
Virtual Screening, Structural Analysis, and Formation Thermodynamics of Carbamazepine Cocrystals
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Compounds and Solvents
2.2. Cocrystal Synthesis
2.3. Solution Crystallization
2.4. Single Crystal and Powder X-Ray Diffraction
2.5. Thermal Analysis
2.6. Solubility Experiments
2.7. High-Performance Liquid Chromatography (HPLC)
2.8. Virtual Cocrystal Screening Methods
2.8.1. Hydrogen Bond Propensity (HBP)
2.8.2. Co-Crystal Graph Network (CCGNet)
2.8.3. Molecular Electrostatic Potential Surface (MEP)
2.9. Computational Methods
2.9.1. Periodic DFT Calculations
2.9.2. Noncovalent Interaction Energies and Lattice Energy Calculation
3. Results and Discussion
3.1. Experimental Screening via the Mechanochemical Method
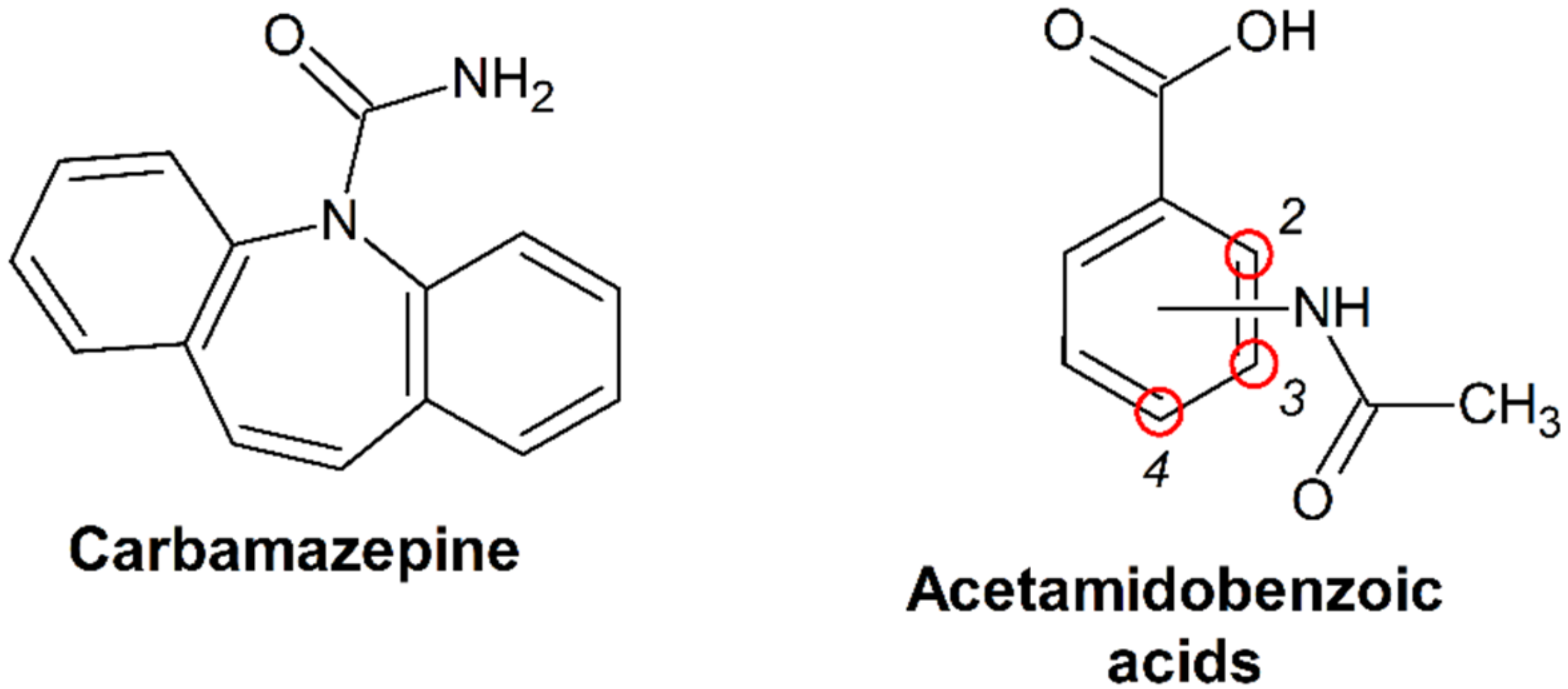
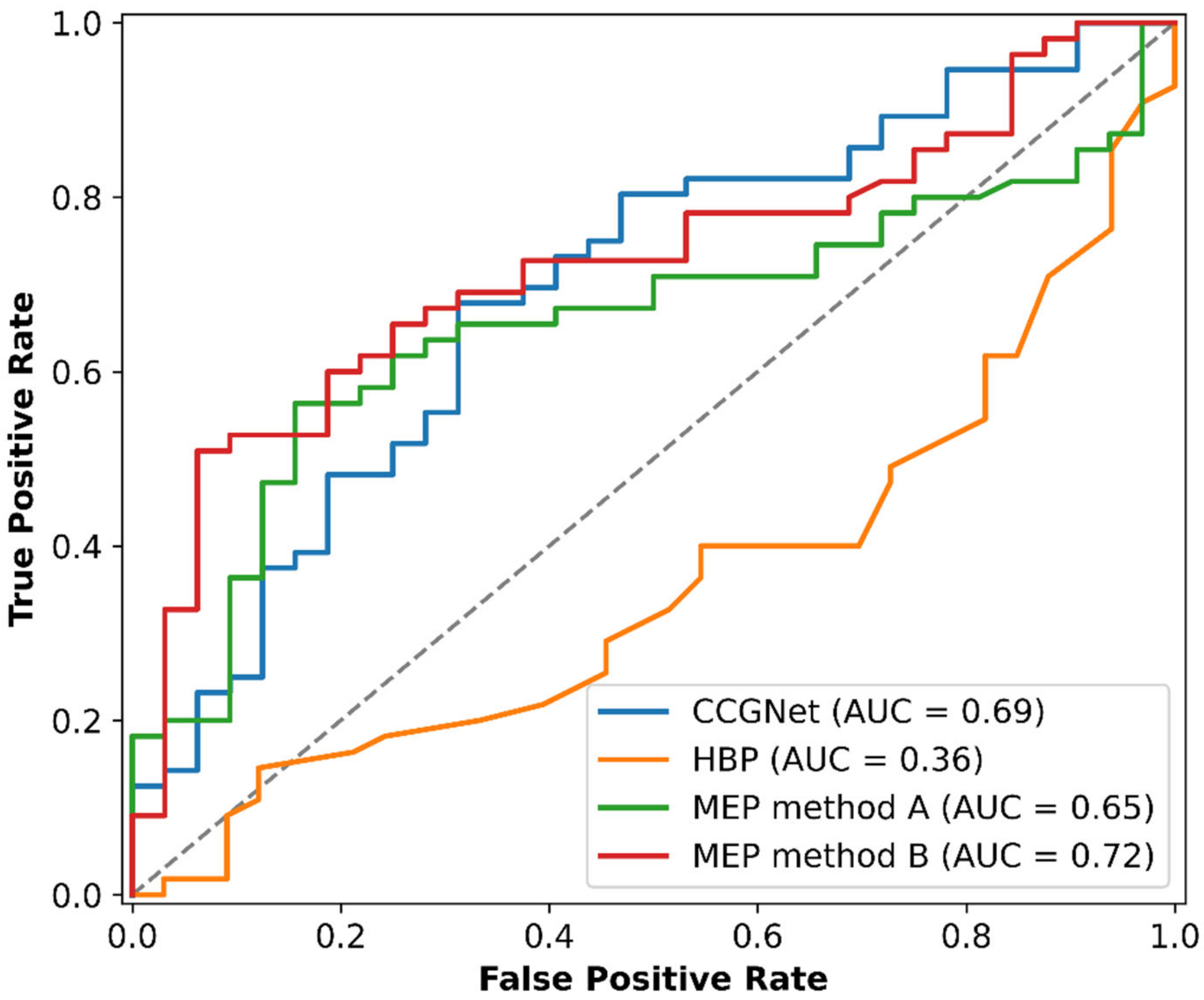
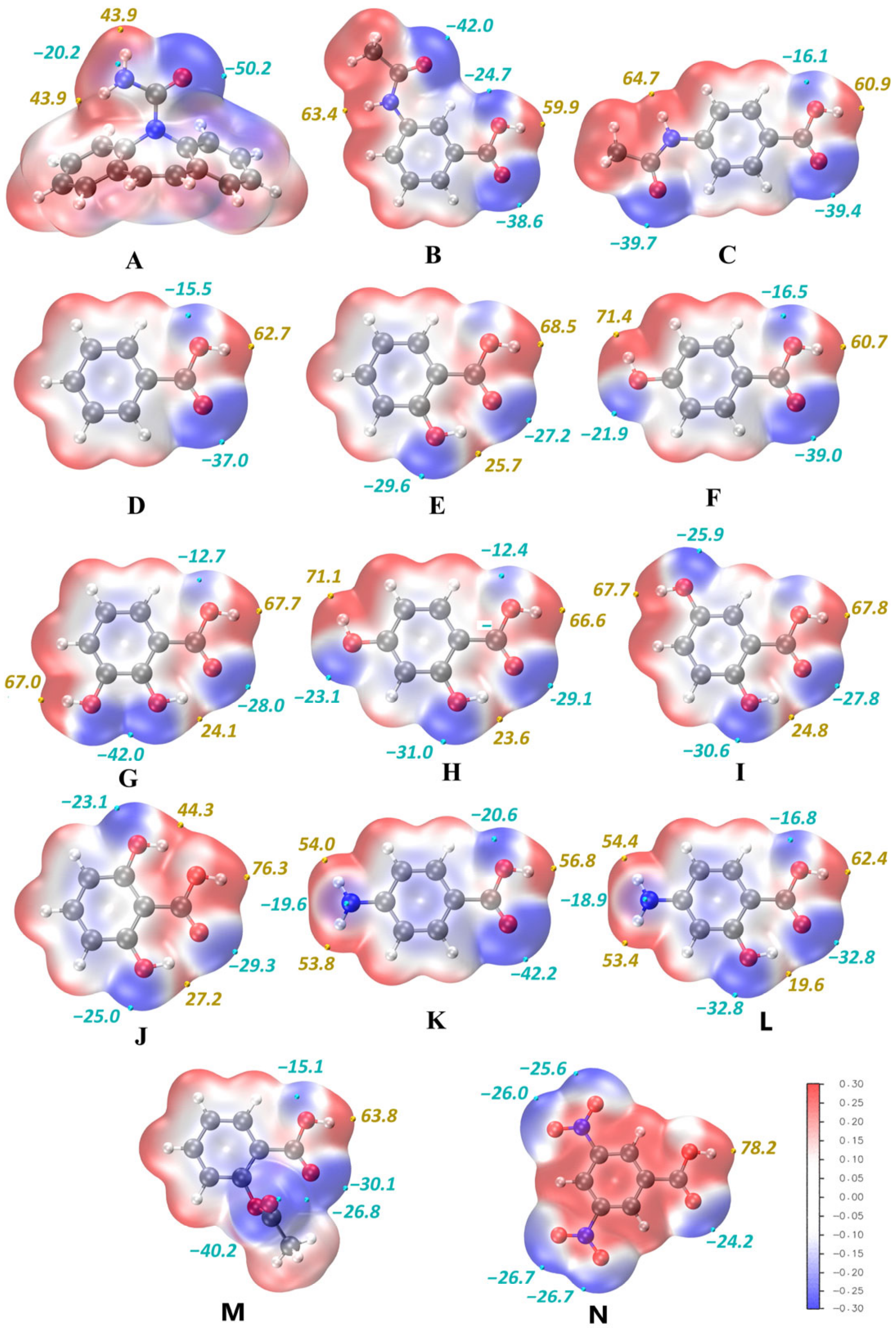
3.2. Virtual Screening of CBZ Cocrystals
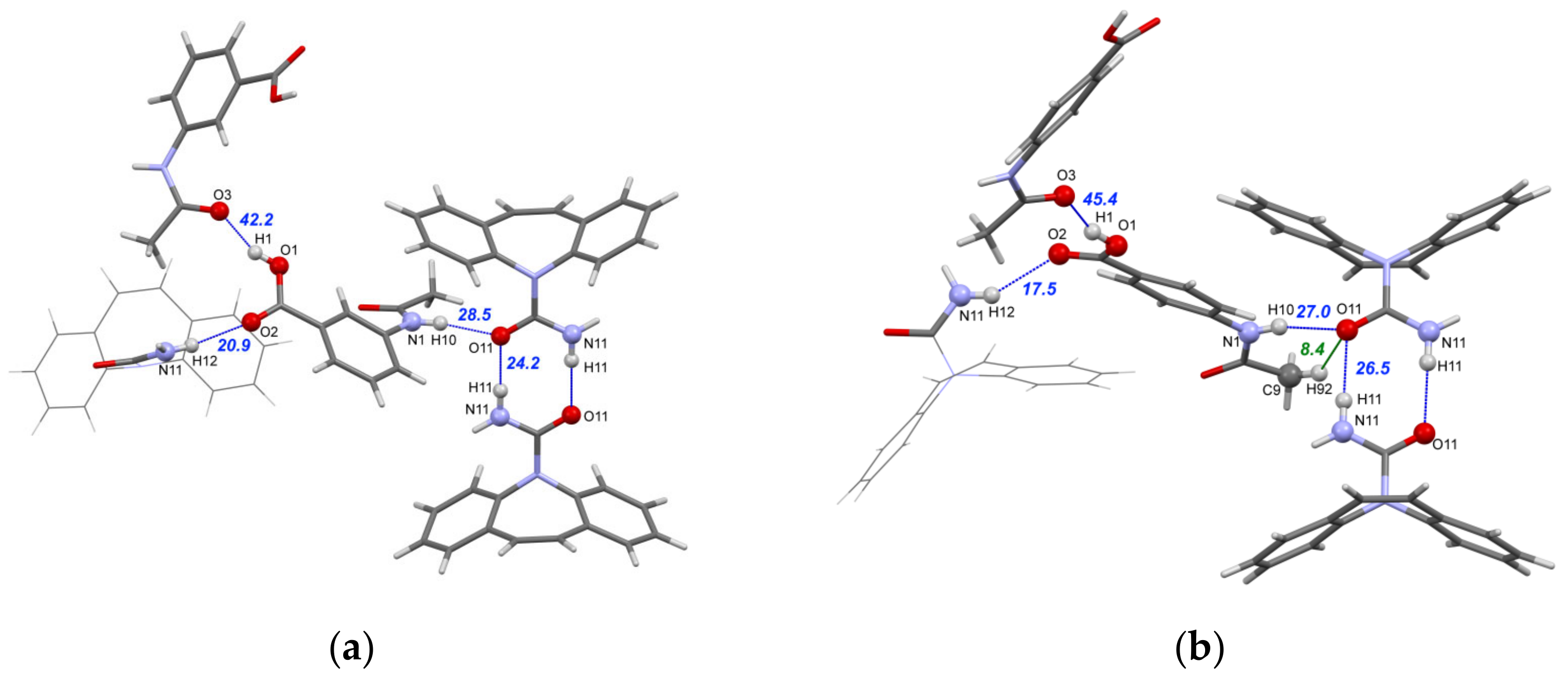
3.3. Crystal Structure Analysis and Intermolecular Interaction Energies in [CBZ + 3AcAmBA] (1:1) and [CBZ + 4AcAmBA] (1:1)
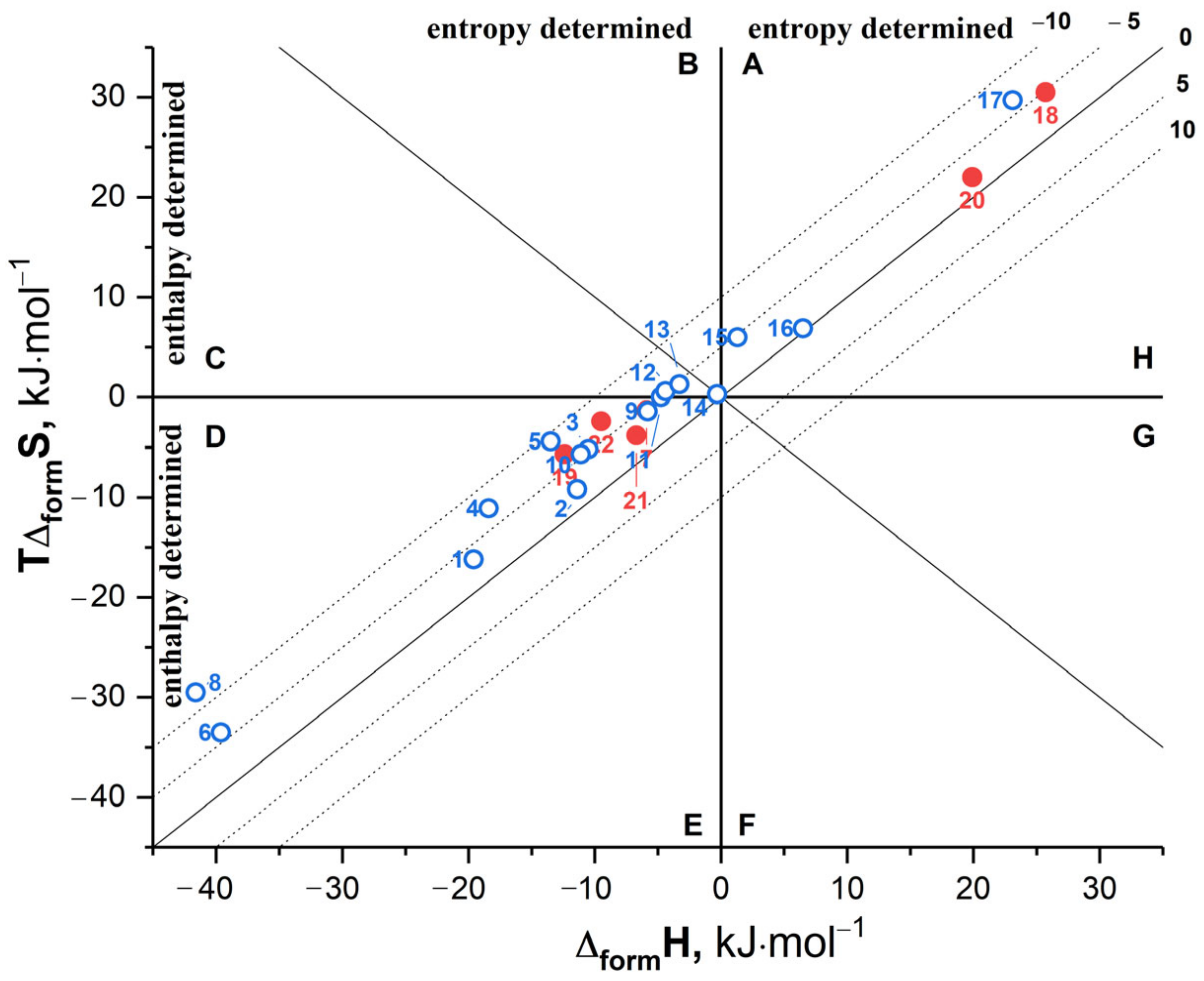
3.4. Formation Thermodynamics of the Carbamazepine Cocrystals
3.5. Stability and Solubility of Carbamazepine Cocrystals in Aqueous Media
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sun, L.; Wang, Y.; Yang, F.; Zhang, X.; Hu, W. Cocrystal Engineering: A Collaborative Strategy toward Functional Materials. Adv. Mater. 2019, 31, 1902328. [Google Scholar] [CrossRef]
- Sun, L.; Zhu, W.; Zhang, X.; Li, L.; Dong, H.; Hu, W. Creating Organic Functional Materials beyond Chemical Bond Synthesis by Organic Cocrystal Engineering. J. Am. Chem. Soc. 2021, 143, 19243–19256. [Google Scholar] [CrossRef]
- Kavanagh, O.N.; Croker, D.M.; Walker, G.M.; Zaworotko, M.J. Pharmaceutical cocrystals: From serendipity to design to application. Drug Discov. Today 2019, 24, 796–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duggirala, N.K.; Perry, M.L.; Almarsson, Ö.; Zaworotko, M.J. Pharmaceutical cocrystals: Along the path to improved medicines. Chem. Commun. 2016, 52, 640–655. [Google Scholar] [CrossRef]
- Bolla, G.; Nangia, A. Pharmaceutical cocrystals: Walking the talk. Chem. Commun. 2016, 52, 8342–8360. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.J.; Steed, J.W. Pharmaceutical cocrystals, salts and multicomponent systems; intermolecular interactions and property based design. Adv. Drug Del. Rev. 2017, 117, 3–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuminek, G.; Cao, F.; Bahia de Oliveira da Rocha, A.; Gonçalves Cardoso, S.; Rodríguez-Hornedo, N. Cocrystals to facilitate delivery of poorly soluble compounds beyond-rule-of-5. Adv. Drug Del. Rev. 2016, 101, 143–166. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.N.; Chen, Y.C.S.; Xuan, B.; Sun, C.C.; Chow, S.F. Cocrystal engineering of pharmaceutical solids: Therapeutic potential and challenges. CrystEngComm 2021, 23, 7005–7038. [Google Scholar] [CrossRef]
- Karimi-Jafari, M.; Padrela, L.; Walker, G.M.; Croker, D.M. Creating Cocrystals: A Review of Pharmaceutical Cocrystal Preparation Routes and Applications. Cryst. Growth Des. 2018, 18, 6370–6387. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Nanda, A. In-silico methods of cocrystal screening: A review on tools for rational design of pharmaceutical cocrystals. J. Drug Deliv. Sci. Technol. 2021, 63, 102527. [Google Scholar] [CrossRef]
