
Effect of N-Terminal Peptide Modifications on In Vitro and In Vivo Properties of 177Lu-Labeled Peptide Analogs Targeting CCK2R
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Peptide Synthesis
2.3. Radiolabeling and Characterization In Vitro
2.4. Cell Uptake and Receptor Binding Studies
2.5. In Vivo Stability
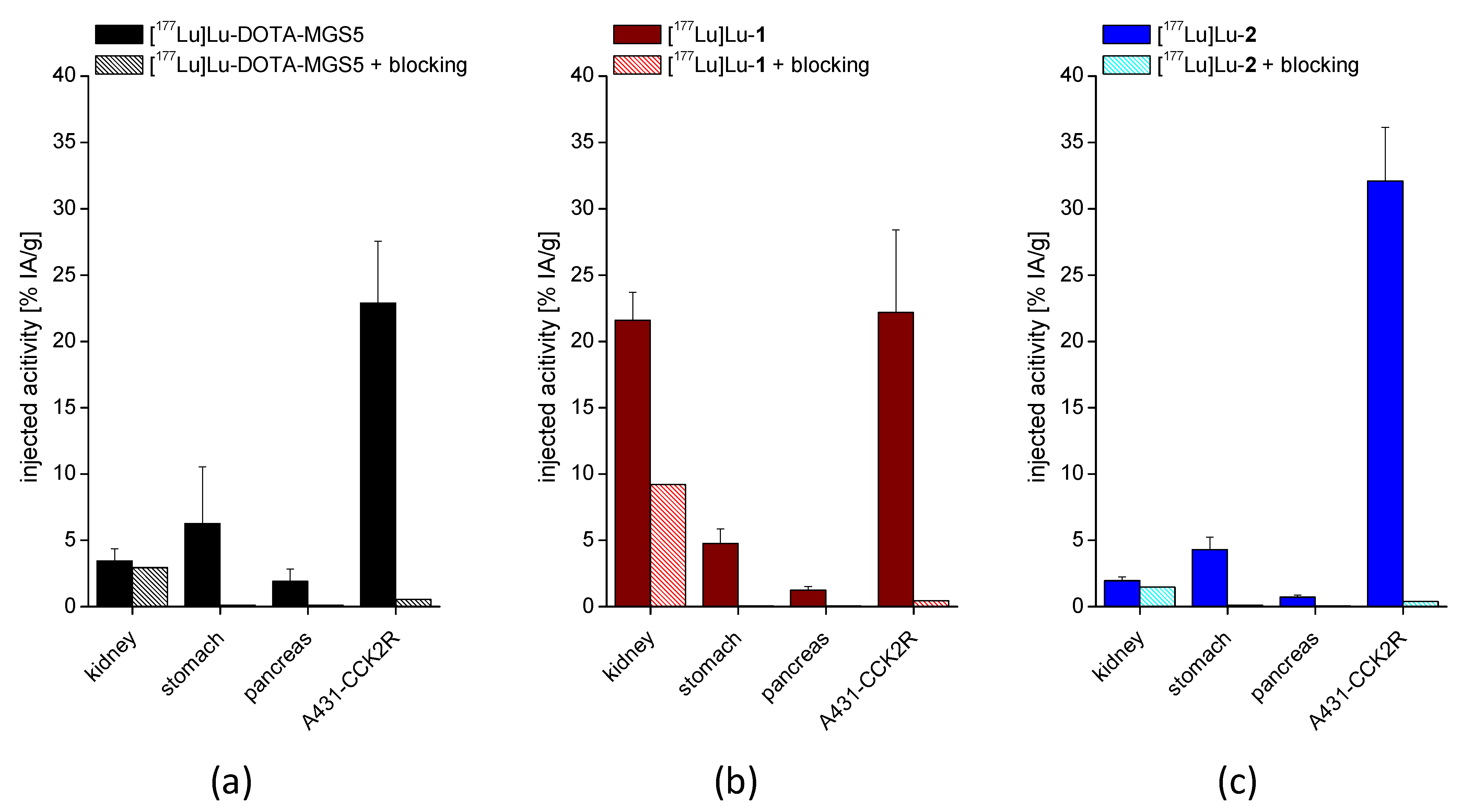
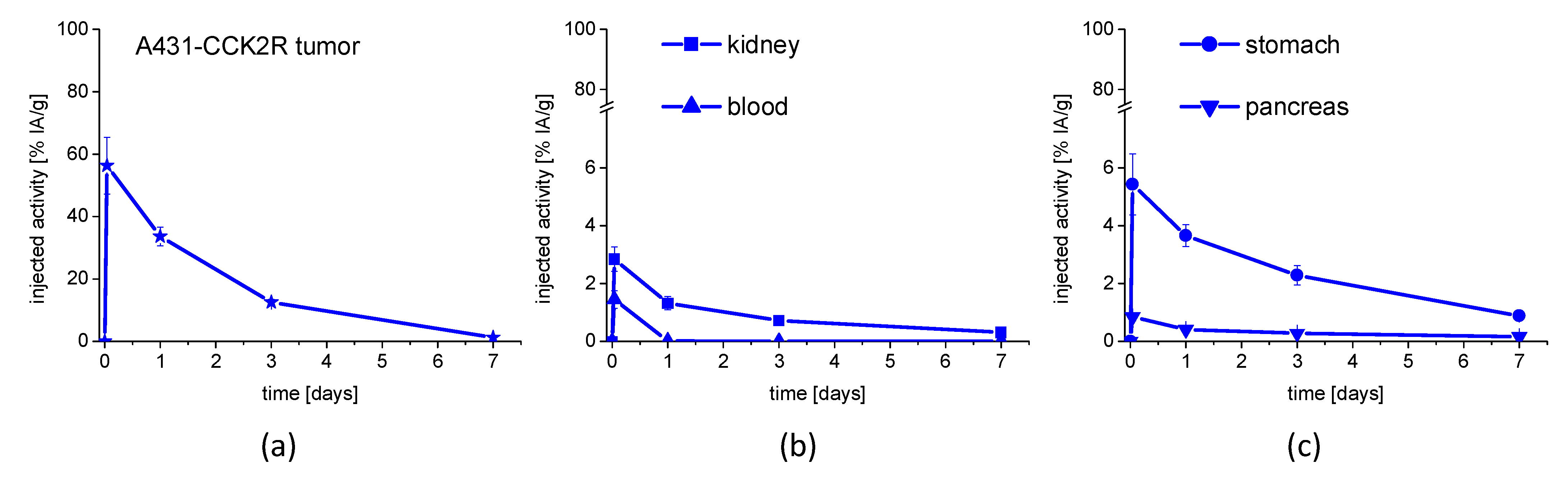
2.6. Biodistribution and Tumor Uptake
3. Results
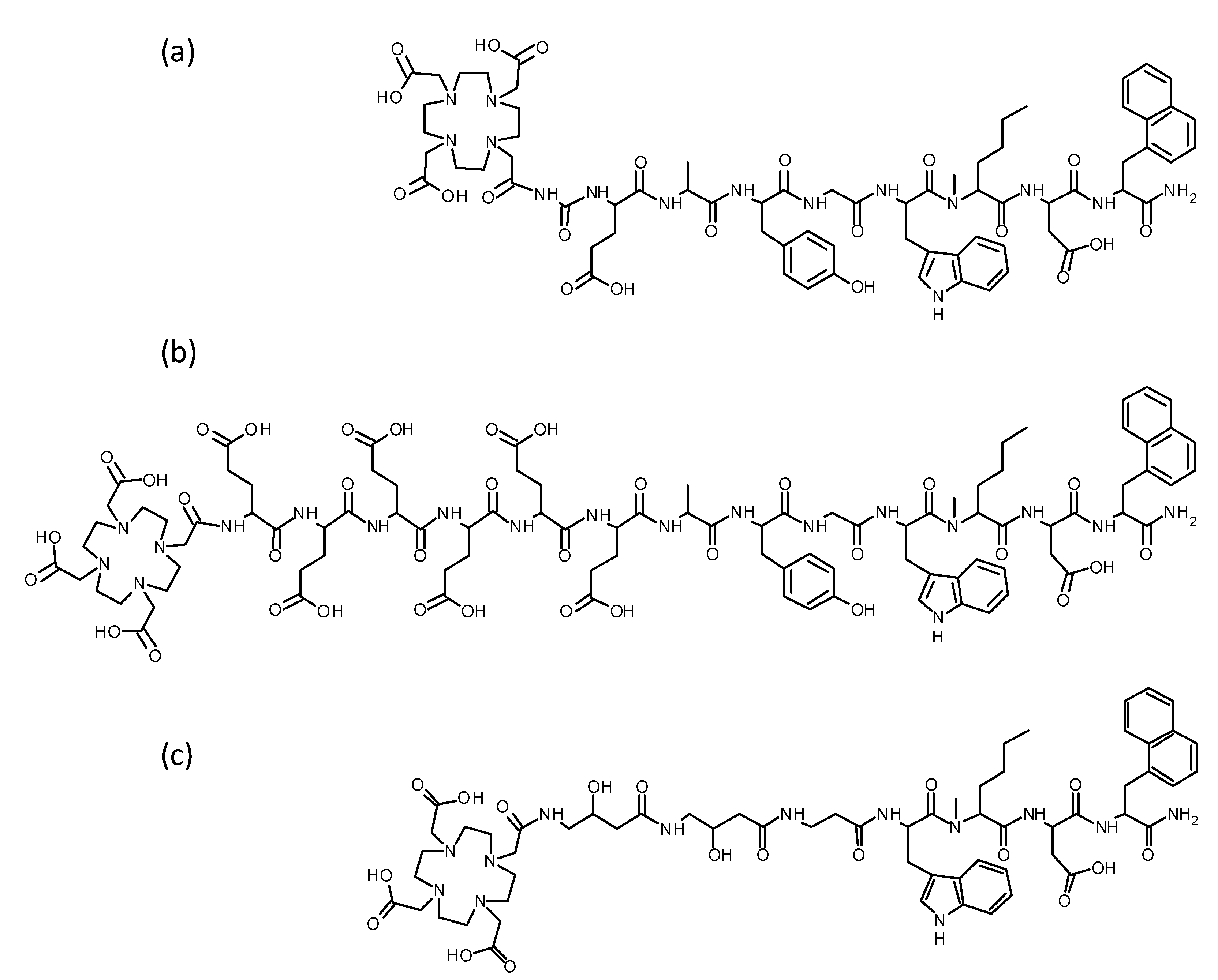
3.1. Peptide Synthesis
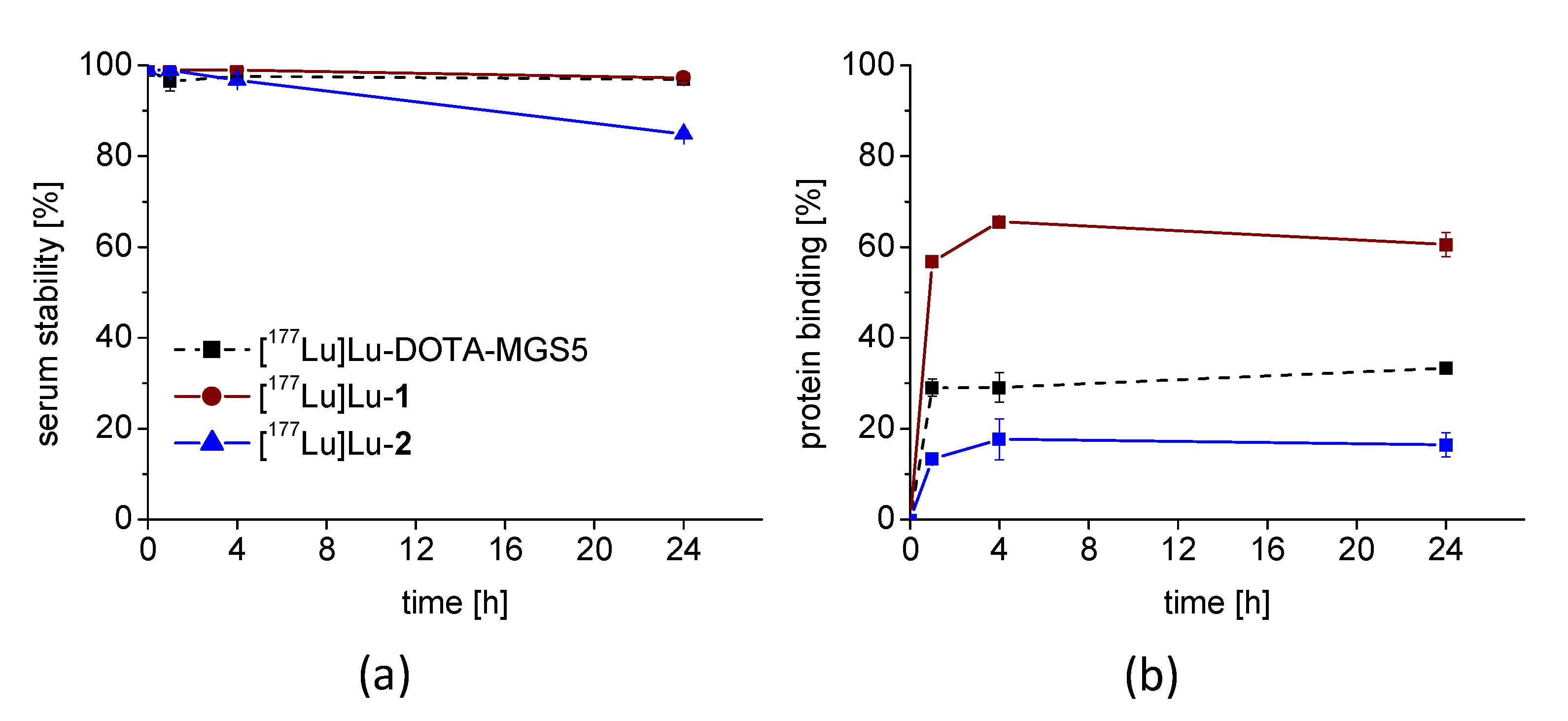
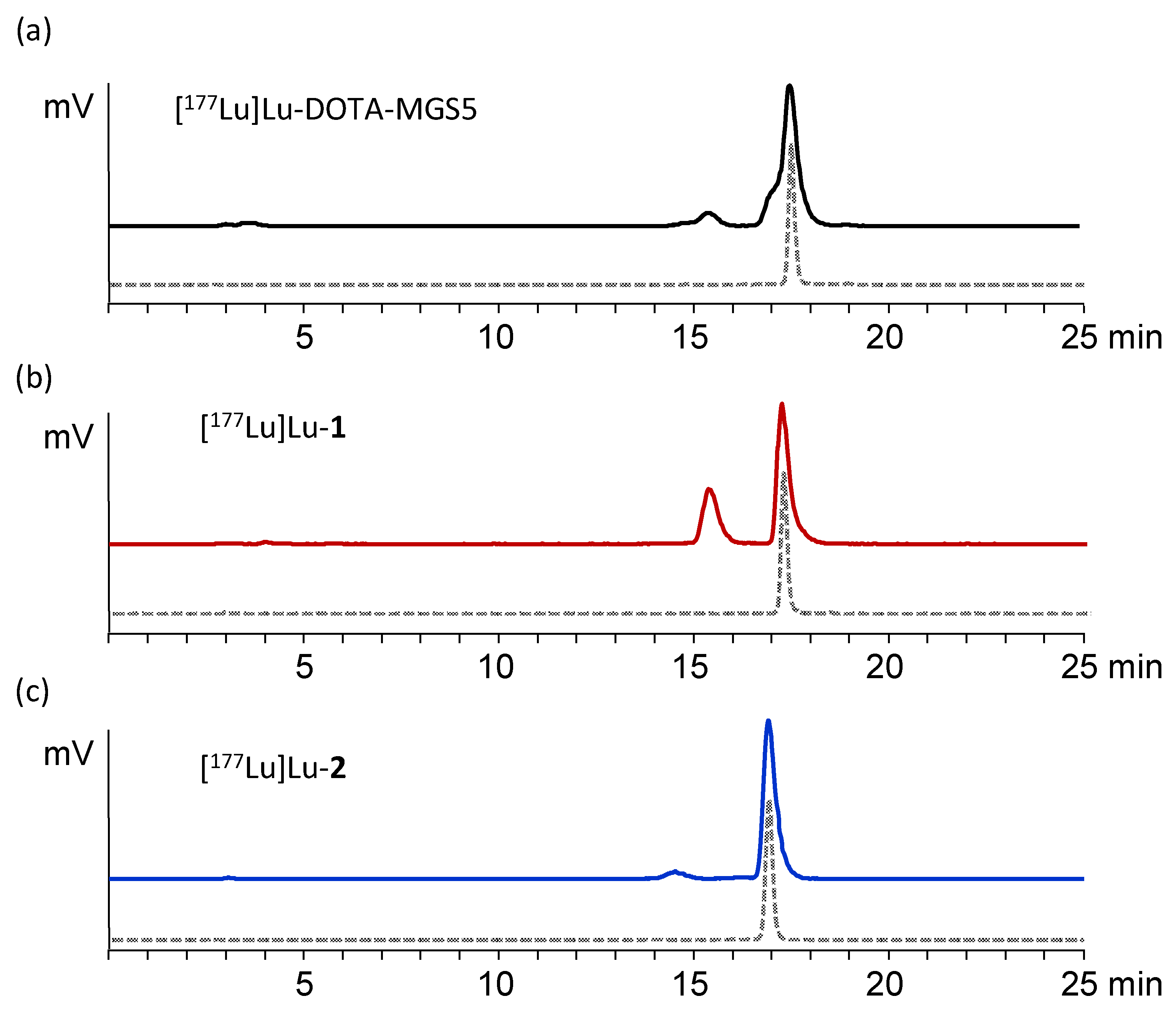
3.2. Radiolabeling and Characterization In Vitro
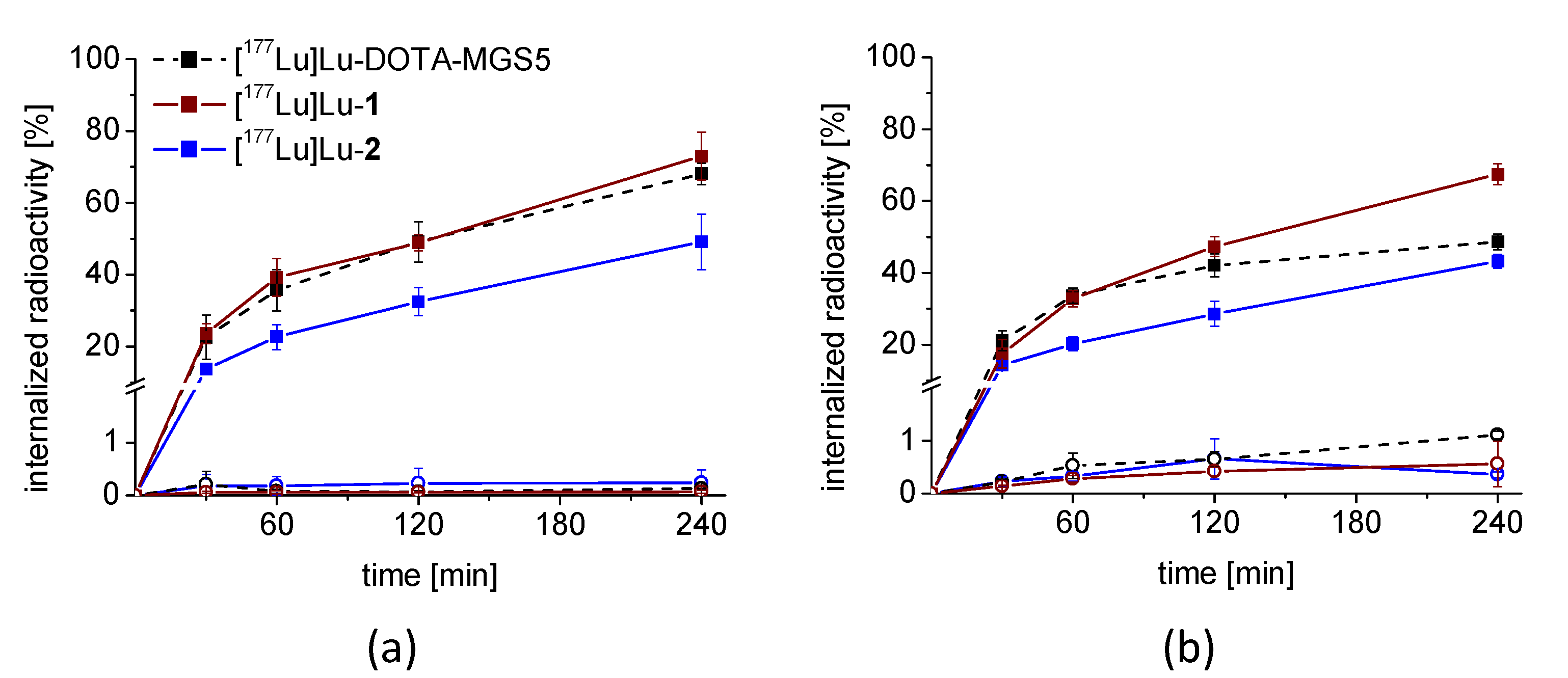
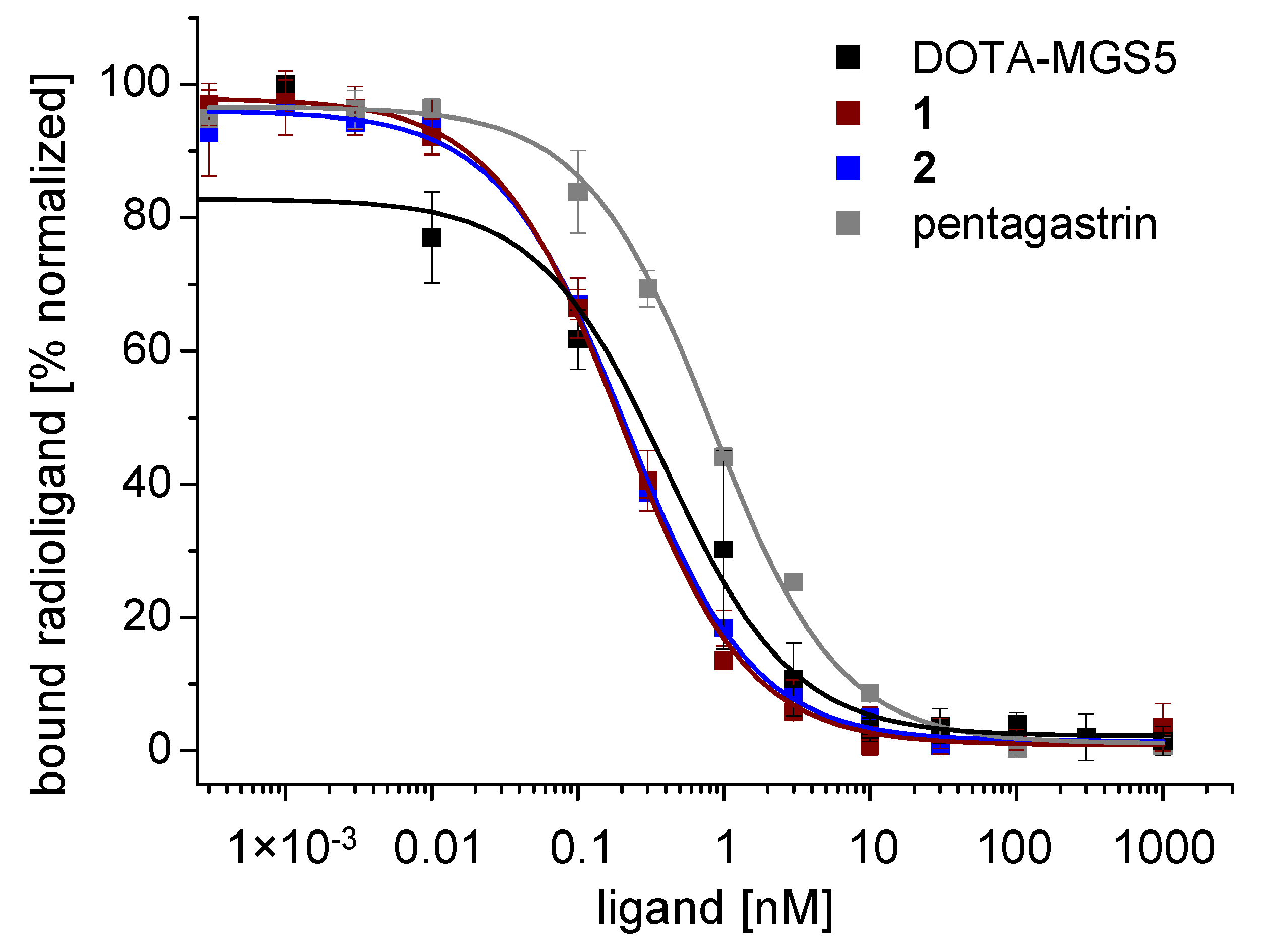
3.3. Cell Uptake and Receptor Binding Studies
3.4. In Vivo Stability and Biodistribution Studies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Noble, F.; Wank, S.A.; Crawley, J.N.; Bradwejn, J.; Seroogy, K.B.; Hamon, M.; Roques, B.P. International Union of Pharmacology. XXI. Structure, Distribution, and Functions of Cholecystokinin Receptors. Pharmacol. Rev. 1999, 51, 745–781. [Google Scholar]
- Reubi, J.C. Targeting CCK receptors in human cancers. Curr. Top. Med. Chem. 2007, 7, 1239–1242. [Google Scholar] [CrossRef]
- Reubi, J.C.; Schaer, J.C.; Waser, B. Cholecystokinin(CCK)-A and CCK-B/Gastrin Receptors in Human Tumors. Cancer Res. 1997, 57, 1377–1386. [Google Scholar]
