
Bacterial Lectin FimH and Its Aggregation Hot-Spots: An Alternative Strategy against Uropathogenic Escherichia coli
 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Aggregation Propensity Analysis of FimH
2.2. Peptide Design and Synthesis
2.3. In Vitro Amyloid Fibril Formation
2.4. Negative Staining and Transmission Electron Microscopy (TEM)
2.5. X-ray Diffraction from Oriented Protein Fibers
2.6. Attenuated Total Reflectance Fourier-Transform Infrared Spectroscopy (ATR FT-IR) and Post-Run Spectra Calculations
2.7. Congo Red Birefringence Assay
2.8. Molecular Dynamics (MD) Simulations
2.8.1. Data Retrieval
2.8.2. Molecular Docking
2.8.3. MD Simulations
2.8.4. Analysis of Simulation Results
2.9. Redundancy of the Selected Peptide-analogues
3. Results
3.1. Aggregation Assays of FimH Peptide-Analogues
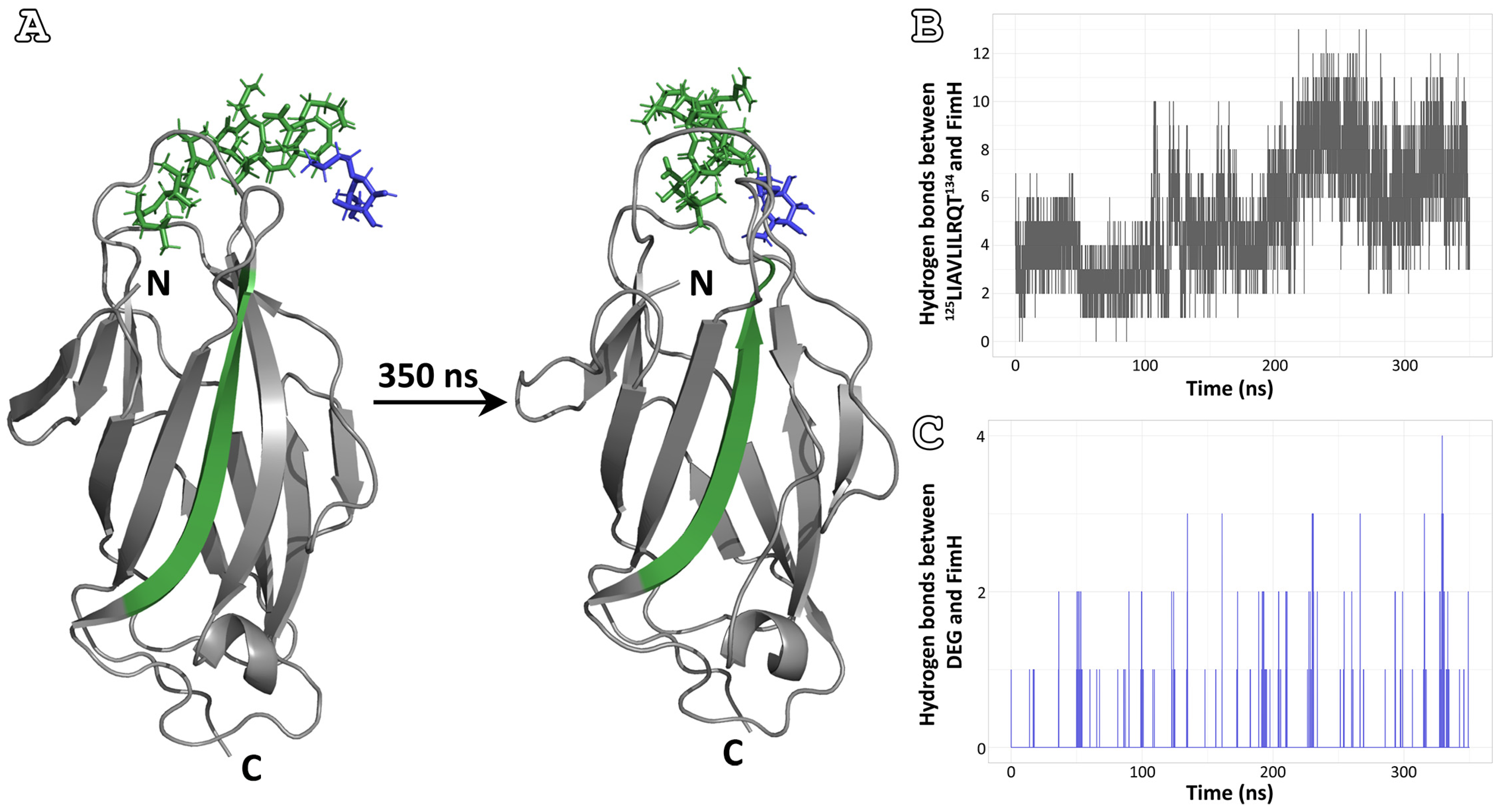
3.2. Self-Oligomerization Simulations
3.3. Affinity for the Mannose-Binding Pocket
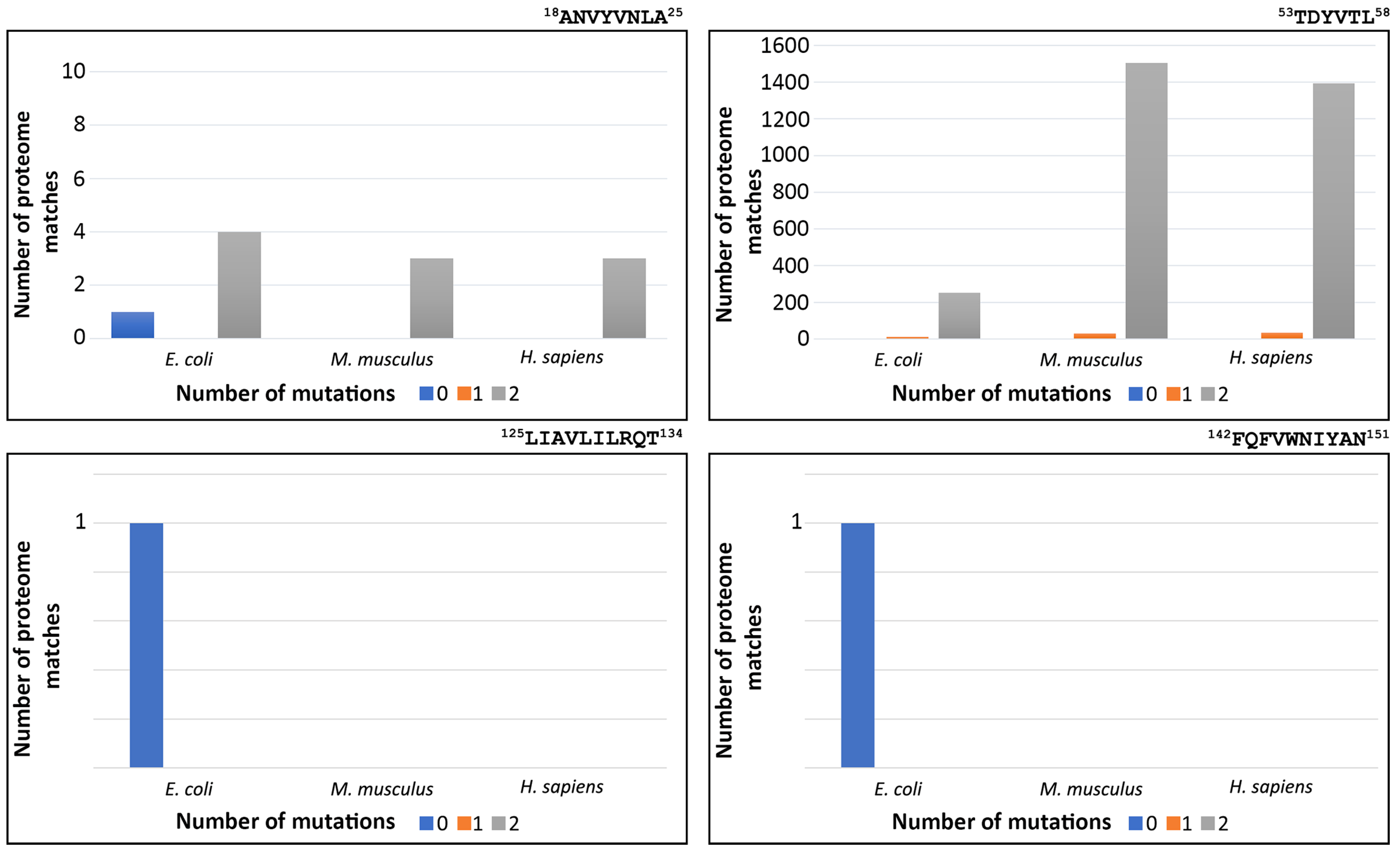
3.4. Redundancy of FimH Peptide-Analogues
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Tandogdu, Z.; Wagenlehner, F.M. Global epidemiology of urinary tract infections. Curr. Opin. Infect. Dis. 2016, 29, 73–79. [Google Scholar] [CrossRef]
- Ulett, G.C.; Totsika, M.; Schaale, K.; Carey, A.J.; Sweet, M.J.; Schembri, M.A. Uropathogenic Escherichia coli virulence and innate immune responses during urinary tract infection. Curr. Opin. Microbiol. 2013, 16, 100–107. [Google Scholar] [CrossRef] [Green Version]
- McLellan, L.K.; Hunstad, D.A. Urinary Tract Infection: Pathogenesis and Outlook. Trends Mol. Med. 2016, 22, 946–957. [Google Scholar] [CrossRef] [Green Version]
