Hosseinkhani, H.; Abdulamir, A.S.; Domb, A.J.; Ickowicz, D.; Rosli, R. Dynamics of PEGylated-Dextran-Spermine Nanoparticles for Gene Delivery to Leukemic Cells. Appl. Biochem. Biotechnol. 2013, 170, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Subramani, K.; Hosseinkhani, H.; Khraisat, A.; Hosseinkhani, M.; Pathak, Y. Targeting nanoparticles as drug delivery systems for cancer treatment. Curr. Nanosci. 2009, 5, 134–140. [Google Scholar] [CrossRef]
- Hosseinkhani, H.; Hosseinkhani, M. Biodegradable polymer-metal complexes for gene and drug delivery. Curr. Drug Saf. 2009, 4, 79–83. [Google Scholar] [CrossRef]
- Hosseinkhani, H.; Aoyama, T.; Ogawa, O.; Tabata, T. Ultrasound enhances the transfection of plasmid DNA by non-viral vector. Curr. Pharm. Biotechnol. 2002, 4, 109–122. [Google Scholar] [CrossRef]
- Hosseinkhani, H.; Aoyama, T.; Ogawa, O.; Tabata, T. Liver targeting of plasmid DNA by pullulan conjugation based on metal coordination. J. Control. Release 2002, 83, 287–302. [Google Scholar] [CrossRef]
- Hosseinkhani, H. siRNA Delivery Systems in Cancer Therapy. Org. Med. Chem. Int. J. 2020, 10, 1–3. [Google Scholar] [CrossRef]
- Malek, A.; Merkel, O.; Fink, L.; Czubayko, F.; Kissel, T.; Aigner, A. In vivo pharmacokinetics, tissue distribution and underlying mechanisms of various PEI(-PEG)/siRNA complexes. Toxicol. Appl. Pharmacol. 2009, 236, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirisala, A.; Uchida, S.; Tockary, T.A.; Yoshinaga, N.; Yoshinaga, N.; Li, J.; Osawa, S.; Gorantla, L.; Fukushima, S.; Osada, K.; et al. Precise tuning of disulphide crosslinking in mRNA polyplex micelles for optimising extracellular and intracellular nuclease tolerability. J. Drug Target. 2019, 27, 670–680. [Google Scholar] [CrossRef]
- Yen, A.; Cheng, Y.; Sylvestre, M.; Gustafson, H.H.; Puri, S.; Pun, S.H. Serum Nuclease Susceptibility of mRNA Cargo in Condensed Polyplexes. Mol. Pharm. 2018, 15, 2268–2276. [Google Scholar] [CrossRef] [PubMed]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Uchida, S.; Itaka, K.; Uchida, H.; Hayakawa, K.; Ogata, T.; Ishii, T.; Fukushima, S.; Osada, K.; Kataoka, K. In vivo messenger RNA introduction into the central nervous system using polyplex nanomicelle. PLoS ONE 2013, 8, e56220. [Google Scholar] [CrossRef]
- Dirisala, A.; Uchida, S.; Li, J.; Van Guyse, J.F.R.; Hayashi, K.; Vummaleti, S.V.C.; Kaur, S.; Mochida, Y.; Fukushima, S.; Kataoka, K. Effective mRNA Protection by Poly(l-ornithine) Synergizes with Endosomal Escape Functionality of a Charge-Conversion Polymer toward Maximizing mRNA Introduction Efficiency. Macromol. Rapid Commun. 2022, 43, e2100754. [Google Scholar] [CrossRef]
- Paramasivam, P.; Franke, C.; Stöter, M.; Höijer, A.; Bartesaghi, S.; Sabirsh, A.; Lindfors, L.; Arteta, M.Y.; Dahlén, A.; Bak, A.; et al. Endosomal escape of delivered mRNA from endosomal recycling tubules visualized at the nanoscale. J. Cell Biol. 2022, 221, e202110137. [Google Scholar] [CrossRef]
- Hajj, K.A.; Whitehead, K.A. Tools for translation: Non-viral materials for therapeutic mRNA delivery. Nat. Rev. Mater. 2017, 2, 17056. [Google Scholar] [CrossRef]
- Martins, I.M.; Reis, R.L.; Azevedo, H.S. Phage display technology in biomaterials engineering: Progress and opportunities for applications in regenerative medicine. ACS Chem. Biol. 2016, 11, 2962–2980. [Google Scholar] [CrossRef] [Green Version]
- Mao, A.S.; Mooney, D.J. Regenerative medicine: Current therapies and future directions. Proc. Natl. Acad. Sci. USA 2015, 112, 14452–14459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseinkhani, H. Developing 3D Technology for Drug Discovery. Curr. Drug Deliv. 2022, 19, 813–814. [Google Scholar] [CrossRef] [PubMed]
- Khalaji, S.; Ebrahimi, N.G.; Hosseinkhani, H. Enhancement of biocompatibility of PVA/HTCC blend polymer with collagen for skin care application. Int. J. Polym. Mater. Polym. Biomater. 2021, 70, 459–468. [Google Scholar] [CrossRef]
- Hosseini, V.; Maroufi, N.F.; Saghati, S.; Asadi, N.; Darabi, M.; Nazari Soltan Ahmad, S.; Hosseinkhani, H.; Rahbarghazi, R. Current progress in hepatic tissue regeneration by tissue engineering. J. Transl. Med. 2019, 17, 383. [Google Scholar] [CrossRef] [Green Version]
- Saberianpour, S.; Heidarzadeh, M.; Geranmayeh, M.H.; Hosseinkhani, H.; Rahbarghazi, R.; Nouri, M. Tissue engineering strategies for the induction of angiogenesis using biomaterials. J. Biol. Eng. 2018, 12, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Toosi, S.; Naderi-Meshkin, H.; Kalalinia, F.; Peivandi, M.T.; Hosseinkhani, H.; Bahrami, A.R.; Heirani-Tabasi, A.; Mirahmadi, M.; Behravan, J. Long bone mesenchymal stem cells (Lb-MSCs): Clinically reliable cells for osteo-diseases. Cell Tissue Bank. 2017, 18, 489–500. [Google Scholar] [CrossRef]
- Abassi, N.; Hashemei, S.M.; Salehi, M.; Jahani, H.; Mowla, S.J.; Soleimani, M.; Hosseinkhani, H. Influence of oriented nanofibrous PCL scaffolds on quantitative gene expression during neural differentiation of mouse embryonic stem cells. J. Biomed. Mater. Res. Part A 2016, 104, 155–164. [Google Scholar] [CrossRef]
- Farokhi, M.; Mottaghitalab, F.; Shokrghozar, M.A.; Ou, K.L.; Mao, C.; Hosseinkhani, H. Importance of dual delivery systems for bone tissue engineering. J. Control. Release 2016, 225, 152–169. [Google Scholar] [CrossRef]
- Toosi, S.; Naderi-Meshkin, H.; Kalalinia, F.; Peivandi, M.T.; Hosseinkhani, H.; Bahrami, A.R.; Heirani-Tabasi, A.; Mirahmadi, M.; Behravan, J. PLGA-incorporated collagen: Toward a Biodegradable Composite Scaffold for Bone-tissue Engineering. J. Biomed. Mater. Res. Part A 2016, 104, 2020–2028. [Google Scholar] [CrossRef]
- Toosi, S.; Naderi-Meshkin, H.; Kalalinia, F.; Peivandi, M.T.; Hosseinkhani, H.; Bahrami, A.R.; Heirani-Tabasi, A.; Mirahmadi, M.; Behravan, J. Comparative characteristics of mesenchymal stem cells derived from reamer-irrigator-aspirator, iliac crest bone marrow, and adipose tissue. Cell. Mol. Biol. 2016, 62, 68–74. [Google Scholar] [PubMed]
- Mottaghitalab, F.; Hosseinkhani, H.; Shokrghozar, M.A.; Mao, C.; Farokhi, M. Silk as potential candidate for bone tissue engineering. J. Control. Release 2015, 215, 112–128. [Google Scholar] [CrossRef]
- Jahani, H.; Azizi Jalilian, F.; Kaviani, S.; Soleimani, M.; Abassi, N.; Hosseinkhani, H. Controlled surface morphology and hydrophilicity of polycaprolactone towards differentiation of mesenchymal stem cells into neural cells. J. Biomed. Mater. Res. Part A 2015, 103, 1875–1881. [Google Scholar] [CrossRef]
- Rahbarghazi, R.; Nassiri, S.M.; Ahmadi, S.H.; Mohammadi, E.; Rabbani, S.; Araghi, A.; Hosseinkhani, H. Dynamic induction of pro-angiogenic milieu after transplantation of marrow-derived mesenchymal stem cells in experimental myocardial infarction. Int. J. Cardiol. 2014, 173, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Ghodsizadeh, A.; Hosseinkhani, H.; Piryaei, A.; Pournasr, B.; Najarasl, M.; Hiraoka, Y.; Baharvand, H. Galactosylated Collagen Matrix Enhanced In vitro Maturation of Human Embryonic Stem Cell-derived Hepatocyte-like Cells. Biotechnol. Lett. 2014, 36, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Tatu, R.; Liu, Q.; Hosseinkhani, H. Stem Cells Based Tissue Engineering for Regenerative Medicine. Nano LIFE 2014, 4, 1–13. [Google Scholar] [CrossRef]
- Ou, K.L.; Hosseinkhani, H. Development of 3D in vitro Technology for Medical Applications. Int. J. Mol. Sci. 2014, 15, 17938–17962. [Google Scholar] [CrossRef] [Green Version]
- Hosseinkhani, H.; Hong, P.D.; Yu, D.S. Self-assembled Proteins and Peptides for Regenerative Medicine. Chem. Rev. 2013, 113, 4837–4861. [Google Scholar] [CrossRef]
- Hosseinkhani, H.; Hiraoka, Y.; Li, C.H.; Chen, Y.R.; Yu, D.S.; Hong, P.D.; Ou, K.L. Engineering Three-Dimensional Collagen-IKVAV Matrix to Mimic Neural Microenvironment. ACS Chem. Neurosci. 2013, 4, 1229–1235. [Google Scholar] [CrossRef] [Green Version]
- Hosseinkhani, H. 3D in vitro technology for drug discovery. Curr. Drug Saf. 2012, 7, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Subramani, K.; Mathew, R.; Hosseinkhani, H.; Hosseinkhani, M. Bone regeneration around dental implants as a treatment for peri-implantitis: A review of the literature. J. Biomim. Biomater. Tissue Eng. 2011, 11, 21–33. [Google Scholar] [CrossRef]
- Hosseinkhani, H.; Hosseinkhani, M.; Hattori, S.; Matsuoka, R.; Kawaguchi, N. Micro and nanoscale in vitro 3D culture system for cardiac stem cells. J. Biomed. Mater. Res. Part A 2010, 94, 1–8. [Google Scholar] [CrossRef]