- Ocak, M.; Helbok, A.; Rangger, C.; Peitl, P.K.; Nock, B.A.; Morelli, G.; Eek, A.; Sosabowski, J.K.; Breeman, W.A.P.; Reubi, J.C.; et al. Comparison of Biological Stability and Metabolism of CCK2 Receptor Targeting Peptides, a Collaborative Project under COST BM0607. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1426–1435. [Google Scholar] [CrossRef] [Green Version]
- Aloj, L.; Aurilio, M.; Rinaldi, V.; D’ambrosio, L.; Tesauro, D.; Peitl, P.K.; Maina, T.; Mansi, R.; von Guggenberg, E.; Joosten, L.; et al. Comparison of the Binding and Internalization Properties of 12 DOTA-Coupled and 111In-Labelled CCK2/Gastrin Receptor Binding Peptides: A Collaborative Project under COST Action BM0607. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1417–1425. [Google Scholar] [CrossRef] [Green Version]
- Roosenburg, S.; Laverman, P.; van Delft, F.L.; Boerman, O.C. Radiolabeled CCK/Gastrin Peptides for Imaging and Therapy of CCK2 Receptor-Expressing Tumors. Amino Acids 2011, 41, 1049–1058. [Google Scholar] [CrossRef] [Green Version]
- Laverman, P.; Joosten, L.; Eek, A.; Roosenburg, S.; Peitl, P.K.; Maina, T.; Mäcke, H.; Aloj, L.; von Guggenberg, E.; Sosabowski, J.K.; et al. Comparative Biodistribution of 12 111In-Labelled Gastrin/CCK2 Receptor-Targeting Peptides. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1410–1416. [Google Scholar] [CrossRef] [Green Version]
- Kaloudi, A.; Nock, B.A.; Krenning, E.P.; Maina, T.; Jong, M.D. Radiolabeled Gastrin/CCK Analogs in Tumor Diagnosis: Towards Higher Stability and Improved Tumor Targeting. Q. J. Nucl. Med. Mol. Imaging 2015, 59, 287–302. [Google Scholar]
