

Understanding the Mechanisms and Treatment of Heart Failure: Quantitative Systems Pharmacology Models with a Focus on SGLT2 Inhibitors and Sex-Specific Differences

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pathophysiology of Heart Failure
2.1. Pathophysiological Differences between HF-pEF and HF-rEF
2.2. Compensatory Mechanisms Impairement
2.3. Regulatory Mechanisms
2.3.1. Vasopressin
2.3.2. Pressure Natriuresis
2.3.3. Renin Angiotensin Aldosterone System
2.3.4. Myogenic Response and Tubuloglomerular Feedback
2.3.5. Renal Sympathetic Nerve Activity
2.3.6. Natriuretic Peptides
3. Treating Heart Failure
3.1. Treatment History
3.2. Next Generation of Heart Failure Treatments: SGLT2 Inhibitors
4. Mathematical Modelling of the Cardiorenal Function
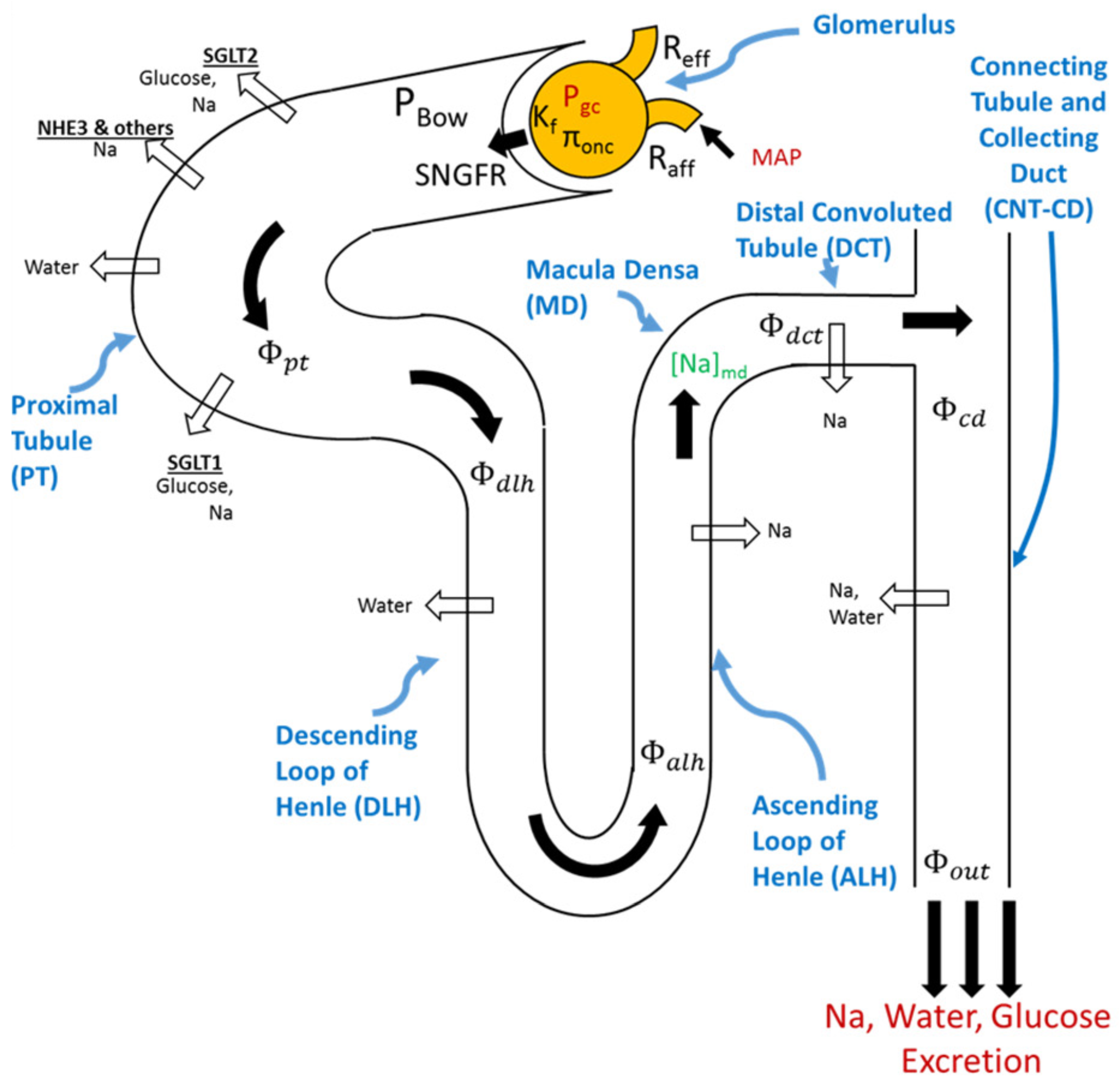
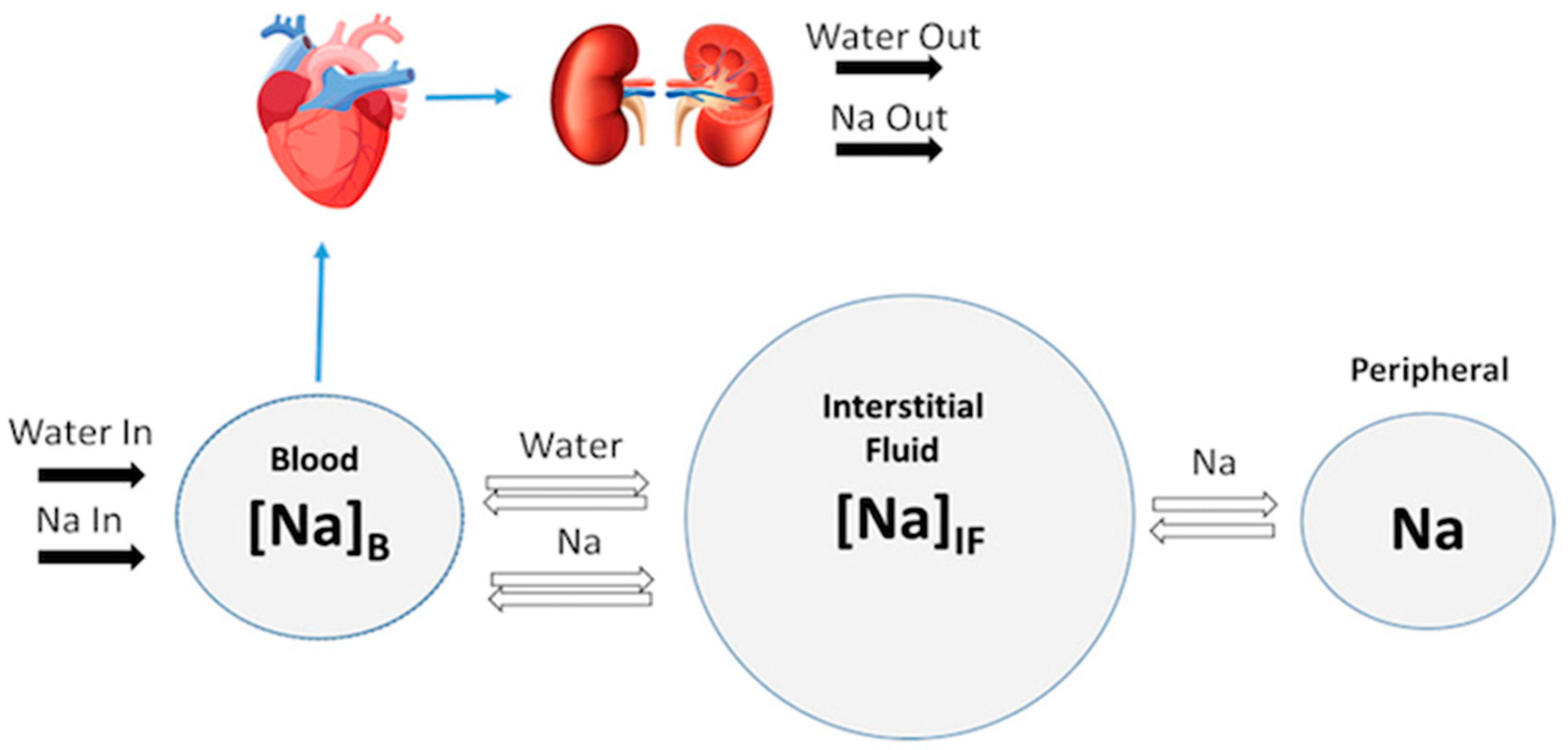
4.1. Modeling Renal Function
4.1.1. Glucose Transport along the Nephron
4.1.2. Sodium Transport along the Nephron
4.2. Modeling Cardiovascular Function


5. Modeling and Optimizing Treatments
6. Considering Sex-Specific Differences in Heart Failure
7. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Roger, V.L. Epidemiology of Heart Failure. Circ. Res. 2021, 128, 1421–1434. [Google Scholar] [CrossRef] [PubMed]
- Savarese, G.; Becher, P.M.; Lund, L.H.; Seferovic, P.; Rosano, G.M.C.; Coats, A.J.S. Global burden of heart failure: A comprehensive and updated review of epidemiology. Cardiovasc. Res. 2023, 118, 3272–3287. [Google Scholar] [CrossRef] [PubMed]
- Kemp, C.D.; Conte, J.V. The pathophysiology of heart failure. Cardiovasc. Pathol. 2012, 21, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Inamdar, A.A.; Inamdar, A.C. Heart Failure: Diagnosis, Management and Utilization. J. Clin. Med. 2016, 5, 62. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.; Brito, D.; Vaqar, S.; Chhabra, L. Congestive Heart Failure; StatPearls Publishing: Treasure Island, FL, USA, 2022; Available online: http://www.ncbi.nlm.nih.gov/books/NBK430873/ (accessed on 18 November 2022).