- Desiraju, G.R. Supramolecular Synthons in Crystal Engineering—A New Organic Synthesis. Angew. Chem. Int. Ed. Engl. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal Engineering: A Holistic View. Angew. Chem. Int. Ed. 2007, 46, 8342–8356. [Google Scholar] [CrossRef] [PubMed]
- Mapp, L.K.; Coles, S.J.; Aitipamula, S. Design of Cocrystals for Molecules with Limited Hydrogen Bonding Functionalities: Propyphenazone as a Model System. Cryst. Growth Des. 2017, 17, 163–174. [Google Scholar] [CrossRef] [Green Version]
- Corpinot, M.K.; Stratford, S.A.; Arhangelskis, M.; Anka-Lufford, J.; Halasz, I.; Judaš, N.; Jones, W.; Bučar, D.-K. On the predictability of supramolecular interactions in molecular cocrystals–the view from the bench. CrystEngComm 2016, 18, 5434–5439. [Google Scholar] [CrossRef] [Green Version]
- Cappuccino, C.; Cusack, D.; Flanagan, J.; Harrison, C.; Holohan, C.; Lestari, M.; Walsh, G.; Lusi, M. How Many Cocrystals Are We Missing? Assessing Two Crystal Engineering Approaches to Pharmaceutical Cocrystal Screening. Cryst. Growth Des. 2022, 22, 1390–1397. [Google Scholar] [CrossRef]
- Abramov, Y.A.; Sun, G.; Zeng, Q. Emerging Landscape of Computational Modeling in Pharmaceutical Development. J. Chem. Inf. Model. 2022, 62, 1160–1171. [Google Scholar] [CrossRef]
- Fábián, L. Cambridge Structural Database Analysis of Molecular Complementarity in Cocrystals. Cryst. Growth Des. 2009, 9, 1436–1443. [Google Scholar] [CrossRef]
- Delori, A.; Galek, P.T.A.; Pidcock, E.; Patni, M.; Jones, W. Knowledge-based hydrogen bond prediction and the synthesis of salts and cocrystals of the anti-malarial drug pyrimethamine with various drug and GRAS molecules. CrystEngComm 2013, 15, 2916–2928. [Google Scholar] [CrossRef]
- Perlovich, G.L. Two-component molecular crystals: Evaluation of the formation thermodynamics based on melting points and sublimation data. CrystEngComm 2017, 19, 2870–2883. [Google Scholar] [CrossRef] [Green Version]
- Perlovich, G.L. Formation Thermodynamics of Two-Component Molecular Crystals: Polymorphism, Stoichiometry, and Impact of Enantiomers. Cryst. Growth Des. 2020, 20, 5526–5537. [Google Scholar] [CrossRef]
- Mohammad, M.A.; Alhalaweh, A.; Velaga, S.P. Hansen solubility parameter as a tool to predict cocrystal formation. Int. J. Pharm. 2011, 407, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, D.; Hunter, C.A.; Prohens, R.; Scuderi, S.; McCabe, J.F. Virtual cocrystal screening. Chem. Sci. 2011, 2, 883–890. [Google Scholar] [CrossRef]
- Grecu, T.; Hunter, C.A.; Gardiner, E.J.; McCabe, J.F. Validation of a Computational Cocrystal Prediction Tool: Comparison of Virtual and Experimental Cocrystal Screening Results. Cryst. Growth Des. 2014, 14, 165–171. [Google Scholar] [CrossRef]
- Abramov, Y.A.; Loschen, C.; Klamt, A. Rational Coformer or Solvent Selection for Pharmaceutical Cocrystallization or Desolvation. J. Pharm. Sci. 2012, 101, 3687–3697. [Google Scholar] [CrossRef] [Green Version]
- Chan, H.C.S.; Kendrick, J.; Neumann, M.A.; Leusen, F.J.J. Towards ab initio screening of co-crystal formation through lattice energy calculations and crystal structure prediction of nicotinamide, isonicotinamide, picolinamide and paracetamol multi-component crystals. CrystEngComm 2013, 15, 3799–3807. [Google Scholar] [CrossRef] [Green Version]