- Foxman, B.; Brown, P. Epidemiology of urinary tract infections: Transmission and risk factors, incidence, and costs. Infect. Dis. Clin. North Am. 2003, 17, 227–241. [Google Scholar] [CrossRef]
- Ronald, A.R.; Nicolle, L.E.; Stamm, E.; Krieger, J.; Warren, J.; Schaeffer, A.; Naber, K.G.; Hooton, T.M.; Johnson, J.; Chambers, S.; et al. Urinary tract infection in adults: Research priorities and strategies. Int. J. Antimicrob. Agents 2001, 17, 343–348. [Google Scholar] [CrossRef]
- Bader, M.S.; Loeb, M.; Brooks, A.A. An update on the management of urinary tract infections in the era of antimicrobial resistance. Postgrad. Med. 2017, 129, 242–258. [Google Scholar] [CrossRef]
- Cole, S.T. Who will develop new antibacterial agents? Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2014, 369, 20130430. [Google Scholar] [CrossRef]
- Martinez, J.J.; Mulvey, M.A.; Schilling, J.D.; Pinkner, J.S.; Hultgren, S.J. Type 1 pilus-mediated bacterial invasion of bladder epithelial cells. EMBO J. 2000, 19, 2803–2812. [Google Scholar] [CrossRef]
- Beachey, E.H. Bacterial adherence: Adhesin-receptor interactions mediating the attachment of bacteria to mucosal surface. J. Infect. Dis. 1981, 143, 325–345. [Google Scholar] [CrossRef]
- Jones, C.H.; Pinkner, J.S.; Roth, R.; Heuser, J.; Nicholes, A.V.; Abraham, S.N.; Hultgren, S.J. FimH adhesin of type 1 pili is assembled into a fibrillar tip structure in the Enterobacteriaceae. Proc. Natl. Acad. Sci. USA 1995, 92, 2081–2085. [Google Scholar] [CrossRef] [Green Version]