- Yu, H.; Basu, S.; Hallow, K.M. Cardiac and renal function interactions in heart failure with reduced ejection fraction: A mathematical modeling analysis. PLoS Comput. Biol. 2020, 16, e1008074. [Google Scholar] [CrossRef]
- Messerli, F.H.; Rimoldi, S.F.; Bangalore, S. The Transition From Hypertension to Heart Failure: Contemporary Update. JACC: Heart Fail. 2017, 5, 543–551. [Google Scholar] [CrossRef]
- Silverberg, D.; Wexler, D.; Blum, M.; Schwartz, D.; Iaina, A. The association between congestive heart failure and chronic renal disease. Curr. Opin. Nephrol. Hypertens. 2004, 13, 163. Available online: https://journals.lww.com/co-nephrolhypertens/Fulltext/2004/03000/The_association_between_congestive_heart_failure.4.aspx (accessed on 3 January 2023). [CrossRef]
- Brown, N.J.; Vaughan, D.E. Angiotensin-Converting Enzyme Inhibitors. Circulation 1998, 97, 1411–1420. [Google Scholar] [CrossRef] [Green Version]
- Riegger, A.J. ACE inhibitors in congestive heart failure. Cardiology 1989, 76 (Suppl. S2), 42–49. [Google Scholar] [CrossRef]
- Lonn, E. Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers in atherosclerosis. Curr. Atheroscler. Rep. 2002, 4, 363–372. [Google Scholar] [CrossRef]
- Faris, R.F.; Flather, M.; Purcell, H.; Poole-Wilson, P.A.; Coats, A.J. Diuretics for heart failure. Cochrane Database Syst. Rev. 2016, 2016, CD003838. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Gu, J. Computational methods and applications for quantitative systems pharmacology. Quant. Biol. 2019, 7, 1. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Straube, R.; Alnaif, A.E.; Huang, L.; Leil, T.A.; Schmidt, B.J. Virtual Populations for Quantitative Systems Pharmacology Models. In Systems Medicine; Bai, J.P.F., Hur, J., Eds.; Springer: New York, NY, USA, 2022; pp. 129–179. [Google Scholar] [CrossRef]
- Tanai, E.; Frantz, S. Pathophysiology of Heart Failure. In Comprehensive Physiology; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2015; pp. 187–214. [Google Scholar] [CrossRef]
- Schwinger, R.H.G. Pathophysiology of heart failure. Cardiovasc. Diagn. Ther. 2021, 11, 263–276. [Google Scholar] [CrossRef]
- CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). N. Engl. J. Med. 1987, 316, 1429–1435. [Google Scholar] [CrossRef]
- Henein, M.Y. (Ed.) Heart Failure in Clinical Practice; Springer: London, UK, 2010. [Google Scholar] [CrossRef]
- Venkatesh, A.; Mohankumar, S. A mathematical model for the secretion of vasopressin using fuzzy truncated normal distribution. Int. J. Pure Appl. Math. 2015, 104, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Cuzzo, B.; Padala, S.A.; Lappin, S.L. Physiology, Vasopressin; StatPearls Publishing: Treasure Island, FL, USA, 2022; Available online: http://www.ncbi.nlm.nih.gov/books/NBK526069/ (accessed on 11 January 2023).
- Baek, E.J.; Kim, S. Current Understanding of Pressure Natriuresis. Electrolytes Blood Press. 2021, 19, 38–45. [Google Scholar] [CrossRef]
- Granger, J.P.; Alexander, B.T.; Llinas, M. Mechanisms of pressure natriuresis. Curr. Hypertens. Rep. 2002, 4, 152–159. [Google Scholar] [CrossRef]
- Fountain, J.H.; Lappin, S.L. Physiology, Renin Angiotensin System; StatPearls Publishing: Treasure Island, FL, USA, 2022; Available online: http://www.ncbi.nlm.nih.gov/books/NBK470410/ (accessed on 13 January 2023).
- Laghlam, D.; Jozwiak, M.; Nguyen, L.S. Renin-Angiotensin-Aldosterone System and Immunomodulation: A State-of-the-Art Review. Cells 2021, 10, 1767. [Google Scholar] [CrossRef]
- Ren, L.; Lu, X.; Danser, A.H.J. Revisiting the Brain Renin-Angiotensin System-Focus on Novel Therapies. Curr. Hypertens. Rep. 2019, 21, 28. [Google Scholar] [CrossRef] [Green Version]
- Nehme, A.; Zouein, F.A.; Zayeri, Z.D.; Zibara, K. An Update on the Tissue Renin Angiotensin System and Its Role in Physiology and Pathology. J. Cardiovasc. Dev. Dis. 2019, 6, 14. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, K.E.; Khan, Z.; Giani, J.F.; Cao, D.-Y.; Bernstein, E.A.; Shen, X.Z. Angiotensin-converting enzyme in innate and adaptive immunity. Nat. Rev. Nephrol. 2018, 14, 325–336. [Google Scholar] [CrossRef]
- Chappell, M.C. Nonclassical Renin-Angiotensin System and Renal Function. In Comprehensive Physiology; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2012; pp. 2733–2752. [Google Scholar] [CrossRef] [Green Version]
- Scott, J.H.; Menouar, M.A.; Dunn, R.J. Physiology, Aldosterone; StatPearls Publishing: Treasure Island, FL, USA, 2022; Available online: http://www.ncbi.nlm.nih.gov/books/NBK470339/ (accessed on 13 January 2023).
- Ocaranza, M.P.; Riquelme, J.A.; García, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-regulatory renin–angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129. [Google Scholar] [CrossRef] [Green Version]
- Just, A. Mechanisms of renal blood flow autoregulation: Dynamics and contributions. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1–R17. [Google Scholar] [CrossRef] [Green Version]
- Sgouralis, I.; Layton, A.T. Mathematical modeling of renal hemodynamics in physiology and pathophysiology. Math. Biosci. 2015, 264, 8–20. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; He, Q.; Wu, W.; Li, Q.; Huang, R.; Pan, X.; Lai, W. Role of the renal sympathetic nerves in renal sodium/potassium handling and renal damage in spontaneously hypertensive rats. Exp. Ther. Med. 2016, 12, 2547–2553. [Google Scholar] [CrossRef] [Green Version]
- DiBona, G.F.; Sawin, L.L. Reflex regulation of renal nerve activity in cardiac failure. Am. J. Physiol. 1994, 266 Pt 2, R27–R39. [Google Scholar] [CrossRef]
- DiBona, G.F.; Jones, S.Y.; Sawin, L.L. Angiotensin receptor antagonist improves cardiac reflex control of renal sodium handling in heart failure. Am. J. Physiol. 1998, 274, H636–H641. [Google Scholar] [CrossRef]
- Sata, Y.; Head, G.A.; Denton, K.; May, C.N.; Schlaich, M.P. Role of the Sympathetic Nervous System and Its Modulation in Renal Hypertension. Front. Med. 2018, 5, 82. [Google Scholar] [CrossRef] [Green Version]
- Kiuchi, M.G.; Esler, M.D.; Fink, G.D.; Osborn, J.W.; Banek, C.T.; Böhm, M.; Denton, K.M.; DiBona, G.F.; Everett, T.H.; Grassi, G.; et al. Renal Denervation Update From the International Sympathetic Nervous System Summit. J. Am. Coll. Cardiol. 2019, 73, 3006–3017. [Google Scholar] [CrossRef] [PubMed]
- Potter, L.R.; Yoder, A.R.; Flora, D.R.; Antos, L.K.; Dickey, D.M. Natriuretic peptides: Their structures, receptors, physiologic functions and therapeutic applications. Handb. Exp. Pharmacol. 2009, 191, 341–366. [Google Scholar] [CrossRef] [Green Version]
- Sandefur, C.C.; Jialal, I. Atrial Natriuretic Peptide; StatPearls Publishing: Treasure Island, FL, USA, 2022; Available online: http://www.ncbi.nlm.nih.gov/books/NBK562257/ (accessed on 16 January 2023).