- Sun, G.; Jin, Y.; Li, S.; Yang, Z.; Shi, B.; Chang, C.; Abramov, Y.A. Virtual Coformer Screening by Crystal Structure Predictions: Crucial Role of Crystallinity in Pharmaceutical Cocrystallization. J. Phys. Chem. Lett. 2020, 11, 8832–8838. [Google Scholar] [CrossRef]
- Sugden, I.J.; Braun, D.E.; Bowskill, D.H.; Adjiman, C.S.; Pantelides, C.C. Efficient Screening of Coformers for Active Pharmaceutical Ingredient Cocrystallization. Cryst. Growth Des. 2022, 22, 4513–4527. [Google Scholar] [CrossRef]
- Wicker, J.G.P.; Crowley, L.M.; Robshaw, O.; Little, E.J.; Stokes, S.P.; Cooper, R.I.; Lawrence, S.E. Will they co-crystallize? CrystEngComm 2017, 19, 5336–5340. [Google Scholar] [CrossRef] [Green Version]
- Devogelaer, J.-J.; Meekes, H.; Tinnemans, P.; Vlieg, E.; de Gelder, R. Co-crystal Prediction by Artificial Neural Networks**. Angew. Chem. Int. Ed. 2020, 59, 21711–21718. [Google Scholar] [CrossRef]
- Wang, D.; Yang, Z.; Zhu, B.; Mei, X.; Luo, X. Machine-Learning-Guided Cocrystal Prediction Based on Large Data Base. Cryst. Growth Des. 2020, 20, 6610–6621. [Google Scholar] [CrossRef]
- Yuan, J.; Liu, X.; Wang, S.; Chang, C.; Zeng, Q.; Song, Z.; Jin, Y.; Zeng, Q.; Sun, G.; Ruan, S.; et al. Virtual coformer screening by a combined machine learning and physics-based approach. CrystEngComm 2021, 23, 6039–6044. [Google Scholar] [CrossRef]
- Jiang, Y.; Yang, Z.; Guo, J.; Li, H.; Liu, Y.; Guo, Y.; Li, M.; Pu, X. Coupling complementary strategy to flexible graph neural network for quick discovery of coformer in diverse co-crystal materials. Nat. Commun. 2021, 12, 5950. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, S.; Mondal, P.K.; Wu, Y.; Gong, W.; Mirmehrabi, M.; Rohani, S. Virtual Multicomponent Crystal Screening: Hydrogen Bonding Revisited. Cryst. Growth Des. 2021, 21, 5862–5872. [Google Scholar] [CrossRef]
- Salem, A.; Nagy, S.; Pál, S.; Széchenyi, A. Reliability of the Hansen solubility parameters as co-crystal formation prediction tool. Int. J. Pharm. 2019, 558, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Roca-Paixão, L.; Correia, N.T.; Affouard, F. Affinity prediction computations and mechanosynthesis of carbamazepine based cocrystals. CrystEngComm 2019, 21, 6991–7001. [Google Scholar] [CrossRef]
- Habgood, M.; Deij, M.A.; Mazurek, J.; Price, S.L.; ter Horst, J.H. Carbamazepine Co-crystallization with Pyridine Carboxamides: Rationalization by Complementary Phase Diagrams and Crystal Energy Landscapes. Cryst. Growth Des. 2010, 10, 903–912. [Google Scholar] [CrossRef]
- Schartman, R.R. On the thermodynamics of cocrystal formation. Int. J. Pharm. 2009, 365, 77–80. [Google Scholar] [CrossRef]
- Zhang, S.; Rasmuson, Å.C. Thermodynamics and Crystallization of the Theophylline–Glutaric Acid Cocrystal. Cryst. Growth Des. 2013, 13, 1153–1161. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, H.; Rasmuson, Å.C. Thermodynamics and crystallization of a theophylline–salicylic acid cocrystal. CrystEngComm 2015, 17, 4125–4135. [Google Scholar] [CrossRef]
- Évora, A.O.L.; Bernardes, C.E.S.; Piedade, M.F.M.; Conceição, A.C.L.; Minas da Piedade, M.E. Energetics of Glycine Cocrystal or Salt Formation with Two Regioisomers: Fumaric Acid and Maleic Acid. Cryst. Growth Des. 2019, 19, 5054–5064. [Google Scholar] [CrossRef]
- Oliveira, M.A.; Peterson, M.L.; Davey, R.J. Relative Enthalpy of Formation for Co-Crystals of Small Organic Molecules. Cryst. Growth Des. 2011, 11, 449–457. [Google Scholar] [CrossRef]