- Connell, I.; Agace, W.; Klemm, P.; Schembri, M.; Marild, S.; Svanborg, C. Type 1 fimbrial expression enhances Escherichia coli virulence for the urinary tract. Proc. Natl. Acad. Sci. USA 1996, 93, 9827–9832. [Google Scholar] [CrossRef] [Green Version]
- Brinton, C.C., Jr. The structure, function, synthesis and genetic control of bacterial pili and a molecular model for DNA and RNA transport in gram negative bacteria. Trans. N. Y. Acad. Sci. 1965, 27, 1003–1054. [Google Scholar] [CrossRef] [PubMed]
- Snyder, J.A.; Lloyd, A.L.; Lockatell, C.V.; Johnson, D.E.; Mobley, H.L. Role of phase variation of type 1 fimbriae in a uropathogenic Escherichia coli cystitis isolate during urinary tract infection. Infect. Immun. 2006, 74, 1387–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ofek, I.; Mosek, A.; Sharon, N. Mannose-specific adherence of Escherichia coli freshly excreted in the urine of patients with urinary tract infections, and of isolates subcultured from the infected urine. Infect. Immun. 1981, 34, 708–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waksman, G.; Hultgren, S.J. Structural biology of the chaperone-usher pathway of pilus biogenesis. Nat. Rev. Microbiol. 2009, 7, 765–774. [Google Scholar] [CrossRef] [Green Version]
- Schembri, M.A.; Sokurenko, E.V.; Klemm, P. Functional flexibility of the FimH adhesin: Insights from a random mutant library. Infect. Immun. 2000, 68, 2638–2646. [Google Scholar] [CrossRef] [Green Version]