- Mohageri, S.; Hosseinkhani, H.; Ebrahimi, N.G.; Solimani, M.; Kajbafzadeh, A.M. Proliferation and differentiation of mesenchymal stem cell on collagen sponge reinforced with polypropylene/polyethylene terephathalate blend fibers. Tissue Eng. Part A 2010, 16, 3821–3830. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Hosseinkhani, H.; Hosseinkhani, M.; Khademhosseini, A.; Yokoyama, Y.; Estrada, G.; Kobayashi, H. Quantitative analytical of cell adhesion on aligned micro- and nanofibers. J. Biomed. Mater. Res. Part A 2008, 84, 291–299. [Google Scholar] [CrossRef]
- Hosseinkhani, H.; Hosseinkhani, M.; Khademhosseini, A.; Gabrielson, N.P.; Pack, D.W.; Kobayashi, H. DNA nanoparticles encapsulated in 3-D tissue engineered scaffold enhance osteogenic differentiation of mesenchymal stem cells. J. Biomed. Mater. Res. Part A 2008, 85, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Hosseinkhani, H.; Hosseinkhani, M.; Tian, F.; Kobayashi, H.; Tabata, Y. Osteogenic differentiation of mesenchymal stem cells in self-assembled-peptide amphiphile nanofibers. Biomaterials 2006, 27, 4079–4086. [Google Scholar] [CrossRef]
- Hosseinkhani, H.; Azzam, T.; Kobayashi, H.; Hiraoka, Y.; Shimokawa, H.; Domb, A.J.; Tabata, Y. Combination of 3-D tissue engineered scaffold and non-viral gene carrier enhance in vitro DNA expression of mesenchymal stem cells. Biomaterials 2006, 27, 4269–4278. [Google Scholar] [CrossRef]
- Hosseinkhani, H.; Hosseinkhani, M.; Tian, F.; Kobayashi, H.; Tabata, Y. Ectopic bone formation in collagen sponge-self assembled peptide amphiphile nanofibers hybrid scaffold in a perfusion culture bioreactor. Biomaterials 2006, 27, 5089–5098. [Google Scholar] [CrossRef]
- Hosseinkhani, H.; Hosseinkhani, M.; Khademhosseini, A.; Kobayashi, H.; Tabata, Y. Enhanced angiogenesis through controlled release of basic fibroblast growth factor from peptide amphiphile for tissue regeneration. Biomaterials 2006, 27, 5836–5844. [Google Scholar] [CrossRef] [PubMed]
- Hosseinkhani, H.; Hosseinkhani, M.; Kobayashi, H. Proliferation and differentiation of mesenchymal stem cells by using self-assembly of peptide-amphiphile nanofibers. Biomed. Mater. 2006, 1, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Hosseinkhani, H.; Hosseinkhani, M.; Kobayashi, H. Design of tissue-engineered nanoscaffold through self-assembly of peptide amphiphile. J. Bioact. Compat. Polym. 2006, 21, 277–296. [Google Scholar] [CrossRef]
- Hosseinkhani, H.; Hosseinkhani, M.; Khademhosseini, A. Emerging Applications of Hydrogels and Microscale Technologies in Drug Discovery. Drug Discov. 2006, 1, 32–34. [Google Scholar]
- Hosseinkhani, H.; Inatsugu, Y.; Hiraoka, Y.; Inoue, S.; Shimokawa, H.; Tabata, Y. Impregnation of plasmid DNA into 3-D scaffold and medium perfusion enhance in vitro DNA expression of mesenchymal stem cells. Tissue Eng. 2005, 11, 1459–1475. [Google Scholar] [CrossRef]
- Hosseinkhani, H.; Inatsugu, Y.; Hiraoka, Y.; Inoue, S.; Tabata, Y. Perfusion culture enhances osteogenic differentiation of rat mesenchymal stem cells in collagen sponge reinforced with poly (glycolic acid) fiber. Tissue Eng. 2005, 11, 1476–1488. [Google Scholar] [CrossRef]
- Bleich, N.K.; Kallai, I.; Lieberman, J.R.; Schwarz, E.M.; Pelled, G.; Gazit, D. Gene therapy approaches to regenerating bone. Adv. Drug Deliv. Rev. 2012, 64, 1320–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, C.H.; Huard, J. Gene therapy approaches to regenerating the musculoskeletal system. Nat. Rev. Rheumatol. 2015, 11, 234–242. [Google Scholar] [CrossRef] [Green Version]
- Evans, C.H. Facilitated endogenous repair: Making tissue engineering simple, practical, and economical. Tissue Eng. 2007, 13, 1987–1993. [Google Scholar] [CrossRef]
- Ricks, D.M.; Kutner, R.; Zhang, X.Y.; Welsh, D.A.; Reiser, J. Optimized lentiviral transduction of mouse bone marrow-derived mesenchymal stem cells. Stem Cells Dev. 2008, 17, 441–450. [Google Scholar] [CrossRef]
- Williams, D.A.; Thrasher, A.J. Concise review: Lessons learned from clinical trials of gene therapy in monogenic immunodeficiency diseases. Stem Cells Transl. Med. 2014, 3, 636–642. [Google Scholar] [CrossRef]
- Folkman, J. Angiogenesis: An organizing principle for drug discovery? Nat. Rev. Drug Discov. 2007, 6, 273–286. [Google Scholar] [CrossRef]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar]
- Risau, W. Mechanisms of angiogenesis. Nature 1997, 386, 671–674. [Google Scholar] [CrossRef] [PubMed]
- McMahon, G. Vegf receptor signaling in tumor angiogenesis. Oncologist 2000, 5, 3–10. [Google Scholar] [CrossRef]
- O’Donnell, A.; Padhani, A.; Hayes, C.; Kakkar, A.J.; Leach, M.; Trigo, J.M.; Scurr, M.; Raynaud, F.; Phillips, S.; Aherne, W.; et al. A Phase I study of the angiogenesis inhibitor SU5416 (semaxanib) in solid tumours, incorporating dynamic contrast MR pharmacodynamic end points. Br. J. Cancer 2005, 93, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Ray, J.M.; Stetler-Stevenson, W.G. The role of matrix metalloproteases and their inhibitors in tumour invasion, metastasis and angiogenesis. Eur. Respir. J. 1994, 7, 2062–2072. [Google Scholar] [CrossRef]
- Moses, M.A. The regulation of neovascularization by matrix metalloproteinases and their inhibitors. Stem Cells 1997, 15, 180–189. [Google Scholar] [CrossRef]
- Isner, J.M.; Pieczek, A.; Schainfeld, R.; Blair, R.; Haley, L.; Asahara, T.; Rosenfield, K.; Razvi, S.; Walsh, K.; Symes, J.F. Clinical evidence of angiogenesis after arterial gene transfer of phVEGF165 in patient with ischaemic limb. Lancet 1996, 348, 370–374. [Google Scholar] [CrossRef]
- Losordo, D.W.; Vale, P.R.; Symes, J.F.; Dunnington, C.H.; Esakof, D.D.; Maysky, M.; Ashare, A.B.; Lathi, K.; Isner, J.M. Gene therapy for myocardial angiogenesis: Initial clinical results with direct myocardial injection of phVEGF165 as sole therapy for myocardial ischemia. Circulation 1998, 98, 2800–2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335−2342. [Google Scholar] [CrossRef] [Green Version]
- Gragoudas, E.S.; Adamis, A.P.; Cunningham, E.T., Jr.; Feinsod, M.; Guyer, D.R. Pegaptanib for neovascular age-related macular degeneration. N. Engl. J. Med. 2004, 351, 2805−2816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, E.L.; Ting, C.Y.; Hsu, P.H.; Lin, Y.C.; Liao, E.C.; Huang, C.Y.; Chang, Y.C.; Chan, H.L.; Chiang, C.S.; Liu, H.L.; et al. Angiogenesis-targeting microbubbles combined with ultrasound-mediated gene therapy in brain tumors. J. Control. Release 2017, 255, 164–175. [Google Scholar] [CrossRef]
- Yamakuchi, M.; Lotterman, C.D.; Bao, C.; Hruban, R.H.; Karim, B.; Mendell, J.T.; Huso, D.; Lowenstein, C.J. P53-induced microRNA-107 inhibits HIF-1 and tumor angiogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 6334–6339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, X.; Wang, W.; Wang, C.; Wang, Y.; Zhou, J.; Ding, Y.; Wang, X.; Jin, Y. A chitosan-graft-PEI-candesartan conjugate for targeted co-delivery of drug and gene in anti-angiogenesis cancer therapy. Biomaterials 2014, 35, 8450–8466. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Mohler, E.R., III; Lederman, R.J.; Mendelsohn, F.O.; Saucedo, J.F.; Goldman, C.K.; Blebea, J.; Macko, J.; Kessler, P.D.; Rasmussen, H.S.; et al. Regional angiogenesis with vascular endothelial growth factor in peripheral arterial disease: A phase II randomized, double-blind, controlled study of adenoviral delivery of vascular endothelial growth factor 121 in patients with disabling intermittent claudication. Circulation 2003, 108, 1933–1938. [Google Scholar]
- Kusumanto, Y.H.; Weel, V.V.; Mulder, N.H.; Smit, A.J.; Dungen, J.J.A.M.V.D.; Hooymans, J.M.M.; Sluiter, W.J.; Tio, R.A.; Quax, P.H.A.; Gans, R.O.B.; et al. Treatment with intramuscular vascular endothelial growth factor gene compared with placebo for patients with diabetes mellitus and critical limb ischemia: A double-blind randomized trial. Hum. Gene Ther. 2006, 17, 683–691. [Google Scholar] [CrossRef] [Green Version]
- Makinen, K.; Manninen, H.; Hedman, M.; Matsi, P.; Mussalo, H.; Alhava, E.; Yla-Herttuala, S. Increased vascularity detected by digital subtraction angiography after VEGF gene transfer to human lower limb artery: A randomized, placebo-controlled, double-blinded phase II study. Mol. Ther. 2002, 6, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Hammer, A.; Steiner, S. Gene therapy for therapeutic angiogenesis in peripheral arterial disease- a systematic review and meta-analysis of randomized, controlled trials. Vasa 2013, 42, 331–339. [Google Scholar] [CrossRef]
- Deev, R.V.; Bozo, I.Y.; Mzhavanadze, N.D.; Voronov, D.A.; Gavrilenko, A.V.; Chervyakov, Y.V.; Staroverov, I.N.; Kalinin, R.E.; Shvalb, P.G.; Isaev, A.A. pCMV-vegf165 intramuscular gene transfer is an effective method of treatment for patients with chronic lower limb ischemia. J. Cardiovasc. Pharmacol. Ther. 2015, 20, 473–482. [Google Scholar] [CrossRef]