- Cannone, V.; Cabassi, A.; Volpi, R.; Burnett, J.C., Jr. Atrial Natriuretic Peptide: A Molecular Target of Novel Therapeutic Approaches to Cardio-Metabolic Disease. Int. J. Mol. Sci. 2019, 20, 3265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, Y.; Nishikimi, T.; Kuwahara, K. Atrial and brain natriuretic peptides: Hormones secreted from the heart. Peptides 2019, 111, 18–25. [Google Scholar] [CrossRef]
- Walter, E.M.D.; Hetzer, R. Surgical treatment concepts for heart failure. HSR Proc. Intensive Care Cardiovasc. Anesth. 2013, 5, 69–75. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3722336/ (accessed on 15 December 2022).
- Allen, L.A.; Felker, G.M. Advances in the surgical treatment of heart failure. Curr. Opin. Cardiol. 2008, 23, 249–253. [Google Scholar] [CrossRef] [Green Version]
- Haji, S.A.; Movahed, A. Update on Digoxin Therapy in Congestive Heart Failure. Am. Fam. Physician 2000, 62, 409–416. Available online: https://www.aafp.org/pubs/afp/issues/2000/0715/p409.html (accessed on 15 December 2022).
- Hauptman, P.J.; Kelly, R.A. Digitalis. Circulation 1999, 99, 1265–1270. [Google Scholar] [CrossRef]
- David, M.N.V.; Shetty, M. Digoxin; StatPearls Publishing: Treasure Island, FL, USA, 2022; Available online: http://www.ncbi.nlm.nih.gov/books/NBK556025/ (accessed on 15 December 2022).
- Caldwell, J.E. Diuretic therapy and exercise performance. Sports Med. 1987, 4, 290–304. [Google Scholar] [CrossRef]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Colvin, M.M.; Drazner, M.H.; Filippatos, G.S.; Fonarow, G.C.; Givertz, M.M.; et al. 2017 ACC/AHA/HFSA Focused Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation 2017, 136, e137–e161. [Google Scholar] [CrossRef]
- Digitalis Investigation Group. The effect of digoxin on mortality and morbidity in patients with heart failure. N. Engl. J. Med. 1997, 336, 525–533. [Google Scholar] [CrossRef]
- Masarone, D.; Martucci, M.L.; Errigo, V.; Pacileo, G. The Use of β-Blockers in Heart Failure with Reduced Ejection Fraction. J. Cardiovasc. Dev. Dis. 2021, 8, 101. [Google Scholar] [CrossRef]
- Chavey, I.W.E. The Importance of Beta Blockers in the Treatment of Heart Failure. afp 2000, 62, 2453–2462. Available online: https://www.aafp.org/pubs/afp/issues/2000/1201/p2453.html (accessed on 15 December 2022).
- Farzam, K.; Jan, A. Beta Blockers; StatPearls Publishing: Treasure Island, FL, USA, 2022; Available online: http://www.ncbi.nlm.nih.gov/books/NBK532906/ (accessed on 28 February 2023).