- Surov, A.O.; Solanko, K.A.; Bond, A.D.; Bauer-Brandl, A.; Perlovich, G.L. Cocrystals of the antiandrogenic drug bicalutamide: Screening, crystal structures, formation thermodynamics and lattice energies. CrystEngComm 2016, 18, 4818–4829. [Google Scholar] [CrossRef]
- Zhang, S.-W.; Brunskill, A.P.J.; Schwartz, E.; Sun, S. Celecoxib–Nicotinamide Cocrystal Revisited: Can Entropy Control Cocrystal Formation? Cryst. Growth Des. 2017, 17, 2836–2843. [Google Scholar] [CrossRef]
- Ahuja, D.; Svärd, M.; Rasmuson, Å.C. Investigation of solid–liquid phase diagrams of the sulfamethazine–salicylic acid co-crystal. CrystEngComm 2019, 21, 2863–2874. [Google Scholar] [CrossRef]
- Surov, A.O.; Voronin, A.P.; Drozd, K.V.; Volkova, T.V.; Vasilev, N.; Batov, D.; Churakov, A.V.; Perlovich, G.L. Extending the Range of Nitrofurantoin Solid Forms: Effect of Molecular and Crystal Structure on Formation Thermodynamics and Physicochemical Properties. Cryst. Growth Des. 2022, 22, 2569–2586. [Google Scholar] [CrossRef]
- Manin, A.N.; Boycov, D.E.; Simonova, O.R.; Volkova, T.V.; Churakov, A.V.; Perlovich, G.L. Formation Thermodynamics of Carbamazepine with Benzamide, Para-Hydroxybenzamide and Isonicotinamide Cocrystals: Experimental and Theoretical Study. Pharmaceutics 2022, 14, 1881. [Google Scholar] [CrossRef]
- Gavezzotti, A.; Colombo, V.; Lo Presti, L. Facts and Factors in the Formation and Stability of Binary Crystals. Cryst. Growth Des. 2016, 16, 6095–6104. [Google Scholar] [CrossRef]
- Taylor, C.R.; Day, G.M. Evaluating the Energetic Driving Force for Cocrystal Formation. Cryst. Growth Des. 2018, 18, 892–904. [Google Scholar] [CrossRef] [Green Version]
- Desiraju, G.R. Crystal Engineering: From Molecule to Crystal. J. Am. Chem. Soc. 2013, 135, 9952–9967. [Google Scholar] [CrossRef]
- Chrzanowski, F.A.; Ahmad, K. The preparation and evaluation of salt forms of linogliride with reduced solubilities as candidates for extended release. Drug Dev. Ind. Pharm. 2017, 43, 421–431. [Google Scholar] [CrossRef]
- Dull, G.M.; Carr, A.; Sharp, E. Nicotine salts, Co-Crystals, and Salt Co-Crystal Complexes. U.S. Patent 2015183801 A1, 3 December 2015. [Google Scholar]
- Mattei, A. Cocrystals of Upadacitinib. U.S. Patent 2022217257 A1, 13 October 2022. [Google Scholar]
- Nortje, C.; van Rensburg, P.J.; Cooke, C.; Erasmus, E. The simultaneous detection and quantification of p-aminobenzoic acid and its phase 2 biotransformation metabolites in human urine using LC–MS/MS. Bioanalysis 2015, 7, 1211–1224. [Google Scholar] [CrossRef] [PubMed]
- Szoke, A.; Hayton, W.L.; Schultz, I.R. Quantification of benzocaine and its metabolites in channel catfish tissues and fluids by HPLC. J. Pharm. Biomed. Anal. 1997, 16, 69–75. [Google Scholar] [CrossRef]
- Sliva, J.; Pantzartzi, C.N.; Votava, M. Inosine Pranobex: A Key Player in the Game Against a Wide Range of Viral Infections and Non-Infectious Diseases. Adv. Ther. 2019, 36, 1878–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beran, J.; Špajdel, M.; Slíva, J. Inosine Pranobex Deserves Attention as a Potential Immunomodulator to Achieve Early Alteration of the COVID-19 Disease Course. Viruses 2021, 13, 2246. [Google Scholar] [CrossRef]
- Stahl, P.H.; Wermuth, C.G.; Pure, I.U.o.; Chemistry, A. (Eds.) Handbook of Pharmaceutical Salts Properties, Selection, and Use; Wiley: Hoboken, NJ, USA, 2008. [Google Scholar]