- Belch, J.; Hiatt, W.R.; Baumgartner, I.; Driver, I.V.; Nikol, S.; Norgren, L.; Belle, E.V. Effect of fibroblast growth factor NV1FGF on amputation and death: A randomised placebo-controlled trial of gene therapy in critical limb ischaemia. Lancet 2011, 377, 1929–1937. [Google Scholar] [CrossRef] [Green Version]
- Nikol, S.; Baumgartner, I.; Belle, E.V.; Diehm, C.; Visona, A.; Capogrossi, M.C.; Ferreira-Maldent, N.; Gallino, A.; Wyatt, M.G.; Wijesinghe, L.D.; et al. Therapeutic angiogenesis with intramuscular NV1FGF improves amputation-free survival in patients with critical limb ischemia. Mol. Ther. 2008, 16, 972–978. [Google Scholar] [CrossRef]
- Creager, M.A.; Olin, J.W.; Belch, J.J.F.; Moneta, G.L.; Henry, T.D.; Rajagopalan, S.; Annex, B.H.; Hiatt, W.R. Effect of hypoxia-inducible factor-1alpha gene therapy on walking performance in patients with intermittent claudication. Circulation 2011, 124, 1765–1773. [Google Scholar] [CrossRef] [Green Version]
- Powell, R.J.; Simons, M.; Mendelsohn, F.O.; Daniel, G.; Henry, T.D.; Koga, M.; Morishita, R.; Annex, B.H. Results of a double-blind, placebo-controlled study to assess the safety of intramuscular injection of hepatocyte growth factor plasmid to improve limb perfusion in patients with critical limb ischemia. Circulation 2008, 118, 58–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigematsu, H.; Yasuda, K.; Iwai, T.; Sasajima, T.; Ishimaru, S.; Ohashi, Y.; Yamaguchi, T.; Ogihara, T.; Morishita, R. Randomized, double-blind, placebo-controlled clinical trial of hepatocyte growth factor plasmid for critical limb ischemia. Gene Ther. 2010, 17, 1152–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anghel, A.; Taranu, G.; Seclaman, E.; Rata, A.; Tamas, L.; Moldovan, H.; Ursoniu, S.; Samoila, C.; Ionac, M.; Popa-Wagner, A. Safety of vascular endothelial and hepatocyte growth factor gene therapy in patients with critical limb ischemia. Curr. Neurovasc. Res. 2011, 8, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Rosengart, T.K.; Bishawi, M.M.; Halbreiner, M.S.; Halbreiner, M.S.; Fakhoury, M.; Finnin, E.; Hollmann, C.; Shroyer, A.L.; Crystal, R.G. Long-term follow-up of a phase 1 trial of angiogenic gene therapy using direct intramyocardial administration of an adenoviral vector expressing the VEGF121 cDNA for the treatment of diffuse coronary artery disease. Hum. Gene Ther. 2013, 24, 203–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartikainen, J.; Hassinen, I.; Hedman, A.; Kivela, A.; Saraste, A.; Knuuti, J.; Husso, M.; Mussalo, H.; Hedman, M.; Rissanen, T.T.; et al. Adenoviral intramyocardial VEGF-DΔNΔC gene transfer increases myocardial perfusion reserve in refractory angina patients: A phase I/IIa study with 1-year follow-up. Eur. Heart J. 2017, 38, 2547–2555. [Google Scholar] [CrossRef] [Green Version]
- Kaminsky, S.M.; Quach, L.; Chen, S.; Pierre-Destine, L.; Van De Graaf, B.; Monette, S.; Rosenberg, J.B.; Sondhi, D.; Hackett, N.R.; Crystal, R.G. Safety of direct cardiac administration of AdVEGF-All6A+, a replication-deficient adenovirus vector cDNA/genomic hybrid expressing all three major isoforms of human vascular endothelial growth factor, to the ischemic myocardium of rats. Hum. Gene Ther. Clin. Dev. 2013, 24, 38–46. [Google Scholar] [CrossRef]
- Miyazaki, M.; Zuk, P.A.; Zou, J.; Yoon, S.H.; Wei, F.; Morishita, Y.; Sintuu, C.; Wang, J.C. Comparison of human mesenchymal stem cells derived from adipose tissue and bone marrow for ex vivo gene therapy in rat spinal fusion model. Spine (Phila Pa1976) 2008, 33, 863–869. [Google Scholar] [CrossRef]
- Steinhardt, Y.; Aslan, H.; Regev, E.; Zilberman, Y.; Kallai, I.; Gazit, D.; Gazit, Z. Maxillofacial- derived stem cells regenerate critical mandibular bone defect. Tissue Eng. Part A 2008, 14, 1763–1773. [Google Scholar] [CrossRef]
- Chang, S.C.; Lin, T.M.; Chung, H.Y.; Chen, P.K.; Lin, F.H.; Lou, J.; Jeng, L.B. Large-scale bicortical skull bone regeneration using ex vivo replication-defective adenoviral-mediated bone morphogenetic protein-2 gene-transferred bone marrow stromal cells and composite biomaterials. Neurosurgery 2009, 65, 75–81 discussion 81–73. [Google Scholar] [CrossRef] [PubMed]
- Virk, M.S.; Sugiyama, O.; Park, S.H.; Gambhir, S.S.; Adams, D.J.; Drissi, H.; Lieberman, J.R. “Same day” ex vivo regional gene therapy: A novel strategy to enhance bone repair. Mol. Ther. 2011, 19, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Tu, Q.; Valverde, P.; Li, S.; Zhang, J.; Yang, P.; Chen, J. Osterix overexpression in mesenchymal stem cells stimulates healing of critical-sized defects in murine calvarial bone. Tissue Eng. 2007, 13, 2431–2440. [Google Scholar] [CrossRef] [PubMed]
- Menendez, M.I.; Clark, D.J.; Carlton, M.; Flanigan, D.C.; Jia, G.; Sammet, S.; Weisbrode, S.E.; Knopp, M.V.; Bertone, A.L. Direct delayed human adenoviral BMP-2 or BMP-6 gene therapy for bone and cartilage regeneration in a pony osteochondral model. Osteoarthr. Cartil. 2011, 19, 1066–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazici, C.; Takahata, M.; Reynolds, D.G.; Xie, C.; Samulski, R.J.; Samulski, J.; Beecham, E.J.; Gertzman, A.A.; Spilker, M.; Zhang, X.; et al. Self- complementary AAV2.5-BMP2-coated femoral allografts mediated superior bone healing versus live autografts in mice with equivalent biomechanics to unfractured femur. Mol. Ther. 2011, 19, 1416–1425. [Google Scholar] [CrossRef]
- Rundle, C.H.; Miyakoshi, N.; Kasukawa, Y.; Chen, S.T.; Sheng, M.H.; Wergedal, J.E.; Lau, K.H.; Baylink, D.J. In vivo bone formation in fracture repair induced by direct retroviral-based gene therapy with bone morphogenetic protein-4. Bone 2003, 32, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Rundle, C.H.; Strong, D.D.; Chen, S.T.; Linkhart, T.A.; Sheng, M.H.; Wergedal, J.E.; Lau, K.H.; Baylink, D.J. Retroviral-based gene therapy with cyclooxygenase-2 promotes the union of bony callus tissues and accelerates fracture healing in the rat. J. Gene Med. 2008, 10, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Hosseinkhani, H.; Yamamoto, M.; Inatsugu, Y.; Hiraoka, Y.; Inoue, S.; Shimokawa, H.; Tabata, Y. Enhanced ectopic bone formation using a combination of plasmid DNA impregnation into 3-D scaffold and bioreactor perfusion culture. Biomaterials 2006, 27, 1387–1398. [Google Scholar] [CrossRef]
- Wehrhan, F.; Amann, K.; Molenberg, A.; Lutz, R.; Neukam, F.W.; Schlegel, K.A. PEG Matrix Enables Cell-Mediated Local BMP-2 Gene Delivery and Increased Bone Formation in a Porcine Critical Size Defect Model of Craniofacial Bone Regeneration. Clin. Oral Implants Res. 2012, 23, 805–813. [Google Scholar] [CrossRef]
- Lai, Q.G.; Yuan, K.F.; Xu, X.; Li, D.R.; Li, G.J.; Wei, F.L.; Yang, Z.J.; Luo, S.L.; Tang, X.P.; Li, S. Transcription factor osterix modified bone marrow mesenchymal stem cells enhance callus formation during distraction osteogenesis. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2011, 111, 412–419. [Google Scholar] [CrossRef]
- Lee, S.J.; Kang, S.W.; Do, H.J.; Han, I.; Shin, D.A.; Kim, J.H.; Lee, S.H. Enhancement of bone regeneration by gene delivery of BMP2/Runx2 bicistronic vector into adipose-derived stromal cells. Biomaterials 2010, 31, 5652–5659. [Google Scholar] [CrossRef] [PubMed]
- Sheyn, D.; Kallai, I.; Tawackoli, W.; Cohn Yakubovich, D.; Oh, A.; Su, S.; Da, X.; Lavi, A.; Kimelman-Bleich, N.; Zilberman, Y.; et al. Gene-Modified adult stem cells regenerate vertebral bone defect in a rat model. Mol. Pharm. 2011, 8, 1592–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osawa, K.; Okubo, Y.; Nakao, K.; Koyama, N.; Bessho, K. Osteoinduction by repeat plasmid injection of human bone morphogenetic protein-2. J. Gene Med. 2010, 12, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Zhu, Y.Y.; Smiley, E.; Bonadio, J.; Rouleau, J.P.; Goldstein, S.A.; McCauley, L.K.; Davidson, B.L.; Roessler, B.J. Stimulation of new bone formation by direct transfer of osteogenic plasmid genes. Proc. Natl. Acad. Sci. USA 1996, 93, 5753–5758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plonka, A.B.; Khorsand, B.; Yu, N.; Sugai, J.V.; Salem, A.K.; Giannobile, W.V.; Elangovan, S. Effect of sustained PDGF nonviral gene delivery on repair of tooth-supporting bone defects. Gene Ther. 2017, 24, 31–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elangovan, S.; D’Mello, S.R.; Hong, L.; Ross, R.D.; Allamargot, C.; Dawson, D.V.; Stanford, C.M.; Johnson, G.K.; Sumner, D.R.; Salem, A.K. The enhancement of bone regeneration by gene activated matrix encoding for platelet derived growth factor. Biomaterials 2014, 35, 737–747. [Google Scholar] [CrossRef] [Green Version]
