


Lentiviral Vectors as a Vaccine Platform against Infectious Diseases

 ,
,
Abstract
:1. Introduction
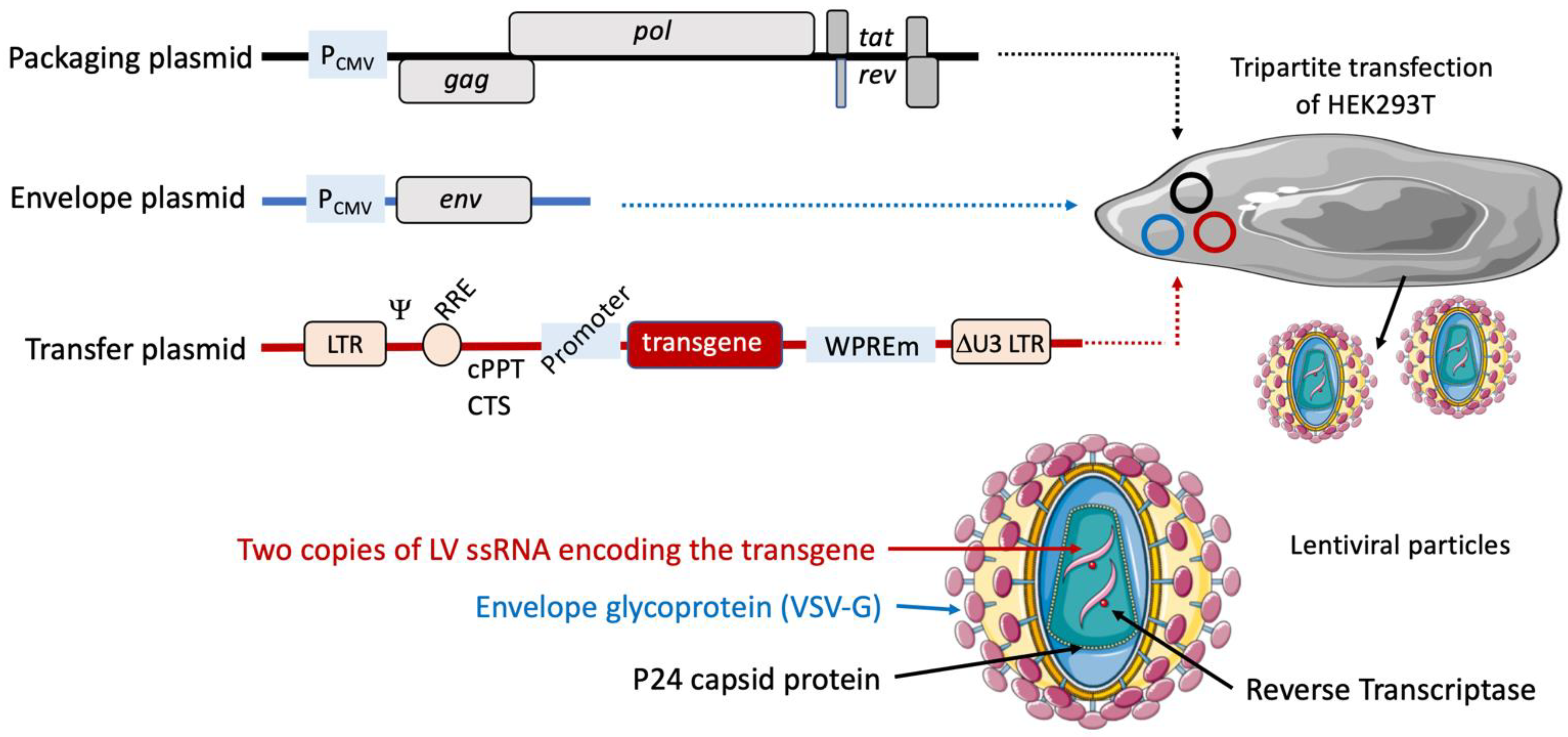
2. Lentiviral Vector-Based Vaccine Platform
3. Tropism of Lentiviral Vectors for Dendritic Cells
4. Weak Inflammatory Properties of Lentiviral Vectors
5. Biosafety of Lentiviral Vectors
6. Lentiviral Vector-Based Vaccination in Preclinical Models
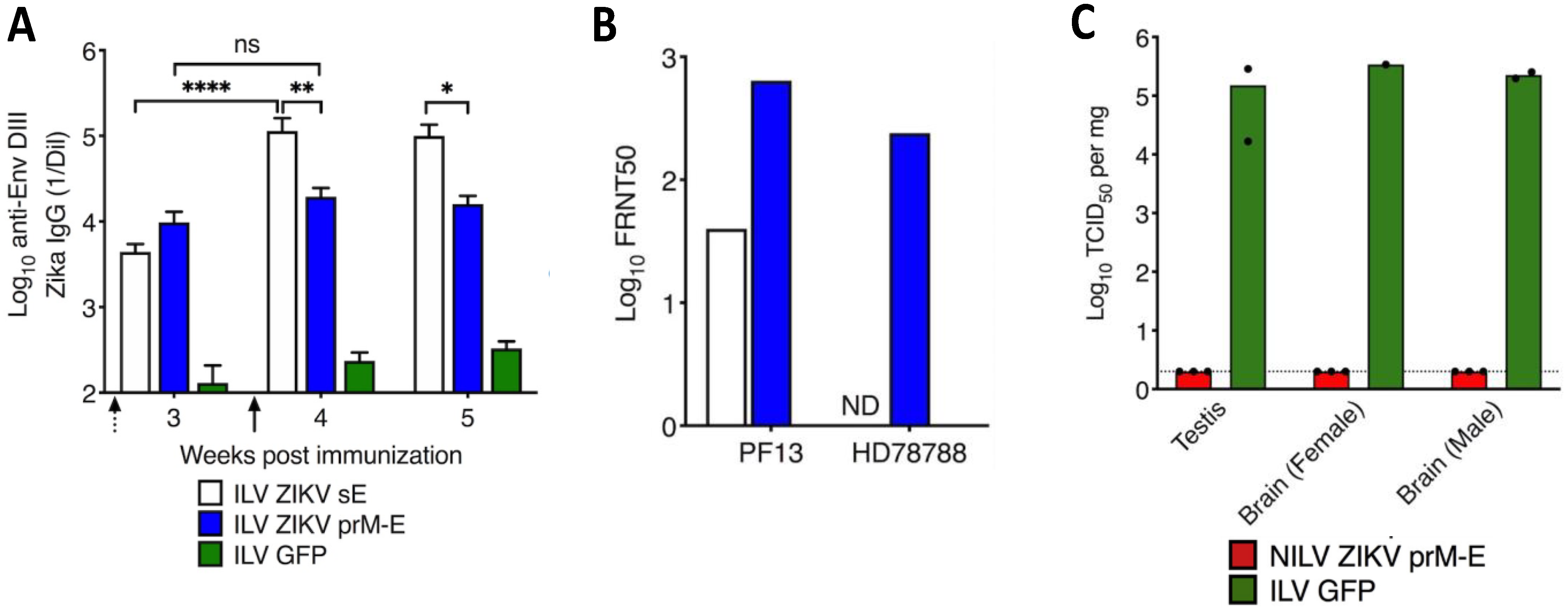
6.1. LV-Based Vaccine Candidates against Flaviviruses
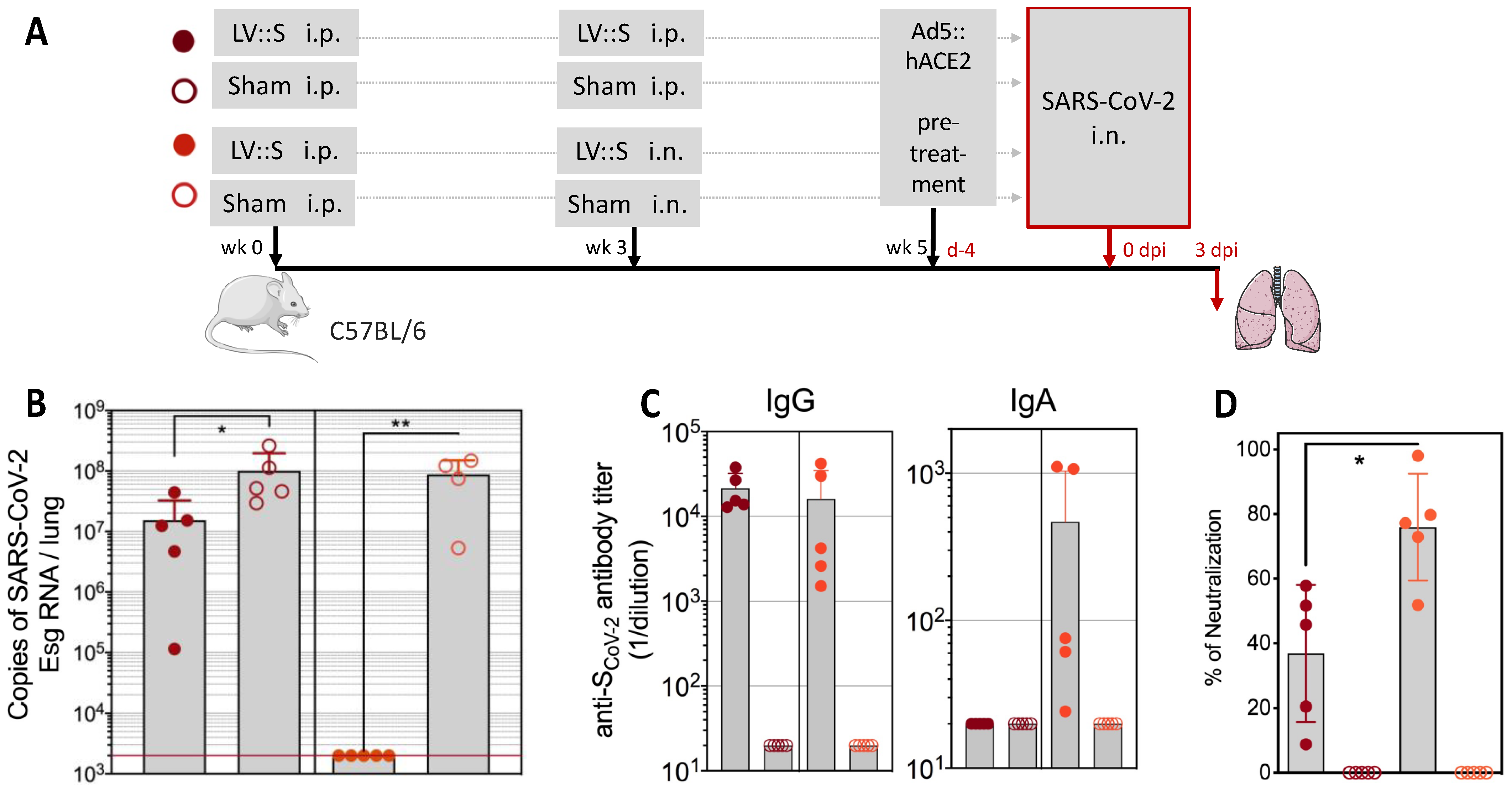
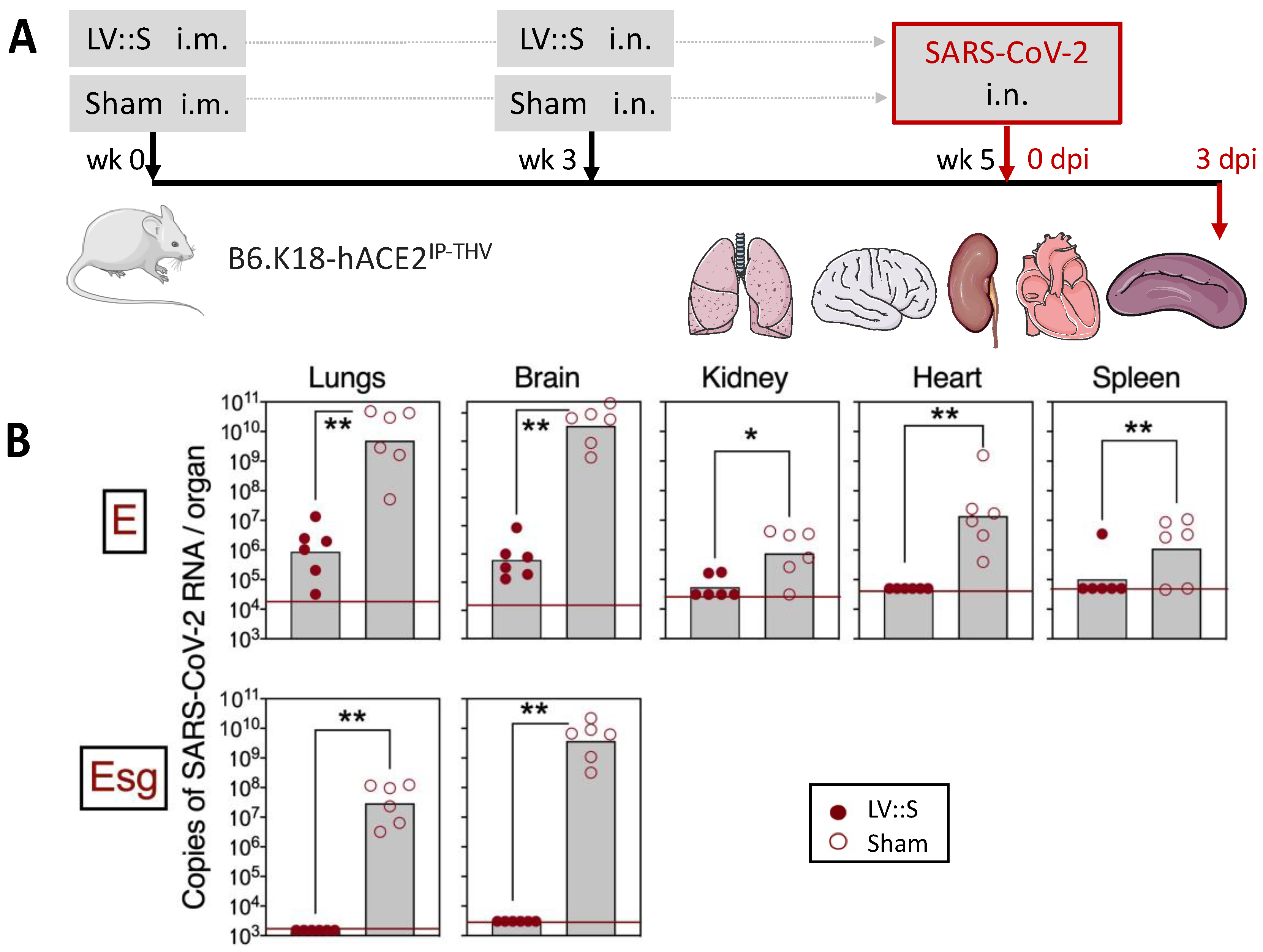
6.2. LV-Based COVID-19 Vaccine Candidate
6.3. LV-Based Tuberculosis Vaccine Candidate
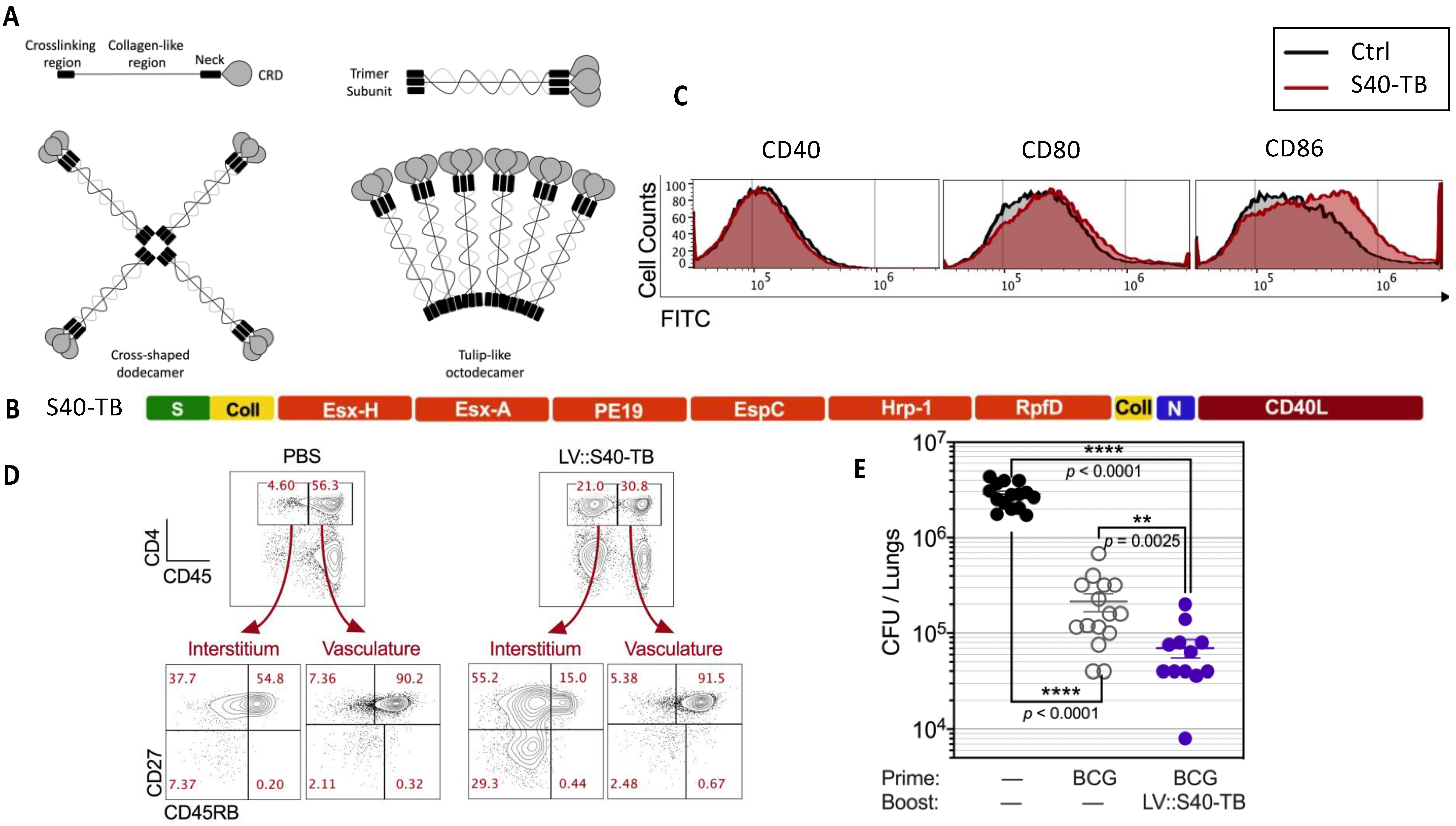
6.4. LV Encoding Secreted Protein Cargo That Targets Dendritic Cells to Induce Anti-Mycobacterial Immunity
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Charneau, P.; Alizon, M.; Clavel, F. A second origin of DNA plus-strand synthesis is required for optimal human immunodeficiency virus replication. J. Virol. 1992, 66, 2814–2820. [Google Scholar] [CrossRef] [Green Version]
