
Regulatory Requirements for the Development of Second-Entry Semisolid Topical Products in the European Union

 , and
, and 
Abstract
:1. Introduction
2. Legal Basis
3. Composition Sameness (Q1/Q2) for the Development of Second-Entry Topical Products
4. Physicochemical and Structural (Q3) Characterization of Second-Entry Topical Products
4.1. General Requirements
- (a)
- Appearance and texture, including the look, feel and smell of the dispensed product;The EMA draft guideline [5] has indicated only appearance but the scope is the same.
- (b)
- Phase states, including high-resolution micrographs to show the absence or presence of undissolved particles to identify single-phase and multiple-phase products;
- (c)
- Structural organization of the matter, including particle size distribution and crystal habit, and/or emulsion globule size distribution and identification of the type of emulsion, e.g., oil in water or water in oil;
- (d)
- Polymorphic form of the active substance within the product for products with a suspended active substance.The EMA draft guideline [5] indicates the particle size distribution and polymorphism for suspensions and the globule size distribution for emulsions, but crystal habit is not mentioned; however, it is mentioned as a factor that may affect bioavailability and should be included in stability studies;
- (e)
- Rheological behavior of liquid and semisolid dosage forms, including
- Complete flow curves plotting shear stress vs. shear rate, and viscosity vs. shear rate until low or high-shear plateaus are achieved. Apparent viscosity should be reported at least at low, middle, and high shear rates.The EMA draft guideline specifies that these shear stress flow sweep experiments should comprise multiple data points across the range of increasing and decreasing shear rates so that any linear portions of the up-curves or down-curves are clearly identified. In contrast to the US FDA, for the EMA, the resulting curves should be characterized by fitting to (modified) power law equations so that numerical data can be produced [15]; however, the minimum of the three share rates to estimate apparent viscosities is not defined, only the need to measure the apparent viscosity at specified shear rates across the rheograms, e.g., η100, and the quantification of the thixotropic relative area (SR) if appropriate.
- Yield stress, if the products exhibit plastic flow behavior.
- Linear viscoelastic response (storage and loss moduli vs. frequency).The EMA draft guideline specifies that viscoelastic storage and loss moduli (G’ and G”), and loss tangent (tan δ) should be determined in these oscillatory strain sweep (shear strain oscillatory amplitude sweep) experiments [15] to obtain parameters that can be compared objectively. The EMA draft guideline also mentions the creep test.
- (f)
- Water activity and/or drying rate, relevant for products with volatile excipients including water;
- (g)
- pH and buffering for products with an aqueous component;
- (h)
- Oleaginous components, which should be characterized according to the tests listed in the US Pharmacopeia;
- (i)
- Specific gravity (density);
- (j)
- Metamorphosis of the product when dispensed from the containers.
4.2. Product-Specific Requirements
4.2.1. Solutions
4.2.2. Suspensions
- (a)
- the drug has a narrow therapeutic index (NTI), but none have been classified as NTI in this route of administration;
- (b)
- the drug exhibits dose-related systemic toxicity, but this can be addressed by comparing systemic exposure with conventional PK BE studies;
- (c)
- the means by which the active substance reaches the local site of action is not established or understood; this is not expected presently and, moreover, it might be claimed that if the formulation is considered to be simple and an extended pharmaceutical equivalence is met, the applied product will be therapeutically equivalent in any case;
- (d)
- the method of administration is not the same, which might be a limitation only if the application device/method is patented;
- (e)
- the product cannot be fully characterized with respect to quality attributes, but this is not foreseen if the reference product has been authorized recently;
- (f)
- it is not possible to measure a quantifiable permeation kinetic or PD event for the product; however, a stratum corneum sampling/tape stripping (TS) study might be used; and
- (g)
- in vitro and in vivo permeation kinetic and PD studies are not applicable or are considered insufficiently predictive of clinical response, e.g., products indicated for the treatment of open wounds and ulcers, which would apply only for complex formulations as explained above. If the formulation is considered simple and extended pharmaceutical equivalence is met, the applied product will be therapeutically equivalent in any case. Therefore, the main limitation is the possibility of developing sensitive IVRT or an in vitro permeation test (IVPT), or the reproducibility of the TS technique.