- Nomikou, N.; Feichtinger, G.A.A.; Saha, S.; Nuernberger, S.; Heimel, P.; Redl, H.; Mchale, A.P.P. Ultrasound-responsive gene-activated matrices for osteogenic gene therapy using matrix-assisted sonoporation. J. Tissue Eng. Regen. Med. 2018, 12, 250. [Google Scholar] [CrossRef]
- Bez, M.; Sheyn, D.; Tawackoli, W.; Avalos, P.; Shapiro, G.; Giaconi, J.C.; Da, X.; Ben David, S.; Gavrity, J.; Awad, H.A.; et al. In situ bone tissue engineering via ultrasound-mediated gene delivery to endogenous progenitor cells in mini-pigs. Sci. Transl. Med. 2017, 9, 3128. [Google Scholar] [CrossRef] [Green Version]
- Kimelman-Bleich, N.; Pelled, G.; Zilberman, Y.; Kallai, I.; Mizrahi, O.; Tawackoli, W.; Gazit, Z.; Gazit, D. Targeted gene-and-host progenitor cell therapy for non-union bone fracture repair. Mol. Ther. 2011, 19, 53–59. [Google Scholar] [CrossRef]
- Park, J.; Lutz, R.; Felszeghy, E.; Wiltfang, J.; Nkenke, E.; Neukam, F.W.; Schlegel, K.A. The effect on bone regeneration of a liposomal vector to deliver BMP-2 gene to bone grafts in peri-implant bone defects. Biomaterials 2007, 28, 2772–2782. [Google Scholar] [CrossRef]
- Lieberman, J.R.; Daluiski, A.; Stevenson, S.; Wu, L.; McAllister, P.; Lee, Y.P.; Kabo, J.M.; Finerman, G.A.; Berk, A.J.; Witte, O.N. The effect of regional gene therapy with bone morphogenetic protein-2-producing bone-marrow cells on the repair of segmental femoral defects in rats. J. Bone Jt. Surg. Am. 1999, 81, 905–917. [Google Scholar] [CrossRef]
- Turgeman, G.; Pittman, D.D.; Muller, R.; Kurkalli, B.G.; Zhou, S.; Pelled, G.; Peyser, A.; Zilberman, Y.; Moutsatsos, I.K.; Gazit, D. Engineered human mesenchymal stem cells: A novel platform for skeletal cell mediated gene therapy. J. Gene Med. 2001, 3, 240–251. [Google Scholar] [CrossRef]
- Park, J.; Ries, J.; Gelse, K.; Kloss, F.; von der Mark, K.; Wiltfang, J.; Neukam, F.W.; Schneider, H. Bone regeneration in critical size defects by cell-mediated BMP-2 gene transfer: A comparison of adenoviral vectors and liposomes. Gene Ther. 2003, 10, 1089–1098. [Google Scholar] [CrossRef] [Green Version]
- Gates, C.B.; Karthikeyan, T.; Fu, F.; Huard, J. Regenerative medicine for the musculoskeletal system based on muscle-derived stem cells. J. Am. Acad. Orthop. Surg. 2008, 16, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Sun, X.; Zhang, X.; Tang, T.; Dai, K. Ectopic osteogenesis by ex vivo gene therapy using beta tricalcium phosphate as a carrier. Connect. Tissue Res. 2008, 49, 343–350. [Google Scholar] [CrossRef]
- Zhu, L.; Chuanchang, D.; Wei, L.; Yilin, C.; Jiasheng, D. Enhanced healing of goat femur-defect using BMP7 gene-modified BMSCs and load-bearing tissue-engineered bone. J. Orthop. Res. 2010, 28, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, A.; Zekas, L.J.; Litsky, A.S.; Weisbrode, S.E.; Bertone, A.L. Dermal fibroblast-mediated BMP2 therapy to accelerate bone healing in an equine osteotomy model. J. Orthop. Res. 2010, 28, 403–411. [Google Scholar] [CrossRef]
- Gysin, R.; Wergedal, J.E.; Sheng, M.H.; Kasukawa, Y.; Miyakoshi, N.; Chen, S.T.; Peng, H.; Lau, K.H.; Mohan, S.; Baylink, D.J. Ex vivo gene therapy with stromal cells transduced with a retroviral vector containing the BMP4 gene completely heals critical size calvarial defect in rats. Gene Ther. 2002, 9, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Virk, M.S.; Conduan, A.; Park, S.H.; Liu, N.; Sugiyama, O.; Cuomo, A.; Kang, C.; Lieberman, J.R. Influence of short-term adenoviral vector and prolonged lentiviral vector mediated bone morphogenetic protein- 2 expression on the quality of bone repair in a rat femoral defect model. Bone 2008, 42, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Baltzer, A.W.A.; Lattermann, C.; Whalen, J.D.; Ghivizzani, S.; Wooley, P.; Krauspe, R.; Robbins, P.D.; Evans, C.H. Potential role of direct adenoviral gene transfer in enhancing facture repair. Clin. Orthop. Relat. Res. 2000, 379, S120–S125. [Google Scholar] [CrossRef]
- Betz, V.M.; Betz, O.B.; Glatt, V.; Gerstenfeld, L.C.; Einhorn, T.A.; Bouxsein, M.L.; Vrahas, M.S.; Evans, C.H. Healing of segmental bone defects by direct percutaneous gene delivery: Effect of vector dose. Hum. Gene Ther. 2007, 18, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Bertone, A.L.; Pittman, D.D.; Bouxsein, M.L.; Li, J.; Clancy, B.; Seehrman, H.J. Adenoviral-mediated transfer of human BMP- 6 gene accelerates healing in a rabbit ulnar osteotomy model. J. Orthop. Res. 2004, 22, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Baltzer, A.W.; Lattermann, C.; Whalen, J.D.; Braunstein, S.; Robbins, P.D.; Evans, C.H. A gene therapy approach to accelerating bone healing. Evaluation of gene expression in a New Zealand white rabbit model. Knee Surg. Sports Traumatol. Arthrosc. 1999, 7, 197–202. [Google Scholar] [CrossRef]
- Egermann, M.; Lill, C.A.; Griesbeck, K.; Evans, C.H.; Robbins, P.D.; Schneider, E.; Baltzer, A.W. Effect of BMP- 2 gene transfer on bone healing in sheep. Gene Ther. 2006, 13, 1290–1299. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Koefoed, M.; Tiyapatanaputi, P.; Gromov, K.; Goater, J.J.; Carmouche, J.; Zhang, X.; Rubery, P.T.; Rabinowitz, J.; Samulski, R.J.; et al. Remodeling of cortical bone allografts mediated by adherent rAAV-RANKL and VEGF gene therapy. Nat. Med. 2005, 11, 291–297. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Zheng, Q.; Kulbatski, I.; Yuan, Q.; Yang, S.; Shao, Z.; Wang, H.; Xiao, B.; Pan, Z.; Tang, S. Bone regeneration with active angiogenesis by basic fibroblast growth factor gene transfected mesenchymal stem cells seeded on porous beta-TCP ceramic scaffolds. Biomed. Mater. 2006, 1, 93–99. [Google Scholar] [CrossRef]
- Aslan, H.; Zilberman, Y.; Arbeli, V.; Sheyn, D.; Matan, Y.; Liebergall, M.; Li, J.Z.; Helm, G.A.; Gazit, D.; Gazit, Z. Nucleofection-based ex vivo nonviral gene delivery to human stem cells as a platform for tissue regeneration. Tissue Eng. 2006, 12, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Wegman, F.; Bijenhof, A.; Schuijff, L.; Oner, F.C.; Dhert, W.J.; Alblas, J. Osteogenic differentiation as a result of BMP-2 plasmid DNA based gene therapy in vitro and In vivo. Eur. Cell Mater. 2011, 21, 230–242. [Google Scholar] [CrossRef]
- Evans, C. Using genes to facilitate the endogenous repair and regeneration of orthopaedic tissues. Int. Orthop. 2014, 38, 1761–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohmander, L.S.; Hellot, S.; Dreher, D.; Krantz, E.F.; Guermazi, A.; Eckstein, F. Intraarticular sprifermin (recombinant human fibroblast growth factor 18) in knee osteoarthritis: A randomized, double- blind, placebo-controlled trial. Arthritis Rheumatol. 2014, 66, 1820–1831. [Google Scholar] [CrossRef]
- Cucchiarini, M.; Madry, H. Biomaterial- guided delivery of gene vectors for targeted articular cartilage repair. Nat. Rev. Rheumatol. 2019, 15, 18–29. [Google Scholar] [CrossRef]
- Adkar, S.S.; Brunger, J.M.; Willard, V.P.; Wu, C.L.; Gersbach, C.A.; Guilak, F. Genome engineering for personalized arthritis therapeutics. Trends Mol. Med. 2017, 23, 917–931. [Google Scholar] [CrossRef]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.M.; Guo, X.M.; Tan, H.S.; Hui, J.H.; Lim, B.; Lee, E.H. Zinc- finger protein 145, acting as an upstream regulator of SOX9, improves the differentiation potential of human mesenchymal stem cells for cartilage regeneration and repair. Arthritis Rheum. 2011, 63, 2711–2720. [Google Scholar] [CrossRef] [PubMed]
- Sieker, J.T.; Kunz, M.; Weißenberger, M.; Gilbert, F.; Frey, S.; Rudert, M.; Steinert, A.F. Direct bone morphogenetic protein 2 and Indian hedgehog gene transfer for articular cartilage repair using bone marrow coagulates. Osteoarthr. Cartil. 2015, 23, 433–442. [Google Scholar] [CrossRef] [Green Version]
- Morisset, S.; Frisbie, D.D.; Robbins, P.D.; Nixon, A.J.; McIlwraith, C.W. IL-1ra/IGF-1 gene therapy modulates repair of microfractured chondral defects. Clin. Orthop. Relat. Res. 2007, 462, 221–228. [Google Scholar] [CrossRef]
- Ivkovic, A.; Pascher, A.; Hudetz, D.; Maticic, D.; Jelic, M.; Dickinson, S.; Loparic, M.; Haspl, M.; Windhager, R.; Pecina, M. Articular cartilage repair by genetically modified bone marrow aspirate in sheep. Gene Ther. 2010, 17, 779–789. [Google Scholar] [CrossRef] [Green Version]
- Madry, H.; Kaul, G.; Cucchiarini, M.; Stein, U.; Zurakowski, D.; Remberger, K.; Menger, M.D.; Kohn, D.; Trippel, S.B. Enhanced repair of articular cartilage defects In vivo by transplanted chondrocytes overexpressing insulin-like growth factor I (IGF- I). Gene Ther. 2005, 12, 1171–1179. [Google Scholar] [CrossRef]
- Cucchiarini, M.; Orth, P.; Madry, H. Direct rAAV SOX9 administration for durable articular cartilage repair with delayed terminal differentiation and hypertrophy In vivo. J. Mol. Med. 2013, 91, 625–636. [Google Scholar] [CrossRef]
