
Delivery Systems for Mitochondrial Gene Therapy: A Review


Abstract
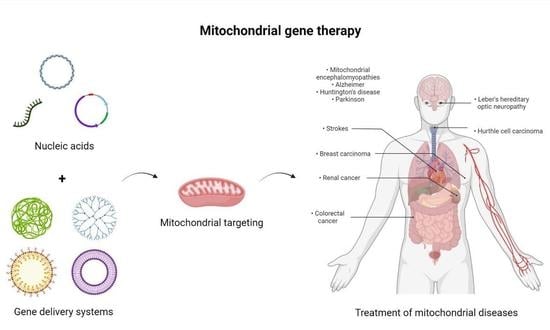
:
1. Introduction
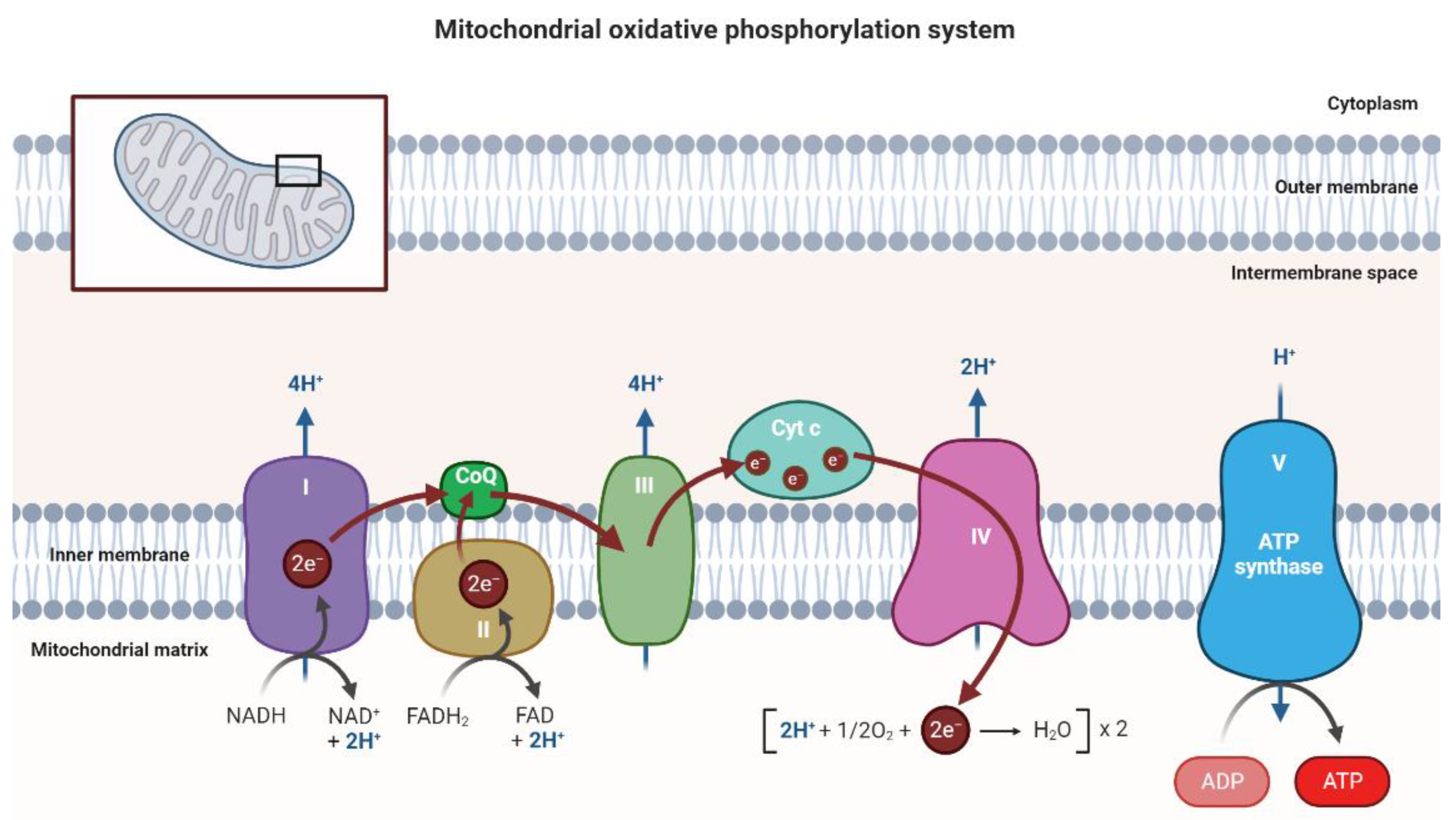
1.1. Mitochondria
1.2. Mitochondrial Genome
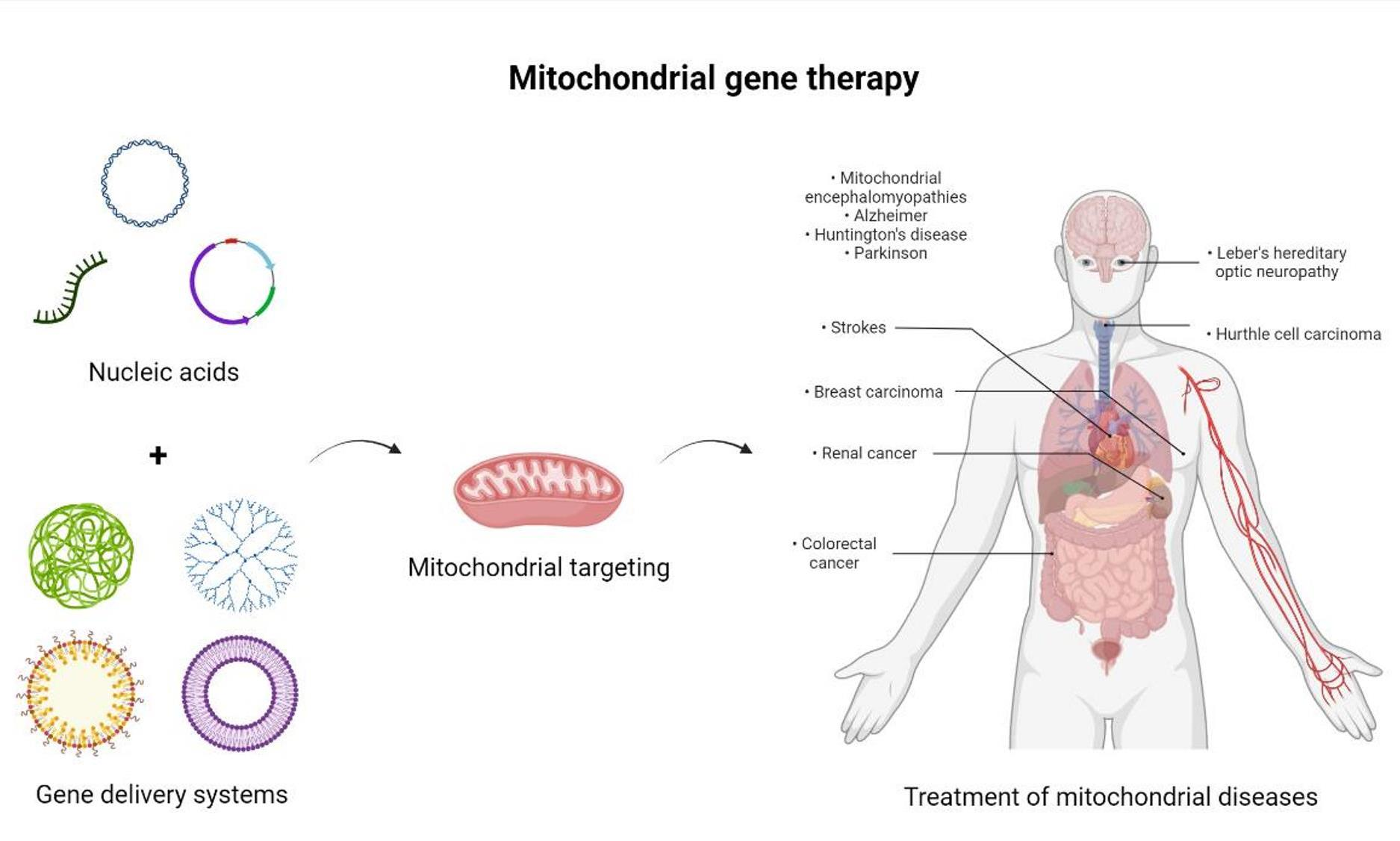
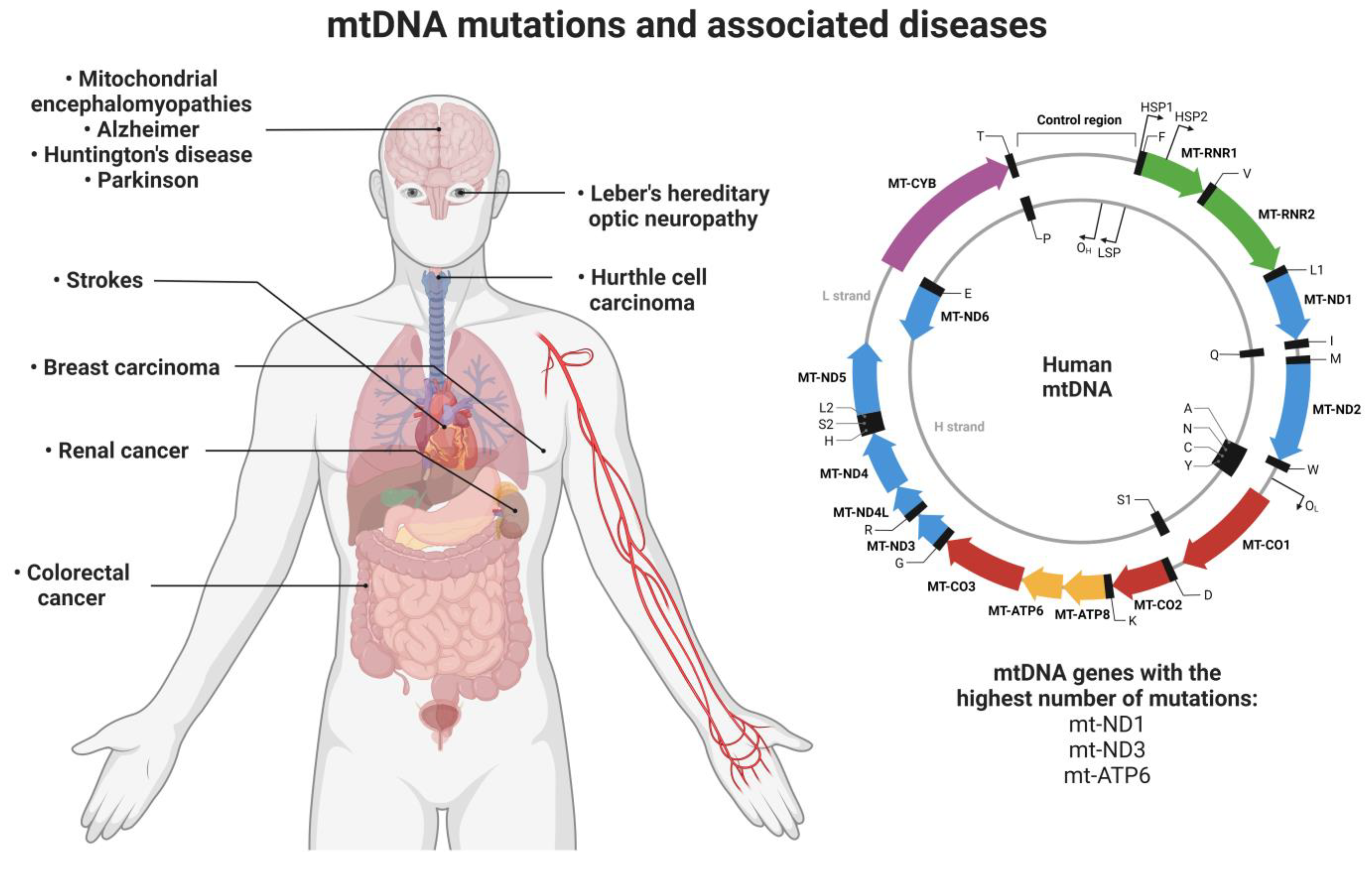
1.3. Mitochondrial Mutations and Associated Diseases
1.4. Conventional Treatments for Mitochondrial Dysfunctions
2. Nanotechnology in Mitochondrial Gene Therapy
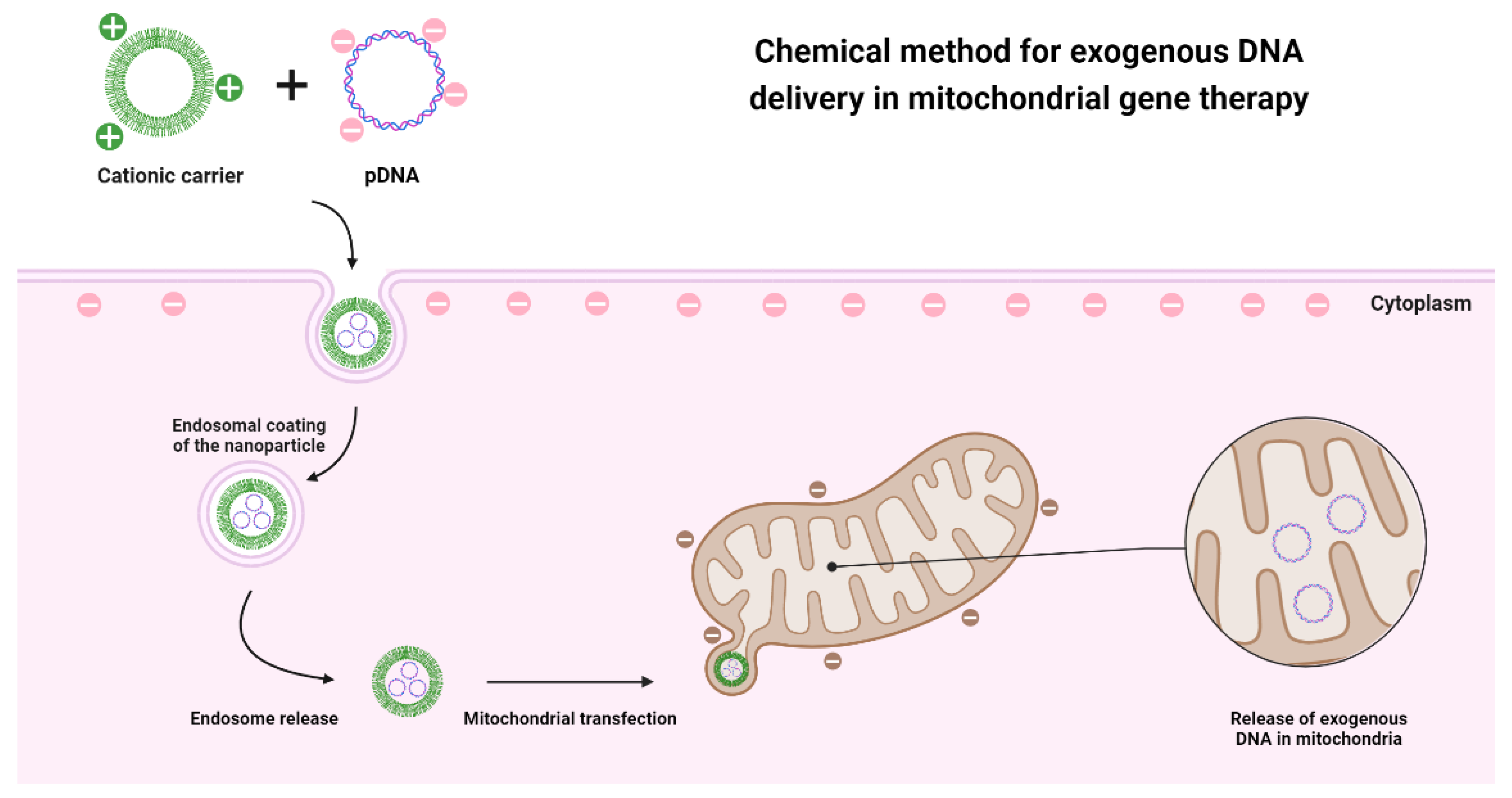
2.1. Mitochondrial Gene Therapy
2.2. Nanotechnology
2.3. Delivery Systems in Mitochondrial Gene Therapy
2.3.1. Polymers
2.3.2. Dendrimers
2.3.3. Lipids
2.3.4. Inorganic Nanoparticles
2.4. Strategies to Be Further Explored for Progress in Mitochondrial Gene Therapy
2.4.1. Dendrigraftpoly-L-lysines Based Delivery Systems
2.4.2. ROS-Responsive PLGA-Based Delivery Systems
2.4.3. MITO-Porter-Based Systems
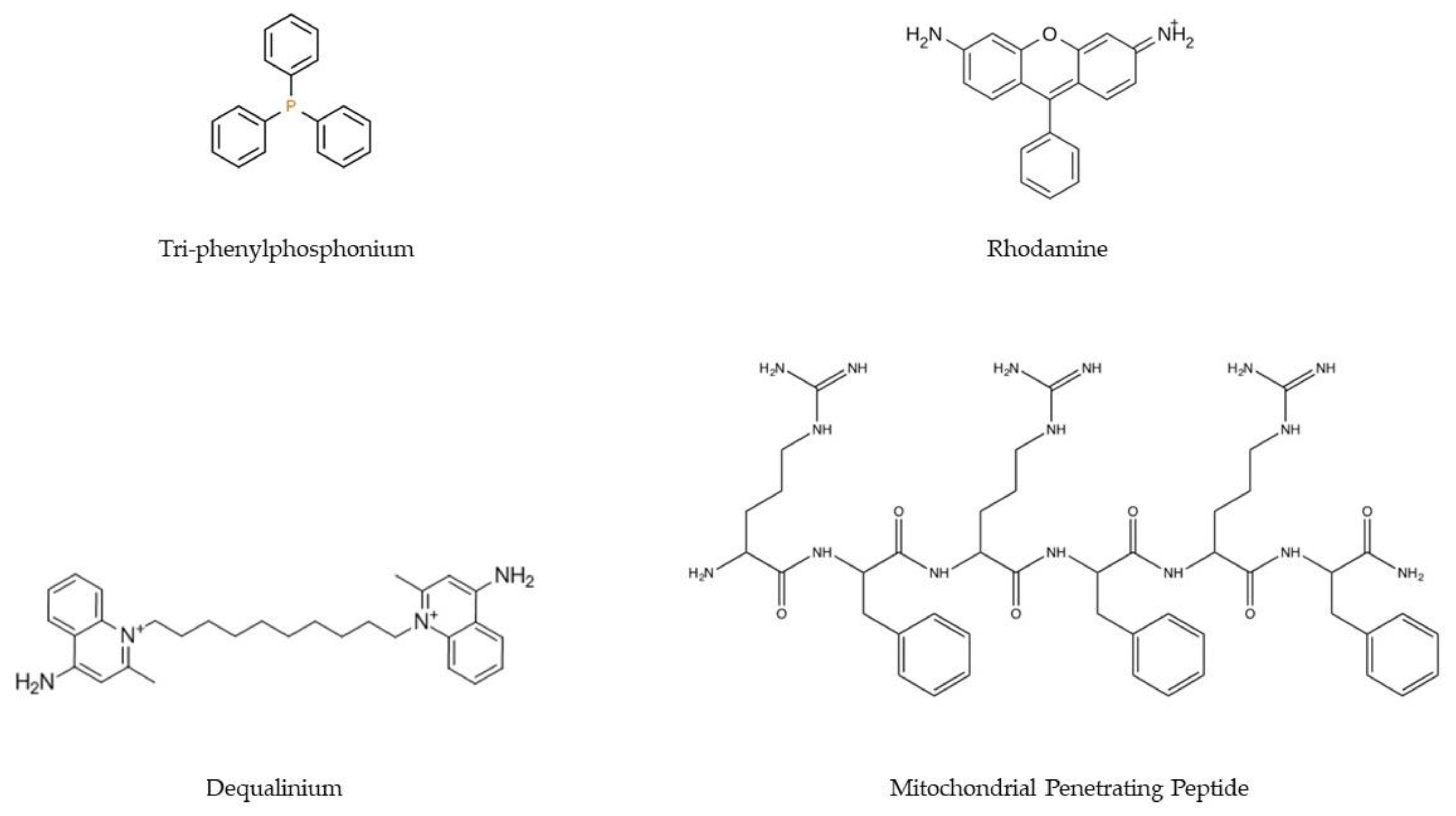
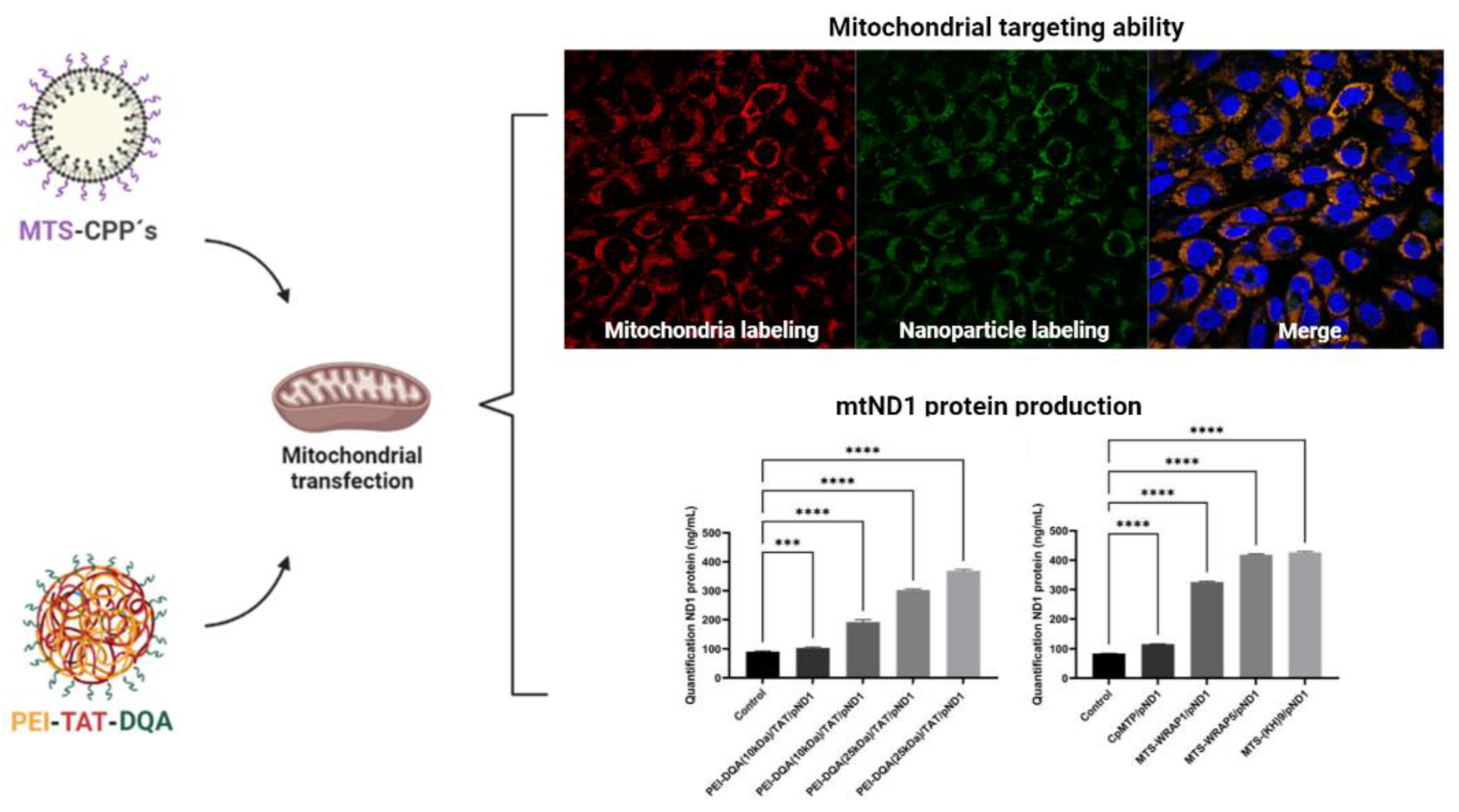
2.4.4. Upgraded TPP-Based Systems
