
Targeting IKKβ Activity to Limit Sterile Inflammation in Acetaminophen-Induced Hepatotoxicity in Mice
 , , , , , and
, , , , , and 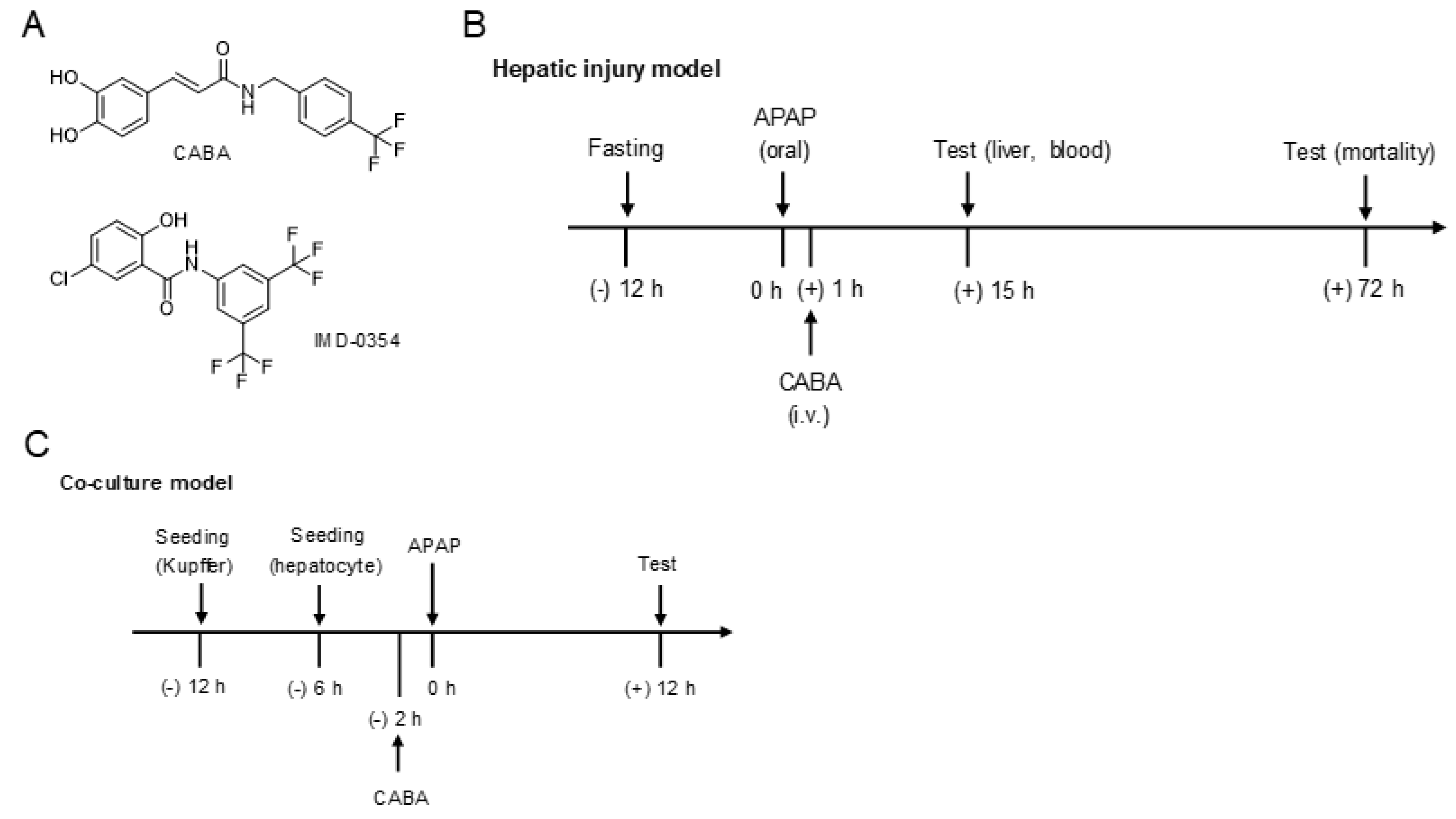{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical Synthesis of CABA
2.2. Antibodies and Pharmacological Agents
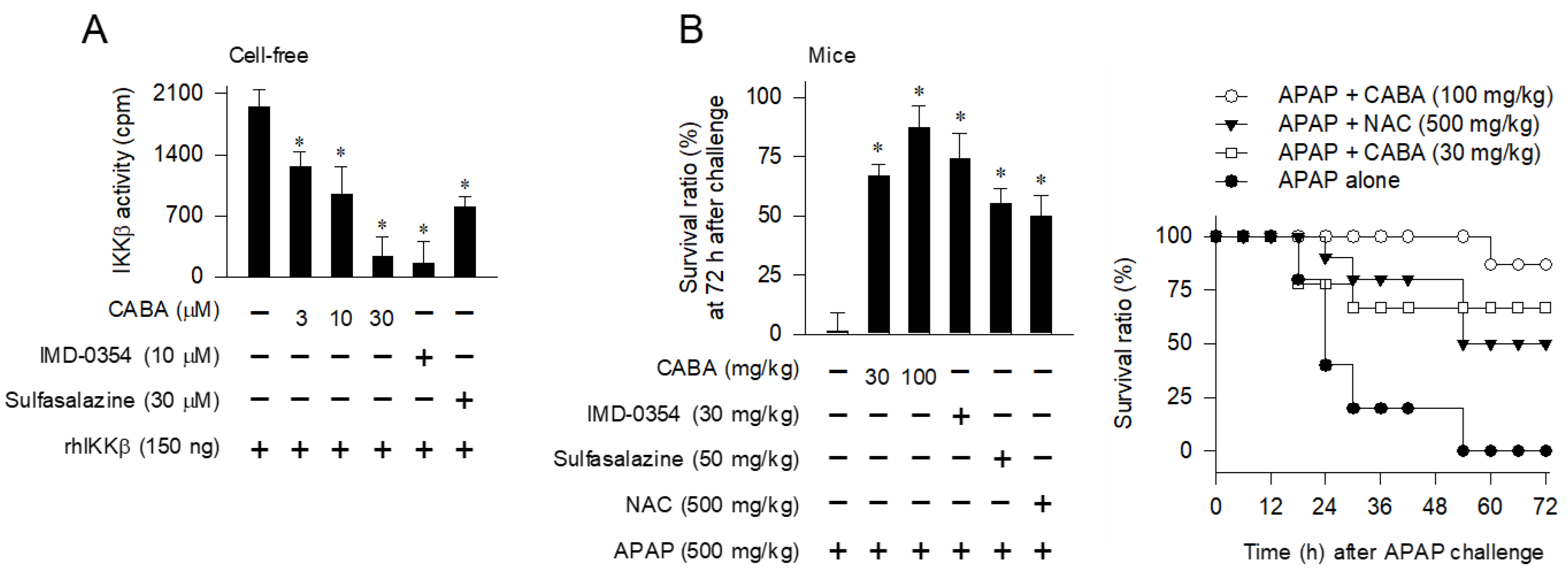
2.3. Cell-Free Kinase Assay of IKKβ or TAK1
2.4. Hepatic Injury Model in Mice
2.5. Co-Culture Model between Primary Hepatocytes and Kupffer Cells
2.6. Western Blot Analysis
2.7. Luciferase Reporter Assay
2.8. RT-PCR Analysis
2.9. ELISA
2.10. Data Analysis
3. Results
3.1. Rescue of APAP-Induced Mortality in Mice by Small-Molecule Inhibitors of IKKβ Activity
3.2. Protection against Necrotic Injury in the Liver by IKKβ Inhibitors
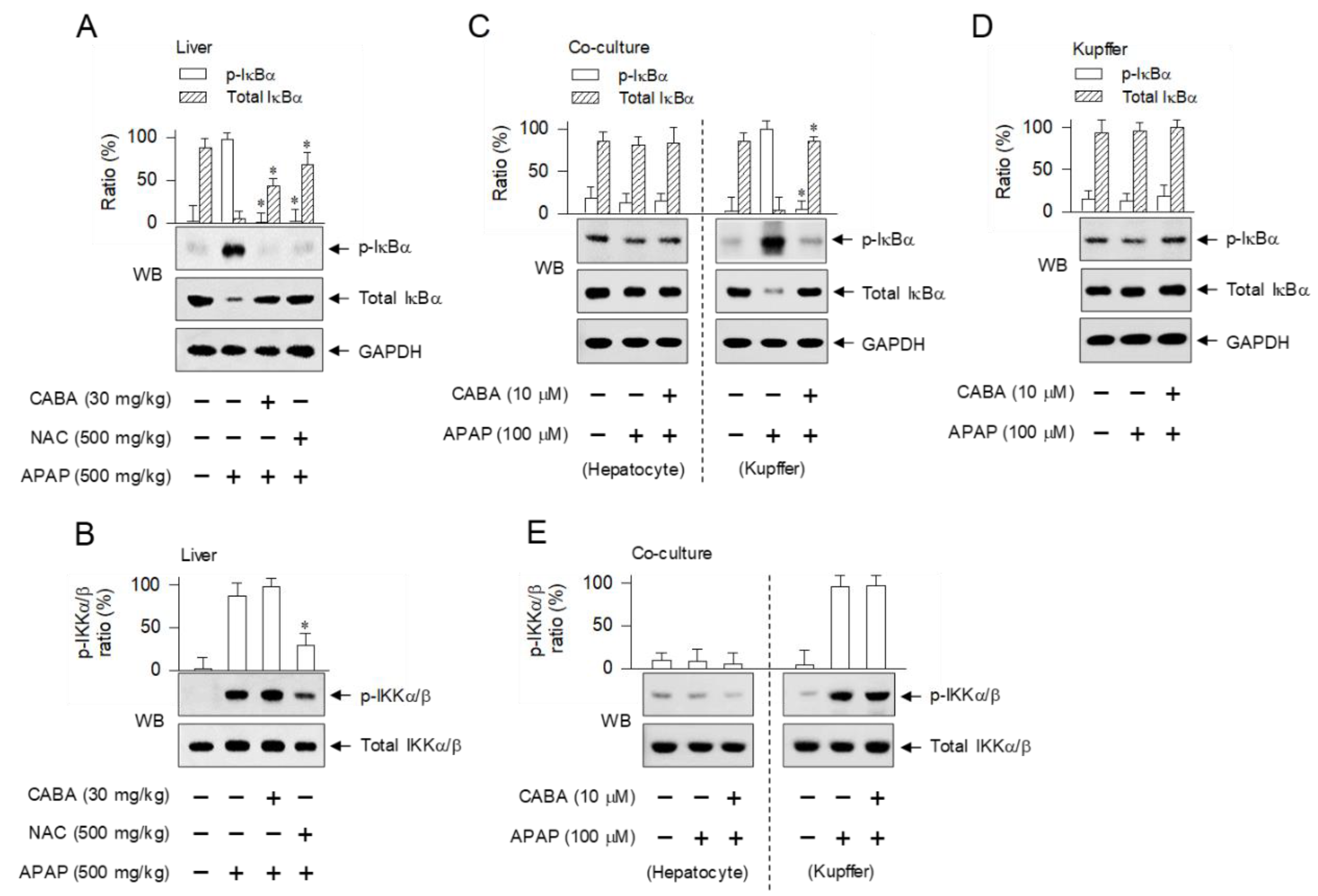
3.3. Inhibition of IKKβ-Catalyzed Phosphorylation of IκBα in Kupffer Cells but Not in Hepatocytes by CABA
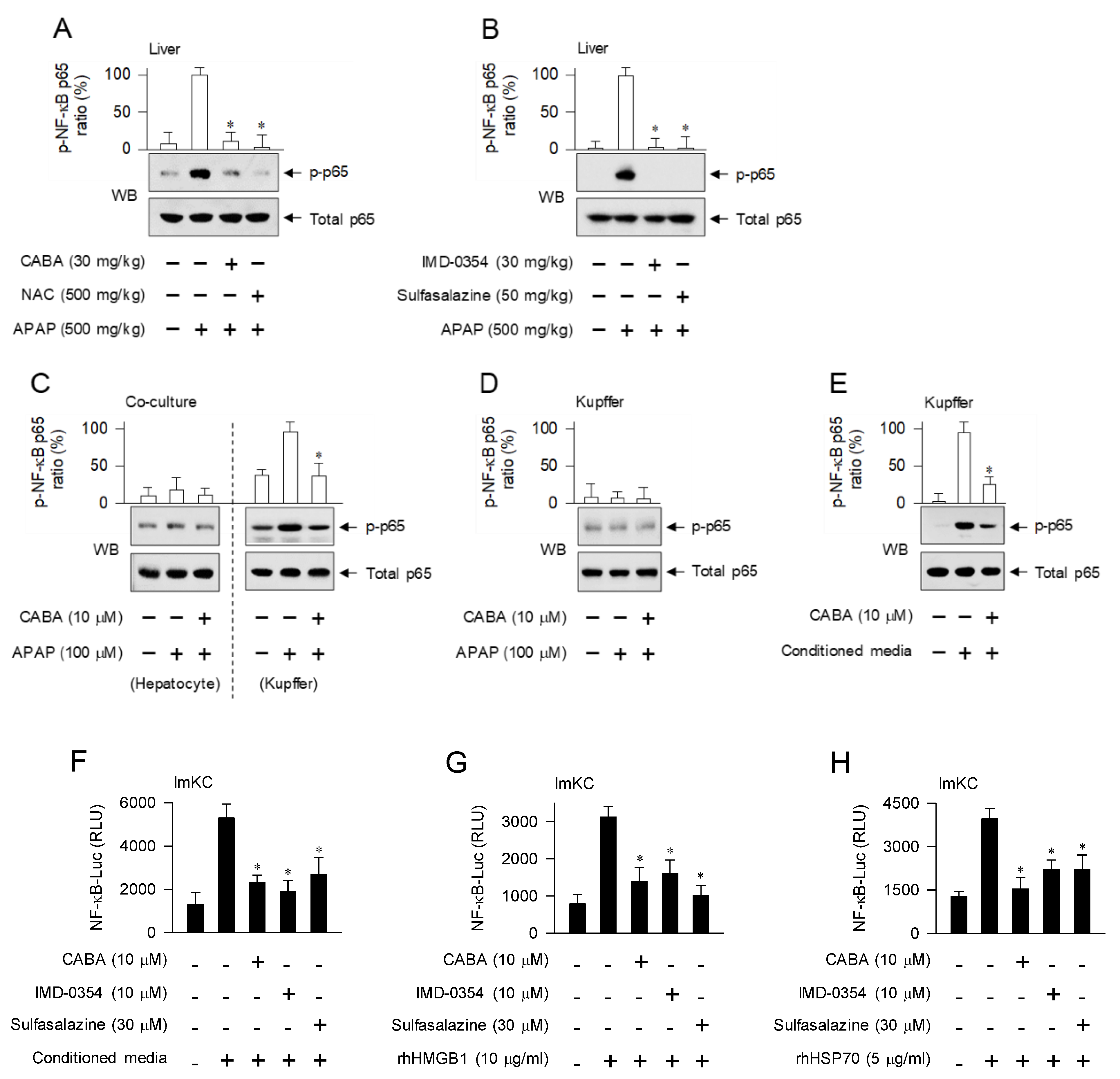
3.4. Interruption of DAMP-Induced Activation of NF-κB Activity in Kupffer Cells by IKKβ Inhibitors
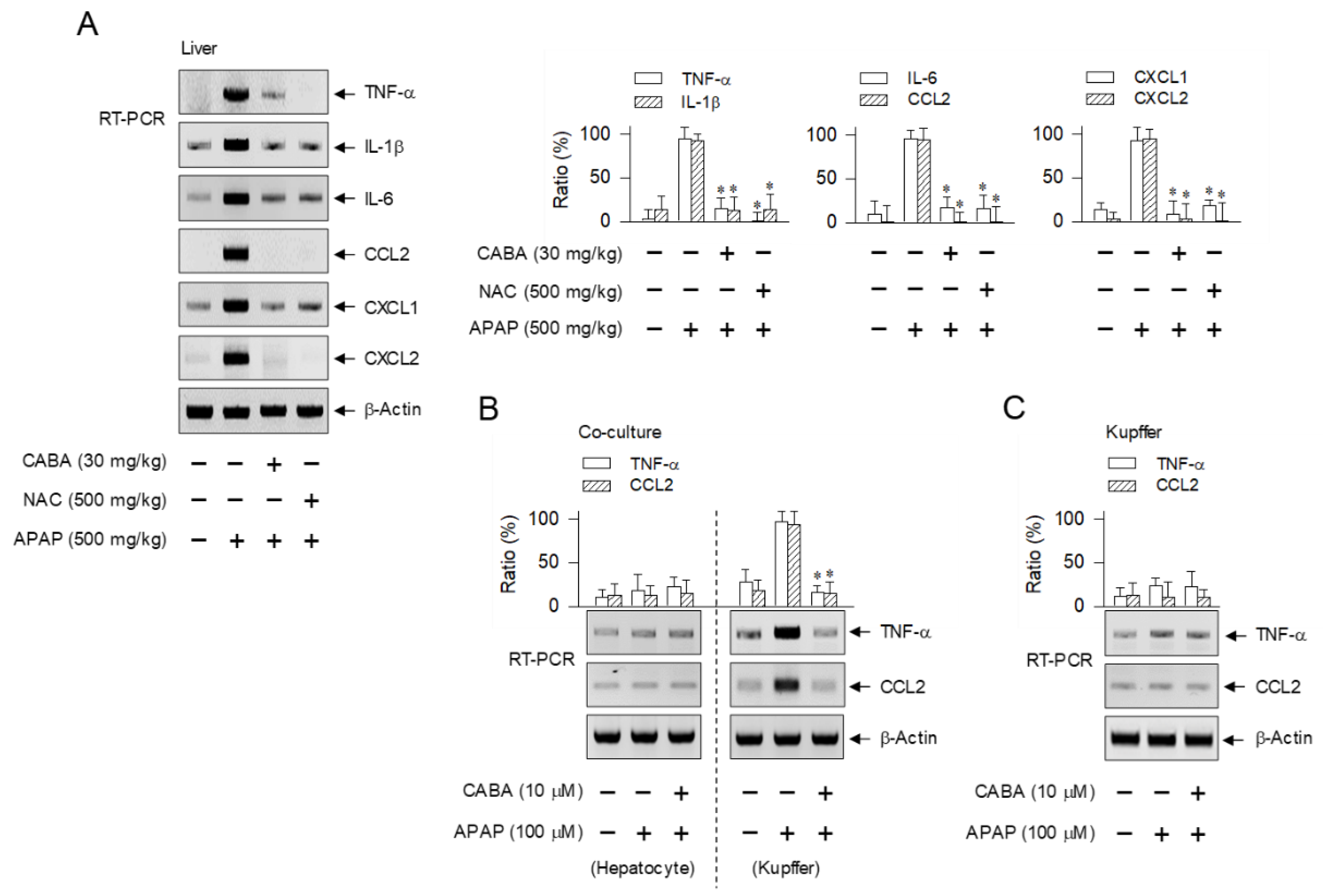
3.5. Suppression of Cytokine and Chemokine Expression during Sterile Inflammation by CABA
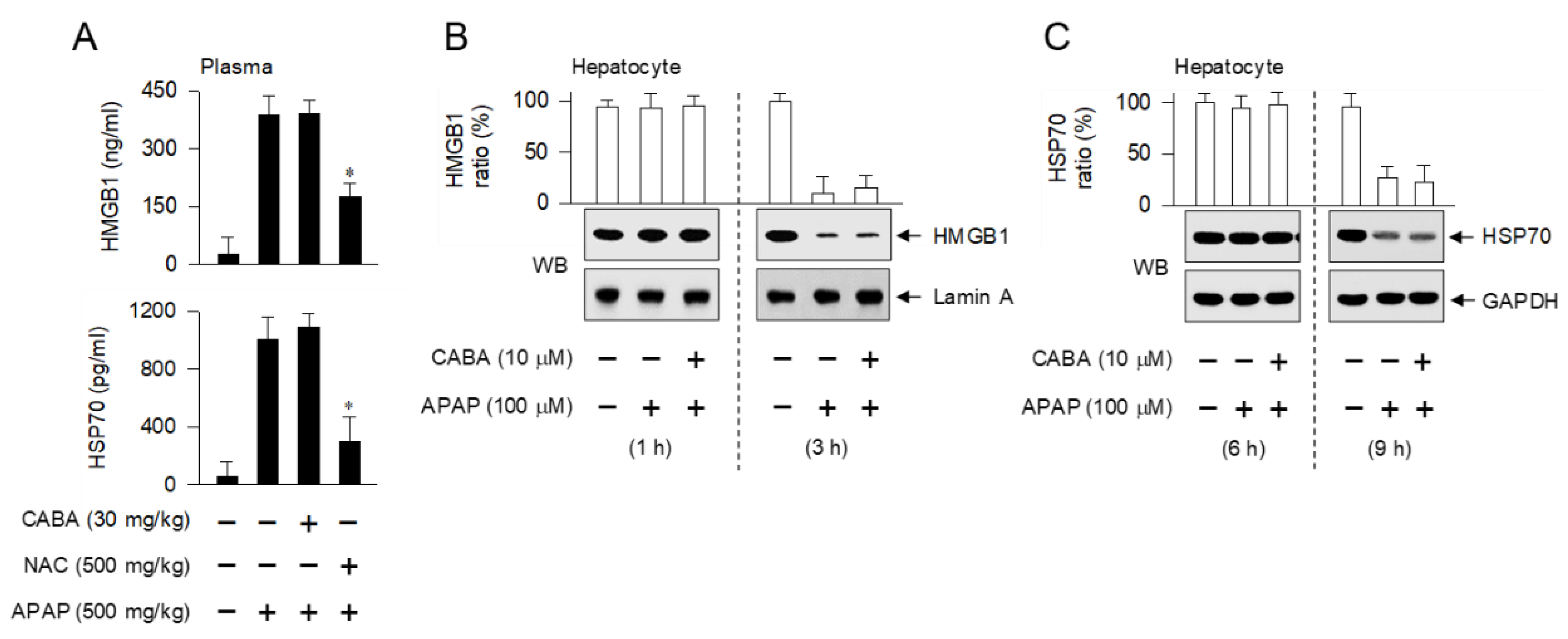
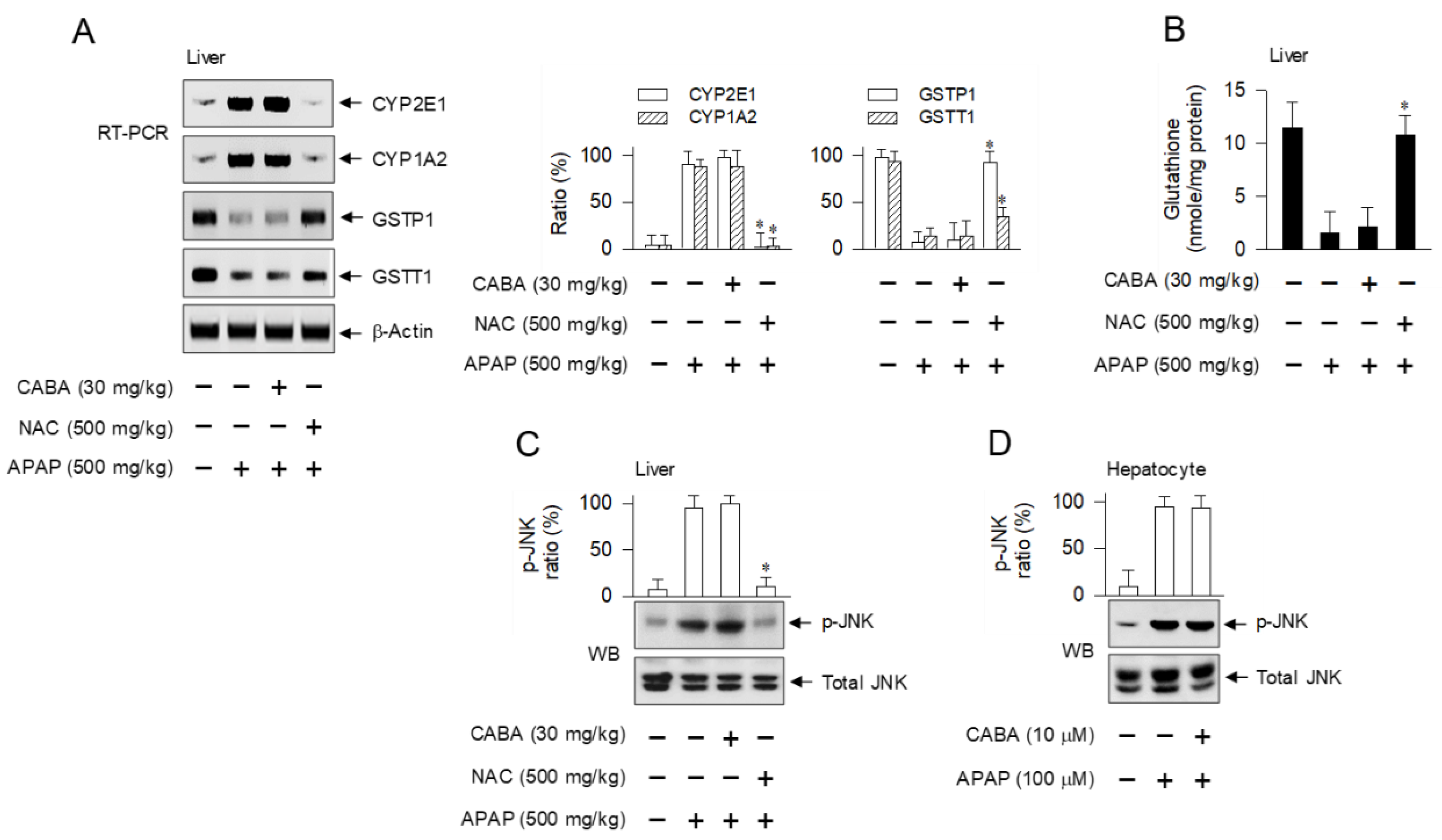
3.6. Inert Effectiveness on DAMP Release and Oxidative Stress in Hepatocytes by CABA
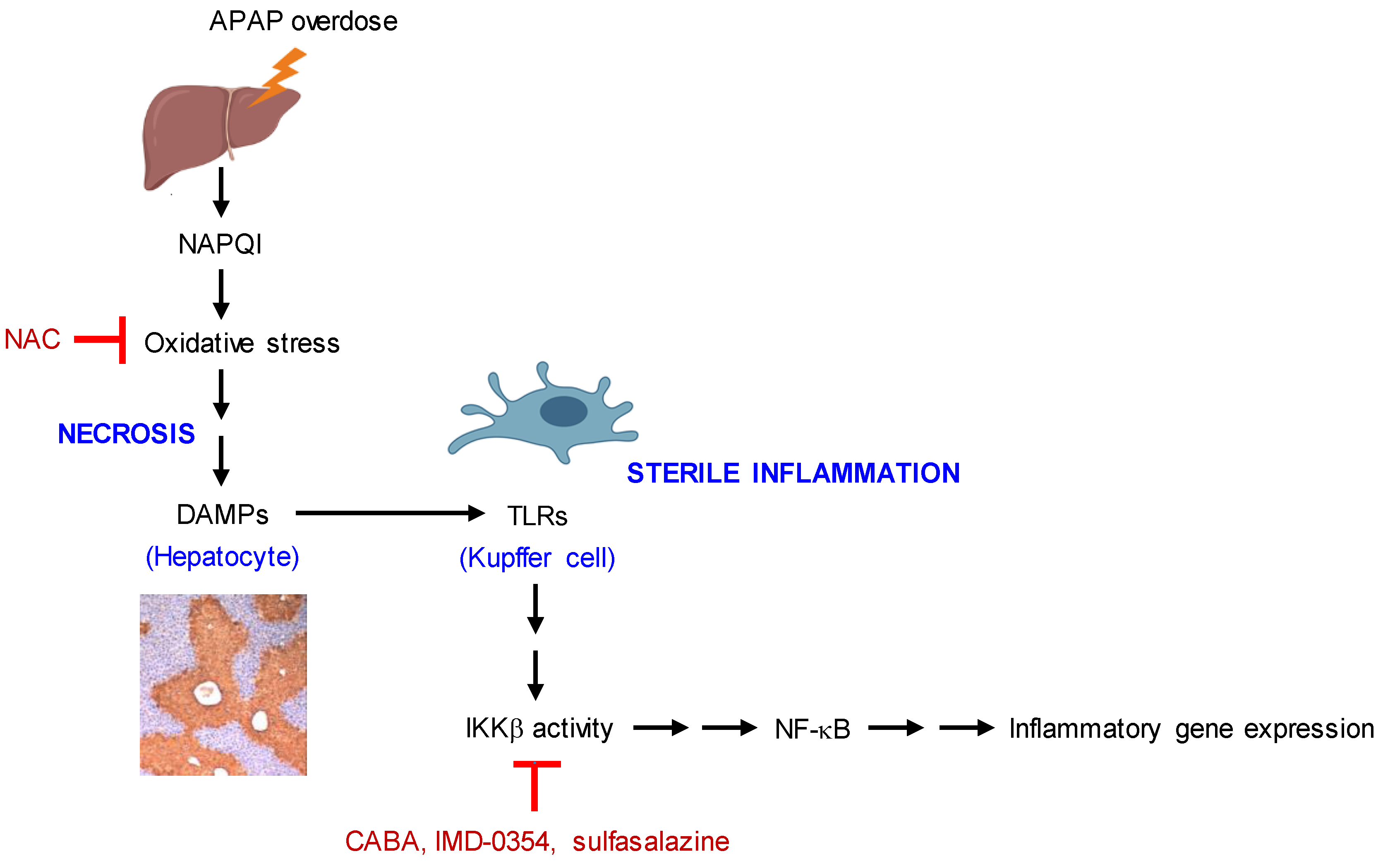