- Le Trong, I.; Aprikian, P.; Kidd, B.A.; Forero-Shelton, M.; Tchesnokova, V.; Rajagopal, P.; Rodriguez, V.; Interlandi, G.; Klevit, R.; Vogel, V.; et al. Structural basis for mechanical force regulation of the adhesin FimH via finger trap-like beta sheet twisting. Cell 2010, 141, 645–655. [Google Scholar] [CrossRef] [Green Version]
- Magala, P.; Klevit, R.E.; Thomas, W.E.; Sokurenko, E.V.; Stenkamp, R.E. RMSD analysis of structures of the bacterial protein FimH identifies five conformations of its lectin domain. Proteins 2020, 88, 593–603. [Google Scholar] [CrossRef]
- Schembri, M.A.; Pallesen, L.; Connell, H.; Hasty, D.L.; Klemm, P. Linker insertion analysis of the FimH adhesin of type 1 fimbriae in an Escherichia coli fimH-null background. FEMS Microbiol. Lett. 1996, 137, 257–263. [Google Scholar] [CrossRef]
- Klemm, P.; Christiansen, G. Three fim genes required for the regulation of length and mediation of adhesion of Escherichia coli type 1 fimbriae. Mol. Gen. Genet. MGG 1987, 208, 439–445. [Google Scholar] [CrossRef]
- Langermann, S.; Palaszynski, S.; Barnhart, M.; Auguste, G.; Pinkner, J.S.; Burlein, J.; Barren, P.; Koenig, S.; Leath, S.; Jones, C.H.; et al. Prevention of mucosal Escherichia coli infection by FimH-adhesin-based systemic vaccination. Science 1997, 276, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Tsolis, A.C.; Papandreou, N.C.; Iconomidou, V.A.; Hamodrakas, S.J. A consensus method for the prediction of ‘aggregation-prone’ peptides in globular proteins. PLoS ONE 2013, 8, e54175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- CrysAlisPRO Agilent Technologies; Version 1.171.37.31; Agilent Technologies UK Ltd.: Oxford, UK, 2014.