- Charneau, P.; Mirambeau, G.; Roux, P.; Paulous, S.; Buc, H.; Clavel, F. HIV-1 reverse transcription. A termination step at the center of the genome. J. Mol. Biol. 1994, 241, 651–662. [Google Scholar] [CrossRef]
- Dragic, T.; Charneau, P.; Clavel, F.; Alizon, M. Complementation of murine cells for human immunodeficiency virus envelope/CD4-mediated fusion in human/murine heterokaryons. J. Virol. 1992, 66, 4794–4802. [Google Scholar] [CrossRef] [Green Version]
- Ku, M.W.; Charneau, P.; Majlessi, L. Use of lentiviral vectors in vaccination. Expert Rev. Vaccines 2021, 20, 1571–1586. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Chavan, R.; Feinberg, M.B. Dendritic cells are preferentially targeted among hematolymphocytes by Modified Vaccinia Virus Ankara and play a key role in the induction of virus-specific T cell responses in vivo. BMC Immunol. 2008, 9, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Muruve, D.A. Molecular basis of the inflammatory response to adenovirus vectors. Gene Ther. 2003, 10, 935–940. [Google Scholar] [CrossRef] [Green Version]
- Ku, M.W.; Authie, P.; Nevo, F.; Souque, P.; Bourgine, M.; Romano, M.; Charneau, P.; Majlessi, L. Lentiviral vector induces high-quality memory T cells via dendritic cells transduction. Commun. Biol. 2021, 4, 713. [Google Scholar] [CrossRef]
- Anna, F.; Lopez, J.; Moncoq, F.; Blanc, C.; Authie, P.; Noirat, A.; Fert, I.; Souque, P.; Nevo, F.; Pawlik, A.; et al. A lentiviral vector expressing a dendritic cell-targeting multimer induces mucosal anti-mycobacterial CD4(+) T-cell immunity. Mucosal Immunol. 2022, 15, 1389–1404. [Google Scholar] [CrossRef]
- Lopez, J.; Anna, F.; Authie, P.; Pawlik, A.; Ku, M.W.; Blanc, C.; Souque, P.; Moncoq, F.; Noirat, A.; Hardy, D.; et al. A lentiviral vector encoding fusion of light invariant chain and mycobacterial antigens induces protective CD4(+) T cell immunity. Cell Rep. 2022, 40, 111142. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://old.abmgood.com/marketing/knowledge_base/The_Lentivirus_System.php#3 (accessed on 15 February 2023).