4.2.3. Gels
4.2.4. Ointments

{kind=link}
| Drug | RS | Q | AT | M | PS | GS | AV | FC | YS | LVR | WA | pH | SG | DR | OC | IVRT | IVPT |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Acyclovir [75] | 18604 | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||||||
| Betamethasone Dipropionate; Calcipotriene [68] | 21852 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||||
| Calcipotriene [76] | 20273 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||
| Crisaborole [77] | 207695 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||
| Gentamicin Sulfate [70] | 62351 | ||||||||||||||||
| Mupirocin [69] | 50591 | √ | √ | √ | √ | √ | √ | √ | √ | ||||||||
| Nitroglycerin [78] | 21359 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||||
| Nystatin [71] | 62124 | ||||||||||||||||
| Nystatin; Triamcinolone Acetonide [74] | 63305 | ||||||||||||||||
| Tacrolimus [79] | 50777 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||
| Tacrolimus [80] | 50777 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||
| Tirbanibulin [81] | 213189 | √ | √ | √ | √ | √ | √ | √ | √ | ||||||||
| Triamcinolone Acetonide [71] | 087385 | ||||||||||||||||
| Triamcinolone Acetonide [73] | 08995 |
4.2.5. Creams
4.2.6. Lotions
4.2.7. Other Topical Dosage Forms
- (a)
- Microscopic birefringence analysis of the dispensed foam after complete collapse, in order to determine whether any crystals of undissolved active substance form during dispensing.
- (b)
- Time to break (from dispensing to complete foam collapse) analysis, conducted at 30 °C, 33 °C, 35 °C, and 40 °C under controlled relative humidity conditions.
- (c)
- Weight per volume of uncollapsed foam.
5. Statistical Comparison of Physicochemical Properties
6. In vitro Release Tests (IVRT)
7. Kinetic Studies to Address Local Availability
8. Pharmacodynamic and Clinical Endpoints
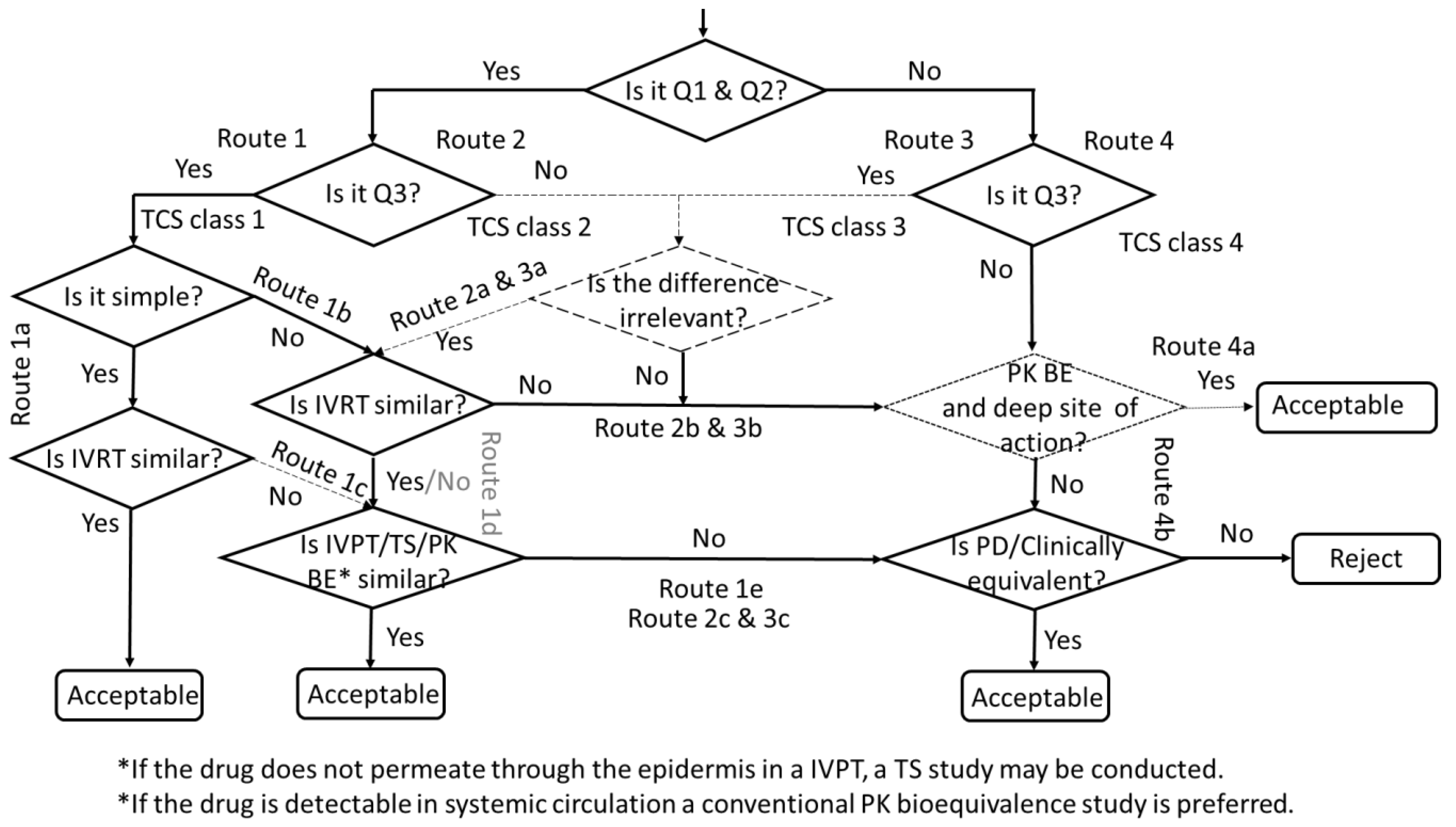
9. Decision Tree for the Development of Second-Entry Topical Products
10. Development of Additional Strengths
11. Virtual Bioequivalence
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). Guideline on the Investigation of Bioequivalence (CPMP/EWP/QWP/1401/98 Rev. 1/ Corr **). London, 20 January 2010. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf (accessed on 13 December 2022).