- Cucchiarini, M.; Madry, H. Overexpression of human IGF-I via direct rAAV-mediated gene transfer improves the early repair of articular cartilage defects In vivo. Gene Ther. 2014, 21, 811–819. [Google Scholar] [CrossRef]
- Wehling, P.; Reinecke, J.; Reinecke, J.; Baltzer, A.W.A.; Granrath, M.; Schulitz, K.P.; Schultz, C.; Krauspe, R.; Whiteside, T.W.; Elder, E.; et al. Clinical responses to gene therapy in joints of two subjects with rheumatoid arthritis. Hum. Gene Ther. 2009, 20, 97–101. [Google Scholar] [CrossRef]
- Kim, M.K.; Ha, C.W.; In, Y.; Cho, S.D.; Choi, E.S.; Ha, J.K.; Lee, J.H.; Yoo, J.D.; Bin, S.I.; Choi, C.H.; et al. A multicenter, double-blind, phase III clinical trial to evaluate the efficacy and safety of a cell and gene therapy in knee osteoarthritis patients. Hum. Gene Ther. Clin. Dev. 2018, 29, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.H.; Ghivizzani, S.C.; Robbins, P.D. Arthritis gene therapy is becoming a reality. Nat. Rev. Rheumatol. 2018, 14, 381–382. [Google Scholar] [CrossRef] [PubMed]
- Knutsen, G.; Drogset, J.O.; Engebretsen, L.; Grøntvedt, T.; Ludvigsen, T.C.; Løken, S.; Solheim, E.; Strand, T.; Johansen, O. A randomized multicenter trial comparing autologous chondrocyte implantation with microfracture: Long-term follow-up at 14 to 15 years. J. Bone Jt. Surg. Am. 2016, 98, 1332–1339. [Google Scholar] [CrossRef]
- Baragi, V.M.; Renkiewicz, R.R.; Qiu, L.; Brammer, D.; Riley, J.M.; Sigler, R.E.; Frenkel, S.R.; Amin, A.; Abramson, S.B.; Roessler, B.J. Transplantation of adenovirally transduced allogeneic chondrocytes into articular cartilage defects In vivo. Osteoarthr. Cartil. 1997, 5, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Madry, H.; Trippel, B.S. Efficient lipid-mediated gene transfer to articular chondrocytes. Gene Ther. 2000, 7, 286–291. [Google Scholar] [CrossRef] [Green Version]
- Madry, H.; Cucchiarini, M.; Terwilliger, E.F.; Trippel, S.B. Recombinant adeno-associated virus vectors efficiently and persistently transduce chondrocytes in normal and osteoarthritic human articular cartilage. Hum. Gene Ther. 2003, 14, 393–402. [Google Scholar] [CrossRef]
- Mingozzi, F.; Anguela, X.M.; Pavani, G.; Chen, Y.; Davidson, R.J.; Hui, D.J.; Yazicioglu, M.; Elkouby, L.; Hinderer, C.J.; Faella, A.; et al. Overcoming preexisting humoral immunity to AAV using capsid decoys. Sci. Transl. Med. 2013, 5, 194ra92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mingozzi, F.; High, K.A. Immune responses to AAV vectors: Overcoming barriers to successful gene therapy. Blood 2013, 122, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, C.J.; Friedel, T.; Buning, H. Surface engineered viral vectors for selective and cell type specific gene delivery. Trends Biotechnol. 2015, 33, 777–790. [Google Scholar] [CrossRef]
- Buning, H.; Huber, A.; Zhang, L.; Meumann, N.; Hacker, U. Engineering the AAV capsid to optimize vector-host-interactions. Curr. Opin. Pharmacol. 2015, 24, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, H.; Olson, E.N.; Bassel-Duby, R. Therapeutic approaches for cardiac regeneration and repair. Nat. Rev. Cardiol. 2018, 15, 585–600. [Google Scholar] [CrossRef]
- Cohn, J.N.; Ferrari, R.; Sharpe, N. Cardiac remodelling-concepts and clinical implications: A consensus paper from an international forum on cardiac remodelling. J. Am. Coll. Cardiol. 2000, 35, 569–582. [Google Scholar] [CrossRef] [Green Version]
- Bongianino, R.; Priori, S.G. Gene therapy to treat cardiac arrhythmias. Nat. Rev. Cardiol. 2015, 12, 531–546. [Google Scholar] [CrossRef]
- Jaski, B.E.; Jessup, M.L.; Mancini, D.M.; Cappola, T.P.; Pauly, D.F.; Greenberg, B.; Borow, K.; Dittrich, H.; Zsebo, K.M.; Hajjar, R.J. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID trial), a first- human phase 1/2 clinical trial. J. Card. Fail. 2009, 15, 171–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jessup, M.; Greenberg, B.; Mancini, D.; Cappola, T.; Pauly, D.F.; Jaski, B.; Yaroshinsky, A.; Zsebo, K.M.; Dittrich, H.; Hajjar, R.J. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): A phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation 2011, 124, 304–313. [Google Scholar] [CrossRef] [Green Version]
- Katz, M.G.; Fargnoli, A.S.; Pritchette, L.A.; Bridges, C.R. Gene delivery technologies for cardiac applications. Gene Ther. 2012, 19, 659–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igarashi, T.; Finet, J.E.; Takeuchi, A.; Fujino, Y.; Strom, M.; Greener, I.D.; Rosenbaum, D.S.; Donahue, J.K. Connexin gene transfer preserves conduction velocity and prevents atrial fibrillation. Circulation 2012, 125, 216–225. [Google Scholar] [CrossRef] [Green Version]
- Guiraud, S.; Aartsma-Rus, A.; Vieira, N.M.; Davies, K.E.; van Ommen, G.J.; Kunkel, L.M. The pathogenesis and therapy of muscular dystrophies. Annu. Rev. Genom. Hum. Genet. 2015, 16, 281–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, T.; Okada, T.; Athanasopoulos, T.; Foster, H.; Takeda, S.; Dickson, G. Long-term functional adeno-associated virus-microdystrophin expression in the dystrophic CXMDj dog. J. Gene Med. 2011, 13, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Foster, H.; Sharp, P.S.; Athanasopoulos, T.; Trollet, C.; Graham, I.R.; Foster, K.; Wells, D.J.; Dickson, G. Codon and mRNA sequence optimization of microdystrophin transgenes improves expression and physiological outcome in dystrophic mdx mice following AAV2/8 gene transfer. Mol. Ther. 2008, 16, 1825–1832. [Google Scholar] [CrossRef] [Green Version]
- Athanasopoulos, T.; Foster, H.; Foster, K.; Dickson, G. Codon optimization of the microdystrophin gene for Duchene muscular dystrophy gene therapy. Methods Mol. Biol. 2011, 709, 21–37. [Google Scholar] [PubMed]
- Koo, T.; Malerba, A.; Athanasopoulos, T.; Trollet, C.; Boldrin, L.; Ferry, A.; Popplewell, L.; Foster, H.; Foster, K.; Dickson, G. Delivery of AAV2/9-microdystrophin genes incorporating helix 1 of the coiled-coil motif in the C-terminal domain of dystrophin improves muscle pathology and restores the level of alpha1-syntrophin and alpha-dystrobrevin in skeletal muscles of mdx mice. Hum. Gene Ther. 2011, 22, 1379–1388. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.H.; Pan, X.; Hakim, C.H.; Yang, H.T.; Yue, Y.; Zhang, K.; Terjung, R.L.; Duan, D. Microdystrophin ameliorates muscular dystrophy in the canine model of duchenne muscular dystrophy. Mol. Ther. 2013, 21, 750–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Zhong, L.; Nahid, M.A.; Gao, G. The potential of adeno-associated viral vectors for gene delivery to muscle tissue. Expert Opin. Drug Deliv. 2014, 11, 345–364. [Google Scholar] [CrossRef] [PubMed]
- Hayashita-Kinoh, H.; Yugeta, N.; Okada, H.; Nitahara-Kasahara, Y.; Chiyo, T.; Okada, T.; Takeda, S. Intra-amniotic rAAV-mediated microdystrophin gene transfer improves canine X-linked muscular dystrophy and may induce immune tolerance. Mol. Ther. 2015, 23, 627–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Guiner, C.; Servais, L.; Montus, M.; Larcher, T.; Fraysse, B.; Moullec, S.; Allais, M.; François, V.; Dutilleul, M.; Malerba, A.; et al. Long-term microdystrophin gene therapy is effective in a canine model of Duchenne muscular dystrophy. Nat. Commun. 2017, 8, 16105. [Google Scholar] [CrossRef] [Green Version]
- Zincarelli, C.; Soltys, S.; Rengo, G.; Rabinowitz, J.E. Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 2008, 16, 1073–1080. [Google Scholar] [CrossRef]
- Prasad, K.M.R.; Xu, Y.; Yang, Z.; Acton, S.T.; French, B.A. Robust cardiomyocyte-specific gene expression following systemic injection of AAV: In vivo gene delivery follows a Poisson distribution. Gene Ther. 2011, 18, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Piras, B.A.; Tian, Y.; Xu, Y.; Thomas, N.A.; O’Connor, D.M.; French, B.A. Systemic injection of AAV9 carrying a periostin promoter targets gene expression to a myofibroblast-like lineage in mouse hearts after reperfused myocardial infarction. Gene Ther. 2016, 23, 469–478. [Google Scholar] [CrossRef]
- Shapiro, S.D.; Ranjan, A.K.; Kawase, Y.; Cheng, R.K.; Kara, R.J.; Bhattacharya, R.; Guzman-Martinez, G.; Sanz, J.; Garcia, M.J.; Chaudhry, H.W. Cyclin A2 induces cardiac regeneration after myocardial infarction through cytokinesis of adult cardiomyocytes. Sci. Transl. Med. 2014, 6, 224ra27. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, T.A.; Son, Y.J. Extrinsic and Intrinsic Determinants of Nerve Regeneration. J. Tissue Eng. 2011, 2, 204173141141839. [Google Scholar] [CrossRef] [PubMed]