- Bolam, H.; Morton, G.; Kalra, P.R. Drug therapies in chronic heart failure: A focus on reduced ejection fraction. Clin. Med. 2018, 18, 138–145. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [Green Version]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Zelniker, T.A.; Wiviott, S.D.; Raz, I.; Im, K.; Goodrich, E.; Bonaca, M.P.; Mosenzon, O.; Kato, E.; Cahn, A.; Furtado, R.H.M.; et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet 2019, 393, 31–39. [Google Scholar] [CrossRef]
- Neuen, B.L.; Young, T.; Heerspink, H.J.L.; Neal, B.; Perkovic, V.; Billot, L.; Mahaffey, K.W.; Charytan, D.M.; Wheeler, D.C.; Arnott, C.; et al. SGLT2 inhibitors for the prevention of kidney failure in patients with type 2 diabetes: A systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2019, 7, 845–854. [Google Scholar] [CrossRef]
- Butler, J.; Handelsman, Y.; Bakris, G.; Verma, S. Use of sodium–glucose co-transporter-2 inhibitors in patients with and without type 2 diabetes: Implications for incident and prevalent heart failure. Eur. J. Heart Fail. 2020, 22, 604–617. [Google Scholar] [CrossRef]
- Packer, M. SGLT2 Inhibitors Produce Cardiorenal Benefits by Promoting Adaptive Cellular Reprogramming to Induce a State of Fasting Mimicry: A Paradigm Shift in Understanding Their Mechanism of Action. Diabetes Care 2020, 43, 508–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anker, S.D.; Butler, J.; Filippatos, G.; Khan, M.S.; Marx, N.; Lam, C.S.; Schnaidt, S.; Ofstad, A.P.; Brueckmann, M.; Jamal, W.; et al. Effect of Empagliflozin on Cardiovascular and Renal Outcomes in Patients With Heart Failure by Baseline Diabetes Status: Results From the EMPEROR-Reduced Trial. Circulation 2021, 143, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Verma, S. Mechanisms of Cardiovascular Benefits of Sodium Glucose Co-Transporter 2 (SGLT2) Inhibitors. JACC Basic Transl. Sci. 2020, 5, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Tanna, M.S.; Goldberg, L.R. The pleiotropic cardiovascular effects of sodium-glucose cotransporter-2 inhibitors. Curr. Opin. Cardiol. 2021, 36, 764–768. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.M.; Yellon, D.M. SGLT2 inhibitors: Hypotheses on the mechanism of cardiovascular protection. Lancet Diabetes Endocrinol. 2018, 6, 435–437. [Google Scholar] [CrossRef] [PubMed]
- Cherney, D.Z.; Odutayo, A.; Aronson, R.; Ezekowitz, J.; Parker, J.D. Sodium Glucose Cotransporter-2 Inhibition and Cardiorenal Protection: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 74, 2511–2524. [Google Scholar] [CrossRef]
- Baartscheer, A.; Schumacher, C.A.; Wüst, R.C.I.; Fiolet, J.W.T.; Stienen, G.J.M.; Coronel, R.; Zuurbier, C.J. Empagliflozin decreases myocardial cytoplasmic Na+ through inhibition of the cardiac Na+/H+ exchanger in rats and rabbits. Diabetologia 2017, 60, 568–573. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.; Kuvin, J.T.; Pandian, N.G.; Smith, J.J.; E Udelson, J.; E Mendelsohn, M.; A Konstam, M.; Karas, R.H. Heart failure etiology affects peripheral vascular endothelial function after cardiac transplantation. J. Am. Coll. Cardiol. 2001, 37, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Gaspari, T.; Spizzo, I.; Liu, H.; Hu, Y.; Simpson, R.W.; Widdop, R.E.; Dear, A.E. Dapagliflozin attenuates human vascular endothelial cell activation and induces vasorelaxation: A potential mechanism for inhibition of atherogenesis. Diabetes Vasc. Dis. Res. 2018, 15, 64–73. [Google Scholar] [CrossRef] [Green Version]
- Juni, R.P.; Kuster, D.W.; Goebel, M.; Helmes, M.; Musters, R.J.; van der Velden, J.; Koolwijk, P.; Paulus, W.J.; van Hinsbergh, V.W. Cardiac Microvascular Endothelial Enhancement of Cardiomyocyte Function Is Impaired by Inflammation and Restored by Empagliflozin. JACC Basic Transl. Sci. 2019, 4, 575–591. [Google Scholar] [CrossRef]
- Sano, M. A new class of drugs for heart failure: SGLT2 inhibitors reduce sympathetic overactivity. J. Cardiol. 2018, 71, 471–476. [Google Scholar] [CrossRef] [Green Version]
- Musante, C.; Ramanujan, S.; Schmidt, B.; Ghobrial, O.; Lu, J.; Heatherington, A. Quantitative Systems Pharmacology: A Case for Disease Models. Clin. Pharmacol. Ther. 2017, 101, 24–27. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.E. Guyton and Hall Textbook of Medical Physiology, 12th ed.; Saunders/Elsevier: Philadelphia, PA, USA, 2011. [Google Scholar]
- How Your Kidneys Work. National Kidney Foundation, 24 December 2015. Available online: https://www.kidney.org/atoz/content/howkidneyswork (accessed on 29 November 2022).
- Bertram, J.F.; Douglas-Denton, R.N.; Diouf, B.; Hughson, M.D.; Hoy, W.E. Human nephron number: Implications for health and disease. Pediatr. Nephrol. 2011, 26, 1529–1533. [Google Scholar] [CrossRef]
- Kanzaki, G.; Tsuboi, N.; Shimizu, A.; Yokoo, T. Human nephron number, hypertension, and renal pathology. Anat. Rec. 2020, 303, 2537–2543. [Google Scholar] [CrossRef]
- Hallow, K.M.; Lo, A.; Beh, J.; Rodrigo, M.; Ermakov, S.; Friedman, S.; De Leon, H.; Sarkar, A.; Xiong, Y.; Sarangapani, R.; et al. A model-based approach to investigating the pathophysiological mechanisms of hypertension and response to antihypertensive therapies: Extending the Guyton model. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 306, R647–R662. [Google Scholar] [CrossRef]
- Guyton, A.C.; Coleman, T.G.; Granger, H.J. Circulation: Overall regulation. Annu. Rev. Physiol. 1972, 34, 13–46. [Google Scholar] [CrossRef]
- Karaaslan, F.; Denizhan, Y.; Kayserilioglu, A.; Gulcur, H.O. Long-Term Mathematical Model Involving Renal Sympathetic Nerve Activity, Arterial Pressure, and Sodium Excretion. Ann. Biomed. Eng. 2005, 33, 1607–1630. [Google Scholar] [CrossRef]
- Uttamsingh, R.J.; Leaning, M.S.; Bushman, J.A.; Carson, E.R.; Finkelstein, L. Mathematical model of the human renal system. Med. Biol. Eng. Comput. 1985, 23, 525–535. [Google Scholar] [CrossRef]
- Kriz, W.; Bankir, L. A standard nomenclature for structures of the kidney-The Renal Commission of the International Union of Physiological Sciences (IUPS). Pflügers Arch. Eur. J. Physiol. 1988, 411, 113–120. [Google Scholar] [CrossRef]
- Douglas College Biology Department. Douglas College Human Anatomy & Physiology II, 2nd ed.; OpenStax: Houston, TX, USA, 2019; Available online: https://pressbooks.bccampus.ca/dcbiol120312092nded/ (accessed on 29 November 2022).