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- McGill, M.R.; Jaeschke, H. Metabolism and disposition of acetaminophen: Recent advances in relation to hepatotoxicity and diagnosis. Pharm. Res. 2013, 30, 2174–2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, M.; Huo, Y.; Yin, S.; Hu, H. Mechanisms of acetaminophen-induced liver injury and its implications for therapeutic interventions. Redox Biol. 2018, 17, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Jaeschke, H. Acetaminophen hepatotoxicity: A mitochondrial perspective. Adv. Pharmacol. 2019, 85, 195–219. [Google Scholar] [CrossRef] [PubMed]
- Hinson, J.A.; Reid, A.B.; McCullough, S.S.; James, L.P. Acetaminophen-induced hepatotoxicity: Role of metabolic activation, reactive oxygen/nitrogen species, and mitochondrial permeability transition. Drug Metab. Rev. 2004, 36, 805–822. [Google Scholar] [CrossRef]
- Hanawa, N.; Shinohara, M.; Saberi, B.; Gaarde, W.A.; Han, D.; Kaplowitz, N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J. Biol. Chem. 2008, 283, 13565–13577. [Google Scholar] [CrossRef] [Green Version]
- Bajt, M.L.; Cover, C.; Lemasters, J.J.; Jaeschke, H. Nuclear translocation of endonuclease G and apoptosis-inducing factor during acetaminophen-induced liver cell injury. Toxicol. Sci. 2006, 94, 217–225. [Google Scholar] [CrossRef]