3. Concluding Remarks/Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abate, M.; Festa, A.; Falco, M.; Lombardi, A.; Luce, A.; Grimaldi, A.; Zappavigna, S.; Sperlongano, P.; Irace, C.; Caraglia, M.; et al. Mitochondria as playmakers of apoptosis, autophagy and senescence. Semin. Cell Dev. Biol. 2019, 98, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Saki, M.; Prakash, A. DNA damage related crosstalk between the nucleus and mitochondria. Free. Radic. Biol. Med. 2017, 107, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Alston, C.L.; Stenton, S.L.; Hudson, G.; Prokisch, H.; Taylor, R.W. The genetics of mitochondrial disease: Dissecting mitochondrial pathology using multi-omic pipelines. J. Pathol. 2021, 254, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Aversa, R.; Petrescu, R.V.; Apicella, A.; Petrescu, F.I. Mitochondria are Naturally Micro Robots—A review. Am. J. Eng. Appl. Sci. 2016, 9, 991–1002. [Google Scholar] [CrossRef]
- Herrera, A.S.; del CAEsparza, M.; AZamyatnin, A.; Aliev, G. Beyond Mitochondria, What Would be the Energy Source of the Cell? Cent. Nerv. Syst. Agents Med. Chem. 2015, 15, 32–41. [Google Scholar] [CrossRef]