- Berkhin, E.B.; Humphreys, M.H. Regulation of renal tubular secretion of organic compounds. Kidney Int. 2001, 59, 17–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suchy-Dicey, A.M.; Laha, T.; Hoofnagle, A.; Newitt, R.; Sirich, T.L.; Meyer, T.W.; Thummel, K.E.; Yanez, N.D.; Himmelfarb, J.; Weiss, N.S.; et al. Tubular Secretion in CKD. J. Am. Soc. Nephrol. 2016, 27, 2148–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Kestenbaum, B. Proximal Tubular Secretory Clearance: A Neglected Partner of Kidney Function. Clin. J. Am. Soc. Nephrol. 2018, 13, 1291–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Layton, A.T.; Layton, H.E. A computational model of epithelial solute and water transport along a human nephron. PLoS Comput. Biol. 2019, 15, e1006108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallow, K.M.; Greasley, P.J.; Helmlinger, G.; Chu, L.; Heerspink, H.J.; Boulton, D.W. Evaluation of renal and cardiovascular protection mechanisms of SGLT2 inhibitors: Model-based analysis of clinical data. Am. J. Physiol. Renal. Physiol. 2018, 315, F1295–F1306. [Google Scholar] [CrossRef] [Green Version]
- Hallow, K.M.; Gebremichael, Y.; Helmlinger, G.; Vallon, V. Primary proximal tubule hyperreabsorption and impaired tubular transport counterregulation determine glomerular hyperfiltration in diabetes: A modeling analysis. Am. J. Physiol. Renal. Physiol. 2017, 312, F819–F835. [Google Scholar] [CrossRef] [Green Version]
- Hallow, K.M.; Gebremichael, Y. A quantitative systems physiology model of renal function and blood pressure regulation: Model description. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 383–392. [Google Scholar] [CrossRef]
- Wang, X.; Armando, I.; Upadhyay, K.; Pascua, A.; Jose, P.A. The regulation of proximal tubular salt transport in hypertension: An update. Curr. Opin. Nephrol. Hypertens. 2009, 18, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Mather, A.; Pollock, C. Glucose handling by the kidney. Kidney Int. 2011, 79, S1–S6. [Google Scholar] [CrossRef] [Green Version]
- Vallon, V.; Platt, K.A.; Cunard, R.; Schroth, J.; Whaley, J.; Thomson, S.C.; Koepsell, H.; Rieg, T. SGLT2 Mediates Glucose Reabsorption in the Early Proximal Tubule. J. Am. Soc. Nephrol. 2011, 22, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Vallon, V. The proximal tubule in the pathophysiology of the diabetic kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R1009–R1022. [Google Scholar] [CrossRef] [Green Version]
- Coady, M.J.; El Tarazi, A.; Santer, R.; Bissonnette, P.; Sasseville, L.J.; Calado, J.; Lussier, Y.; Dumayne, C.; Bichet, D.G.; Lapointe, J.-Y. MAP17 Is a Necessary Activator of Renal Na+/Glucose Cotransporter SGLT2. JASN 2017, 28, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Horita, S.; Nakamura, M.; Suzuki, M.; Satoh, N.; Suzuki, A.; Homma, Y.; Nangaku, M. The role of renal proximal tubule transport in the regulation of blood pressure. Kidney Res. Clin. Pract. 2017, 36, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Rieg, T.; Vallon, V. Development of SGLT1 and SGLT2 inhibitors. Diabetologia 2018, 61, 2079–2086. [Google Scholar] [CrossRef] [Green Version]
- Titze, J. Sodium balance is not just a renal affair. Curr. Opin. Nephrol. Hypertens. 2014, 23, 101–105. [Google Scholar] [CrossRef]
- Hammon, M.; Grossmann, S.; Linz, P.; Kopp, C.W.; Dahlmann, A.; Garlichs, C.D.; Janka, R.; Cavallaro, A.; Luft, F.C.; Uder, M.; et al. 23Na Magnetic Resonance Imaging of the Lower Leg of Acute Heart Failure Patients during Diuretic Treatment. PLoS ONE 2015, 10, e0141336. [Google Scholar] [CrossRef]
- Titze, J. Water-Free Sodium Accumulation. Semin. Dial. 2009, 22, 253–255. [Google Scholar] [CrossRef]
- Hallow, K.M.; Helmlinger, G.; Greasley, P.J.; McMurray, J.J.V.; Boulton, D.W. Why do SGLT2 inhibitors reduce heart failure hospitalization? A differential volume regulation hypothesis. Diabetes Obes. Metab. 2018, 20, 479–487. [Google Scholar] [CrossRef] [Green Version]
- Arts, T.; Delhaas, T.; Bovendeerd, P.; Verbeek, X.; Prinzen, F.W. Adaptation to mechanical load determines shape and properties of heart and circulation: The CircAdapt model. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1943–H1954. [Google Scholar] [CrossRef]