- Jaeschke, H.; Ramachandran, A. Mechanisms and pathophysiological significance of sterile inflammation during acetaminophen hepatotoxicity. Food Chem. Toxicol. 2020, 138, 111240. [Google Scholar] [CrossRef]
- Mihm, S. Danger-Associated Molecular Patterns (DAMPs): Molecular Triggers for Sterile Inflammation in the Liver. Int. J. Mol. Sci. 2018, 19, 3104. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Tonnesseen, T.I. DAMPs and sterile inflammation in drug hepatotoxicity. Hepatol. Int. 2019, 13, 42–50. [Google Scholar] [CrossRef]
- Krenkel, O.; Mossanen, J.C.; Tacke, F. Immune mechanisms in acetaminophen-induced acute liver failure. Hepatobiliary Surg. Nutr. 2014, 3, 331–343. [Google Scholar] [CrossRef]
- Jaeschke, H.; Williams, C.D.; Ramachandran, A.; Bajt, M.L. Acetaminophen hepatotoxicity and repair: The role of sterile inflammation and innate immunity. Liver Int. 2012, 32, 8–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnham, K.; Yang, T.; Smith, H.; Knight, S. A review of alternative intravenous acetylcysteine regimens for acetaminophen overdose. Expert Rev. Clin. Pharmacol. 2021, 14, 1267–1278. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFκB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, X.; Zhang, Z.; Shen, H.; Xiong, Y.; Shah, Y.M.; Liu, Y.; Fan, X.G.; Rui, L. Hepatic NF-κB-Inducing Kinase and Inhibitor of NF-κB Kinase Subunit α Promote Liver Oxidative Stress, Ferroptosis, and Liver Injury. Hepatol. Commun. 2021, 5, 1704–1720. [Google Scholar] [CrossRef]