- Reubi, J.C.; Waser, B. Unexpected High Incidence of Cholecystokinin-B/Gastrin Receptors in Human Medullary Thyroid Carcinomas. Int. J. Cancer 1996, 67, 644–647. [Google Scholar] [CrossRef]
- Behr, T.M.; Jenner, N.; Bã, M. Radiolabeled Peptides for Targeting Cholecystokinin-B/Gastrin Receptor-Expressing Tumors. J. Nucl. Med. 1999, 40, 1029–1044. [Google Scholar]
- Dufresne, M.; Seva, C.; Fourmy, D. Cholecystokinin and Gastrin Receptors. Physiol. Rev. 2006, 86, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Béhé, M.; Becker, W.; Gotthardt, M.; Angerstein, C.; Behr, T.M. Improved Kinetic Stability of DTPA-DGlu as Compared with Conventional Monofunctional DTPA in Chelating Indium and Yttrium: Preclinical and Initial Clinical Evaluation of Radiometal Labelled Minigastrin Derivatives. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 1140–1146. [Google Scholar] [CrossRef]
- Behr, T.M.; Béhé, M. Cholecystokinin-B/Gastrin Receptor-Targeting Peptides for Staging and Therapy of Medullary Thyroid Cancer and Other Cholecystokinin-B Receptor-Expressing Malignancies. Semin. Nucl. Med. 2002, 32, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Béhé, M.; Kluge, G.; Becker, W.; Gotthardt, M.; Behr, T.M. Use of Polyglutamic Acids to Reduce Uptake of Radiometal-Labeled Minigastrin in the Kidneys. J. Nucl. Med. 2005, 46, 1012–1015. [Google Scholar] [PubMed]