- European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). Guideline on the Requirements for Clinical Documentation for Orally Inhaled Products Including Requirements for Demonstration of Therapeutic Equivalence between Two Inhaled Products for Use in Treatment of Asthma & Chronic Obstructive Pulmonary Disease (CPMP/EWP/4151/00 Rev. 1). London, 22 January 2009. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-requirements-clinical-documentation-orally-inhaled-products-including-requirements_en.pdf (accessed on 13 December 2022).
- European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). Guideline on Equivalence Studies for the Demonstration of Therapeutic Equivalence for Locally Applied, Locally Acting Products in the Gastrointestinal Tract (CPMP/EWP/239/95 Rev. 1, Corr.1*). London, 18 October 2018. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-equivalence-studies-demonstration-therapeutic-equivalence-locally-applied-locally-acting_en.pdf (accessed on 13 December 2022).
- The European Agency for the Evaluation of Medicinal Products (EMEA). Committee of Proprietary Medicinal Products (CHMP). Note for Guidance on the Clinical Requirements for Locally Applied Locally Acting Products Containing Known Constituents. CPMP/EWP/239/95 final. London, November 1995. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/note-guidance-clinical-requirements-locally-applied-locally-acting-products-containing-known_en.pdf (accessed on 13 December 2022).
- European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). Draft guideline on quality and equivalence of topical products (CHMP/QWP/708282/2018). London, 18 October 2018. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-quality-equivalence-topical-products_en.pdf (accessed on 13 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Physicochemical and Structural (Q3) Characterization of Topical Drug Products Submitted in ANDAs. Draft Guidance for Industry. October 2022. Available online: https://www.fda.gov/media/162471/download (accessed on 13 December 2022).
- Consolidated Text of Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community Code Relating to Medicinal Products for Human Use. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A02001L0083-20220101 (accessed on 13 December 2022).
- García-Arieta, A. Interactions between active pharmaceutical ingredients and excipients affecting bioavailability: Impact on bioequivalence. Eur. J. Pharm. Sci. 2014, 65, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Picazo, A.; Colón-Useche, S.; Perez-Amorós, B.; González-Álvarez, M.; Molina-Martínez, I.; González-Álvarez, I.; García-Arieta, A.; Bermejo, M. Investigation to Explain Bioequivalence Failure in Pravastatin Immediate-Release Products. Pharmaceutics 2019, 11, 663. [Google Scholar] [CrossRef] [PubMed]
- Eisenbud, D.; Hunter, H.; Kessler, L.; Zulkowski, K. Hydrogel wound dressings: Where do we stand in 2003? Ostomy Wound Manag. 2003, 49, 52–57. [Google Scholar]
- von Grote, E.C.; Palaniswarmy, K.; Meckfessel, M.H. Managing Occupational Irritant Contact Dermatitis Using a Two-Step Skincare Regimen Designed to Prevent Skin Damage and Support Skin Recovery. J. Drugs Dermatol. 2016, 15, 1504–1510. [Google Scholar] [PubMed]
- Hadgraft, J.; Lane, M.E. Drug crystallization—Implications for topical and transdermal delivery. Expert Opin. Drug Deliv. 2016, 13, 817–830. [Google Scholar] [CrossRef]
- Goh, C.F.; Boyd, B.J.; Craig, D.Q.M.; Lane, M.E. Profiling of drug crystallization in the skin. Expert Opin. Drug Deliv. 2020, 17, 1321–1334. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Guidance for Industry. ANDA Submissions—Refuse-to-Receive Standards. Available online: https://www.fda.gov/files/drugs/published/ANDA-Submissions----Refuse-to-Receive-Standards-Rev.2.pdf (accessed on 17 December 2022).
- Dabbaghi, M.; Namjoshi, S.; Panchal, B.; Grice, J.E.; Prakash, S.; Roberts, M.S.; Mohammed, Y. Viscoelastic and Deformation Characteristics of Structurally Different Commercial Topical Systems. Pharmaceutics 2021, 13, 1351. [Google Scholar] [CrossRef]
- European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). Reflection paper on statistical methodology for the comparative assessment of quality attributes in drug development (EMA/CHMP/138502/2017). Amsterdam, 26 July 2021. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/reflection-paper-statistical-methodology-comparative-assessment-quality-attributes-drug-development_en.pdf (accessed on 13 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Product-Specific Guidances for Generic Drug Development. Available online: https://www.accessdata.fda.gov/scripts/cder/psg/index.cfm (accessed on 13 December 2022).