- Tanaka, A.; Muto, S.; Jung, K.; Itai, A.; Matsuda, H. Topical application with a new NF-κB inhibitor improves atopic dermatitis in NC/NgaTnd mice. J. Investig. Dermatol. 2007, 127, 855–863. [Google Scholar] [CrossRef]
- Weber, C.K.; Liptay, S.; Wirth, T.; Adler, G.; Schmid, R.M. Suppression of NF-κB activity by sulfasalazine is mediated by direct inhibition of IκB kinases α and β. Gastroenterology 2000, 119, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Ruoß, M.; Vosough, M.; Königsrainer, A.; Nadalin, S.; Wagner, S.; Sajadian, S.; Huber, D.; Heydari, Z.; Ehnert, S.; Hengstler, J.G.; et al. Towards improved hepatocyte cultures: Progress and limitations. Food Chem. Toxicol. 2020, 138, 111188. [Google Scholar] [CrossRef]
- Mohar, I.; Brempelis, K.J.; Murray, S.A.; Ebrahimkhani, M.R.; Crispe, I.N. Isolation of Non-parenchymal Cells from the Mouse Liver. Methods Mol. Biol. 2015, 1325, 3–17. [Google Scholar] [CrossRef]
- Li, Y.R.; Lin, C.C.; Huang, C.Y.; Wong, Y.H.; Hsieh, C.H.; Wu, H.W.; Chen, J.J.W.; Wu, Y.S. Study of the inhibitory effects on TNF-α-induced NF-κB activation of IMD0354 analogs. Chem. Biol. Drug Des. 2017, 90, 1307–1311. [Google Scholar] [CrossRef]
- Lala, V.; Goyal, A.; Minter, D.A. Liver Function Tests; StatPearls © 2022; StatPearls Publishing LLC: Treasure Island, FL, USA, 2022. [Google Scholar]