- Battye, T.G.; Kontogiannis, L.; Johnson, O.; Powell, H.R.; Leslie, A.G. iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruysschaert, J.-M.; Raussens, V. ATR-FTIR Analysis of Amyloid Proteins. In Peptide Self-Assembly: Methods and Protocols; Nilsson, B.L., Doran, T.M., Eds.; Springer: New York, NY, USA, 2018; pp. 69–81. [Google Scholar]
- Savitzky, A.; Golay, M.J.E. Smoothing and Differentiation of Data by Simplified Least Squares Procedures. Anal. Chem. 1964, 36, 1627–1639. [Google Scholar] [CrossRef]
- Bely, M.; Makovitzky, J. Sensitivity and specificity of Congo red staining according to Romhanyi. Comparison with Puchtler’s or Bennhold’s methods. Acta Histochem. 2006, 108, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Romhanyi, G. Selective differentiation between amyloid and connective tissue structures based on the collagen specific topo-optical staining reaction with congo red. Virchows Archiv. A Pathology. Pathol. Anat. 1971, 354, 209–222. [Google Scholar] [CrossRef]
- Bouckaert, J.; Berglund, J.; Schembri, M.; De Genst, E.; Cools, L.; Wuhrer, M.; Hung, C.S.; Pinkner, J.; Slattegard, R.; Zavialov, A.; et al. Receptor binding studies disclose a novel class of high-affinity inhibitors of the Escherichia coli FimH adhesin. Mol. Microbiol. 2005, 55, 441–455. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Delano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific LLC: South San Francisco, CA, USA, 2005. [Google Scholar]
- Alekseenko, A.; Ignatov, M.; Jones, G.; Sabitova, M.; Kozakov, D. Protein-Protein and Protein-Peptide Docking with ClusPro Server. Methods Mol. Biol. 2020, 2165, 157–174. [Google Scholar] [CrossRef]
- Kozakov, D.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Hall, D.R.; Vajda, S. How good is automated protein docking? Proteins Struct. Funct. Bioinform. 2013, 81, 2159–2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Tsiolaki, P.L.; Nasi, G.I.; Baltoumas, F.A.; Fishman, S.; Tu, H.C.; Iconomidou, V.A. Delving into the amyloidogenic core of human leukocyte chemotactic factor 2. J. Struct. Biol. 2019, 207, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Tsiolaki, P.L.; Nasi, G.I.; Baltoumas, F.A.; Louros, N.N.; Magafa, V.; Hamodrakas, S.J.; Iconomidou, V.A. alphaCGRP, another amyloidogenic member of the CGRP family. J. Struct. Biol. 2018, 203, 27–36. [Google Scholar] [CrossRef]
- Honorato, R.V.; Koukos, P.I.; Jimenez-Garcia, B.; Tsaregorodtsev, A.; Verlato, M.; Giachetti, A.; Rosato, A.; Bonvin, A. Structural Biology in the Clouds: The WeNMR-EOSC Ecosystem. Front. Mol. Biosci. 2021, 8, 729513. [Google Scholar] [CrossRef] [PubMed]
- van Zundert, G.C.P.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [Green Version]
- Kutzner, C.; Pall, S.; Fechner, M.; Esztermann, A.; de Groot, B.L.; Grubmuller, H. More bang for your buck: Improved use of GPU nodes for GROMACS 2018. J. Comput. Chem. 2019, 40, 2418–2431. [Google Scholar] [CrossRef] [Green Version]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Carballo-Pacheco, M.; Strodel, B. Comparison of force fields for Alzheimer’s A beta42: A case study for intrinsically disordered proteins. Protein Sci. A Publ. Protein Soc. 2017, 26, 174–185. [Google Scholar] [CrossRef] [Green Version]