- European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). Product-Specific Bioequivalence Guidance. Available online: https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-guidelines/clinical-pharmacology-pharmacokinetics/product-specific-bioequivalence-guidance (accessed on 13 December 2022).
- Angamuthu, M.; Shankar, V.K.; Murthy, S.N. Water Activity and Its Significance in Topical Dosage Forms. J. Pharm. Sci. 2018, 107, 1656–1666. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Calcipotriene. Solution/Topical. August 2010. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Calcipotriene_sol_20611_RC08-10.pdf (accessed on 14 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Clobetasol Propionate. Solution/Topical. August 2010. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Clobetasol_Propionate_sol_19966_RC08-10.pdf (accessed on 14 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Diclofenac Sodium. Solution/Topical. June 2011. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Diclofenac_%20Sodium_sol_top_20947_RC04-11.pdf (accessed on 14 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Erythromycin. Solution/Topical 2%. February 2010. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Erythromycin_SolTopical_%2064187_RC2-10.pdf (accessed on 14 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Fluorouracil. Solution/Topical 5%. June 2011. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Fluorouracil_sol5_16831_RC03-11.pdf (accessed on 14 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Fluorouracil. Solution/Topical 2%. June 2011. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Fluorouracil_sol2_16831_RC03-11.pdf (accessed on 14 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Hydrocortisone. Solution; Topical. June 2016. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Hydrocortisone_topical%20solution_RLD%2081271_RC06-16.pdf (accessed on 14 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Minoxidil. Solution/Topical 2%. May 2010. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Minoxidil_sol_%2019501_OTC_RC5-10.pdf (accessed on 14 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Minoxidil. Solution/Topical 5%. May 2010. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Minoxidil_sol_%2020834_OTC_RC5-10.pdf (accessed on 14 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Podofilox. Solution/Topical. August 2010. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Podofilox_sol_19795_2_RC08-11.pdf (accessed on 14 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Clindamycin Phosphate. Solution; Topical. June 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_050537-Sol.pdf (accessed on 14 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Clotrimazole. Solution; Topical. February 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Clotrimazole%20Topical%20Solution%20NDA%20018181%20RV%20Feb%202019.pdf (accessed on 14 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Diclofenac Sodium. Solution; Topical. July 2017. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Diclofenac%20sodium_topical%20solution_NDA%20204623_RC05-17.pdf (accessed on 14 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Tretinoin. Solution; Topical. September 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_016921.pdf (accessed on 14 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Ciclopirox. Solution/Topical. February 2011. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Ciclopirox_soln_21022_%20RC2-10.pdf (accessed on 14 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Efinaconazole. Solution; Topical. September 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Efinaconazole_draft_Topical%20solution_RLD%20203567_RC09-18.pdf (accessed on 14 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Tavaborole. Solution; Topical. September 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Tavaborole%20topical%20solution%20NDA%20204427%20RC%2009-2018.pdf (accessed on 14 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Hydrogen Peroxide. Solution; Topical. September 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_209305.pdf (accessed on 14 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Betamethasone Dipropionate; Calcipotriene. Suspension; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_022185.pdf (accessed on 14 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). In Vitro Release Test Studies for Topical Drug Products Submitted in ANDAs. Draft Guidance for Industry. October 2022. Available online: https://www.fda.gov/media/162476/download (accessed on 14 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Spinosad. Suspension; Topical. October. 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_022408.pdf (accessed on 14 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Ciclopirox. Suspension, Topical. February 2012. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Ciclopirox%20Topical%20Suspension%20NDA%20019824%20RV%20Feb%202019.pdf (accessed on 14 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Ketoconazole Shampoo (Suspension)/ Topical. February 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Ketoconazole_sham_1_%2020310_RC09-12.pdf (accessed on 14 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Adapalene. Gel; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_020380.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Adapalene. Gel; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_021753.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Adapalene; Benzoyl Peroxide. Gel; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_022320.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Adapalene; Benzoyl Peroxide. Gel; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_207917.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Bexarotene. Gel; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_021056.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Clindamycin Phosphate. Gel; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_050782.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Clindamycin Phosphate. Gel; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_050615.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Clindamycin Phosphate; Tretinoin. Gel; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_050802.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Clindamycin Phosphate; Tretinoin. Gel; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_050803.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Dapsone. Gel; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_207154.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Dapsone. Gel; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_021794.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Diclofenac Sodium. Gel; Topical. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_022122.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Diclofenac Sodium. Gel; Topical. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_021005.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Ketokonazole. Gel; Topical. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_021946.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Metronidazole. Gel; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_019737.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Metronidazole. Gel; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_021789.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Podofilox. Gel; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_020529.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Tazarotene. Gel; Topical. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_020600-Gel-0.1P.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Tretinoin. Gel; Topical. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_017579.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Tretinoin. Gel; Topical. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_022070.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Tretinoin. Gel; Topical. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_017955.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Benzoyl peroxide; Clindamycin phosphate. Gel; Topical. November 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_050819.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Brominidine tartrate. Gel; Topical. September 2015. 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Brimonidine%20tartrate_topical%20gel_204708_RC09-15.pd (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Clobetasol propionate. Gel; Topical. October 2016. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Clobetasol%20Propionate_topical%20gel%200.05_RLD%2075368_RC09-16.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Desonide. Gel; Topical. September 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Desonide_draft_Topical%20gel_RLD%20021844_RC09-18.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Betamethasone Dipropionate; Calcipotriene. Ointment; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_021852.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Mupirocin. Ointment; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_018604.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Gentamicin Sulfate. Ointment; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_060463.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Nystatin. Ointment; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_060571.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Triamcinolone Acetonide. Ointment; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_011600.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Triamcinolone Acetonide. Ointment; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_011600-Oin-0.05P.