- Breeman, W.A.P.; Fröberg, A.C.; de Blois, E.; van Gameren, A.; Melis, M.; de Jong, M.; Maina, T.; Nock, B.A.; Erion, J.L.; Mäcke, H.R.; et al. Optimised Labeling, Preclinical and Initial Clinical Aspects of CCK-2 Receptor-Targeting with 3 Radiolabeled Peptides. Nucl. Med. Biol. 2008, 35, 839–849. [Google Scholar] [CrossRef]
- Good, S.; Walter, M.A.; Waser, B.; Wang, X.; Müller-Brand, J.; Béhé, M.; Reubi, J.-C.; Maecke, H.R. Macrocyclic Chelator-Coupled Gastrin-Based Radiopharmaceuticals for Targeting of Gastrin Receptor-Expressing Tumours. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 1868–1877. [Google Scholar] [CrossRef] [Green Version]
- Klingler, M.; Summer, D.; Rangger, C.; Haubner, R.; Foster, J.; Sosabowski, J.; Decristoforo, C.; Virgolini, I.; von Guggenberg, E. DOTA-MGS5, a New Cholecystokinin-2 Receptor-Targeting Peptide Analog with an Optimized Targeting Profile for Theranostic Use. J. Nucl. Med. 2019, 60, 1010–1016. [Google Scholar] [CrossRef] [Green Version]
- Klingler, M.; Hörmann, A.A.; Rangger, C.; Desrues, L.; Castel, H.; Gandolfo, P.; von Guggenberg, E. Stabilization Strategies for Linear Minigastrin Analogues: Further Improvements via the Inclusion of Proline into the Peptide Sequence. J. Med. Chem. 2020, 63, 14668–14679. [Google Scholar] [CrossRef]
- Rottenburger, C.; Nicolas, G.P.; McDougall, L.; Kaul, F.; Cachovan, M.; Vija, A.H.; Schibli, R.; Geistlich, S.; Schumann, A.; Rau, T.; et al. Cholecystokinin 2 Receptor Agonist 177Lu-PP-F11N for Radionuclide Therapy of Medullary Thyroid Carcinoma: Results of the Lumed Phase 0a Study. J. Nucl. Med. 2020, 61, 520–526. [Google Scholar] [CrossRef]