- Obrador, E.; Salvador-Palmer, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.; Estrela, J. The Link between Oxidative Stress, Redox Status, Bioenergetics and Mitochondria in the Pathophysiology of ALS. Int. J. Mol. Sci. 2021, 22, 6352. [Google Scholar] [CrossRef]
- Jerome, M.S.; Kuthethur, R.; Kabekkodu, S.P.; Chakrabarty, S. Regulation of mitochondrial function by forkhead transcription factors. Biochimie 2022, 198, 96–108. [Google Scholar] [CrossRef]
- Prasai, K. Regulation of mitochondrial structure and function by protein import: A current review. Pathophysiology 2017, 24, 107–122. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nature 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Roger, A.J.; Muñoz-Gómez, S.A.; Kamikawa, R. The Origin and Diversification of Mitochondria. Curr. Biol. 2017, 27, R1177–R1192. [Google Scholar] [CrossRef] [Green Version]
- Fontanesi, F. Mitochondria: Structure and Role in Respiration. eLS 2015, 1–13. [Google Scholar] [CrossRef]
- Picard, M.; McEwen, B.S. Psychological Stress and Mitochondria: A Systematic Review. Psychosom. Med. 2018, 80, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Johnston, I.G.; Williams, B.P. Evolutionary Inference across Eukaryotes Identifies Specific Pressures Favoring Mitochondrial Gene Retention. Cell Syst. 2016, 2, 101–111. [Google Scholar] [CrossRef]
- Reznik, E.; Miller, M.L.; Şenbabaoğlu, Y.; Riaz, N.; Sarungbam, J.; Tickoo, S.K.; A Al-Ahmadie, H.; Lee, W.; E Seshan, V.; Hakimi, A.A.; et al. Mitochondrial DNA copy number variation across human cancers. Elife 2016, 5, e10769. [Google Scholar] [CrossRef] [PubMed]
- Jedynak-Slyvka, M.; Jabczynska, A.; Szczesny, R. Human Mitochondrial RNA Processing and Modifications: Overview. Int. J. Mol. Sci. 2021, 22, 7999. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, A.R.; Minczuk, M. Mitochondrial transcription and translation: Overview. Essays Biochem. 2018, 62, 309–320. [Google Scholar] [CrossRef]
- Fox, T.D. Mitochondrial protein synthesis, import, and assembly. Genetics 2012, 192, 1203–1234. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, D.; Zhang, D.; Li, P.; Gao, Y. Mitochondrial Protein Translation: Emerging Roles and Clinical Significance in Disease. Front. Cell Dev. Biol. 2021, 9, 675465. [Google Scholar] [CrossRef]