- Mahar, M.; Cavalli, V. Intrinsic mechanisms of neuronal axon regeneration. Nat. Rev. Neurosci. 2018, 19, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Homs, J.; Ariza, L.; Pagès, G.; Udina, E.; Navarro, X.; Chillón, M.; Bosch, A. Schwann cell targeting via intrasciatic injection of AAV8 as gene therapy strategy for peripheral nerve regeneration. Gene Ther. 2011, 18, 622–630. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Sandner, B.; Schackel, T.; Nicholson, L.; Chtarto, A.; Tenenbaum, L.; Puttagunta, R.; Müller, R.; Weidner, N.; Blesch, A. Regulated viral BDNF delivery in combination with Schwann cells promotes axonal regeneration through capillary alginate hydrogels after spinal cord injury. Acta Biomater. 2017, 60, 167–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquardt, L.M.; Ee, X.; Iyer, N.; Hunter, D.; Mackinnon, S.E.; Wood, M.D.; Sakiyama-Elbert, S.E. Finely tuned temporal and spatial delivery of GDNF promotes enhanced nerve regeneration in a long nerve defect model. Tissue Eng. Part A 2015, 21, 2852–2864. [Google Scholar] [CrossRef]
- Shakhbazau, A.; Mohanty, C.; Shcharbin, D.; Bryszewska, M.; Caminade, A.M.; Majoral, J.P.; Alant, J.; Midha, R. Doxycycline-regulated GDNF expression promotes axonal regeneration and functional recovery in transected peripheral nerve. J. Control. Release 2013, 172, 841–851. [Google Scholar] [CrossRef]
- Markusic, D.M.; de Waart, D.R.; Seppen, J. Separating lentiviral vector injection and induction of gene expression in time, does not prevent an immune response to rtTA in rats. PLoS ONE 2010, 5, e9974. [Google Scholar] [CrossRef] [PubMed]
- Le Guiner, C.; Stieger, K.; Toromanoff, A.; Guilbaud, M.; Mendes-Madeira, A.; Devaux, M.; Guigand, L.; Cherel, Y.; Moullier, P.; Rolling, F.; et al. Transgene regulation using the tetracycline-inducible TetR-KRAB system after AAV-mediated gene transfer in rodents and nonhuman primates. PLoS ONE 2014, 9, e102538. [Google Scholar] [CrossRef] [PubMed]
- Eggers, R.; de Winter, F.; Hoyng, S.A.; Hoeben, R.C.; Malessy, M.J.A.; Tannemaat, M.R.; Verhaagen, J. Timed GDNF gene therapy using an immune-evasive gene switch promotes long distance axon regeneration. Brain 2019, 142, 295–311. [Google Scholar] [CrossRef]
- Yungher, B.J.; Luo, X.; Salgueiro, Y.; Blackmore, M.G.; Park, K.K. Viral vector-based improvement of optic nerve regeneration: Characterization of individual axons’ growth patterns and synaptogenesis in a visual target. Gene Ther. 2015, 22, 811–821. [Google Scholar] [CrossRef] [Green Version]
- Gascón, S.; Murenu, E.; Masserdotti, G.; Ortega, F.; Russo, G.L.; Petrik, D.; Deshpande, A.; Heinrich, C.; Karow, M.; Robertson, S.P.; et al. Identification and successful negotiation of a metabolic checkpoint in direct neuronal reprogramming. Cell Stem Cell 2016, 18, 396–409. [Google Scholar] [CrossRef] [Green Version]
- Torper, O.; Ottosson, D.R.; Pereira, M.; Lau, S.; Cardoso, T.; Grealish, S.; Parmar, M. In vivo reprogramming of striatal NG2 glia into functional neurons that integrate into local host circuitry. Cell Rep. 2015, 12, 474–481. [Google Scholar] [CrossRef] [Green Version]
- Su, Z.; Niu, W.; Liu, M.L.; Zou, Y.; Zhang, C.L. In vivo conversion of astrocytes to neurons in the injured adult spinal cord. Nat. Commun. 2014, 5, 3338. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.C.; Ma, N.X.; Pei, Z.F.; Wu, Z.; Do-Monte, F.H.; Keefe, S.; Yellin, E.; Chen, M.S.; Yin, J.C.; Lee, G.; et al. A neuroD1 AAV-based gene therapy for functional brain repair after ischemic injury through In vivo astrocyte-to-neuron conversion. Mol. Ther. 2020, 28, 217–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, R.T.; Nares, S.; Reynolds, M.A. Periodontal regeneration-intrabony defects: A systematic review from the AAP Regeneration Workshop. J. Periodontol. 2015, 86, S77–S104. [Google Scholar] [CrossRef] [PubMed]
- Ramseier, C.A.; Abramson, Z.R.; Jin, Q.; Giannobile, W.V. Gene therapeutics for periodontal regenerative medicine. Dent. Clin. N. Am. 2006, 50, 245–263, ix. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Lee, C.S.; Tejeda, K.M.; Giannobile, W.V. Gene transfer and expression of platelet-derived growth factors modulate periodontal cellular activity. J. Dent. Res. 2001, 80, 892–897. [Google Scholar] [CrossRef] [Green Version]
- Giannobile, W.V.; Lee, C.S.; Tomala, M.P.; Tejeda, K.M.; Zhu, Z. Platelet-derived growth factor (PDGF) gene delivery for application in periodontal tissue engineering. J. Periodontol. 2001, 72, 815–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Q.; Anusaksathien, O.; Webb, S.A.; Printz, M.A.; Giannobile, W.V. Engineering of tooth-supporting structures by delivery of PDGF gene therapy vectors. Mol. Ther. 2004, 9, 519–526. [Google Scholar] [CrossRef]
- Chang, P.C.; Cirelli, J.A.; Jin, Q.; Seol, Y.J.; Sugai, J.V.; D’Silva, N.J.; Danciu, T.E.; Chandler, L.A.; Sosnowski, B.A.; Giannobile, W.V. Adenovirus encoding human platelet-derived growth factor-B delivered to alveolar bone defects exhibits safety and biodistribution profiles favorable for clinical use. Hum. Gene Ther. 2009, 20, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Chen, P.K.T.; Jeng, L.B.; Huang, C.S.; Yang, L.C.; Chung, H.Y.; Chang, S.C.N. Periodontal regeneration using ex vivo autologous stem cells engineered to express the BMP-2 gene: An alternative to alveolaplasty. Gene Ther. 2008, 15, 1469–1477. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, K.H.; Gwak, E.H.; Rhee, S.H.; Lee, J.C.; Shin, S.Y.; Koo, K.T.; Lee, Y.M.; Seol, Y.J. Ex vivo bone morphogenetic protein 2 gene delivery using periodontal ligament stem cells for enhanced re-osseointegration in the regenerative treatment of peri-implantitis. J. Biomed. Mater. Res. A 2015, 103, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, M.; Mizunuma, K.; Murakami, T.; Akamine, A. Induction of dental pulp stem cell differentiation into odontoblasts by electroporation-mediated gene delivery of growth/differentiation factor 11 (Gdf11). Gene Ther. 2002, 9, 814–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickles, J.O. An Introduction to the Physiology of Hearing, 4th ed.; Emerald Group Publishing Limited: Bingley, UK, 2012. [Google Scholar]
- Chien, W.W.; Monzack, E.L.; McDougald, D.S.; Cunningham, L.L. Gene therapy for sensorineural hearing loss. Ear Hear. 2015, 36, 1–7. [Google Scholar] [CrossRef]
- Isgrig, K.; Shteamer, J.W.; Belyantseva, I.A.; Drummond, M.C.; Fitzgerald, T.S.; Vijayakumar, S.; Jones, S.M.; Griffith, A.J.; Friedman, T.B.; Cunningham, L.L.; et al. Gene therapy restores balance and auditory functions in a mouse model of Usher syndrome. Mol. Ther. 2017, 25, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Emptoz, A.; Michel, V.; Lelli, A.; Akil, O.; Boutet de Monvel, J.; Lahlou, G.; Meyer, A.; Dupont, T.; Nouaille, S.; Ey, E.; et al. Local gene therapy durably restores vestibular function in a mouse model of Usher syndrome type 1G. Proc. Natl. Acad. Sci. USA 2017, 114, 9695–9700. [Google Scholar] [CrossRef] [Green Version]
- Pan, B.; Askew, C.; Askew, C.; Heman-Ackah, S.; Asai, Y.; Indzhykulian, A.A.; Jodelka, F.M.; Hastings, M.L.; Lentz, J.J.; Vandenberghe, L.H.; et al. Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c. Nat. Biotechnol. 2017, 35, 264–272. [Google Scholar] [CrossRef]
- Gyorgy, B.; Meijer, E.J.; Ivanchenko, M.V.; Tenneson, K.; Emond, F.; Hanlon, K.S.; Indzhykulian, A.A.; Volak, A.; Karavitaki, K.D.; Tamvakologos, P.I.; et al. Gene transfer with AAV9-PHP.B rescues hearing in a mouse model of Usher syndrome 3A and transduces hair cells in a non-human primate. Mol. Ther. Methods Clin. Dev. 2019, 13, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Askew, C.; Rochat, C.; Pan, B.; Asai, Y.; Ahmed, H.; Child, E.; Schneider, B.L.; Aebischer, P.; Holt, J.R. Tmc gene therapy restores auditory function in deaf mice. Sci. Transl. Med. 2015, 7, 295ra108. [Google Scholar] [CrossRef]
- Chien, W.W.; McDougald, D.S.; Roy, S.; Fitzgerald, T.S.; Cunningham, L.L. Cochlear gene transfer mediated by adeno-associated virus: Comparison of two surgical approaches. Laryngoscope 2015, 125, 2557–2564. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Huang, M.; Shu, Y.; Ruprecht, A.; Wang, H.; Tang, Y.; Vandenberghe, L.H.; Wang, Q.; Gao, G.; Kong, W.J.; et al. Delivery of adeno-associated virus vectors in adult mammalian inner-ear cell subtypes without auditory dysfunction. Hum. Gene Ther. 2018, 29, 492–506. [Google Scholar] [CrossRef]
- Isgrig, K.; McDougald, D.S.; Zhu, J.; Wang, H.J.; Bennett, J.; Chien, W.W. AAV2.7m8 is a powerful viral vector for inner ear gene therapy. Nat. Commun. 2019, 10, 427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, F.; Chu, C.; Qi, J.; Li, W.; You, D.; Li, K.; Chen, X.; Zhao, W.; Cheng, C.; Liu, X.; et al. AAV-ie enables safe and efficient gene transfer to inner ear cells. Nat. Commun. 2019, 10, 3733. [Google Scholar] [CrossRef] [Green Version]