- Dorbes, S.; Mestre-Voegtlé, B.; Coulais, Y.; Picard, C.; Silvente-Poirot, S.; Poirot, M.; Benoist, E. Synthesis, Characterization and in Vitro Evaluation of New Oxorhenium- and Oxotechnetium-CCK4 Derivatives as Molecular Imaging Agents for CCK2-Receptor Targeting. Eur. J. Med. Chem. 2010, 45, 423–429. [Google Scholar] [CrossRef]
- Brillouet, S.; Dorbes, S.; Courbon, F.; Picard, C.; Delord, J.P.; Benoist, E.; Poirot, M.; Mestre-Voegtlé, B.; Silvente-Poirot, S. Development of a New Radioligand for Cholecystokinin Receptor Subtype 2 Scintigraphy: From Molecular Modeling to in Vivo Evaluation. Bioorg. Med. Chem. 2010, 18, 5400–5412. [Google Scholar] [CrossRef] [PubMed]
- Scemama, J.L.; Fourmy, D.; Zahidi, A.; Pradayrol, L.; Susini, C.; Ribet, A. Characterisation of Gastrin Receptors on a Rat Pancreatic Acinar Cell Line (AR42J). A Possible Model for Studying Gastrin Mediated Cell Growth and Proliferation. Gut 1987, 28, 233–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aloj, L.; Caraco, C.; Panico, M.; Zannetti, A.; Vecchio, S.D.; Tesauro, D.; Luca, S.D.; Arra, C.; Pedone, C.; Morelli, G.; et al. In Vitro and In Vivo Evaluation of 111In-DTPA-Glu-G-CCK8 for Cholecystokinin-B Receptor Imaging. J. Nucl. Med. 2004, 45, 485–494. [Google Scholar] [PubMed]
- Hörmann, A.A.; Klingler, M.; Rezaeianpour, M.; Hörmann, N.; Gust, R.; Shahhosseini, S.; von Guggenberg, E. Initial In Vitro and In Vivo Evaluation of a Novel CCK2R Targeting Peptide Analog Labeled with Lutetium-177. Molecules 2020, 25, 4585. [Google Scholar] [CrossRef] [PubMed]