- Sheldrick, G. SADABS, Program for Scaling and Correction of Area Detector Data; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Spek, A. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. Sect. C 2015, 71, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galek, P.T.A.; Allen, F.H.; Fábián, L.; Feeder, N. Knowledge-based H-bond prediction to aid experimental polymorph screening. CrystEngComm 2009, 11, 2634–2639. [Google Scholar] [CrossRef]
- Taylor, R.; Wood, P.A. A Million Crystal Structures: The Whole Is Greater than the Sum of Its Parts. Chem. Rev. 2019, 119, 9427–9477. [Google Scholar] [CrossRef]
- Sarkar, N.; Sinha, A.S.; Aakeröy, C.B. Systematic investigation of hydrogen-bond propensities for informing co-crystal design and assembly. CrystEngComm 2019, 21, 6048–6055. [Google Scholar] [CrossRef]
- Sandhu, B.; McLean, A.; Sinha, A.S.; Desper, J.; Sarjeant, A.A.; Vyas, S.; Reutzel-Edens, S.M.; Aakeröy, C.B. Evaluating Competing Intermolecular Interactions through Molecular Electrostatic Potentials and Hydrogen-Bond Propensities. Cryst. Growth Des. 2018, 18, 466–478. [Google Scholar] [CrossRef]
- Murray, J.S.; Politzer, P. Hydrogen Bonding: A Coulombic σ-Hole Interaction. J. Indian Inst. Sci. 2020, 100, 21–30. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Grecu, T.; Adams, H.; Hunter, C.A.; McCabe, J.F.; Portell, A.; Prohens, R. Virtual Screening Identifies New Cocrystals of Nalidixic Acid. Cryst. Growth Des. 2014, 14, 1749–1755. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S. Quantum-mechanical condensed matter simulations with CRYSTAL. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Vener, M.V.; Levina, E.O.; Koloskov, O.A.; Rykounov, A.A.; Voronin, A.P.; Tsirelson, V.G. Evaluation of the lattice energy of the two-component molecular crystals using solid-state density functional theory. Cryst. Growth Des. 2014, 14, 4997–5003. [Google Scholar] [CrossRef]
- Manin, A.N.; Voronin, A.P.; Drozd, K.V.; Perlovich, G.L. Thermodynamic properties of Nalidixic and Oxolinic acids: Experimental and computational study. Thermochim. Acta 2019, 682, 178411. [Google Scholar] [CrossRef]
- Surov, A.O.; Manin, A.N.; Voronin, A.P.; Boycov, D.E.; Magdysyuk, O.V.; Perlovich, G.L. New Pharmaceutical Cocrystal Forms of Flurbiprofen: Structural, Physicochemical, and Thermodynamic Characterization. Cryst. Growth Des. 2019, 19, 5751–5761. [Google Scholar] [CrossRef]
- Gatti, C.; Saunders, V.R.; Roetti, C. Crystal field effects on the topological properties of the electron density in molecular crystals: The case of urea. J. Chem. Phys. 1994, 101, 10686–10696. [Google Scholar] [CrossRef]
- Mata, I.; Alkorta, I.; Espinosa, E.; Molins, E. Relationships between interaction energy, intermolecular distance and electron density properties in hydrogen bonded complexes under external electric fields. Chem. Phys. Lett. 2011, 507, 185–189. [Google Scholar] [CrossRef]
- Vener, M.V.; Egorova, A.N.; Churakov, A.V.; Tsirelson, V.G. Intermolecular hydrogen bond energies in crystals evaluated using electron density properties: DFT computations with periodic boundary conditions. J. Comput. Chem. 2012, 33, 2303–2309. [Google Scholar] [CrossRef] [PubMed]
- Shishkina, A.V.; Zhurov, V.V.; Stash, A.I.; Vener, M.V.; Pinkerton, A.A.; Tsirelson, V.G. Noncovalent interactions in crystalline picolinic acid N-oxide: Insights from experimental and theoretical charge density analysis. Cryst. Growth Des. 2013, 13, 816–828. [Google Scholar] [CrossRef]