- Bovendeerd, P.H.M.; Borsje, P.; Arts, T.; van De Vosse, F.N. Dependence of Intramyocardial Pressure and Coronary Flow on Ventricular Loading and Contractility: A Model Study. Ann. Biomed. Eng. 2006, 34, 1833–1845. [Google Scholar] [CrossRef] [Green Version]
- Hallow, K.M.; Van Brackle, C.H.; Anjum, S.; Ermakov, S. Cardiorenal Systems Modeling: Left Ventricular Hypertrophy and Differential Effects of Antihypertensive Therapies on Hypertrophy Regression. Front. Physiol. 2021, 12, 679930. Available online: https://www.frontiersin.org/articles/10.3389/fphys.2021.679930 (accessed on 22 December 2022). [CrossRef] [PubMed]
- Sorger, P.K.; Allerheiligen, S.R.B. Quantitative and Systems Pharmacology in the Post-Genomic Era: New Approaches to Discovering Drugs and Understanding Therapeutic Mechanisms; An NIH White Paper by the QSP Workshop Group; NIH: Bethesda, MD, USA, 2011; Volume 48, pp. 1–47. [Google Scholar]
- Cardinal, O.; Burlot, C.; Fu, Y.; Crosley, P.; Hitt, M.; Craig, M.; Jenner, A.L. Establishing combination PAC-1 and TRAIL regimens for treating ovarian cancer based on patient-specific pharmacokinetic profiles using in silico clinical trials. Comp. Syst. Oncol. 2022, 2, e1035. [Google Scholar] [CrossRef]
- Jenner, A.L.; Cassidy, T.; Belaid, K.; Bourgeois-Daigneault, M.-C.; Craig, M. In silico trials predict that combination strategies for enhancing vesicular stomatitis oncolytic virus are determined by tumor aggressivity. J. Immunother. Cancer 2021, 9, e001387. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, T.; Craig, M. Determinants of combination GM-CSF immunotherapy and oncolytic virotherapy success identified through in silico treatment personalization. PLoS Comput. Biol. 2019, 15, e1007495. [Google Scholar] [CrossRef]
- Jenner, A.L.; Aogo, R.A.; Alfonso, S.; Crowe, V.; Deng, X.; Smith, A.P.; Morel, P.A.; Davis, C.L.; Smith, A.M.; Craig, M. COVID-19 virtual patient cohort suggests immune mechanisms driving disease outcomes. PLOS Pathog. 2021, 17, e1009753. [Google Scholar] [CrossRef]
- Wang, H.; Ma, H.; Sové, R.J.; Emens, L.A.; Popel, A.S. Quantitative systems pharmacology model predictions for efficacy of atezolizumab and nab-paclitaxel in triple-negative breast cancer. J. Immunother. Cancer 2021, 9, e002100. [Google Scholar] [CrossRef]
- Mathur, D.; Barnett, E.; Scher, H.I.; Xavier, J.B. Optimizing the future: How mathematical models inform treatment schedules for cancer. Trends Cancer 2022, 8, 506–516. [Google Scholar] [CrossRef]
- Brater, D.C. Drug dosing in patients with impaired renal function. Clin. Pharmacol. Ther. 2009, 86, 483–489. [Google Scholar] [CrossRef]
- Zhang, L.; Boulton, D.W.; Pfister, M. A pharmacometric approach to quantify the impact of chronic kidney disease and hemodialysis on systemic drug exposure: Application to saxagliptin. J. Clin. Pharmacol. 2012, 52 (Suppl. S1), 126S–133S. [Google Scholar] [CrossRef]
- Gerlach, A.T.; Pickworth, K.K.; Seth, S.K.; Tanna, S.B.; Barnes, J.F. Enoxaparin and bleeding complications: A review in patients with and without renal insufficiency. Pharmacotherapy 2000, 20, 771–775. [Google Scholar] [CrossRef]
- Voelker, J.R.; Cartwright–Brown, D.; Anderson, S.; Leinfelder, J.; Sica, D.A.; Kokko, J.P.; Brater, D.C. Comparison of loop diuretics in patients with chronic renal insufficiency. Kidney Int. 1987, 32, 572–578. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Ng, C.M.; List, J.F.; Pfister, M. Synergy between scientific advancement and technological innovation, illustrated by a mechanism-based model characterizing sodium-glucose cotransporter-2 inhibition. J. Clin. Pharmacol. 2010, 50 (Suppl. S9), 113S–120S. [Google Scholar] [CrossRef]
- Center for Drug Evaluation and Research. Pharmacokinetics in Patients with Impaired Renal Function—Study Design, Data Analysis, and Impact on Dosing and Labeling. U.S. Food and Drug Administration, 9 March 2020. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/pharmacokinetics-patients-impaired-renal-function-study-design-data-analysis-and-impact-dosing-and (accessed on 18 January 2023).