- Khavari, P.A.; Rollman, O.; Vahlquist, A. Cutaneous gene transfer for skin and systemic diseases. J. Intern. Med. 2002, 252, 1–10. [Google Scholar] [CrossRef]
- Jeschke, M.G.; Richter, G.; Herndon, D.N.; Geissler, E.K.; Hartl, M.; Hofstatter, F.; Jauch, K.W.; Perez-Polo, J.R. Therapeutic success and efficacy of nonviral liposomal cDNA gene transfer to the skin In vivo is dose dependent. Gene Ther. 2001, 8, 1777–1784. [Google Scholar] [CrossRef] [Green Version]
- Jeschke, M.G.; Herndon, D.N. The combination of IGF-I and KGF cDNA improves dermal and epidermal regeneration by increased VEGF expression and neovascularization. Gene Ther. 2007, 14, 1235–1242. [Google Scholar] [CrossRef]
- Branski, L.K.; Masters, O.E.; Herndon, D.N.; Mittermayr, R.; Redl, H.; Traber, D.L.; Cox, R.A.; Kita, K.; Jeschke, M.G. Pre-clinical evaluation of liposomal gene transfer to improve dermal and epidermal regeneration. Gene Ther. 2010, 17, 770–778. [Google Scholar] [CrossRef] [Green Version]
- Peng, L.H.; Wei, W.; Qi, X.T.; Shan, Y.H.; Zhang, F.J.; Chen, X.; Zhu, Q.Y.; Yu, L.; Liang, W.Q.; Gao, J.Q. Epidermal stem cells manipulated by pDNA-VEGF165/CYD-PEI nanoparticles loaded gelatin/β-TCP matrix as a therapeutic agent and gene delivery vehicle for wound healing. Mol. Pharm. 2013, 10, 3090–3102. [Google Scholar] [CrossRef]
- Keeley, E.C.; Mehrad, B.; Strieter, R.M. Chemokines as mediators of neovascularization. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1928–1936. [Google Scholar] [CrossRef] [Green Version]
- Conget, P.; Rodriguez, F.; Kramer, S.; Allers, C.; Simon, V.; Palisson, F.; Gonzalez, S.; Yubero, M.J. Replenishment of type VII collagen and re-epithelialization of chronically ulcerated skin after intradermal administration of allogeneic mesenchymal stromal cells in two patients with recessive dystrophic epidermolysis bullosa. Cytotherapy 2010, 12, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Chen, L.; Scott, P.G.; Tredget, E.E. Mesenchymal stem cells enhance wound healing through differentiation and angiogenesis. Stem Cells 2007, 25, 2648–2659. [Google Scholar] [CrossRef] [Green Version]
- Dhoke, N.R.; Kaushik, K.; Das, A. Cxcr6-based mesenchymal stem cell gene therapy potentiates skin regeneration in murine diabetic wounds. Mol. Ther. 2020, 28, 1314–1326. [Google Scholar] [CrossRef] [PubMed]
- Luoma, K.; Riihimaki, H.; Luukkonen, R.; Raininko, R.; Viikari-Juntura, E.; Lamminen, A. Low back pain in relation to lumbar disc degeneration. Spine 2000, 25, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Frith, J.E.; Menzies, D.J.; Cameron, A.R.; Ghosh, P.; Whitehead, D.L.; Gronthos, S.; Zannettino, A.C.; Cooper-White, J.J. Effects of bound versus soluble pentosan polysulphate in PEG/HA-based hydrogels tailored for intervertebral disc regeneration. Biomaterials 2014, 35, 1150–1162. [Google Scholar] [CrossRef]
- Feng, G.; Chen, H.; Li, J.; Huang, Q.; Gupte, M.J.; Liu, H.; Song, Y.; Ge, Z. Gene therapy for nucleus pulposus regeneration by heme oxygenase-1 plasmid DNA carried by mixed polyplex micelles with thermo-responsive heterogeneous coronas. Biomaterials 2015, 52, 1–13. [Google Scholar] [CrossRef]
- Feng, G.; Zhang, Z.; Dang, M.; Zhang, X.; Doleyres, Y.; Song, Y.; Chen, D.; Ma, P.X. Injectable nanofibrous spongy microspheres for NR4A1 plasmid DNA transfection to reverse fibrotic degeneration and support disc regeneration. Biomaterials 2017, 131, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Sharifzadeh, G.; Hosseinkhani, H. Biomolecule–responsive hydrogels in medicine. Adv. Healthc. Mater. 2017, 6, 1700801. [Google Scholar] [CrossRef]
- Vega, S.L.; Kwon, M.Y.; Burdick, J.A. Recent advances in hydrogels for cartilage tissue engineering. Eur. Cell. Mater. 2017, 33, 59–75. [Google Scholar] [CrossRef]
- Filardo, G.; Kon, E.; Roffi, A.; Di Martino, A.; Marcacci, M. Scaffold-based repair for cartilage healing: A systematic review and technical note. Arthroscopy 2013, 29, 174–186. [Google Scholar] [CrossRef]
- Girotto, D.; Urbani, S.; Brun, P.; Renier, D.; Barbucci, R.; Abatangelo, G. Tissue-specific gene expression in chondrocytes grown on three- dimensional hyaluronic acid scaffolds. Biomaterials 2003, 24, 3265–3275. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.K.; Tharp, K.M.; Browne, S.; Ye, J.; Stahl, A.; Yeghiazarians, Y.; Healy, K.E. Matrix metalloproteinase-13 mediated degradation of hyaluronic acid- based matrices orchestrates stem cell engraftment through vascular integration. Biomaterials 2016, 89, 136–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, A.; Hasan, A.; Al Kindi, H.; Gaharwar, A.K.; Rao, V.T.S.; Nikkhah, M.; Shin, S.R.; Krafft, D.; Dokmeci, M.R.; Shum-Tim, D.; et al. Injectable graphene oxide/ hydrogel-based angiogenic gene delivery system for vasculogenesis and cardiac repair. ACS Nano 2014, 8, 8050–8062. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.H.; Schaffer, D.V.; Shea, L.D. Engineering biomaterial systems to enhance viral vector gene delivery. Mol. Ther. 2011, 19, 1407–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, H.J.; Liu, J.; Riddle, K.; Matsumoto, T.; Leach, K.; Mooney, D.J. Non-viral gene delivery regulated by stiffness of cell adhesion substrates. Nat. Mater. 2005, 4, 460–464. [Google Scholar] [CrossRef]
- Rey-Rico, A.; Venkatesan, J.K.; Schmitt, G.; Concheiro, A.; Madry, H.; Alvarez-Lorenzo, C.; Cucchiarini, M. rAAV-mediated overexpression of TGF- beta via vector delivery in polymeric micelles stimulates the biological and reparative activities of human articular chondrocytes in vitro and in a human osteochondral defect model. Int. J. Nanomed. 2017, 12, 6985–6996. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.H.; Haleem, A.M.; Yao, V.; Li, J.; Xiao, X.; Chu, C.R. Release of bioactive adeno-associated virus from fibrin scaffolds: Effects of fibrin glue concentrations. Tissue Eng. Part A 2017, 17, 1969–1978. [Google Scholar] [CrossRef]
- Chen, W.; Chen, H.; Zheng, D.; Zhang, H.; Deng, L.; Cui, W.; Zhang, Y.; Santos, H.A.; Shen, H. Gene-hydrogel microenvironment regulates extracellular matrix metabolism balance in nucleus pulposus. Adv. Sci. 2020, 7, 1902099. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Chen, Q.; He, S.; Yang, M.; Maguire, E.M.; An, W.; Afzal, T.A.; Luong, L.A.; Zhang, L.; Xiao, Q. miR-22 is a novel mediator of vascular smooth muscle cell phenotypic modulation and neointima formation. Circulation 2018, 137, 1824–1841. [Google Scholar] [CrossRef] [PubMed]
- Brunger, J.M.; Huynh, N.P.T.; Guenther, C.M.; Pinera, P.P.; Moutos, F.T.; Sanchez-Adams, J.; Gersbach, C.A.; Guilak, F. Scaffold-mediated lentiviral transduction for functional tissue engineering of cartilage. Proc. Natl. Acad. Sci. USA 2014, 11, E798–E806. [Google Scholar] [CrossRef] [Green Version]
- Saito, E.; Suarez-Gonzalez, D.; Murphy, W.L.; Hollister, S.J. Biomineral coating increases bone formation by ex vivo BMP-7 gene therapy in rapid prototyped poly(L-lactic acid) (PLLA) and poly(ε-caprolactone) (PCL) porous scaffolds. Adv. Healthc. Mater. 2015, 4, 621–632. [Google Scholar] [CrossRef] [Green Version]
- King, W.J.; Krebsbach, P.H. Growth factor delivery: How surface interactions modulate release in vitro and In vivo. Adv. Drug Delivery Rev. 2012, 64, 1239–1256. [Google Scholar] [CrossRef] [Green Version]
- Pilipchuk, S.P.; Fretwurst, T.; Yu, N.; Larsson, L.; Kavanagh, N.M.; Asa’ad, F.; Cheng, K.C.K.; Lahann, J.; Giannobile, W.V. Micropatterned scaffolds with immobilized growth factor genes regenerate bone and periodontal ligament-like tissues. Adv. Healthc. Mater. 2018, 7, 1800750. [Google Scholar] [CrossRef]
- Tierney, E.G.; Duffy, G.P.; Hibbitts, A.J.; Cryan, S.A.; O’Brien, F.J. The development of non-viral gene-activated matrices for bone regeneration using polyethyleneimine (PEI) and collagen-based scaffolds. J. Control. Release 2012, 158, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Curtin, C.M.; Tierney, E.G.; McSorley, K.; Cryan, S.A.; Duffy, G.P.; O’Brien, F.J. Combinatorial gene therapy accelerates bone regeneration: Non-viral dual delivery of VEGF and BMP2 in a collagen-nanohydroxyapatite scaffold. Adv. Healthc. Mater. 2014, 4, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Elangovan, S.; Khorsand, B.; Do, A.V.; Hong, L.; Dewerth, A.; Kormann, M.; Ross, R.D.; Sumner, D.R.; Allamargot, C.; Salem, A.K. Chemically modified RNA activated matrices enhance bone regeneration. J. Control. Release 2015, 218, 22–28. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.E.; Zuckerman, J.E.; Choi, C.H.; Seligson, D.; Tolcher, A.; Alabi, C.A.; Yen, Y.; Heidel, J.D.; Ribas, A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 2010, 464, 1067–1070. [Google Scholar] [CrossRef] [Green Version]