- Ramaiah, S.K.; Jaeschke, H. Role of neutrophils in the pathogenesis of acute inflammatory liver injury. Toxicol. Pathol. 2007, 35, 757–766. [Google Scholar] [CrossRef]
- Kanarek, N.; Ben-Neriah, Y. Regulation of NF-κB by ubiquitination and degradation of the IκBs. Immunol. Rev. 2012, 246, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.P.; Behar, M.; Birnbaum, H.A.; Hoffmann, A.; Wright, P.E.; Ghosh, G. Analysis of the RelA:CBP/p300 interaction reveals its involvement in NF-κB-driven transcription. PLoS Biol. 2013, 11, e1001647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, K.H.; Staudt, L.M. Toll-like receptor signaling. Cold Spring Harb. Perspect. Biol. 2013, 5, a011247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madrigal, P.; Alasoo, K. AP-1 Takes Centre Stage in Enhancer Chromatin Dynamics. Trends Cell Biol. 2018, 28, 509–511. [Google Scholar] [CrossRef]
- Taniguchi, T.; Ogasawara, K.; Takaoka, A.; Tanaka, N. IRF family of transcription factors as regulators of host defense. Annu. Rev. Immunol. 2001, 19, 623–655. [Google Scholar] [CrossRef]
- Ye, J.; Wang, L.; Zhang, X.; Tantishaiyakul, V.; Rojanasakul, Y. Inhibition of TNF-α gene expression and bioactivity by site-specific transcription factor-binding oligonucleotides. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L386–L394. [Google Scholar] [CrossRef] [Green Version]