- Palazzesi, F.; Prakash, M.K.; Bonomi, M.; Barducci, A. Accuracy of current all-atom force-fields in modeling protein disordered states. J. Chem. Theory Comput. 2015, 11, 2–7. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.; Chandrasekhar, J.; Madura, J.; Impey, R.; Klein, M. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Berendsen, H.; Postma, J.P.M.; van Gunsteren, W.; DiNola, A.D.; Haak, J.R. Molecular-Dynamics with Coupling to An External Bath. J. Chem. Phys. 1984, 81, 3684. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C. Transport Properties Computed by Linear Response through Weak Coupling to a Bath. In Computer Simulation in Materials Science: Interatomic Potentials, Simulation Techniques and Applications; Meyer, M., Pontikis, V., Eds.; Springer: Dordrecht, The Netherlands, 1991; pp. 139–155. [Google Scholar]
- Hess, B.; Bekker, H.; Berendsen, H.; Fraaije, J. LINCS: A Linear Constraint Solver for molecular simulations. J. Comput. Chem. 1998, 18, 1463–1472. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.; Darden, T.; Lee, H.; Pedersen, L. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- O’Nuallain, B.; Williams, A.D.; Westermark, P.; Wetzel, R. Seeding specificity in amyloid growth induced by heterologous fibrils. J. Biol. Chem. 2004, 279, 17490–17499. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Reumers, J.; Couceiro, J.R.; De Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.C.; et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011, 7, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Sunde, M.; Blake, C.C. From the globular to the fibrous state: Protein structure and structural conversion in amyloid formation. Q. Rev. Biophys. 1998, 31, 1–39. [Google Scholar] [CrossRef]
- Huang, T.H.; Yang, D.S.; Fraser, P.E.; Chakrabartty, A. Alternate aggregation pathways of the Alzheimer beta-amyloid peptide. An in vitro model of preamyloid. J. Biol. Chem. 2000, 275, 36436–36440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shivu, B.; Seshadri, S.; Li, J.; Oberg, K.A.; Uversky, V.N.; Fink, A.L. Distinct beta-sheet structure in protein aggregates determined by ATR-FTIR spectroscopy. Biochemistry 2013, 52, 5176–5183. [Google Scholar] [CrossRef]
- Makin, O.S.; Serpell, L.C. X-ray diffraction studies of amyloid structure. Methods Mol. Biol. 2005, 299, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Howie, A.J. The nomenclature committee of the international society of amyloidosis: Back towards “green birefringence”. Amyloid Int. J. Exp. Clin. Investig. Off. J. Int. Soc. Amyloidosis 2019, 26, 96. [Google Scholar] [CrossRef]
- Howie, A.J. Origins of a pervasive, erroneous idea: The “green birefringence” of Congo red-stained amyloid. Int. J. Exp. Pathol. 2019, 100, 208–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Chen, H.; Bai, H.; Lai, L. Molecular dynamics simulations on the oligomer-formation process of the GNNQQNY peptide from yeast prion protein Sup35. Biophys. J. 2007, 93, 1484–1492. [Google Scholar] [CrossRef] [Green Version]
- Hung, C.S.; Bouckaert, J.; Hung, D.; Pinkner, J.; Widberg, C.; DeFusco, A.; Auguste, C.G.; Strouse, R.; Langermann, S.; Waksman, G.; et al. Structural basis of tropism of Escherichia coli to the bladder during urinary tract infection. Mol. Microbiol. 2002, 44, 903–915. [Google Scholar] [CrossRef] [Green Version]
- Nagahori, N.; Lee, R.T.; Nishimura, S.; Page, D.; Roy, R.; Lee, Y.C. Inhibition of adhesion of type 1 fimbriated Escherichia coli to highly mannosylated ligands. Chembiochem A Eur. J. Chem. Biol. 2002, 3, 836–844. [Google Scholar] [CrossRef]