- Vener, M.V.; Shishkina, A.V.; Rykounov, A.A.; Tsirelson, V.G. Cl⋯ Cl interactions in molecular crystals: Insights from the theoretical charge density analysis. J. Phys. Chem. A 2013, 117, 8459–8467. [Google Scholar] [CrossRef]
- Dash, S.G.; Thakur, T.S. Computational Screening of Multicomponent Solid Forms of 2-Aryl-Propionate Class of NSAID, Zaltoprofen, and Their Experimental Validation. Cryst. Growth Des. 2021, 21, 449–461. [Google Scholar] [CrossRef]
- Khalaji, M.; Potrzebowski, M.J.; Dudek, M.K. Virtual Cocrystal Screening Methods as Tools to Understand the Formation of Pharmaceutical Cocrystals—A Case Study of Linezolid, a Wide-Range Antibacterial Drug. Cryst. Growth Des. 2021, 21, 2301–2314. [Google Scholar] [CrossRef]
- Fang, L.; Ma, Y.; Xiao, Y.; Li, Z.; Gao, Z.; Wu, S.; Rohani, S.; Gong, J. Theoretical and Structural Understanding of the Different Factors Influencing the Formation of Multicomponent Crystals of 2,4-Dichlorophenoxyacetic Acid with N-heterocyclic Compounds. Cryst. Growth Des. 2022, 22, 1707–1719. [Google Scholar] [CrossRef]
- Barbas, R.; Portell, A.; Hunter, C.A.; Prohens, R.; Frontera, A. Combined computational/experimental investigation of new cocrystals of the drug bosentan. CrystEngComm 2022, 24, 5105–5111. [Google Scholar] [CrossRef]
- Barbas, R.; Font-Bardia, M.; Paradkar, A.; Hunter, C.A.; Prohens, R. Combined Virtual/Experimental Multicomponent Solid Forms Screening of Sildenafil: New Salts, Cocrystals, and Hybrid Salt–Cocrystals. Cryst. Growth Des. 2018, 18, 7618–7627. [Google Scholar] [CrossRef]
- Sarkar, N.; Aakeröy, C.B. Evaluating hydrogen-bond propensity, hydrogen-bond coordination and hydrogen-bond energy as tools for predicting the outcome of attempted co-crystallisations. Supramol. Chem. 2020, 32, 81–90. [Google Scholar] [CrossRef]
- Sarkar, N.; Gonnella, N.C.; Krawiec, M.; Xin, D.; Aakeröy, C.B. Evaluating the Predictive Abilities of Protocols Based on Hydrogen-Bond Propensity, Molecular Complementarity, and Hydrogen-Bond Energy for Cocrystal Screening. Cryst. Growth Des. 2020, 20, 7320–7327. [Google Scholar] [CrossRef]
- Bryant, M.J.; Maloney, A.G.P.; Sykes, R.A. Predicting mechanical properties of crystalline materials through topological analysis. CrystEngComm 2018, 20, 2698–2704. [Google Scholar] [CrossRef]
- Chisholm, J.A.; Motherwell, S. COMPACK: A program for identifying crystal structure similarity using distances. J. Appl. Crystallogr. 2005, 38, 228–231. [Google Scholar] [CrossRef] [Green Version]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Etter, M.C.; MacDonald, J.C.; Bernstein, J. Graph-set analysis of hydrogen-bond patterns in organic crystals. Acta Crystallogr. Sect. B Struct. Sci. 1990, 46, 256–262. [Google Scholar] [CrossRef]
- Saha, S.; Desiraju, G.R. Acid⋯Amide Supramolecular Synthon in Cocrystals: From Spectroscopic Detection to Property Engineering. J. Am. Chem. Soc. 2018, 140, 6361–6373. [Google Scholar] [CrossRef]
- Gelbrich, T.; Hursthouse, M.B. Systematic investigation of the relationships between 25 crystal structures containing the carbamazepine molecule or a close analogue: A case study of the XPac method. CrystEngComm 2006, 8, 448–460. [Google Scholar] [CrossRef]
- Liu, C.; Wang, C.; Wan, S.; Liu, L.; Sun, C.C.; Qian, F. An Elusive Drug–Drug Cocrystal Prepared Using a Heteroseeding Strategy. Cryst. Growth Des. 2021, 21, 5659–5668. [Google Scholar] [CrossRef]
- Rapeenun, P.; Rarey, J.; Flood, A.E. Shortcut Method for the Prediction of the Cocrystal Solubility Line. Cryst. Growth Des. 2021, 21, 5534–5543. [Google Scholar] [CrossRef]