- Yeagley, D.; Lang, C.H. Endotoxin-Induced IL-6 Promoter Activation in Skeletal Muscle Requires an NF-κB Site. Int. J. Interferon Cytokine Mediat. Res. 2010, 2010, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Ping, D.; Jones, P.L.; Boss, J.M. TNF regulates the in vivo occupancy of both distal and proximal regulatory regions of the MCP-1/JE gene. Immunity 1996, 4, 455–469. [Google Scholar] [CrossRef] [Green Version]
- Son, D.S.; Roby, K.F. Interleukin-1α-induced chemokines in mouse granulosa cells: Impact on keratinocyte chemoattractant chemokine, a CXC subfamily. Mol. Endocrinol. 2006, 20, 2999–3013. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, H.W.; Trautwein, C.; Tacke, F. Functional role of monocytes and macrophages for the inflammatory response in acute liver injury. Front. Physiol. 2012, 3, 56. [Google Scholar] [CrossRef] [Green Version]
- Gum, S.I.; Cho, M.K. Recent updates on acetaminophen hepatotoxicity: The role of nrf2 in hepatoprotection. Toxicol. Res. 2013, 29, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Yin, S.; Yan, M.; Win, S.; Aung Than, T.; Aghajan, M.; Hu, H.; Kaplowitz, N. Protective role of p53 in acetaminophen hepatotoxicity. Free Radic. Biol. Med. 2017, 106, 111–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borude, P.; Bhushan, B.; Gunewardena, S.; Akakpo, J.; Jaeschke, H.; Apte, U. Pleiotropic Role of p53 in Injury and Liver Regeneration after Acetaminophen Overdose. Am. J. Pathol. 