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Nystatin; Triamcinolone Acetonide. Ointment; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_060572.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Acyclovir. Ointment; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_050591.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Calcipotriene. Ointment; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_020273.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Crisaborole. Ointment; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_207695.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Nitroglycerin. Ointment; Intra-Anal. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_021359.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Tacrolimus. Ointment; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_050777-Oin-0.1P.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Tacrolimus. Ointment; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_050777-Oin-0.03P.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Tirbanibulin. Ointment; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_213189.pdf (accessed on 17 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Acyclovir. Cream; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_021478.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Acyclovir; Hydrocortisone. Cream; topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_022436.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Ammonium lactate. Cream; Topical. November 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_020508.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Betamethasone Dipropionate; Calcipotriene. Cream; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_213422.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Butenafine Hydrochloride. Cream; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_020524.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Butenafine Hydrochloride. Cream; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_021307.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Calcipotriene. Cream; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_020554.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Docosanol. Cream; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_020941.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Doxepin Hydrochloride. Cream; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_020126.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Fluocinolone Acetonide. Cream; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_012787.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Fluorouracil. Cream; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_016988.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Fluorouracil. Cream; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_022259.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Gentamicin Sulfate; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_060462.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Ivermectin. Cream; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_206255.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Ketoconazole. Cream; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_019084.pdf (accessed on 26 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Luliconazole. Cream; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_204153.pdf (accessed on 26 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Metronidazole. Cream; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_020531.pdf (accessed on 26 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Metronidazole. Cream; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_020743.pdf (accessed on 26 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Mupirocin Calcium. Cream; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_050746.pdf (accessed on 26 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Nystatin. Cream; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_060575.pdf (accessed on 26 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Nystatin; Triamcinolone Acetonide. Cream; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_060576.pdf (accessed on 26 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Oxymetazoline Hydrochloride. Cream; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_208552.pdf (accessed on 26 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Ozenoxacin. Cream; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_208945.pdf (accessed on 26 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Penciclovir. Cream; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_020629.pdf (accessed on 26 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Pimecrolimus. Cream; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_021302.pdf (accessed on 26 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Silver Sulfadiazine. Cream; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_017381.pdf (accessed on 26 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Tazarotene. Cream; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_021184-Cre-0.05P.pdf (accessed on 26 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Tazarotene. Cream; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_021184-Cre-0.1P.pdf (accessed on 26 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Triamcinolone Acetonide. Cream; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_011601.pdf (accessed on 26 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Fluorouracil. Cream; Topical. February 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Fluorouracil%20Topical%20Cream%20NDA%20020985%20RV%20Feb%202019.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Ammonium Lactate. Lotion; Topical. November 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_019155.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Abametapir. Lotion; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_206966.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Benzyl Alcohol. Lotion; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_022129.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Clindamicin Phosphate. Lotion; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_050600.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Miconazole nitrate; White petrolatum; Zinc oxide. Lotion; Topical. November 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_021026.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Triamcinolone Acetonide. Lotion; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_011602.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Malathion. Lotion; Topical. June 2012. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Malathion_draft_Topical%20lotion_RLD%2018613_RC06-12.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Mometasone furoate. Lotion; Topical. April 2016. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Mometasone%20furoate_topical%20lotion_RLD%2019796_RC04-16.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Halobetasol. Lotion; Topical. October 2016. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Halobetasol_topical%20lotion_RLD%20208183_RC%2009-16.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Ivermectin. Lotion; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_202736.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Metronidazole. Lotion; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_020901.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Tazarotene. Lotion; Topical. October 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_211882.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Clindamycin phosphate. Aerosol, foam; Topical. June 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_050801.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Nystatin. Powder; Topical. August 2021. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_060578.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Ciclopirox. Shampoo/Topical. February 2010. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Ciclopirox_shmp_21159_RC2-10.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Clobetasol Propionate. Shampoo/Topical. February 2011. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Clobetasol_Propionate_shmp_21644_RC2-11.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Clobetasol Propionate. Spray/Topical. February 2010. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Clobetasol_Propionate_spray_21835_RC2-11.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Desoximetasone. Spray; Topical. March 2015. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Desoximetasone_topical%20spray%200.25_204141_RC03-15.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Clindamycin phosphate. Swab; Topical. June 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_050537-Swa.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Erythromycin. Swab; Topical. February 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Erythromycin_swab_64126_RC2-10.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Glycopyrronium tosylate. Cloth; Topical. September 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_210361.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Tretinoin. Gel; Topical. June 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_020475.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Predicarbate. Ointment; Topical. June 2016. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Prednicarbate_topical%20ointment_RLD%2019568_RC06-16.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Calcipotriene. Aerosol, foam; Topical. November 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_022563.pdf (accessed on 23 December 2022).