- Fani, M.; Weingärtner, V.; Kolenc Peitl, P.; Mansi, R.; Gaonkar, R.H.; Garnuszek, P.; Mikolajczak, R.; Novak, D.; Simoncic, U.; Hubalewska-Dydejczyk, A.; et al. Selection of the First 99mTc-Labelled Somatostatin Receptor Subtype 2 Antagonist for Clinical Translation-Preclinical Assessment of Two Optimized Candidates. Pharmaceuticals 2020, 14, 19. [Google Scholar] [CrossRef] [PubMed]
- von Guggenberg, E.; Rangger, C.; Sosabowski, J.; Laverman, P.; Reubi, J.-C.; Virgolini, I.J.; Decristoforo, C. Preclinical Evaluation of Radiolabeled DOTA-Derivatized Cyclic Minigastrin Analogs for Targeting Cholecystokinin Receptor Expressing Malignancies. Mol. Imaging Biol. 2012, 14, 366–375. [Google Scholar] [CrossRef] [PubMed]
- von Guggenberg, E.; Sallegger, W.; Helbok, A.; Ocak, M.; King, R.; Mather, S.J.; Decristoforo, C. Cyclic Minigastrin Analogues for Gastrin Receptor Scintigraphy with Technetium-99m: Preclinical Evaluation. J. Med. Chem. 2009, 52, 4786–4793. [Google Scholar] [CrossRef] [PubMed]
- Sauter, A.W.; Mansi, R.; Hassiepen, U.; Muller, L.; Panigada, T.; Wiehr, S.; Wild, A.-M.; Geistlich, S.; Béhé, M.; Rottenburger, C.; et al. Targeting of the Cholecystokinin-2 Receptor with the Minigastrin Analog 177Lu-DOTA-PP-F11N: Does the Use of Protease Inhibitors Further Improve In Vivo Distribution? J. Nucl. Med. 2019, 60, 393–399. [Google Scholar] [CrossRef] [Green Version]