- Zennou, V.; Petit, C.; Guetard, D.; Nerhbass, U.; Montagnier, L.; Charneau, P. HIV-1 genome nuclear import is mediated by a central DNA flap. Cell 2000, 101, 173–185. [Google Scholar] [CrossRef] [Green Version]
- Donello, J.E.; Loeb, J.E.; Hope, T.J. Woodchuck hepatitis virus contains a tripartite posttranscriptional regulatory element. J. Virol. 1998, 72, 5085–5092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyvaerts, C.; Liechtenstein, T.; Bricogne, C.; Escors, D.; Breckpot, K. Targeted Lentiviral Vectors: Current Applications and Future Potential. In Gene Therapy—Tools and Potential Applications; InTech: London, UK, 2013. [Google Scholar] [CrossRef] [Green Version]
- Iwakuma, T.; Cui, Y.; Chang, L.J. Self-inactivating lentiviral vectors with U3 and U5 modifications. Virology 1999, 261, 120–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arhel, N.J.; Souquere-Besse, S.; Munier, S.; Souque, P.; Guadagnini, S.; Rutherford, S.; Prevost, M.C.; Allen, T.D.; Charneau, P. HIV-1 DNA Flap formation promotes uncoating of the pre-integration complex at the nuclear pore. EMBO J. 2007, 26, 3025–3037. [Google Scholar] [CrossRef]
- Coutant, F.; Sanchez David, R.Y.; Felix, T.; Boulay, A.; Caleechurn, L.; Souque, P.; Thouvenot, C.; Bourgouin, C.; Beignon, A.S.; Charneau, P. A nonintegrative lentiviral vector-based vaccine provides long-term sterile protection against malaria. PLoS ONE 2012, 7, e48644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutner, R.H.; Puthli, S.; Marino, M.P.; Reiser, J. Simplified production and concentration of HIV-1-based lentiviral vectors using HYPERFlask vessels and anion exchange membrane chromatography. BMC Biotechnol. 2009, 9, 10. [Google Scholar] [CrossRef] [Green Version]
- Ricobaraza, A.; Gonzalez-Aparicio, M.; Mora-Jimenez, L.; Lumbreras, S.; Hernandez-Alcoceba, R. High-Capacity Adenoviral Vectors: Expanding the Scope of Gene Therapy. Int. J. Mol. Sci. 2020, 21, 3643. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, H.; Colosi, P. Effect of genome size on AAV vector packaging. Mol. Ther. 2010, 18, 80–86. [Google Scholar] [CrossRef]
- Orlova, O.V.; Glazkova, D.V.; Bogoslovskaya, E.V.; Shipulin, G.A.; Yudin, S.M. Development of Modified Vaccinia Virus Ankara-Based Vaccines: Advantages and Applications. Vaccines 2022, 10, 1516. [Google Scholar] [CrossRef]
- Breckpot, K.; Escors, D.; Arce, F.; Lopes, L.; Karwacz, K.; Van Lint, S.; Keyaerts, M.; Collins, M. HIV-1 lentiviral vector immunogenicity is mediated by Toll-like receptor 3 (TLR3) and TLR7. J. Virol. 2010, 84, 5627–5636. [Google Scholar] [CrossRef] [Green Version]
- Brown, B.D.; Sitia, G.; Annoni, A.; Hauben, E.; Sergi, L.S.; Zingale, A.; Roncarolo, M.G.; Guidotti, L.G.; Naldini, L. In vivo administration of lentiviral vectors triggers a type I interferon response that restricts hepatocyte gene transfer and promotes vector clearance. Blood 2007, 109, 2797–2805. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Zhang, J.; Mi, Z.; Robbins, P.; Falo, L.D., Jr. Immunization with lentiviral vector-transduced dendritic cells induces strong and long-lasting T cell responses and therapeutic immunity. J. Immunol. 2005, 174, 3808–3817. [Google Scholar] [CrossRef] [Green Version]
- Guibinga, G.H.; Miyanohara, A.; Esko, J.D.; Friedmann, T. Cell surface heparan sulfate is a receptor for attachment of envelope protein-free retrovirus-like particles and VSV-G pseudotyped MLV-derived retrovirus vectors to target cells. Mol. Ther. 2002, 5, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Finkelshtein, D.; Werman, A.; Novick, D.; Barak, S.; Rubinstein, M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc. Natl. Acad. Sci. USA 2013, 110, 7306–7311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hastie, E.; Cataldi, M.; Marriott, I.; Grdzelishvili, V.Z. Understanding and altering cell tropism of vesicular stomatitis virus. Virus Res. 2013, 176, 16–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolic, J.; Belot, L.; Raux, H.; Legrand, P.; Gaudin, Y.; Albertini, A.A. Structural basis for the recognition of LDL-receptor family members by VSV glycoprotein. Nat. Commun. 2018, 9, 1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amirache, F.; Levy, C.; Costa, C.; Mangeot, P.E.; Torbett, B.E.; Wang, C.X.; Negre, D.; Cosset, F.L.; Verhoeyen, E. Mystery solved: VSV-G-LVs do not allow efficient gene transfer into unstimulated T cells, B cells, and HSCs because they lack the LDL receptor. Blood 2014, 123, 1422–1424. [Google Scholar] [CrossRef] [PubMed]
- Coutant, F.; Frenkiel, M.P.; Despres, P.; Charneau, P. Protective antiviral immunity conferred by a nonintegrative lentiviral vector-based vaccine. PLoS ONE 2008, 3, e3973. [Google Scholar] [CrossRef]
- Esslinger, C.; Romero, P.; MacDonald, H.R. Efficient transduction of dendritic cells and induction of a T-cell response by third-generation lentivectors. Hum. Gene Ther. 2002, 13, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Ardeshna, K.M.; Pizzey, A.R.; Thomas, N.S.; Orr, S.; Linch, D.C.; Devereux, S. Monocyte-derived dendritic cells do not proliferate and are not susceptible to retroviral transduction. Br. J. Haematol. 2000, 108, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.G.; Adam, M.A.; Miller, A.D. Gene transfer by retrovirus vectors occurs only in cells that are actively replicating at the time of infection. Mol. Cell. Biol. 1990, 10, 4239–4242. [Google Scholar] [CrossRef] [Green Version]
- Ku, M.W.; Authie, P.; Bourgine, M.; Anna, F.; Noirat, A.; Moncoq, F.; Vesin, B.; Nevo, F.; Lopez, J.; Souque, P.; et al. Brain cross-protection against SARS-CoV-2 variants by a lentiviral vaccine in new transgenic mice. EMBO Mol. Med. 2021, 13, e14459. [Google Scholar] [CrossRef]
- Ku, M.W.; Bourgine, M.; Authie, P.; Lopez, J.; Nemirov, K.; Moncoq, F.; Noirat, A.; Vesin, B.; Nevo, F.; Blanc, C.; et al. Intranasal vaccination with a lentiviral vector protects against SARS-CoV-2 in preclinical animal models. Cell Host Microbe 2021, 29, 236–249.e6. [Google Scholar] [CrossRef] [PubMed]
- Vesin, B.; Lopez, J.; Noirat, A.; Authié, P.; Fert, I.; Le Chevalier, F.; Moncoq, F.; Nemirov, N.; Blanc, C.; Planchais, C.; et al. An intranasal lentiviral booster broadens immune recognition of SARS-CoV-2 variants and reinforces the waning mRNA vaccine-induced immunity that it targets to lung mucosa. Mol. Ther. 2022, 30, 2984–2997. [Google Scholar] [CrossRef]
- Funke, S.; Maisner, A.; Muhlebach, M.D.; Koehl, U.; Grez, M.; Cattaneo, R.; Cichutek, K.; Buchholz, C.J. Targeted cell entry of lentiviral vectors. Mol. Ther. 2008, 16, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Humbert, J.M.; Frecha, C.; Amirache Bouafia, F.; N’Guyen, T.H.; Boni, S.; Cosset, F.L.; Verhoeyen, E.; Halary, F. Measles virus glycoprotein-pseudotyped lentiviral vectors are highly superior to vesicular stomatitis virus G pseudotypes for genetic modification of monocyte-derived dendritic cells. J. Virol. 2012, 86, 5192–5203. [Google Scholar] [CrossRef] [Green Version]
- de Swart, R.L.; Yuksel, S.; Osterhaus, A.D. Relative contributions of measles virus hemagglutinin- and fusion protein-specific serum antibodies to virus neutralization. J. Virol. 2005, 79, 11547–11551. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Dai, B.; Wang, P. Vaccines delivered by integration-deficient lentiviral vectors targeting dendritic cells induces strong antigen-specific immunity. Vaccine 2010, 28, 6675–6683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Yang, H.; Rideout, K.; Cho, T.; Joo, K.I.; Ziegler, L.; Elliot, A.; Walls, A.; Yu, D.; Baltimore, D.; et al. Engineered lentivector targeting of dendritic cells for in vivo immunization. Nat. Biotechnol. 2008, 26, 326–334. [Google Scholar] [CrossRef] [Green Version]