- Mangas-Sanjuán, V.; Pleguezuelos-Villa, M.; Merino-Sanjuán, M.; Hernández, M.J.; Nácher, A.; García-Arieta, A.; Peris, D.; Hidalgo, I.; Soler, L.; Sallan, M.; et al. Assessment of the Inter-Batch Variability of Microstructure Parameters in Topical Semisolids and Impact on the Demonstration of Equivalence. Pharmaceutics 2019, 11, 503, Erratum in: Pharmaceutics 2020, 12, 436. [Google Scholar] [CrossRef]
- Xu, Z.; Mangas-Sanjuán, V.; Merino-Sanjuán, M.; Merino, V.; García-Arieta, A. Influence of Inter- and Intra-Batch Variability on the Sample Size Required for Demonstration of Equivalent Microstructure of Semisolid Dosage Forms. Pharmaceutics 2020, 12, 1159. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Scale-up and Postapproval Changes: Chemistry, Manufacturing, and Controls; In Vitro Release Testing and In Vivo Bioequivalence Documentation. Nonsterile Semisolid Dosage Forms. Guidance for Industry. Available online: https://www.fda.gov/media/71141/download (accessed on 23 December 2022).
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Revision of M4E Guidelione on Enhancing the Format and Structure of Benefit-Risk Information in ICH. 15. June 2016. Available online: https://database.ich.org/sites/default/files/M4E_R2__Guideline.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). In Vitro Permeation Test Studies for Topical Drug Products Submitted in ANDAs. Draft Guidance for Industry. October 2022. Available online: https://www.fda.gov/media/162475/download (accessed on 25 December 2022).
- Shah, V.P. IV-IVC for topically applied preparations--a critical evaluation. Eur. J. Pharm. Biopharm. 2005, 60, 309–314. [Google Scholar] [CrossRef]
- Ministry of Health and Wellfare of Japan. Guideline for Bioequivalence Studies of Generic Products for Topcial Use. July 7, 2003. Available online: http://www.nihs.go.jp/drug/be-guide(e)/Topical_BE-E.pdf (accessed on 23 December 2022).
- Russell, L.M.; Guy, R.H. Measurement and prediction of the rate and extent of drug delivery into and through the skin. Expert Opin. Drug Deliv. 2009, 6, 355–369. [Google Scholar] [CrossRef]
- N’Dri-Stempfer, B.; Navidi, W.C.; Guy, R.H.; Bunge, A.L. Improved bioequivalence assessment of topical dermatological drug products using dermatopharmacokinetics. Pharm. Res. 2009, 26, 316–328. [Google Scholar] [CrossRef]
- Cordery, S.F.; Pensado, A.; Chiu, W.S.; Shehab, M.Z.; Bunge, A.L.; Delgado-Charro, M.B.; Guy, R.H. Topical bioavailability of diclofenac from locally-acting, dermatological formulations. Int. J. Pharm. 2017, 529, 55–64. [Google Scholar] [CrossRef]
- de Araujo, T.P.; Fittipaldi, I.M.; Bedor, D.C.G.; Duarte, M.L.; Cordery, S.F.; Guy, R.H.; Delgado-Charro, M.B.; de Santana, D.P.; Leal, L.B. Topical bio(in)equivalence of metronidazole formulations in vivo. Int. J. Pharm. 2018, 541, 167–172. [Google Scholar] [CrossRef]
- Pensado, A.; Chiu, W.S.; Cordery, S.F.; Rantou, E.; Bunge, A.L.; Delgado-Charro, M.B.; Guy, R.H. Stratum Corneum Sampling to Assess Bioequivalence between Topical Acyclovir Products. Pharm. Res. 2019, 36, 180. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Lidocaine; Prilocaine. Cream; Topical. December 2014. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Lidocaine;%20Prilocaine_draft_Topical%20cream_RLD%20019941_RC12-14.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Azelaic acid. Cream; Topical. May 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Azelaic%20Acid%20Cream%2020%20NDA%20020428%20PSG%20Page%20RV%20May%202019.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance on Amcinonide. Lotion; Topical. Hune 2016. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Amcinonide_topical%20lotion_RLD%2076329_RC%2006-16.pdf (accessed on 23 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Topical Dermatologic Corticosteroids: In Vivo Bioequivalence. Guidance for Industry. 2 June 1995. Available online: https://www.fda.gov/media/70931/download (accessed on 26 December 2022).