- Sosabowski, J.K.; Matzow, T.; Foster, J.M.; Finucane, C.; Ellison, D.; Watson, S.A.; Mather, S.J. Targeting of CCK-2 Receptor–Expressing Tumors Using a Radiolabeled Divalent Gastrin Peptide. J. Nucl. Med. 2009, 50, 2082–2089. [Google Scholar] [CrossRef] [Green Version]
- Kolenc-Peitl, P.; Mansi, R.; Tamma, M.; Gmeiner-Stopar, T.; Sollner-Dolenc, M.; Waser, B.; Baum, R.P.; Reubi, J.C.; Maecke, H.R. Highly Improved Metabolic Stability and Pharmacokinetics of Indium-111-DOTA-Gastrin Conjugates for Targeting of the Gastrin Receptor. J. Med. Chem. 2011, 54, 2602–2609. [Google Scholar] [CrossRef]
- Ocak, M.; Helbok, A.; von Guggenberg, E.; Ozsoy, Y.; Kabasakal, L.; Kremser, L.; Decristoforo, C. Influence of Biological Assay Conditions on Stability Assessment of Radiometal-Labelled Peptides Exemplified Using a 177Lu-DOTA-Minigastrin Derivative. Nucl. Med. Biol. 2011, 38, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Konijnenberg, M.W.; Breeman, W.A.P.; de Blois, E.; Chan, H.S.; Boerman, O.C.; Laverman, P.; Kolenc-Peitl, P.; Melis, M.; de Jong, M. Therapeutic Application of CCK2R-Targeting PP-F11: Influence of Particle Range, Activity and Peptide Amount. EJNMMI Res. 2014, 4, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberdiac, P.; Mineur, L. Normal tissue tolerance to external beam radiation therapy: The stomach. Cancer Radiother. 2010, 14, 336–339. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Amino Acid Sequence | Purity | MW Calc m/z [M + H]+ | MW Found m/z [M + H]+ |
|---|---|---|---|---|