2018, 188, 1406–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M.; Fuentes, M.E.; Yamaguchi, K.; Durnin, M.H.; Dalrymple, S.A.; Hardy, K.L.; Goeddel, D.V. Embryonic lethality, liver degeneration, and impaired NF-κB activation in IKK-β-deficient mice. Immunity 1999, 10, 421–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, S.; Kamata, H.; Luo, J.L.; Leffert, H.; Karin, M. IKKβ couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakurai, T.; Maeda, S.; Chang, L.; Karin, M. Loss of hepatic NF-κB activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc. Natl. Acad. Sci. USA 2006, 103, 10544–10551. [Google Scholar] [CrossRef] [Green Version]
- Yau, T.O.; Chan, C.F.; Gee-San Lam, S.; Cheung, O.F.; Ching, Y.P.; Jin, D.Y.; Sham, M.H.; Ng, I.O. Hepatocyte-specific activation of NF-κB does not aggravate chemical hepatocarcinogenesis in transgenic mice. J. Pathol. 2009, 217, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeld, M.E.; Prichard, L.; Shiojiri, N.; Fausto, N. Prevention of hepatic apoptosis and embryonic lethality in RelA/TNFR-1 double knockout mice. Am. J. Pathol. 2000, 156, 997–1007. [Google Scholar] [CrossRef] [Green Version]
- Geisler, F.; Algül, H.; Paxian, S.; Schmid, R.M. Genetic inactivation of RelA/p65 sensitizes adult mouse hepatocytes to TNF-induced apoptosis in vivo and in vitro. Gastroenterology 2007, 132, 2489–2503. [Google Scholar] [CrossRef]