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Topical Dermatologic Corticosteroids: In Vivo Bioequivalence. Draft Guidance for Industry. October 2022. Available online: https://www.fda.gov/media/162457/download (accessed on 26 December 2022).
- European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). Questions and Answers on Guideline Title: Clinical Investigation of Corticosteroids Intended for Use on the Skin. (CPMP/EWP/21441/2006). London, 16 November 2006. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/questions-answer-guideline-title-clinical-investigation-corticosteroids-intended-use-skin_en.pdf (accessed on 23 December 2022).
- Shah, V.P.; Yacobi, A.; Rădulescu, F.Ş.; Miron, D.S.; Lane, M.E. A science based approach to topical drug classification system (TCS). Int. J. Pharm. 2015, 491, 21–25. [Google Scholar] [CrossRef]
- Cristofoletti, R.; Patel, N.; Dressman, J.B. Assessment of Bioequivalence of Weak Base Formulations Under Various Dosing Conditions Using Physiologically Based Pharmacokinetic Simulations in Virtual Populations. Case Examples: Ketoconazole and Posaconazole. J. Pharm. Sci. 2017, 106, 560–569. [Google Scholar] [CrossRef]
- Doki, K.; Darwich, A.S.; Patel, N.; Rostami-Hodjegan, A. Virtual bioequivalence for achlorhydric subjects: The use of PBPK modelling to assess the formulation-dependent effect of achlorhydria. Eur. J. Pharm. Sci. 2017, 109, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Tsakalozou, E.; Alam, K.; Babiskin, A.; Zhao, L. Physiologically-Based Pharmacokinetic Modeling to Support Determination of Bioequivalence for Dermatological Drug Products: Scientific and Regulatory Considerations. Clin. Pharmacol. Ther. 2022, 111, 1036–1049. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Draft Guidance for Industry. The Use of Physiologically Based Pharmacokinetic Analyses—Biopharmaceutics Applications for Oral Drug Product Development, Manufacturing Changes, and Controls. Pharmaceutical Quality/CMC. October 2020. Available online: https://www.fda.gov/media/142500/download (accessed on 13 December 2022).
- Tsakalozou, E.; Babiskin, A.; Zhao, L. Physiologically-based pharmacokinetic modeling to support bioequivalence and approval of generic products: A case for diclofenac sodium topical gel, 1. CPT Pharmacomet. Syst. Pharmacol. 2021, 10, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Pleguezuelos-Villa, M.; Merino-Sanjuán, M.; Hernández, M.J.; Nácher, A.; Peris, D.; Hidalgo, I.; Soler, L.; Sallan, M.; Merino, V. Relationship between rheological properties, in vitro release and in vivo equivalency of topical formulations of diclofenac. Int. J. Pharm. 2019, 572, 118755. [Google Scholar] [CrossRef] [PubMed]

| Drug | RS | Q1Q2 | A | V | pH | SG | DR | ST |
|---|---|---|---|---|---|---|---|---|
| Calcipotriene [20] | 020611 | √ | ||||||
| Clindamycin Phosphate [30] | 050537 | √ | ||||||
| Clobetasol Propionate [21] | 019966 | √ | ||||||
| Clotrimazole [31] | 018181 | √ | ||||||
| Diclofenac sodium [32] | 204623 | √ | ||||||
| Diclofenac sodium [22] | 020947 | √ | ||||||
| Efinaconazole [35] | 203567 | √ | √ | √ | √ | √ | √ | |
| Erythromycin [23] | 064187 | √ | ||||||
| Fluorouracil [24] | 016831 | √ | ||||||
| Fluorouracil [25] | 016831 | √ | ||||||
| Hydrocortisone [26] | 081271 | √ | ||||||
| Hydrogen peroxide [37] | 209305 | √ | √ | √ | √ | √ | √ | |
| Minoxidil [27] | 019501 | √ | ||||||
| Minoxidil [28] | 020834 | √ | ||||||
| Podofilox [29] | 019795 | √ | ||||||
| Tavaborole [36] | 204427 | √ | √ | √ | √ | √ | ||
| Tretinoin [33] | 016921 | √ | √ |
| Drug | RS | Q | AT | M | PS | GS | AV | FC | YS | LVR | WA | pH | SG | DR | OC | IVRT | IVPT |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Betamethasone Dipropionate; Calcipotriene [38] | 22185 | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||||||
| Spinosad [40] | 22408 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ |
| Drug | RS | Q | AT | M | PS | GP | AV | FC | YS | LVR | WA | pH | SG | DR | OC | IVRT | IVPT |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Adapalene [43] | 20380 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||||
| Adapalene [44] | 21753 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||||
| Adapalene, Benzoyl Peroxide [45] | 22320 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||||
| Adapalene, Benzoyl Peroxide [46] | 207917 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||||
| Bexarotene [47] | 21056 | √ | √ | √ | √ | √ | √ | √ | √ | ||||||||
| Clindamycin Phosphate [48] | 50782 | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||||||
| Clindamycin Phosphate [49] | 50615 | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||||||
| Clindamycin Phosphate; Tretinoin [50] | 50802 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||||
| Clindamycin Phosphate; Tretinoin [51] | 50803 | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||||||
| Dapsone [52] | 207154 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||||
| Dapsone [53] | 21794 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||||
| Diclofenac Sodium [54] | 22122 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||