- Goyvaerts, C.; De Groeve, K.; Dingemans, J.; Van Lint, S.; Robays, L.; Heirman, C.; Reiser, J.; Zhang, X.Y.; Thielemans, K.; De Baetselier, P.; et al. Development of the Nanobody display technology to target lentiviral vectors to antigen-presenting cells. Gene Ther. 2012, 19, 1133–1140. [Google Scholar] [CrossRef] [Green Version]
- Merlin, S.; Follenzi, A. Transcriptional Targeting and MicroRNA Regulation of Lentiviral Vectors. Mol. Ther. Methods Clin. Dev. 2019, 12, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Lopes, L.; Dewannieux, M.; Gileadi, U.; Bailey, R.; Ikeda, Y.; Whittaker, C.; Collin, M.P.; Cerundolo, V.; Tomihari, M.; Ariizumi, K.; et al. Immunization with a lentivector that targets tumor antigen expression to dendritic cells induces potent CD8+ and CD4+ T-cell responses. J. Virol. 2008, 82, 86–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gobin, S.J.; Biesta, P.; Van den Elsen, P.J. Regulation of human beta 2-microglobulin transactivation in hematopoietic cells. Blood 2003, 101, 3058–3064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Elsen, P.J. Expression regulation of major histocompatibility complex class I and class II encoding genes. Front. Immunol. 2011, 2, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beignon, A.S.; Mollier, K.; Liard, C.; Coutant, F.; Munier, S.; Riviere, J.; Souque, P.; Charneau, P. Lentiviral vector-based prime/boost vaccination against AIDS: Pilot study shows protection against Simian immunodeficiency virus SIVmac251 challenge in macaques. J. Virol. 2009, 83, 10963–10974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buffa, V.; Negri, D.R.; Leone, P.; Borghi, M.; Bona, R.; Michelini, Z.; Compagnoni, D.; Sgadari, C.; Ensoli, B.; Cara, A. Evaluation of a self-inactivating lentiviral vector expressing simian immunodeficiency virus gag for induction of specific immune responses in vitro and in vivo. Viral Immunol. 2006, 19, 690–701. [Google Scholar] [CrossRef]
- Gallinaro, A.; Borghi, M.; Bona, R.; Grasso, F.; Calzoletti, L.; Palladino, L.; Cecchetti, S.; Vescio, M.F.; Macchia, D.; Morante, V.; et al. Integrase Defective Lentiviral Vector as a Vaccine Platform for Delivering Influenza Antigens. Front. Immunol. 2018, 9, 171. [Google Scholar] [CrossRef] [Green Version]
- Iglesias, M.C.; Frenkiel, M.P.; Mollier, K.; Souque, P.; Despres, P.; Charneau, P. A single immunization with a minute dose of a lentiviral vector-based vaccine is highly effective at eliciting protective humoral immunity against West Nile virus. J. Gene Med. 2006, 8, 265–274. [Google Scholar] [CrossRef]
- Cousin, C.; Oberkampf, M.; Felix, T.; Rosenbaum, P.; Weil, R.; Fabrega, S.; Morante, V.; Negri, D.; Cara, A.; Dadaglio, G.; et al. Persistence of Integrase-Deficient Lentiviral Vectors Correlates with the Induction of STING-Independent CD8(+) T Cell Responses. Cell Rep. 2019, 26, 1242–1257 e1247. [Google Scholar] [CrossRef] [Green Version]
- Fonteneau, J.F.; Larsson, M.; Beignon, A.S.; McKenna, K.; Dasilva, I.; Amara, A.; Liu, Y.J.; Lifson, J.D.; Littman, D.R.; Bhardwaj, N. Human immunodeficiency virus type 1 activates plasmacytoid dendritic cells and concomitantly induces the bystander maturation of myeloid dendritic cells. J. Virol. 2004, 78, 5223–5232. [Google Scholar] [CrossRef] [Green Version]
- Rossetti, M.; Gregori, S.; Hauben, E.; Brown, B.D.; Sergi, L.S.; Naldini, L.; Roncarolo, M.G. HIV-1-derived lentiviral vectors directly activate plasmacytoid dendritic cells, which in turn induce the maturation of myeloid dendritic cells. Hum. Gene Ther. 2011, 22, 177–188. [Google Scholar] [CrossRef]
- Pichlmair, A.; Diebold, S.S.; Gschmeissner, S.; Takeuchi, Y.; Ikeda, Y.; Collins, M.K.; Reis e Sousa, C. Tubulovesicular structures within vesicular stomatitis virus G protein-pseudotyped lentiviral vector preparations carry DNA and stimulate antiviral responses via Toll-like receptor 9. J. Virol. 2007, 81, 539–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hacein-Bey-Abina, S.; von Kalle, C.; Schmidt, M.; Le Deist, F.; Wulffraat, N.; McIntyre, E.; Radford, I.; Villeval, J.L.; Fraser, C.C.; Cavazzana-Calvo, M.; et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2003, 348, 255–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs). 2018. Available online: https://www.fdanews.com/ext/resources/files/2018/07-11-18-GeneTherapy.pdf?1531336820 (accessed on 20 February 2023).
- TheraVectys-Clinical-Trial. Safety, Tolerability and Immunogenicity Induced by the THV01 Treatment in Patients Infected With HIV-1 Clade B and Treated with Highly Active Antiretroviral Therapy (HAART). 2019. Available online: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2011-006260-52 (accessed on 15 December 2022).
- Magnani, A.; Semeraro, M.; Adam, F.; Booth, C.; Dupre, L.; Morris, E.C.; Gabrion, A.; Roudaut, C.; Borgel, D.; Toubert, A.; et al. Long-term safety and efficacy of lentiviral hematopoietic stem/progenitor cell gene therapy for Wiskott-Aldrich syndrome. Nat. Med. 2022, 28, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Magrin, E.; Semeraro, M.; Hebert, N.; Joseph, L.; Magnani, A.; Chalumeau, A.; Gabrion, A.; Roudaut, C.; Marouene, J.; Lefrere, F.; et al. Long-term outcomes of lentiviral gene therapy for the beta-hemoglobinopathies: The HGB-205 trial. Nat. Med. 2022, 28, 81–88. [Google Scholar] [CrossRef]
- Milone, M.C.; O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef] [Green Version]
- Banasik, M.B.; McCray, P.B., Jr. Integrase-defective lentiviral vectors: Progress and applications. Gene Ther. 2010, 17, 150–157. [Google Scholar] [CrossRef] [Green Version]
- Saenz, D.T.; Loewen, N.; Peretz, M.; Whitwam, T.; Barraza, R.; Howell, K.G.; Holmes, J.M.; Good, M.; Poeschla, E.M. Unintegrated lentivirus DNA persistence and accessibility to expression in nondividing cells: Analysis with class I integrase mutants. J. Virol. 2004, 78, 2906–2920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hacein-Bey-Abina, S.; Von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef]
- Olbrich, H.; Slabik, C.; Stripecke, R. Reconstructing the immune system with lentiviral vectors. Virus Genes 2017, 53, 723–732. [Google Scholar] [CrossRef] [Green Version]
- Delville, M.; Soheili, T.; Bellier, F.; Durand, A.; Denis, A.; Lagresle-Peyrou, C.; Cavazzana, M.; Andre-Schmutz, I.; Six, E. A Nontoxic Transduction Enhancer Enables Highly Efficient Lentiviral Transduction of Primary Murine T Cells and Hematopoietic Stem Cells. Mol. Ther. Methods Clin. Dev. 2018, 10, 341–347. [Google Scholar] [CrossRef] [Green Version]
- Jang, Y.; Kim, Y.S.; Wielgosz, M.M.; Ferrara, F.; Ma, Z.; Condori, J.; Palmer, L.E.; Zhao, X.; Kang, G.; Rawlings, D.J.; et al. Optimizing lentiviral vector transduction of hematopoietic stem cells for gene therapy. Gene Ther. 2020, 27, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Clinical-Trial, I.D. Phase 1 Study of Intradermal LV305 in Patients with Locally Advanced, Relapsed or Metastatic Cancer Expressing NY-ESO-1. 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT02122861 (accessed on 8 February 2023).