| DOTA-MGS5 | DOTA-DGlu-Ala-Tyr-Gly-Trp-(N-Me)Nle-Asp-1Nal-NH2 | >95% | 1449.67 | 1450.20 |
| 1 | DOTA-(DGlu)6-Ala-Tyr-Gly-Trp-(N-Me)Nle-Asp-1Nal-NH2 | ≥98% | 2095.13 | 2094.13 |
| 2 | DOTA-(GABOB)2-βAla-Trp-(N-Me)Nle-Asp-1Nal-NH2 | ≥98% | 1302.43 | 1302.34 |
| Time Point p.i. | 1 h | 24 h | 3 Days | 7 Days |
|---|---|---|---|---|
| Tumor-to-blood | 38.9 | 3240.4 | 4856.5 | 241.2 |
| Tumor-to-stomach | 10.4 | 9.2 | 5.5 | 1.4 |
| Tumor-to-kidney | 19.8 | 25.5 | 17.3 | 4.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hörmann, A.A.; Klingler, M.; Rangger, C.; Mair, C.; Joosten, L.; Franssen, G.M.; Laverman, P.; von Guggenberg, E. Effect of N-Terminal Peptide Modifications on In Vitro and In Vivo Properties of 177Lu-Labeled Peptide Analogs Targeting CCK2R. Pharmaceutics 2023, 15, 796. https://doi.org/10.3390/pharmaceutics15030796
Hörmann AA, Klingler M, Rangger C, Mair C, Joosten L, Franssen GM, Laverman P, von Guggenberg E. Effect of N-Terminal Peptide Modifications on In Vitro and In Vivo Properties of 177Lu-Labeled Peptide Analogs Targeting CCK2R. Pharmaceutics. 2023; 15(3):796. https://doi.org/10.3390/pharmaceutics15030796
Chicago/Turabian StyleHörmann, Anton Amadeus, Maximilian Klingler, Christine Rangger, Christian Mair, Lieke Joosten, Gerben M. Franssen, Peter Laverman, and Elisabeth von Guggenberg. 2023. "Effect of N-Terminal Peptide Modifications on In Vitro and In Vivo Properties of 177Lu-Labeled Peptide Analogs Targeting CCK2R" Pharmaceutics 15, no. 3: 796. https://doi.org/10.3390/pharmaceutics15030796