- Chitkara, D.; Mittal, A.; Mahato, R.I. miRNAs in pancreatic cancer: Therapeutic potential, delivery challenges and strategies. Adv. Drug Deliv. Rev. 2015, 81, 34–52. [Google Scholar] [CrossRef]
- Dong, X.; Tian, H.; Chen, L.; Chen, J.; Chen, X. Biodegradable mPEG-b-P(MCC g-OEI) copolymers for efficient gene delivery. J. Control. Release 2011, 152, 135–142. [Google Scholar] [CrossRef]
- Xu, X.; Jian, Y.; Li, Y.; Zhang, X.; Tu, Z.; Gu, Z. Bio-inspired supramolecular hybrid dendrimers self-assembled from low-generation peptide dendrons for highly efficient gene delivery and biological tracking. ACS Nano 2014, 8, 9255–9264. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Zheng, N.; Song, Z.; Yin, L.; Cheng, J. Trigger-responsive, fast degradable poly(β-amino ester)s for enhanced DNA unpackaging and reduced toxicity. Biomaterials 2014, 35, 5006–5015. [Google Scholar] [CrossRef] [Green Version]
- Tai, Z.; Wang, X.; Tian, J.; Gao, Y.; Zhang, L.; Yao, C.; Wu, X.; Zhang, W.; Zhu, Q.; Gao, S. Biodegradable stearylated peptide with internal disulfide bonds for efficient delivery of siRNA in vitro and In vivo. Biomacromolecules 2015, 16, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Pack, D.W.; Hoffman, A.S.; Pun, S.; Stayton, P.S. Design and development of polymers for gene delivery. Nat. Rev. Drug Discov. 2005, 4, 581–593. [Google Scholar] [CrossRef]
- Tian, H.; Chen, J.; Chen, X. Nanoparticles for gene delivery. Small 2013, 9, 2034–2044. [Google Scholar] [CrossRef]
- Tian, H.; Lin, L.; Chen, J.; Chen, X.; Park, T.G.; Maruyama, A. RGD targeting hyaluronic acid coating system for PEI-PBLG polycation gene carriers. J. Control. Release 2011, 155, 47–53. [Google Scholar] [CrossRef]
- Yang, K.; Gong, H.; Shi, X.; Wan, J.; Zhang, Y.; Liu, Z. In vivo biodistribution and toxicology of functionalized nano-graphene oxide in mice after oral and intraperitoneal administration. Biomaterials 2013, 34, 2787−2795. [Google Scholar] [CrossRef] [PubMed]
- Kanakia, S.; Toussaint, J.D.; Mullick Chowdhury, S.; Tembulkar, T.; Lee, S.; Jiang, Y.P.; Lin, R.Z.; Shroyer, K.R.; Moore, W.; Sitharaman, B. Dose ranging, expanded acute toxicity and safety pharmacology studies for intravenously administered functionalized graphene nanoparticle formulations. Biomaterials 2014, 35, 7022−7031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Kim, Y.S.; Lim, M.Y.; Kim, H.Y.; Kong, S.; Kang, M.; Choo, Y.W.; Jun, J.H.; Ryu, S.; Jeong, H.Y.; et al. Dual roles of graphene oxide to attenuate inflammation and elicit timely polarization of macrophage phenotypes for cardiac repair. ACS Nano 2018, 12, 1959–1977. [Google Scholar] [CrossRef] [PubMed]
- Mora-Raimundo, P.; Lozano, D.; Manzano, M.; Vallet-Regí, M. Nanoparticles to knockdown osteoporosis-related gene and promote osteogenic marker expression for osteoporosis treatment. ACS Nano 2019, 13, 5451–5464. [Google Scholar] [CrossRef] [Green Version]
- Atluri, K.; Seabold, D.; Hong, L.; Elangovan, S.; Salem, A.K. Nanoplex-mediated codelivery of fibroblast growth factor and bone morphogenetic protein genes promotes osteogenesis in human adipocyte-derived mesenchymal stem cells. Mol. Pharm. 2015, 12, 3032–3042. [Google Scholar] [CrossRef] [Green Version]
- Khorsand, B.; Nicholson, N.; Do, A.V.; Femino, J.E.; Martin, J.A.; Petersen, E.; Guetschow, B.; Fredericks, D.C.; Salem, A.K. Regeneration of bone using nanoplex delivery of FGF-2 and BMP-2 genes in diaphyseal long bone radial defects in a diabetic rabbit model. J. Control. Release 2017, 248, 53–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buck, J.; Grossen, P.; Cullis, P.R.; Huwyler, J.; Witzigmann, D. Lipid-Based DNA Therapeutics: Hallmarks of Non-Viral Gene Delivery. ACS Nano 2019, 13, 3754–3782. [Google Scholar] [CrossRef] [PubMed]
- Sainz-Ramos, M.; Villate-Beitia, I.; Gallego, I.; AL Qtaish, N.; Menéndez, M.; Lagartera, L.; Grijalvo, S.; Eritja, R.; Puras, G.; Pedraz, J.L. Correlation between Biophysical Properties of NiosomesElaborated with Chloroquine and Different Tensioactives and Their Transfection Efficiency. Pharmaceutics 2021, 13, 1787. [Google Scholar] [CrossRef] [PubMed]
- Quagliarini, E.; Renzi, S.; Digiacomo, L.; Giulimondi, F.; Sartori, B.; Amenitsch, H.; Tassinari, V.; Masuelli, L.; Bei, R.; Cui, L.; et al. Microfluidic Formulation of DNA-Loaded Multicomponent Lipid Nanoparticles for Gene Delivery. Pharmaceutics 2021, 13, 1292. [Google Scholar] [CrossRef] [PubMed]
- What Ingredients Are in the COVID-19 Vaccines? Available online: https://portal.ct.gov/vaccine-portal/Vaccine-Knowledge-Base/Articles/Ingredients-In-Vaccine?language=en_US (accessed on 6 July 2021).
- Zorde Khvalevsky, E.; Gabai, R.; Itzhak-Haim, R.; Horwitz, E.; Brunschwig, Z.; Orbach, A.; Shemi, A.; Golan, T.; Domb, A.J.; Galun, E.; et al. Mutant KRAS—A druggable target for pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20723–20728. [Google Scholar] [CrossRef] [Green Version]









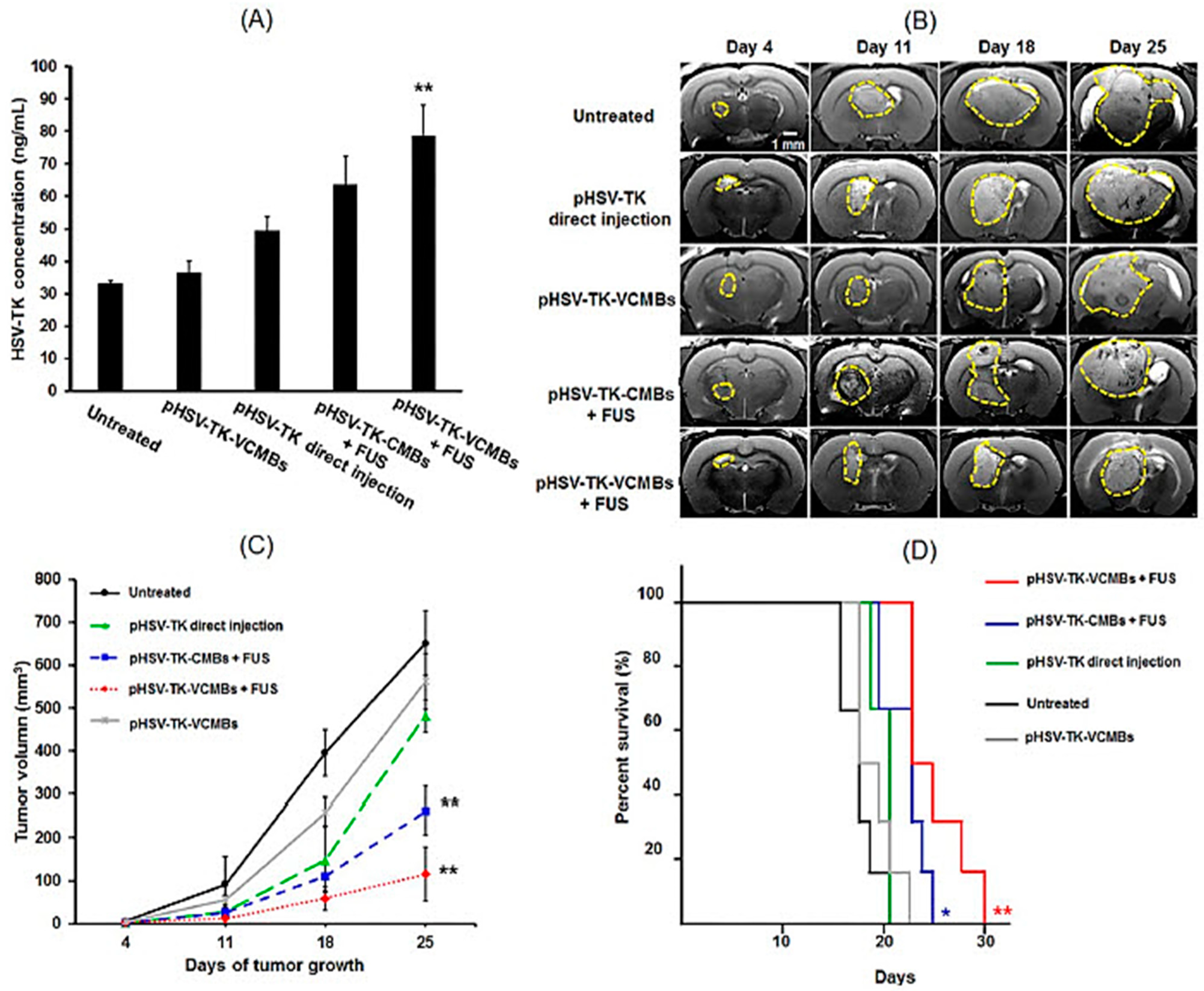{kind=link}
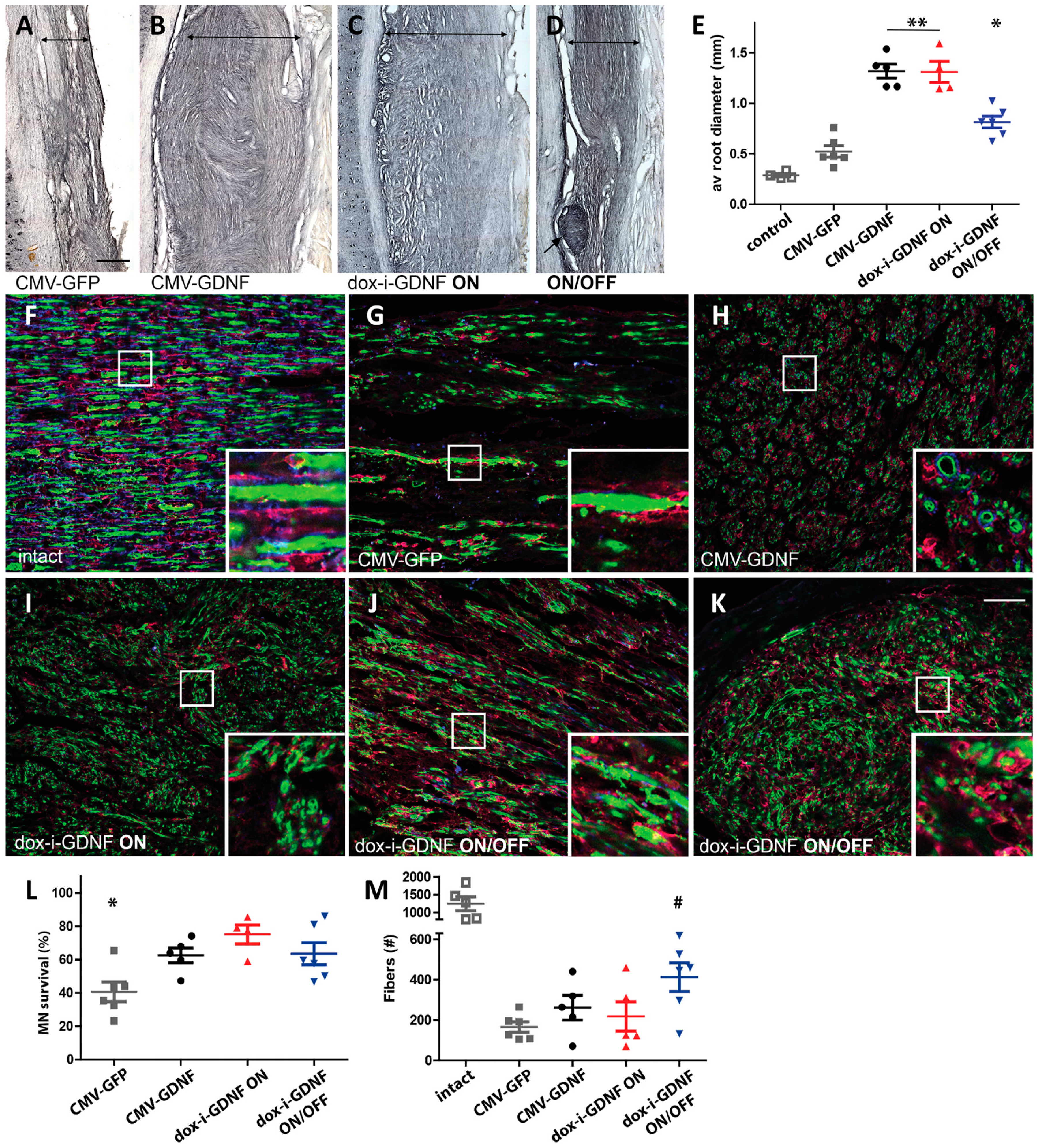{kind=link}
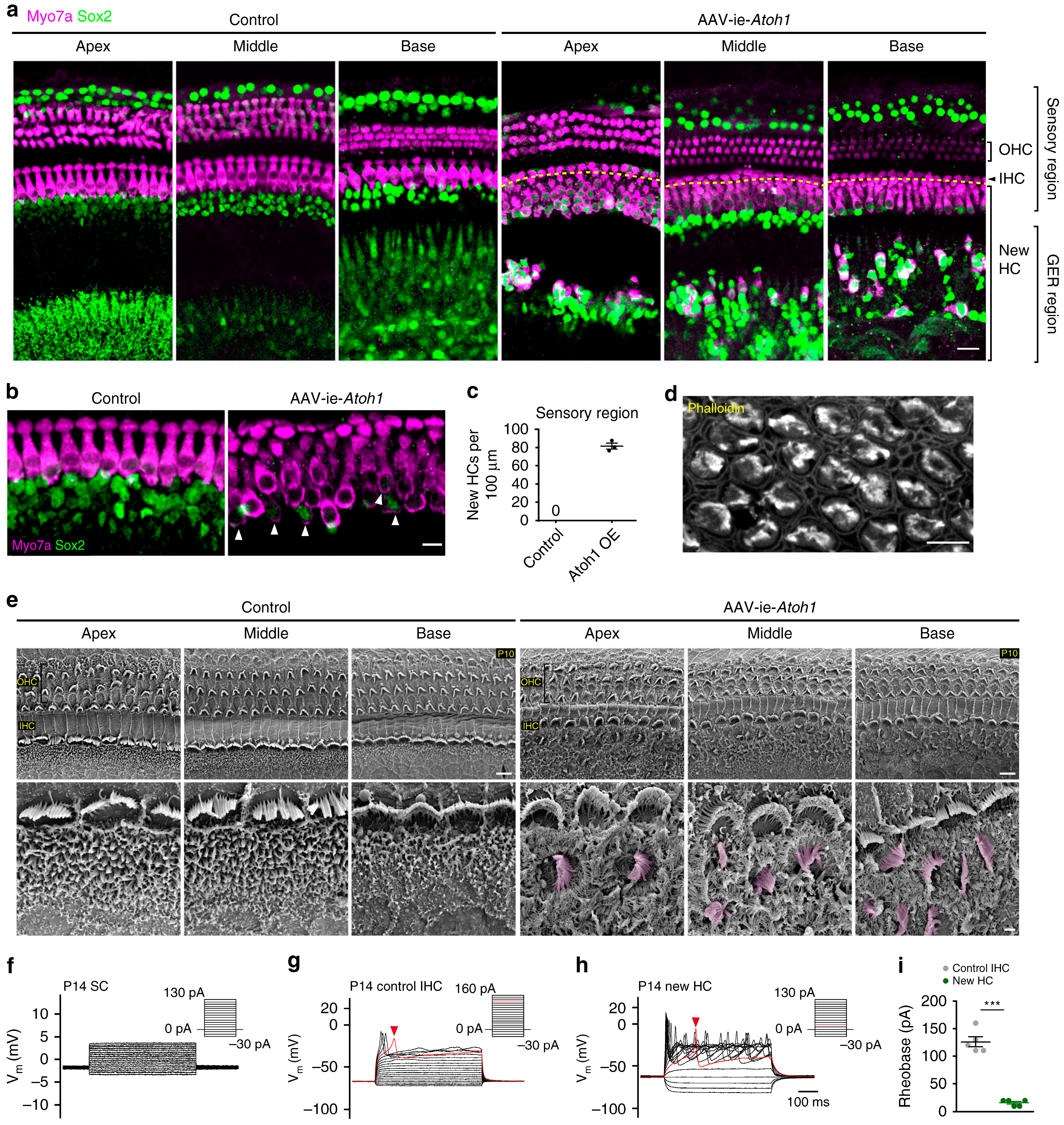{kind=link}
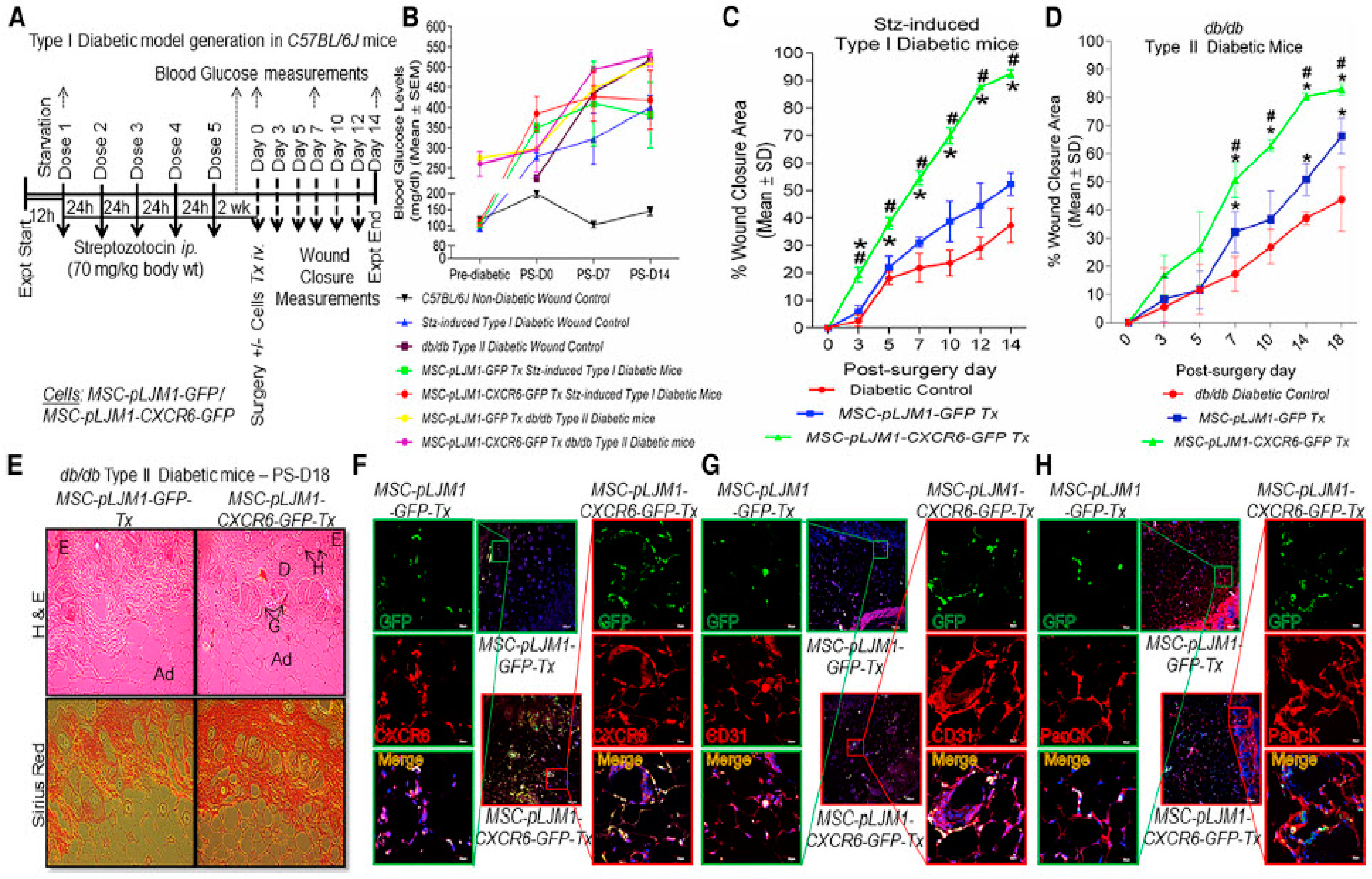{kind=link}
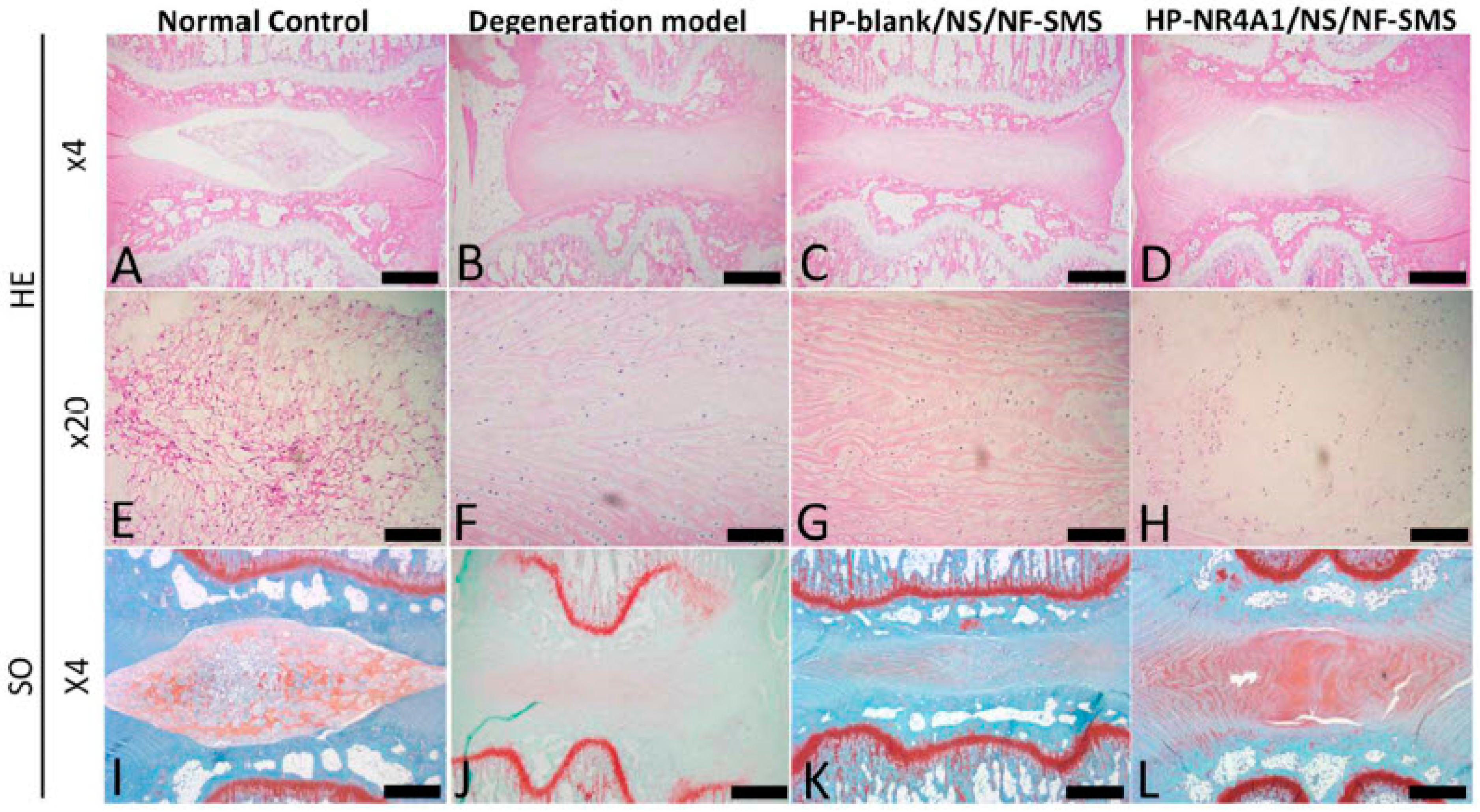{kind=link}
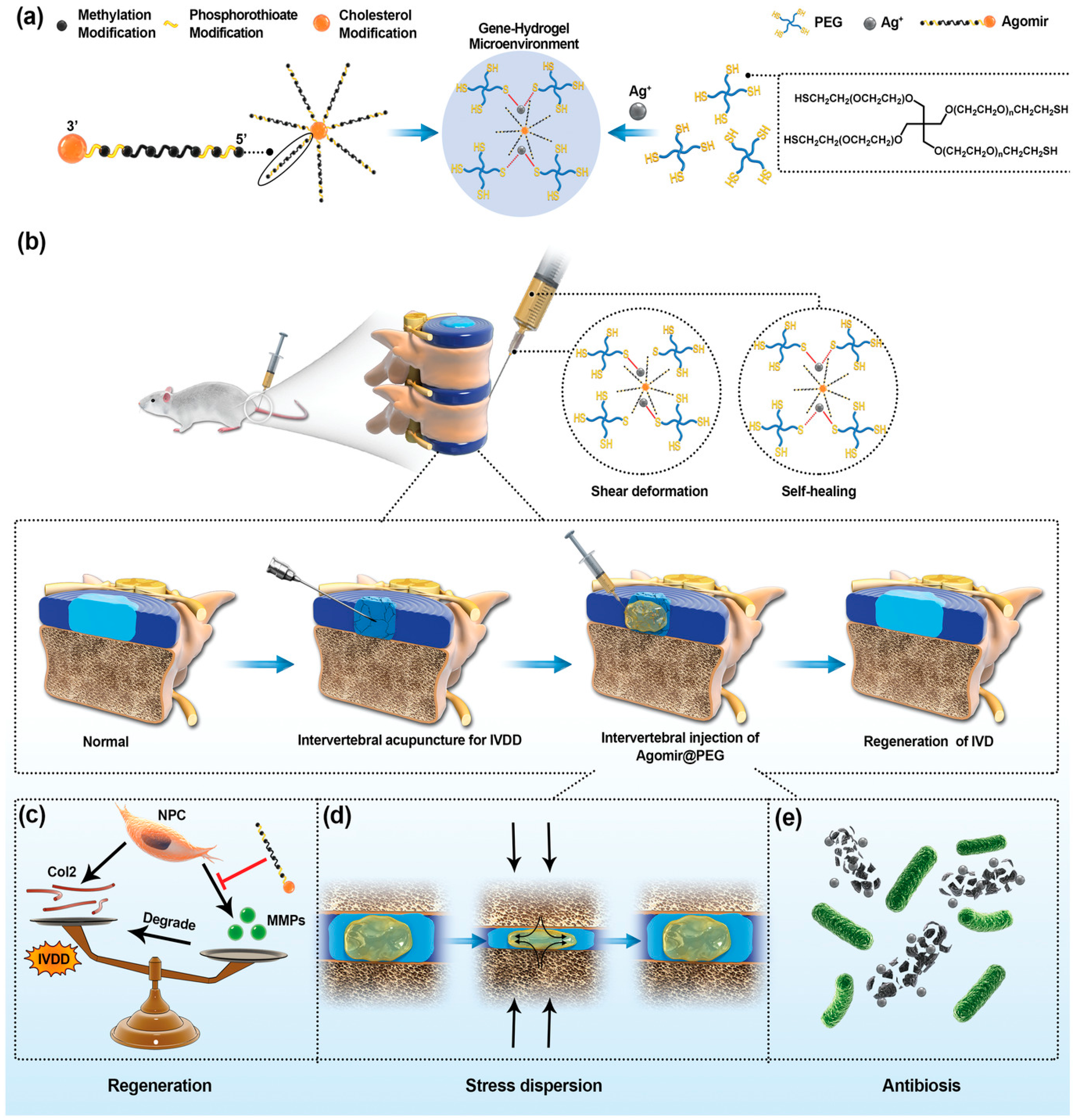{kind=link}
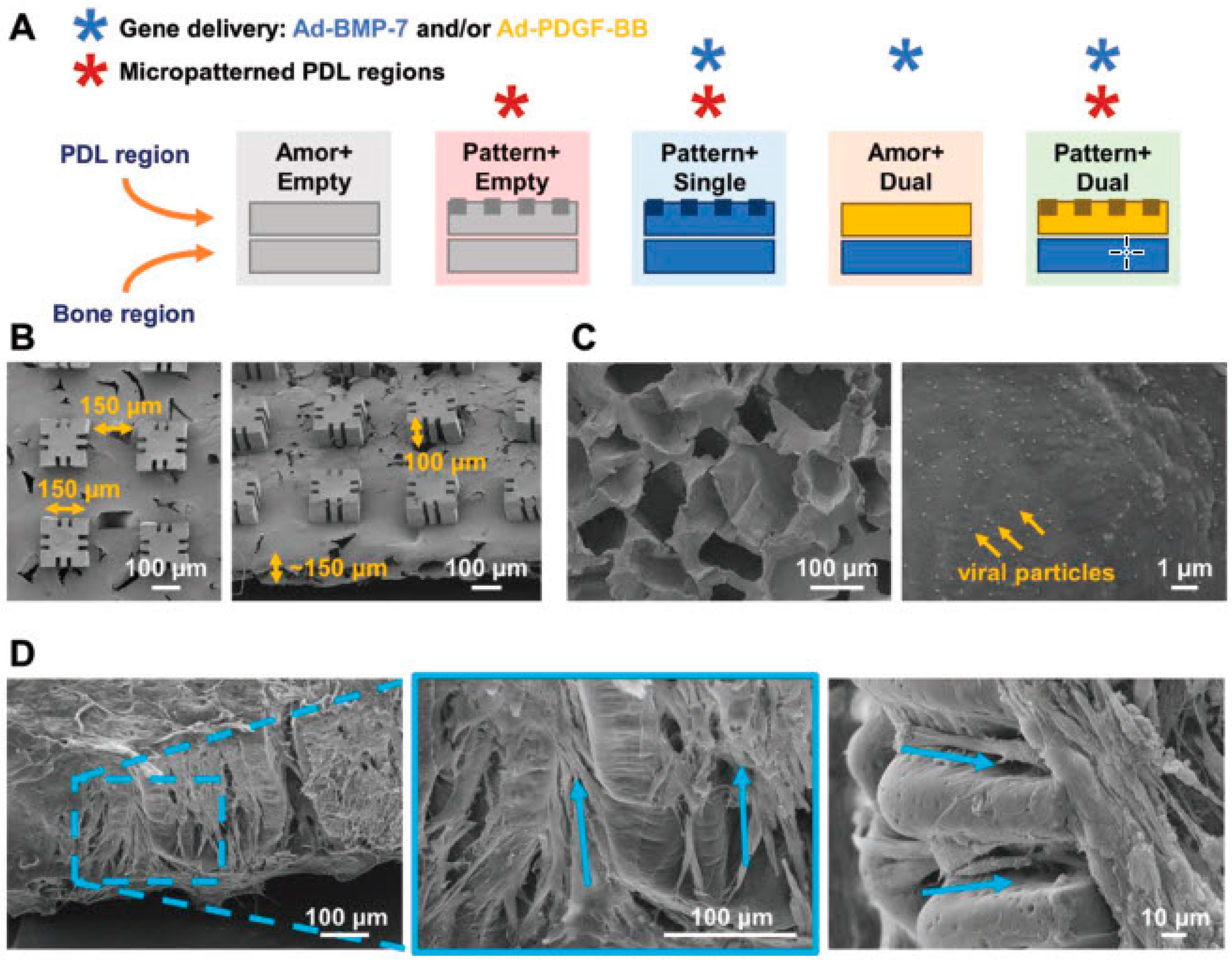{kind=link}
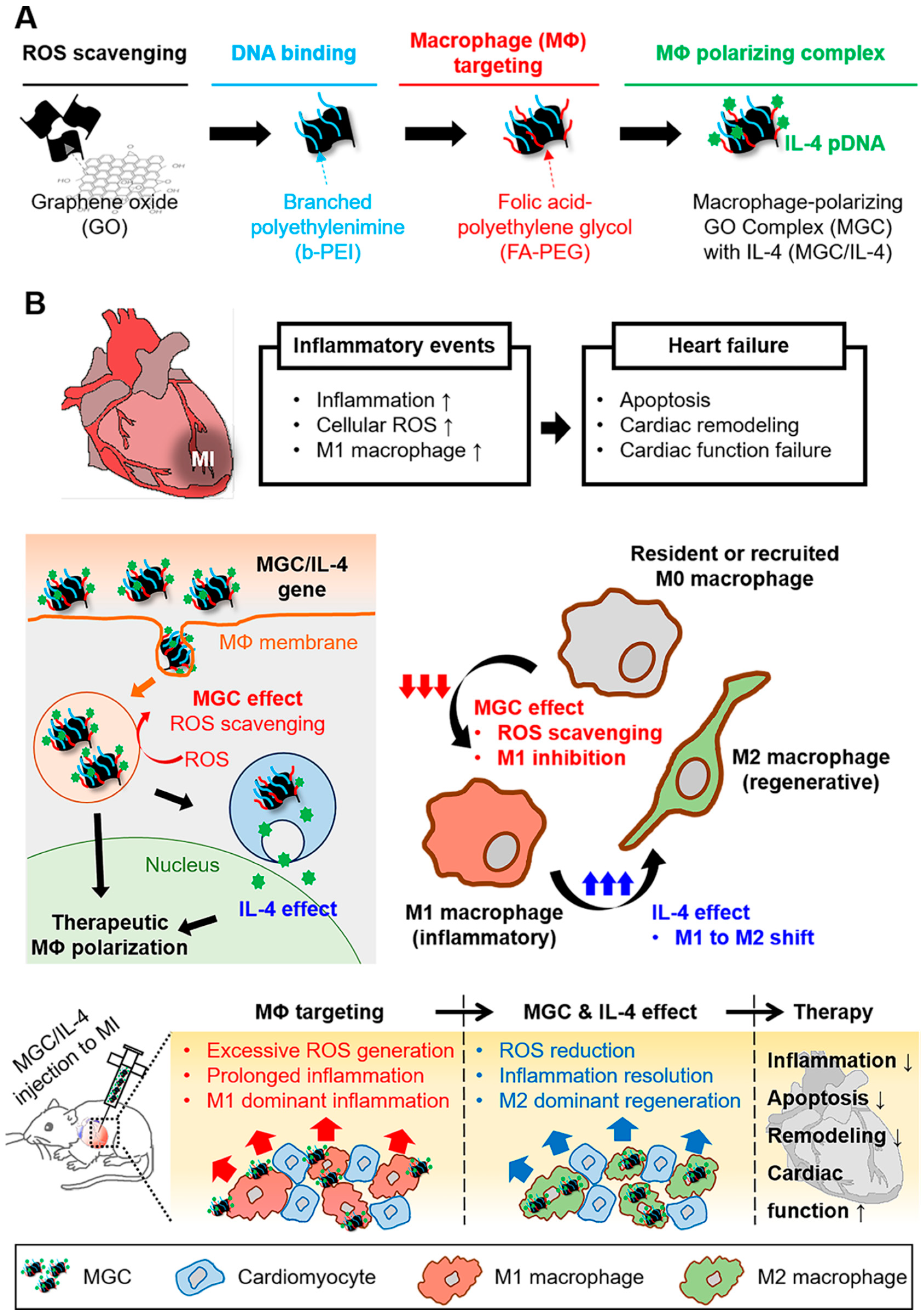{kind=link}
| Gene Delivery Methods | Advantages | Disadvantages | Ref. |
|---|---|---|---|
| Viral-mediated methods |
|
| [55] |
| Non-viral-mediated methods |
|
| [58] |
| Gene Delivery | Approach | Vector | Gene | Model | Remarks | Ref. |
|---|---|---|---|---|---|---|
| Viral | Ex vivo | Adenoviral | BMP-2 | Rat | Bone regeneration was similar in adipose tissue-derived MSCs and human bone marrow | [182] |
| Adenoviral | BMP-2 | Mice | Full bone defect regeneration was attained | [183] | ||
| Adenoviral | BMP-2 | Porcine | After 6 months complete, defect healing was obtained | [184] | ||
| Lentiviral | BMP-2 | Rat | Bone regeneration with a greater bone volume | [185] | ||
| Retroviral | Osterix | Mouse | 85% of regeneration was induced by MSCs | [186] | ||
| In vivo | Adenoviral | BMP-2 or BMP-6 | Pony | Long-term healing was not achieved for bone | [187] | |
| AAV | BMP-2 | Mouse | A new cortical shell was formed by scAAV2.5-BMP2 allografts that was highly similar to that generated via live autografts | [188] | ||
| Retroviral | BMP-2/4 | Rat | Healing rate was similar to untreated controls and was followed by excessive formation of ectopic bone that finally remodelled | [189] | ||
| Retroviral | COX-2 | Rat | Ectopic bone formation avoided with a faster healing rate | [190] | ||
| Non-viral | Ex vivo | Gelatin | BMP-2 | Rat | Homogenous bone regeneration | [191] |
| Liposome | BMP-2 | Porcine | Critical-size bone defects regenerated | [192] | ||
| Liposome | Osterix | Rabbit | Thickness of new trabeculae, the amount of the newly formed bone, and bone mineral density improved | [193] | ||
| Nucleofection | BMP2/Runx2 | Mice | Bone formation was markedly higher in nucleofected cells than BMP-2 alone | [194] | ||
| Nucleofection | BMP-6 | Rat | Bone healing was greater in treated cells | [195] | ||
| In vivo | Naked DNA | BMP-2 | Mice | Increasing the number of injections led to frequent regeneration of bones | [196] | |
| GAM | BMP-4 | Rat | After 18 weeks, an excessive amount of osteogenesis was stimulated | [197] | ||
| GAM | PDGF | Rat | Delayed bone regeneration and prolonged inflammation | [198] | ||
| GAM | PDGF | Rat | Improved bone healing after 4 weeks of implantation | [199] | ||
| Sonoporation | BMP-2/BMP-7 | Mouse | Ectopic osteogenic differentiation was achieved by co-delivery of genes | [200] | ||
| Sonoporation | BMP-6 | Porcine | 40% of cells transfected at the fracture site. 6 weeks after treatment, complete fracture healing was observed | [201] | ||
| Electroporation | BMP-9 | Mouse | 5 weeks after gene delivery led to complete repair of the bone defect | [202] | ||
| Liposome | BMP-2 | Porcine | Enhanced bone repair was monitored for treated groups | [203] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hosseinkhani, H.; Domb, A.J.; Sharifzadeh, G.; Nahum, V. Gene Therapy for Regenerative Medicine. Pharmaceutics 2023, 15, 856. https://doi.org/10.3390/pharmaceutics15030856
Hosseinkhani H, Domb AJ, Sharifzadeh G, Nahum V. Gene Therapy for Regenerative Medicine. Pharmaceutics. 2023; 15(3):856. https://doi.org/10.3390/pharmaceutics15030856
Chicago/Turabian StyleHosseinkhani, Hossein, Abraham J. Domb, Ghorbanali Sharifzadeh, and Victoria Nahum. 2023. "Gene Therapy for Regenerative Medicine" Pharmaceutics 15, no. 3: 856. https://doi.org/10.3390/pharmaceutics15030856