- Takebayashi, Y.; Sue, K.; Furuya, T.; Yoda, S. Polymorphic Solubility Ratio of Famotidine and Sulfathiazole in Various Solvents. Cryst. Growth Des. 2021, 21, 2868–2875. [Google Scholar] [CrossRef]
- Yoshimura, M.; Miyake, M.; Kawato, T.; Bando, M.; Toda, M.; Kato, Y.; Fukami, T.; Ozeki, T. Impact of the Dissolution Profile of the Cilostazol Cocrystal with Supersaturation on the Oral Bioavailability. Cryst. Growth Des. 2017, 17, 550–557. [Google Scholar] [CrossRef]
- Omori, M.; Watanabe, T.; Uekusa, T.; Oki, J.; Inoue, D.; Sugano, K. Effects of Coformer and Polymer on Particle Surface Solution-Mediated Phase Transformation of Cocrystals in Aqueous Media. Mol. Pharm. 2020, 17, 3825–3836. [Google Scholar] [CrossRef] [PubMed]
- Alvani, A.; Shayanfar, A. Solution Stability of Pharmaceutical Cocrystals. Cryst. Growth Des. 2022, 22, 6323–6337. [Google Scholar] [CrossRef]
- Hunter, C.A.; Prohens, R. Solid form and solubility. CrystEngComm 2017, 19, 23–26. [Google Scholar] [CrossRef] [Green Version]
- Kuminek, G.; Cavanagh, K.L.; Rodríguez-Hornedo, N. Measurement and Mathematical Relationships of Cocrystal Thermodynamic Properties. In Pharmaceutical Crystals: Science and Engineering; Li, T., Mattei, A., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2019. [Google Scholar] [CrossRef]
- Manin, A.N.; Drozd, K.V.; Perlovich, G.L. Influence of ionization and position of substituents on the solubility, solvation and transfer processes: A thermodynamic study of hydroxybenzamide and acetamidobenzoic acid isomers. J. Mol. Liq. 2022, 347, 118320. [Google Scholar] [CrossRef]
- Good, D.J.; Rodríguez-Hornedo, N. Solubility Advantage of Pharmaceutical Cocrystals. Cryst. Growth Des. 2009, 9, 2252–2264. [Google Scholar] [CrossRef]
- Qiu, S.; Li, M. Effects of coformers on phase transformation and release profiles of carbamazepine cocrystals in hydroxypropyl methylcellulose based matrix tablets. Int. J. Pharm. 2015, 479, 118–128. [Google Scholar] [CrossRef]
- Huang, D.; Chan, H.C.S.; Wu, Y.; Li, L.; Zhang, L.; Lv, Y.; Yang, X.; Zhou, Z. Phase solubility investigation and theoretical calculations on drug-drug cocrystals of carbamazepine with Emodin, Paeonol. J. Mol. Liq. 2021, 329, 115604. [Google Scholar] [CrossRef]
- Kaur, R.; Cavanagh, K.L.; Rodríguez-Hornedo, N.; Matzger, A.J. Multidrug Cocrystal of Anticonvulsants: Influence of Strong Intermolecular Interactions on Physiochemical Properties. Cryst. Growth Des. 2017, 17, 5012–5016. [Google Scholar] [CrossRef] [PubMed]
- Kuminek, G.; Rodriguez-Hornedo, N.; Siedler, S.; Rocha, H.V.A.; Cuffini, S.L.; Cardoso, S.G. How cocrystals of weakly basic drugs and acidic coformers might modulate solubility and stability. Chem. Commun. 2016, 52, 5832–5835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sathisaran, I.; Dalvi, S. Investigating Cocrystallization of Carbamazepine with Structurally Compatible Coformers: New Cocrystal and Eutectic Phases with Enhanced Dissolution. AAPS PharmSciTech 2021, 22, 29. [Google Scholar] [CrossRef] [PubMed]
- Childs, S.L.; Rodríguez-Hornedo, N.; Reddy, L.; Jayasankar, A.; Maheshwari, C.; McCausland, L.; Shipplett, R.; Stahly, B. Screening strategies based on solubility and solution composition generate pharmaceutically acceptable cocrystals of carbamazepine. CrystEngComm 2008, 10, 856–864. [Google Scholar] [CrossRef]