- Thankavel, K.; Madison, B.; Ikeda, T.; Malaviya, R.; Shah, A.H.; Arumugam, P.M.; Abraham, S.N. Localization of a domain in the FimH adhesin of Escherichia coli type 1 fimbriae capable of receptor recognition and use of a domain-specific antibody to confer protection against experimental urinary tract infection. J. Clin. Investig. 1997, 100, 1123–1136. [Google Scholar] [CrossRef] [Green Version]
- Sauer, M.M.; Jakob, R.P.; Eras, J.; Baday, S.; Eris, D.; Navarra, G.; Berneche, S.; Ernst, B.; Maier, T.; Glockshuber, R. Catch-bond mechanism of the bacterial adhesin FimH. Nat. Commun. 2016, 7, 10738. [Google Scholar] [CrossRef] [Green Version]
- Willmund, F.; del Alamo, M.; Pechmann, S.; Chen, T.; Albanese, V.; Dammer, E.B.; Peng, J.; Frydman, J. The cotranslational function of ribosome-associated Hsp70 in eukaryotic protein homeostasis. Cell 2013, 152, 196–209. [Google Scholar] [CrossRef] [Green Version]
- Vetsch, M.; Puorger, C.; Spirig, T.; Grauschopf, U.; Weber-Ban, E.U.; Glockshuber, R. Pilus chaperones represent a new type of protein-folding catalyst. Nature 2004, 431, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Barnhart, M.M.; Pinkner, J.S.; Soto, G.E.; Sauer, F.G.; Langermann, S.; Waksman, G.; Frieden, C.; Hultgren, S.J. PapD-like chaperones provide the missing information for folding of pilin proteins. Proc. Natl. Acad. Sci. USA 2000, 97, 7709–7714. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, M.; Ishikawa, T.; Rechsteiner, H.; Glockshuber, R. Reconstitution of pilus assembly reveals a bacterial outer membrane catalyst. Science 2008, 320, 376–379. [Google Scholar] [CrossRef]
- Phan, G.; Remaut, H.; Wang, T.; Allen, W.J.; Pirker, K.F.; Lebedev, A.; Henderson, N.S.; Geibel, S.; Volkan, E.; Yan, J.; et al. Crystal structure of the FimD usher bound to its cognate FimC-FimH substrate. Nature 2011, 474, 49–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khodaparast, L.; Khodaparast, L.; Gallardo, R.; Louros, N.N.; Michiels, E.; Ramakrishnan, R.; Ramakers, M.; Claes, F.; Young, L.; Shahrooei, M.; et al. Aggregating sequences that occur in many proteins constitute weak spots of bacterial proteostasis. Nat. Commun. 2018, 9, 866. [Google Scholar] [CrossRef] [Green Version]
- Goldschmidt, L.; Teng, P.K.; Riek, R.; Eisenberg, D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc. Natl. Acad. Sci. USA 2010, 107, 3487–3492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer-Stroh, S.; Debulpaep, M.; Kuemmerer, N.; Lopez de la Paz, M.; Martins, I.C.; Reumers, J.; Morris, K.L.; Copland, A.; Serpell, L.; Serrano, L.; et al. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat. Methods 2010, 7, 237–242. [Google Scholar] [CrossRef]
- Rousseau, F.; Serrano, L.; Schymkowitz, J.W. How evolutionary pressure against protein aggregation shaped chaperone specificity. J. Mol. Biol. 2006, 355, 1037–1047. [Google Scholar] [CrossRef]
- Ventura, S.; Zurdo, J.; Narayanan, S.; Parreno, M.; Mangues, R.; Reif, B.; Chiti, F.; Giannoni, E.; Dobson, C.M.; Aviles, F.X.; et al. Short amino acid stretches can mediate amyloid formation in globular proteins: The Src homology 3 (SH3) case. Proc. Natl. Acad. Sci. USA 2004, 101, 7258–7263. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.J.; Gallo, R.L. Antimicrobial peptides. Curr. Biol. CB 2016, 26, R14–R19. [Google Scholar] [CrossRef] [PubMed]