- Blier, P.U.; Dufresne, F.; Burton, R.S. Natural selection and the evolution of mtDNA-encoded peptides: Evidence for intergenomic co-adaptation. TRENDS Genet. 2001, 17, 400–406. [Google Scholar] [CrossRef]
- Brischigliaro, M.; Zeviani, M. Cytochrome c oxidase deficiency. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148335. [Google Scholar] [CrossRef]
- Buneeva, O.; Fedchenko, V.; Kopylov, A.; Medvedev, A. Mitochondrial Dysfunction in Parkinson’s Disease: Focus on Mitochondrial DNA. Biomedicines 2020, 8, 591. [Google Scholar] [CrossRef] [PubMed]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef] [PubMed]
- Kotrys, A.V.; Szczesny, R.J. Mitochondrial Gene Expression and Beyond—Novel Aspects of Cellular Physiology. Cells 2019, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef]
- Pearce, E.L.; Poffenberger, M.C.; Chang, C.-H.; Jones, R.G. Fueling Immunity: Insights into Metabolism and Lymphocyte Function. Science 2013, 342, 1242454. [Google Scholar] [CrossRef]
- Morrow, R.M.; Picard, M.; Derbeneva, O.; Leipzig, J.; McManus, M.J.; Gouspillou, G.; Barbat-Artigas, S.; Dos Santos, C.; Hepple, R.T.; Murdock, D.G.; et al. Mitochondrial energy deficiency leads to hyperproliferation of skeletal muscle mitochondria and enhanced insulin sensitivity. Proc. Natl. Acad. Sci. USA 2017, 114, 2705–2710. [Google Scholar] [CrossRef]
- Hebert-Chatelain, E.; Desprez, T.; Serrat, R.; Bellocchio, L.; Soria-Gomez, E.; Busquets-Garcia, A.; Zottola, A.C.P.; Delamarre, A.; Cannich, A.; Vincent, P.; et al. A cannabinoid link between mitochondria and memory. Nature 2016, 539, 555–559. [Google Scholar] [CrossRef]
- Azzouz, M.; Ralph, G.S.; Storkebaum, E.; Walmsley, L.E.; Mitrophanous, K.A.; Kingsman, S.M.; Carmeliet, P.; Mazarakis, N.D. VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature 2004, 429, 413–417. [Google Scholar] [CrossRef]
- Latorre-Pellicer, A.; Moreno-Loshuertos, R.; Lechuga-Vieco, A.V.; Sánchez-Cabo, F.; Torroja, C.; Acín-Pérez, R.; Calvo, E.; Aix, E.; González-Guerra, A.; Logan, A.; et al. Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 2016, 535, 561–565. [Google Scholar] [CrossRef]
- Russell, O.M.; Gorman, G.S.; Lightowlers, R.N.; Turnbull, D.M. Mitochondrial Diseases: Hope for the Future. Cell 2020, 181, 168–188. [Google Scholar] [CrossRef]