- Somaiah, N.; Block, M.S.; Kim, J.W.; Shapiro, G.I.; Do, K.T.; Hwu, P.; Eder, J.P.; Jones, R.L.; Lu, H.; Ter Meulen, J.H.; et al. First-in-Class, First-in-Human Study Evaluating LV305, a Dendritic-Cell Tropic Lentiviral Vector, in Sarcoma and Other Solid Tumors Expressing NY-ESO-1. Clin. Cancer Res. 2019, 25, 5808–5817. [Google Scholar] [CrossRef] [Green Version]
- Adotevi, O.; Mollier, K.; Neuveut, C.; Cardinaud, S.; Boulanger, E.; Mignen, B.; Fridman, W.H.; Zanetti, M.; Charneau, P.; Tartour, E.; et al. Immunogenic HLA-B*0702-restricted epitopes derived from human telomerase reverse transcriptase that elicit antitumor cytotoxic T-cell responses. Clin. Cancer Res. 2006, 12, 3158–3167. [Google Scholar] [CrossRef] [Green Version]
- Adotevi, O.; Mollier, K.; Neuveut, C.; Dosset, M.; Ravel, P.; Fridman, W.H.; Tartour, E.; Charneau, P.; Wain-Hobson, S.; Langlade-Demoyen, P. Targeting human telomerase reverse transcriptase with recombinant lentivector is highly effective to stimulate antitumor CD8 T-cell immunity in vivo. Blood 2010, 115, 3025–3032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albershardt, T.C.; Campbell, D.J.; Parsons, A.J.; Slough, M.M.; Ter Meulen, J.; Berglund, P. LV305, a dendritic cell-targeting integration-deficient ZVex(TM)-based lentiviral vector encoding NY-ESO-1, induces potent anti-tumor immune response. Mol. Ther. Oncolytics 2016, 3, 16010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapatte, L.; Colombetti, S.; Cerottini, J.C.; Levy, F. Efficient induction of tumor antigen-specific CD8+ memory T cells by recombinant lentivectors. Cancer Res. 2006, 66, 1155–1160. [Google Scholar] [CrossRef] [Green Version]
- Esslinger, C.; Chapatte, L.; Finke, D.; Miconnet, I.; Guillaume, P.; Levy, F.; MacDonald, H.R. In vivo administration of a lentiviral vaccine targets DCs and induces efficient CD8(+) T cell responses. J. Clin. Investig. 2003, 111, 1673–1681. [Google Scholar] [CrossRef] [Green Version]
- Garcia Casado, J.; Janda, J.; Wei, J.; Chapatte, L.; Colombetti, S.; Alves, P.; Ritter, G.; Ayyoub, M.; Valmori, D.; Chen, W.; et al. Lentivector immunization induces tumor antigen-specific B and T cell responses in vivo. Eur. J. Immunol. 2008, 38, 1867–1876. [Google Scholar] [CrossRef]
- Hu, B.; Tai, A.; Wang, P. Immunization delivered by lentiviral vectors for cancer and infectious diseases. Immunol. Rev. 2011, 239, 45–61. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Majumder, N.; Lin, H.; Watkins, S.; Falo, L.D., Jr.; You, Z. Induction of therapeutic antitumor immunity by in vivo administration of a lentiviral vaccine. Hum. Gene Ther. 2005, 16, 1255–1266. [Google Scholar] [CrossRef]
- Kimura, T.; Koya, R.C.; Anselmi, L.; Sternini, C.; Wang, H.J.; Comin-Anduix, B.; Prins, R.M.; Faure-Kumar, E.; Rozengurt, N.; Cui, Y.; et al. Lentiviral vectors with CMV or MHCII promoters administered in vivo: Immune reactivity versus persistence of expression. Mol. Ther. 2007, 15, 1390–1399. [Google Scholar] [CrossRef]
- Liu, Y.; Peng, Y.; Mi, M.; Guevara-Patino, J.; Munn, D.H.; Fu, N.; He, Y. Lentivector immunization stimulates potent CD8 T cell responses against melanoma self-antigen tyrosinase-related protein 1 and generates antitumor immunity in mice. J. Immunol. 2009, 182, 5960–5969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morante, V.; Borghi, M.; Farina, I.; Michelini, Z.; Grasso, F.; Gallinaro, A.; Cecchetti, S.; Di Virgilio, A.; Canitano, A.; Pirillo, M.F.; et al. Integrase-Defective Lentiviral Vector Is an Efficient Vaccine Platform for Cancer Immunotherapy. Viruses 2021, 13, 355. [Google Scholar] [CrossRef] [PubMed]
- Palmowski, M.J.; Lopes, L.; Ikeda, Y.; Salio, M.; Cerundolo, V.; Collins, M.K. Intravenous injection of a lentiviral vector encoding NY-ESO-1 induces an effective CTL response. J. Immunol. 2004, 172, 1582–1587. [Google Scholar] [CrossRef]
- Pollack, S.M.; Lu, H.; Gnjatic, S.; Somaiah, N.; O’Malley, R.B.; Jones, R.L.; Hsu, F.J.; Ter Meulen, J. First-in-Human Treatment With a Dendritic Cell-targeting Lentiviral Vector-expressing NY-ESO-1, LV305, Induces Deep, Durable Response in Refractory Metastatic Synovial Sarcoma Patient. J. Immunother. 2017, 40, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Joo, K.I.; Lim, M.; Wang, P. Dendritic cell-directed vaccination with a lentivector encoding PSCA for prostate cancer in mice. PLoS ONE 2012, 7, e48866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karwacz, K.; Mukherjee, S.; Apolonia, L.; Blundell, M.P.; Bouma, G.; Escors, D.; Collins, M.K.; Thrasher, A.J. Nonintegrating lentivector vaccines stimulate prolonged T-cell and antibody responses and are effective in tumor therapy. J. Virol. 2009, 83, 3094–3103. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Guan, J.; Wen, B.; Zhu, N.; Chen, H.; Song, J.; Yang, Y.; Wang, Y.; Tan, W. Induction of broadly neutralising HCV antibodies in mice by integration-deficient lentiviral vector-based pseudotyped particles. PLoS ONE 2013, 8, e62684. [Google Scholar] [CrossRef] [Green Version]
- Grasso, F.; Negri, D.R.; Mochi, S.; Rossi, A.; Cesolini, A.; Giovannelli, A.; Chiantore, M.V.; Leone, P.; Giorgi, C.; Cara, A. Successful therapeutic vaccination with integrase defective lentiviral vector expressing nononcogenic human papillomavirus E7 protein. Int. J. Cancer 2013, 132, 335–344. [Google Scholar] [CrossRef]
- Asefa, B.; Korokhov, N.; Lemiale, F. Heterologous HIV-based lentiviral/adenoviral vectors immunizations result in enhanced HIV-specific immunity. Vaccine 2010, 28, 3617–3624. [Google Scholar] [CrossRef]
- Blasi, M.; Negri, D.; Saunders, K.O.; Baker, E.J.; Stadtler, H.; LaBranche, C.; Mildenberg, B.; Morton, G.; Ciarla, A.; Shen, X.; et al. Immunogenicity, safety, and efficacy of sequential immunizations with an SIV-based IDLV expressing CH505 Envs. NPJ Vaccines 2020, 5, 107. [Google Scholar] [CrossRef]