- Gieschke, R.; Carr, R. Conceptual and organizational barriers to quantitative systems pharmacology modeling of pathophysiological systemic drug hypotheses. CPT Pharmacomet. Syst. Pharmacol. 2022, 11, 1556–1559. [Google Scholar] [CrossRef]
- Box, G.E.P.; Draper, N.R. Response Surfaces, Mixtures, and Ridge Analyses; John Wiley & Sons: Hoboken, NJ, USA, 2007. [Google Scholar]
- Chaudhari, S.; Cushen, S.C.; Osikoya, O.; Jaini, P.A.; Posey, R.; Mathis, K.W.; Goulopoulou, S. Mechanisms of Sex Disparities in Cardiovascular Function and Remodeling. Compr. Physiol. 2018, 9, 375–411. [Google Scholar] [CrossRef]
- Rubinow, D.R.; Schmidt, P.J. Sex differences and the neurobiology of affective disorders. Neuropsychopharmacology 2019, 44. [Google Scholar] [CrossRef] [Green Version]
- Chrousos, G.P. Stress and sex versus immunity and inflammation. Sci. Signal. 2010, 3, pe36. [Google Scholar] [CrossRef]
- Munger, K.; Baylis, C. Sex differences in renal hemodynamics in rats. Am. J. Physiol. Ren. Physiol. 1988, 254, F223–F231. [Google Scholar] [CrossRef]
- Sabolić, I.; Asif, A.R.; Budach, W.E.; Wanke, C.; Bahn, A.; Burckhardt, G. Gender differences in kidney function. Pflugers Arch. Eur. J. Physiol. 2007, 455, 397–429. [Google Scholar] [CrossRef]
- Leete, J.; Layton, A.T. Sex-specific long-term blood pressure regulation: Modeling and analysis. Comput. Biol. Med. 2019, 104, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Leete, J.; Gurley, S.; Layton, A.T. Modeling sex differences in the renin angiotensin system and the efficacy of antihypertensive therapies. Comput. Chem. Eng. 2018, 112, 253–264. [Google Scholar] [CrossRef]
- Guillaud, F.; Hannaert, P. A Computational Model of the Circulating Renin-Angiotensin System and Blood Pressure Regulation. Acta Biotheor. 2010, 58, 143–170. [Google Scholar] [CrossRef] [PubMed]
- Sabolić, I.; Vrhovac, I.; Eror, D.B.; Gerasimova, M.; Rose, M.; Breljak, D.; Ljubojević, M.; Brzica, H.; Sebastiani, A.; Thal, S.C.; et al. Expression of Na+-d-glucose cotransporter SGLT2 in rodents is kidney-specific and exhibits sex and species differences. Am. J. Physiol. Cell Physiol. 2012, 302, C1174–C1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veiras, L.C.; Girardi, A.C.; Curry, J.; Pei, L.; Ralph, D.L.; Tran, A.; Castelo-Branco, R.C.; Pastor-Soler, N.; Arranz, C.T.; Yu, A.S.; et al. Sexual Dimorphic Pattern of Renal Transporters and Electrolyte Homeostasis. J. Am. Soc. Nephrol. 2017, 28, 3504–3517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, R.; McDonough, A.A.; Layton, A.T. Functional implications of the sex differences in transporter abundance along the rat nephron: Modeling and analysis. Am. J. Physiol. Ren. Physiol. 2019, 317, F1462–F1474. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; McDonough, A.A.; Layton, H.E.; Layton, A.T. Functional implications of sexual dimorphism of transporter patterns along the rat proximal tubule: Modeling and analysis. Am. J. Physiol. Ren. Physiol. 2018, 315, F692–F700. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; McDonough, A.A.; Layton, A.T. Sex differences in solute transport along the nephrons: Effects of Na+ transport inhibition. Am. J. Physiol. Ren. Physiol. 2020, 319, F487–F505. [Google Scholar] [CrossRef]
- Hu, R.; McDonough, A.A.; Layton, A.T. Sex differences in solute and water handling in the human kidney: Modeling and functional implications. iScience 2021, 24, 102667. [Google Scholar] [CrossRef]
- Layton, A.T. His and her mathematical models of physiological systems. Math. Biosci. 2021, 338, 108642. [Google Scholar] [CrossRef]
- Sullivan, J.C.; Pardieck, J.L.; Hyndman, K.A.; Pollock, J.S. Renal NOS activity, expression, and localization in male and female spontaneously hypertensive rats. Am. J. Physiol. -Regul. Integr. Comp. Physiol. 2010, 298, R61–R69. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Edwards, A.; Layton, A.T. Effects of pH and medullary blood flow on oxygen transport and sodium reabsorption in the rat outer medulla. Am. J. Physiol. Ren. Physiol. 2010, 298, F1369–F1383. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Sullivan, J.C.; Edwards, A.; Layton, A.T. Sex-specific computational models of the spontaneously hypertensive rat kidneys: Factors affecting nitric oxide bioavailability. Am. J. Physiol. Ren. Physiol. 2017, 313, F174–F183. [Google Scholar] [CrossRef] [Green Version]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner–La Rocca, H.-P.; Choi, D.-J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- Holgado, J.L.; Lopez, C.; Fernandez, A.; Sauri, I.; Uso, R.; Trillo, J.L.; Vela, S.; Nuñez, J.; Redon, J.; Ruiz, A. Acute kidney injury in heart failure: A population study. ESC Heart Fail. 2020, 7, 415–422. [Google Scholar] [CrossRef] [Green Version]
- Empagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2023, 388, 117–127. [CrossRef]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic kidney disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ndiaye, J.F.; Nekka, F.; Craig, M. Understanding the Mechanisms and Treatment of Heart Failure: Quantitative Systems Pharmacology Models with a Focus on SGLT2 Inhibitors and Sex-Specific Differences. Pharmaceutics 2023, 15, 1002. https://doi.org/10.3390/pharmaceutics15031002
Ndiaye JF, Nekka F, Craig M. Understanding the Mechanisms and Treatment of Heart Failure: Quantitative Systems Pharmacology Models with a Focus on SGLT2 Inhibitors and Sex-Specific Differences. Pharmaceutics. 2023; 15(3):1002. https://doi.org/10.3390/pharmaceutics15031002
Chicago/Turabian StyleNdiaye, Jean François, Fahima Nekka, and Morgan Craig. 2023. "Understanding the Mechanisms and Treatment of Heart Failure: Quantitative Systems Pharmacology Models with a Focus on SGLT2 Inhibitors and Sex-Specific Differences" Pharmaceutics 15, no. 3: 1002. https://doi.org/10.3390/pharmaceutics15031002