- Gour, S.; Kumar, V.; Singh, A.; Gadhave, K.; Goyal, P.; Pandey, J.; Giri, R.; Yadav, J.K. Mammalian antimicrobial peptide protegrin-4 self assembles and forms amyloid-like aggregates: Assessment of its functional relevance. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2019, 25, e3151. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Arce, F.T.; Mustata, M.; Ramachandran, S.; Capone, R.; Nussinov, R.; Lal, R. Antimicrobial protegrin-1 forms amyloid-like fibrils with rapid kinetics suggesting a functional link. Biophys. J. 2011, 100, 1775–1783. [Google Scholar] [CrossRef] [Green Version]
- Salinas, N.; Tayeb-Fligelman, E.; Sammito, M.D.; Bloch, D.; Jelinek, R.; Noy, D.; Uson, I.; Landau, M. The amphibian antimicrobial peptide uperin 3.5 is a cross-alpha/cross-beta chameleon functional amyloid. Proc. Natl. Acad. Sci. USA 2021, 118, e2014442118. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.K.; King, G.J.; Conibear, A.C.; Ramos, M.C.; Chaousis, S.; Henriques, S.T.; Craik, D.J. Mirror Images of Antimicrobial Peptides Provide Reflections on Their Functions and Amyloidogenic Properties. J. Am. Chem. Soc. 2016, 138, 5706–5713. [Google Scholar] [CrossRef] [Green Version]
- Ambroggio, E.E.; Kim, D.H.; Separovic, F.; Barrow, C.J.; Barnham, K.J.; Bagatolli, L.A.; Fidelio, G.D. Surface behavior and lipid interaction of Alzheimer beta-amyloid peptide 1–42: A membrane-disrupting peptide. Biophys. J. 2005, 88, 2706–2713. [Google Scholar] [CrossRef] [Green Version]
- Mydock-McGrane, L.K.; Hannan, T.J.; Janetka, J.W. Rational design strategies for FimH antagonists: New drugs on the horizon for urinary tract infection and Crohn’s disease. Expert Opin. Drug Discov. 2017, 12, 711–731. [Google Scholar] [CrossRef]
- Kleandrova, V.V.; Ruso, J.M.; Speck-Planche, A.; Dias Soeiro Cordeiro, M.N. Enabling the Discovery and Virtual Screening of Potent and Safe Antimicrobial Peptides. Simultaneous Prediction of Antibacterial Activity and Cytotoxicity. ACS Comb. Sci. 2016, 18, 490–498. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bands (cm−1) | Band Assignments | |||
|---|---|---|---|---|
| 18ANVYVNLA25 | 53TDYVTL58 | 125LIAVLILRQT134 | 142FQFVWNIYAN15 | |
| 1136 | 1138 | 1136 | 1134 | TFA |
| - | - | 1184 | 1182 | TFA |
| 1205 | 1203 | 1205 | 1203 | TFA |
| 1242 | - | - | - | β-sheet (amide III) |
| 1516 | 1516 | 1516 | Tyrosine | |
| 1549 | 1550 | 1543 | 1550 | β-sheet (amide II) |
| - | 1614 | β-sheet (amide I) | ||
| 1631 | 1643 | 1630 | 1632 | β-sheet (amide I) |
| 1676 | 1676 | 1670 | 1665 | TFA * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasi, G.I.; Georgakopoulou, K.I.; Theodoropoulou, M.K.; Papandreou, N.C.; Chrysina, E.D.; Tsiolaki, P.L.; Iconomidou, V.A. Bacterial Lectin FimH and Its Aggregation Hot-Spots: An Alternative Strategy against Uropathogenic Escherichia coli. Pharmaceutics 2023, 15, 1018. https://doi.org/10.3390/pharmaceutics15031018
Nasi GI, Georgakopoulou KI, Theodoropoulou MK, Papandreou NC, Chrysina ED, Tsiolaki PL, Iconomidou VA. Bacterial Lectin FimH and Its Aggregation Hot-Spots: An Alternative Strategy against Uropathogenic Escherichia coli. Pharmaceutics. 2023; 15(3):1018. https://doi.org/10.3390/pharmaceutics15031018
Chicago/Turabian StyleNasi, Georgia I., Konstantina I. Georgakopoulou, Marilena K. Theodoropoulou, Nikos C. Papandreou, Evangelia D. Chrysina, Paraskevi L. Tsiolaki, and Vassiliki A. Iconomidou. 2023. "Bacterial Lectin FimH and Its Aggregation Hot-Spots: An Alternative Strategy against Uropathogenic Escherichia coli" Pharmaceutics 15, no. 3: 1018. https://doi.org/10.3390/pharmaceutics15031018