- Blasi, M.; Wescott, E.C.; Baker, E.J.; Mildenberg, B.; LaBranche, C.; Rountree, W.; Haynes, B.F.; Saunders, K.O.; Moody, M.A.; Negri, D.; et al. Therapeutic vaccination with IDLV-SIV-Gag results in durable viremia control in chronically SHIV-infected macaques. NPJ Vaccines 2020, 5, 36. [Google Scholar] [CrossRef] [PubMed]
- Negri, D.; Blasi, M.; LaBranche, C.; Parks, R.; Balachandran, H.; Lifton, M.; Shen, X.; Denny, T.; Ferrari, G.; Vescio, M.F.; et al. Immunization with an SIV-based IDLV Expressing HIV-1 Env 1086 Clade C Elicits Durable Humoral and Cellular Responses in Rhesus Macaques. Mol. Ther. 2016, 24, 2021–2032. [Google Scholar] [CrossRef] [PubMed]
- Negri, D.R.; Michelini, Z.; Baroncelli, S.; Spada, M.; Vendetti, S.; Buffa, V.; Bona, R.; Leone, P.; Klotman, M.E.; Cara, A. Successful immunization with a single injection of non-integrating lentiviral vector. Mol. Ther. 2007, 15, 1716–1723. [Google Scholar] [CrossRef]
- Iglesias, M.C.; Mollier, K.; Beignon, A.S.; Souque, P.; Adotevi, O.; Lemonnier, F.; Charneau, P. Lentiviral vectors encoding HIV-1 polyepitopes induce broad CTL responses in vivo. Mol. Ther. 2007, 15, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- de Wispelaere, M.; Ricklin, M.; Souque, P.; Frenkiel, M.P.; Paulous, S.; Garcia-Nicolas, O.; Summerfield, A.; Charneau, P.; Despres, P. A Lentiviral Vector Expressing Japanese Encephalitis Virus-like Particles Elicits Broad Neutralizing Antibody Response in Pigs. PLoS Negl. Trop. Dis. 2015, 9, e0004081. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Nicolas, O.; Ricklin, M.E.; Liniger, M.; Vielle, N.J.; Python, S.; Souque, P.; Charneau, P.; Summerfield, A. A Japanese Encephalitis Virus Vaccine Inducing Antibodies Strongly Enhancing In Vitro Infection Is Protective in Pigs. Viruses 2017, 9, 124. [Google Scholar] [CrossRef] [Green Version]
- Ku, M.W.; Anna, F.; Souque, P.; Petres, S.; Prot, M.; Simon-Loriere, E.; Charneau, P.; Bourgine, M. A Single Dose of NILV-Based Vaccine Provides Rapid and Durable Protection against Zika Virus. Mol. Ther. 2020, 28, 1772–1782. [Google Scholar] [CrossRef]
- Majlessi, L.; Charneau, P. An anti-Covid-19 lentiviral vaccine candidate that can be administered by the nasal route. Med. Sci. 2021, 37, 1172–1175. [Google Scholar] [CrossRef]
- Vesin, B.; Authié, P.; Blanc, C.; Fert, I.; Noirat, A.; Le Chevalier, F.; Wei, Y.; Ku, M.; Nemirov, K.; Anna, F.; et al. Full Lung Prophylaxis against SARS-CoV-2 by One Shot or Booster Intranasal Lentiviral Vaccination in Syrian Golden Hamsters. Vaccines 2023, 11, 12. [Google Scholar] [CrossRef]
- Pierson, T.C.; Diamond, M.S. The continued threat of emerging flaviviruses. Nat. Microbiol. 2020, 5, 796–812. [Google Scholar] [CrossRef]
- Lowe, R.; Barcellos, C.; Brasil, P.; Cruz, O.G.; Honorio, N.A.; Kuper, H.; Carvalho, M.S. The Zika Virus Epidemic in Brazil: From Discovery to Future Implications. Int. J. Environ. Res. Public Health 2018, 15, 96. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ling, L.; Zhang, Z.; Marin-Lopez, A. Current Advances in Zika Vaccine Development. Vaccines 2022, 10, 1816. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzman, M.G.; Harris, E. Dengue. Lancet 2015, 385, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Ngono, A.E.; Shresta, S. Immune Response to Dengue and Zika. Annu. Rev. Immunol. 2018, 36, 279–308. [Google Scholar] [CrossRef] [Green Version]
- Mongkolsapaya, J.; Dejnirattisai, W.; Xu, X.N.; Vasanawathana, S.; Tangthawornchaikul, N.; Chairunsri, A.; Sawasdivorn, S.; Duangchinda, T.; Dong, T.; Rowland-Jones, S.; et al. Original antigenic sin and apoptosis in the pathogenesis of dengue hemorrhagic fever. Nat. Med. 2003, 9, 921–927. [Google Scholar] [CrossRef]
- Weiskopf, D.; Angelo, M.A.; de Azeredo, E.L.; Sidney, J.; Greenbaum, J.A.; Fernando, A.N.; Broadwater, A.; Kolla, R.V.; De Silva, A.D.; de Silva, A.M.; et al. Comprehensive analysis of dengue virus-specific responses supports an HLA-linked protective role for CD8+ T cells. Proc. Natl. Acad. Sci. USA 2013, 110, E2046–E2053. [Google Scholar] [CrossRef] [Green Version]
- Zellweger, R.M.; Eddy, W.E.; Tang, W.W.; Miller, R.; Shresta, S. CD8+ T cells prevent antigen-induced antibody-dependent enhancement of dengue disease in mice. J. Immunol. 2014, 193, 4117–4124. [Google Scholar] [CrossRef] [Green Version]
- Zellweger, R.M.; Tang, W.W.; Eddy, W.E.; King, K.; Sanchez, M.C.; Shresta, S. CD8+ T Cells Can Mediate Short-Term Protection against Heterotypic Dengue Virus Reinfection in Mice. J. Virol. 2015, 89, 6494–6505. [Google Scholar] [CrossRef] [Green Version]
- Sabchareon, A.; Wallace, D.; Sirivichayakul, C.; Limkittikul, K.; Chanthavanich, P.; Suvannadabba, S.; Jiwariyavej, V.; Dulyachai, W.; Pengsaa, K.; Wartel, T.A.; et al. Protective efficacy of the recombinant, live-attenuated, CYD tetravalent dengue vaccine in Thai schoolchildren: A randomised, controlled phase 2b trial. Lancet 2012, 380, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Gianchecchi, E.; Manenti, A.; Kistner, O.; Trombetta, C.; Manini, I.; Montomoli, E. How to assess the effectiveness of nasal influenza vaccines? Role and measurement of sIgA in mucosal secretions. Influenza Other Respir. Viruses 2019, 13, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Kawai, A.; Yamamoto, Y.; Nogimori, T.; Takeshita, K.; Yamamoto, T.; Yoshioka, Y. The Potential of Neuraminidase as an Antigen for Nasal Vaccines to Increase Cross-Protection against Influenza Viruses. J. Virol. 2021, 95, e0118021. [Google Scholar] [CrossRef] [PubMed]
- Sterlin, D.; Mathian, A.; Miyara, M.; Mohr, A.; Anna, F.; Claer, L.; Quentric, P.; Fadlallah, J.; Devilliers, H.; Ghillani, P.; et al. IgA dominates the early neutralizing antibody response to SARS-CoV-2. Sci. Transl. Med. 2021, 13, eabd2223. [Google Scholar] [CrossRef]