- Dambach, D.M.; Durham, S.K.; Laskin, J.D.; Laskin, D.L. Distinct roles of NF-κB p50 in the regulation of acetaminophen-induced inflammatory mediator production and hepatotoxicity. Toxicol. Appl. Pharmacol. 2006, 211, 157–165. [Google Scholar] [CrossRef]
- Ringelhan, M.; Schmid, R.M.; Geisler, F. The NF-κB subunit RelA/p65 is dispensable for successful liver regeneration after partial hepatectomy in mice. PLoS ONE 2012, 7, e46469. [Google Scholar] [CrossRef] [PubMed] [Green Version]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.-H.; Jung, D.-E.; Song, J.Y.; Jung, J.; Jung, J.-K.; Lee, H.; Roh, E.; Hong, J.T.; Han, S.-B.; Kim, Y. Targeting IKKβ Activity to Limit Sterile Inflammation in Acetaminophen-Induced Hepatotoxicity in Mice. Pharmaceutics 2023, 15, 710. https://doi.org/10.3390/pharmaceutics15020710
Kim S-H, Jung D-E, Song JY, Jung J, Jung J-K, Lee H, Roh E, Hong JT, Han S-B, Kim Y. Targeting IKKβ Activity to Limit Sterile Inflammation in Acetaminophen-Induced Hepatotoxicity in Mice. Pharmaceutics. 2023; 15(2):710. https://doi.org/10.3390/pharmaceutics15020710
Chicago/Turabian StyleKim, Song-Hee, Da-Eun Jung, Jin Yong Song, Jihye Jung, Jae-Kyung Jung, Heesoon Lee, Eunmiri Roh, Jin Tae Hong, Sang-Bae Han, and Youngsoo Kim. 2023. "Targeting IKKβ Activity to Limit Sterile Inflammation in Acetaminophen-Induced Hepatotoxicity in Mice" Pharmaceutics 15, no. 2: 710. https://doi.org/10.3390/pharmaceutics15020710