| Diclofenac Sodium [55] | 21005 | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||||||
| Ketoconazole [56] | 21946 | √ | √ | √ | √ | √ | √ | √ | √ | ||||||||
| Metronidazole [57] | 19737 | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||||||
| Metronidazole [58] | 21789 | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||||||
| Podofilox [59] | 20529 | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||||||
| Tazarotene [60] | 20600 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||||
| Tretinoin [61] | 17579 | √ | √ | √ | √ | √ | √ | √ | √ | ||||||||
| Tretinoin [62] | 22070 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||||
| Tretinoin [63] | 17955 | √ | √ | √ | √ | √ | √ | √ | √ |
| Drug | RS | Q | AT | M | PS | GS | AV | FC | YS | LVR | WA | pH | SG | DR | OC | IVRT | IVPT |
| Acyclovir [82] | 21478 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||
| Acyclovir, Hydrocortisone [83] | 22436 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||
| Ammonium Lactate [84] | 20508 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||
| Betamethasone Dipropionate; Calcipotriene [85] | 213422 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||
| Butenafine Hydrochloride [86] | 20524 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||
| Butenafine Hydrochloride [87] | 21307 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||
| Calcipotriene [88] | 20554 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||
| Docosanol [89] | 20941 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||
| Doxepin Hydrochloride [90] | 20126 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||
| Fluocinolone Acetonide [91] | 12787 | ||||||||||||||||
| Fluorouracil [92] | 16988 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||
| Fluorouracil [93] | 22259 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||
| Gentamicin Sulfate [94] | 62307 | ||||||||||||||||
| Ivermectin [95] | 206255 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||
| Ketoconazole [96] | 19084 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||
| Luliconazole [97] | 204153 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||
| Metronidazole [98] | 20531 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||
| Metronidazole [99] | 20743 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||
| Mupirocin Calcium [100] | 50746 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||
| Nystatin [101] | 64022 | ||||||||||||||||
| Nystatin; Triamcinolone Acetonide [102] | 62364 | ||||||||||||||||
| Oxymetazoline Hydrochloride [103] | 208552 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||
| Ozenoxacin [104] | 208945 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||
| Penciclovir [105] | 20629 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||
| Pimecrolimus [106] | 21302 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||
| Silver Sulfadiazine [107] | 17381 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||
| Tazarotene [108] | 21184 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||
| Tazarotene [109] | 21184 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||
| Triamcinolone Acetonide [110] | 11601 |
| Drug | RS | Q | AT | M | PS | GS | AV | FC | YS | LVR | WA | pH | SG | DR | OC | IVRT | IVPT |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Abametapir [113] | 206966 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||||
| Ammonium Lactate [112] | 19155 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||
| Benzyl Alcohol [114] | 22129 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||||
| Clindamycin Phosphate [115] | 50600 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||
| Halobetasol Propionate [120] | 209355 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||
| Ivermectin [121] | 202736 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||||
| Malathion [118] | 018613 | √ | |||||||||||||||
| Metronidazole [122] | 20901 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||
| Miconazole Nitrate; White Petrolatum; Zinc Oxide [116] | 21026 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||||
| Mometasone Furoate [119] | 019796 | √ | |||||||||||||||
| Tazarotene [123] | 211882 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||
| Triamcinolone Acetonide [117] | 11602 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Arieta, A.; Gordon, J.; Gwaza, L.; Merino, V.; Mangas-Sanjuan, V. Regulatory Requirements for the Development of Second-Entry Semisolid Topical Products in the European Union. Pharmaceutics 2023, 15, 601. https://doi.org/10.3390/pharmaceutics15020601
García-Arieta A, Gordon J, Gwaza L, Merino V, Mangas-Sanjuan V. Regulatory Requirements for the Development of Second-Entry Semisolid Topical Products in the European Union. Pharmaceutics. 2023; 15(2):601. https://doi.org/10.3390/pharmaceutics15020601
Chicago/Turabian StyleGarcía-Arieta, Alfredo, John Gordon, Luther Gwaza, Virginia Merino, and Víctor Mangas-Sanjuan. 2023. "Regulatory Requirements for the Development of Second-Entry Semisolid Topical Products in the European Union" Pharmaceutics 15, no. 2: 601. https://doi.org/10.3390/pharmaceutics15020601