- van Doremalen, N.; Purushotham, J.N.; Schulz, J.E.; Holbrook, M.G.; Bushmaker, T.; Carmody, A.; Port, J.R.; Yinda, C.K.; Okumura, A.; Saturday, G.; et al. Intranasal ChAdOx1 nCoV-19/AZD1222 vaccination reduces viral shedding after SARS-CoV-2 D614G challenge in preclinical models. Sci. Transl. Med. 2021, 13, eabh0755. [Google Scholar] [CrossRef]
- Chen, B.M.; Cheng, T.L.; Roffler, S.R. Polyethylene Glycol Immunogenicity: Theoretical, Clinical, and Practical Aspects of Anti-Polyethylene Glycol Antibodies. ACS Nano 2021, 15, 14022–14048. [Google Scholar] [CrossRef]
- Aghagoli, G.; Gallo Marin, B.; Katchur, N.J.; Chaves-Sell, F.; Asaad, W.F.; Murphy, S.A. Neurological Involvement in COVID-19 and Potential Mechanisms: A Review. Neurocrit. Care 2020, 34, 1062–1071. [Google Scholar] [CrossRef]
- Ali Awan, H.; Najmuddin Diwan, M.; Aamir, A.; Ali, M.; Di Giannantonio, M.; Ullah, I.; Shoib, S.; De Berardis, D. SARS-CoV-2 and the Brain: What Do We Know about the Causality of ‘Cognitive COVID? J. Clin. Med. 2021, 10, 3441. [Google Scholar] [CrossRef]
- Bourgonje, A.R.; Abdulle, A.E.; Timens, W.; Hillebrands, J.L.; Navis, G.J.; Gordijn, S.J.; Bolling, M.C.; Dijkstra, G.; Voors, A.A.; Osterhaus, A.D.; et al. Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). J. Pathol. 2020, 251, 228–248. [Google Scholar] [CrossRef]
- Global Tuberculosis Report 2022. 2022. Available online: https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2022 (accessed on 15 February 2023).
- Andersen, P.; Scriba, T.J. Moving tuberculosis vaccines from theory to practice. Nat. Rev. Immunol. 2019, 19, 550–562. [Google Scholar] [CrossRef]
- Lewinsohn, D.A.; Lewinsohn, D.M.; Scriba, T.J. Polyfunctional CD4(+) T Cells as Targets for Tuberculosis Vaccination. Front. Immunol. 2017, 8, 1262. [Google Scholar] [CrossRef] [Green Version]
- Cresswell, P.; Roche, P.A. Invariant chain-MHC class II complexes: Always odd and never invariant. Immunol. Cell Biol. 2014, 92, 471–472. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.; Surolia, A. Collectins: Sentinels ofinnate immunity. BioEssays 2007, 29, 452–464. [Google Scholar] [CrossRef]
- Presanis, J.S.; Kojima, M.; Sim, R.B. Biochemistry and genetics of mannan-binding lectin (MBL). Biochem. Soc. Trans. 2003, 31, 748–752. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Termini, J.M.; Raffa, F.N.; Williams, C.A.; Kornbluth, R.S.; Stone, G.W. Vaccination with a fusion protein that introduces HIV-1 gag antigen into a multitrimer CD40L construct results in enhanced CD8+ T cell responses and protection from viral challenge by vaccinia-gag. J. Virol. 2014, 88, 1492–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Termini, J.M.; Rivas, Y.; Otero, M.; Raffa, F.N.; Bhat, V.; Farooq, A.; Stone, G.W. A multi-trimeric fusion of CD40L and gp100 tumor antigen activates dendritic cells and enhances survival in a B16-F10 melanoma DNA vaccine model. Vaccine 2015, 33, 4798–4806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laman, J.D.; Claassen, E.; Noelle, R.J. Functions of CD40 and Its Ligand, gp39 (CD40L). Crit. Rev. Immunol. 2017, 37, 371–420. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathology/Virus | Main Results | Reference |
|---|---|---|
| West Nile Virus | The single administration of a nonintegrative LV encoding a secreted form of the envelope protein of a virulent strain of West Nile virus induces a robust antibody response, as well as full and long-lasting sterilizing protection from a challenge with a lethal dose of West Nile virus in a murine model. | [29,49] |
| AIDS | Induction of broad CD8+ cytotoxic T-cell responses with a LV encoding HIV-1 polyepitopes in humanized mice. LV-based prime-boost vaccination demonstrating protective immunity against simian immunodeficiency virus SIVmac251 challenge in macaques. | [46,90] |
| Japanese Encephalitis Virus | An LV encoding the pre-membrane and envelope protein (prME) of Japanese encephalitis elicits a broad neutralizing antibody response and protection in pigs. | [91,92] |
| Malaria | Long-term sterile protection against malaria in mice immunized with a nonintegrative LV encoding Plasmodium yoelii circumsporozoite protein (CSP) and challenged with sporozoites. | [16] |
| Zika | A single dose of nonintegrative LV-based vaccine candidate provides rapid and durable protection against Zika virus. | [93] |
| COVID-19 | A nonintegrative LV encoding the Spike envelope protein of SARS-CoV-2, used in an i.m. prime dose, followed by an i.n. boost, induces sterilizing protection of the respiratory system and the central nervous system in hACE2 humanized transgenic mice and golden hamsters. The efficacy of this LV-based COVID-19 vaccine candidate, used as a late i.n. booster in initially mRNA-vaccinated individuals, is superior to that of a third mRNA dose, which correlates with its ability to induce mucosal IgA and resident memory B and T cells in the respiratory airways. | [33,34,35,94,95] |
| Tuberculosis | Development of a new-generation LV-based tuberculosis vaccine/booster candidate. Two new generations of LVs have been generated. Their strong immunogenicity for CD4+ T cells correlates with a high protective potential when administered either alone or as a booster in BCG-primed mice. | [8,9] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nemirov, K.; Bourgine, M.; Anna, F.; Wei, Y.; Charneau, P.; Majlessi, L. Lentiviral Vectors as a Vaccine Platform against Infectious Diseases. Pharmaceutics 2023, 15, 846. https://doi.org/10.3390/pharmaceutics15030846
Nemirov K, Bourgine M, Anna F, Wei Y, Charneau P, Majlessi L. Lentiviral Vectors as a Vaccine Platform against Infectious Diseases. Pharmaceutics. 2023; 15(3):846. https://doi.org/10.3390/pharmaceutics15030846
Chicago/Turabian StyleNemirov, Kirill, Maryline Bourgine, François Anna, Yu Wei, Pierre Charneau, and Laleh Majlessi. 2023. "Lentiviral Vectors as a Vaccine Platform against Infectious Diseases" Pharmaceutics 15, no. 3: 846. https://doi.org/10.3390/pharmaceutics15030846