- Sathisaran, I.; Dalvi, S.V. Cocrystallization of carbamazepine with amides: Cocrystal and eutectic phases with improved dissolution. J. Mol. Struct. 2019, 1193, 398–415. [Google Scholar] [CrossRef]
- Yamamoto, K.; Tsutsumi, S.; Ikeda, Y. Establishment of cocrystal cocktail grinding method for rational screening of pharmaceutical cocrystals. Int. J. Pharm. 2012, 437, 162–171. [Google Scholar] [CrossRef]
- Shayanfar, A.; Asadpour-Zeynali, K.; Jouyban, A. Solubility and dissolution rate of a carbamazepine–cinnamic acid cocrystal. J. Mol. Liq. 2013, 187, 171–176. [Google Scholar] [CrossRef]
- Surov, A.O.; Volkova, T.V.; Churakov, A.V.; Proshin, A.N.; Terekhova, I.V.; Perlovich, G.L. Cocrystal formation, crystal structure, solubility and permeability studies for novel 1,2,4-thiadiazole derivative as a potent neuroprotector. Eur. J. Pharm. Sci. 2017, 109, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Surov, A.O.; Voronin, A.P.; Simagina, A.A.; Churakov, A.V.; Skachilova, S.Y.; Perlovich, G.L. Saccharin salts of biologically active hydrazone derivatives. New J. Chem. 2015, 39, 8614–8622. [Google Scholar] [CrossRef]
- Drozd, K.V.; Manin, A.N.; Perlovich, G.L. Comparative analysis of experimental methods for determining thermodynamic parameters of formation of multi-component molecular crystals: Benefits and limitations. J. Mol. Liq. 2019, 295. [Google Scholar] [CrossRef]
- Ma, K.; Zhang, Y.; Kan, H.; Cheng, L.; Luo, L.; Su, Q.; Gao, J.; Gao, Y.; Zhang, J. Thermodynamic and Kinetic Investigation on the Crucial Factors Affecting Adefovir Dipivoxil-Saccharin Cocrystallization. Pharm. Res. 2014, 31, 1766–1778. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | TPR (Sensitivity), % | TNR (Specificity), % | Accuracy, % | Balanced Accuracy, % |
|---|---|---|---|---|
| HPB | 40.0 | 45.5 | 42.0 | 42.7 |
| MEP method A | 78.2 | 28.1 | 59.8 | 53.2 |
| CCGNet | 80.4 | 46.9 | 68.2 | 63.6 |
| Cocrystal | lnKf | ΔformG | ΔformH | T·ΔformS |
|---|---|---|---|---|
| [CBZ + 3AcAmBA] (1:1) | 1.17 ± 0.02 | −2.91 ± 0.05 | −6.7 ± 0.5 | −3.8 ± 0.5 |
| [CBZ + 4AcAmBA] (1:1) | 2.86 ± 0.03 | −7.09 ± 0.08 | −9.5 ± 0.6 | −2.4 ± 0.6 |
| Cocrystal | S0(CF), a Mol·L−1 | [CBZ]eu, Mol·L−1 | [CF]eu, Mol·L−1 | Keu b | Scc, c Mol·L−1 |
|---|---|---|---|---|---|
| [CBZ + 2AcAmBA] (1:1) | 11.5·10−3 | (1.10 ± 0.06)·10−3 | (7.5 ± 0.1)·10−3 | 6.8 ± 0.4 | (2.8 ± 0.1)·10−3 |
| [CBZ + 3AcAmBA] (1:1) | 4.1·10−3 | (9.8 ± 0.2)·10−4 | (1.75 ± 0.08)·10−3 | 1.8 ± 0.1 | (1.30 ± 0.03)·10−3 |
| [CBZ + 4AcAmBA] (1:1) | 4.6·10−3 | (1.00 ± 0.05)·10−3 | (6.4 ± 0.2)·10−4 | 0.64 ± 0.04 | (0.80 ± 0.02)·10−3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Surov, A.O.; Ramazanova, A.G.; Voronin, A.P.; Drozd, K.V.; Churakov, A.V.; Perlovich, G.L. Virtual Screening, Structural Analysis, and Formation Thermodynamics of Carbamazepine Cocrystals. Pharmaceutics 2023, 15, 836. https://doi.org/10.3390/pharmaceutics15030836
Surov AO, Ramazanova AG, Voronin AP, Drozd KV, Churakov AV, Perlovich GL. Virtual Screening, Structural Analysis, and Formation Thermodynamics of Carbamazepine Cocrystals. Pharmaceutics. 2023; 15(3):836. https://doi.org/10.3390/pharmaceutics15030836
Chicago/Turabian StyleSurov, Artem O., Anna G. Ramazanova, Alexander P. Voronin, Ksenia V. Drozd, Andrei V. Churakov, and German L. Perlovich. 2023. "Virtual Screening, Structural Analysis, and Formation Thermodynamics of Carbamazepine Cocrystals" Pharmaceutics 15, no. 3: 836. https://doi.org/10.3390/pharmaceutics15030836