- Tinker, R.J.; Lim, A.Z.; Stefanetti, R.J.; McFarland, R. Current and Emerging Clinical Treatment in Mitochondrial Disease. Mol. Diagn. Ther. 2021, 25, 181–206. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Ahmad, K.; Sue, C.M. The broadening spectrum of mitochondrial disease: Shifts in the diagnostic paradigm. Biochim. et Biophys. Acta (BBA)—Gen. Subj. 2013, 1840, 1360–1367. [Google Scholar] [CrossRef] [PubMed]
- Farrar, G.J.; Chadderton, N.; Kenna, P.F.; Millington-Ward, S. Mitochondrial disorders: Aetiologies, models systems, and candidate therapies. Trends Genet. 2013, 29, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.; Zuryn, S. Mitochondrial Genome (mtDNA) Mutations that Generate Reactive Oxygen Species. Antioxidants 2019, 8, 392. [Google Scholar] [CrossRef] [PubMed]
- Bordoni, L.; Gabbianelli, R. Mitochondrial DNA and Neurodegeneration: Any Role for Dietary Antioxidants? Antioxidants 2020, 9, 764. [Google Scholar] [CrossRef]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 2017, 592, 728–742. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Stefano, G.B.; Kream, R.M. Mitochondrial DNA heteroplasmy in human health and disease. Biomed. Rep. 2016, 4, 259–262. [Google Scholar] [CrossRef]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA heteroplasmy in disease and targeted nuclease-based therapeutic approaches. EMBO Rep. 2020, 21, e49612. [Google Scholar] [CrossRef]
- Elorza, A.A.; Soffia, J.P. mtDNA Heteroplasmy at the Core of Aging-Associated Heart Failure. An Integrative View of OXPHOS and Mitochondrial Life Cycle in Cardiac Mitochondrial Physiology. Front. Cell Dev. Biol. 2021, 9, 625020. [Google Scholar] [CrossRef]
- Schon, E.A.; DiMauro, S.; Hirano, M. Human mitochondrial DNA: Roles of inherited and somatic mutations. Nat. Rev. Genet. 2012, 13, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Lawless, C.; Greaves, L.; Reeve, A.K.; Turnbull, D.M.; Vincent, A.E. The rise and rise of mitochondrial DNA mutations. Open Biol. 2020, 10, 200061. [Google Scholar] [CrossRef] [PubMed]
- Permana Maksum, I.; Saputra, S.R.; Indrayati, N.; Yusuf, M.; Subroto, T. Bioinformatics Study of m.9053G>A Mutation at the ATP6 Gene in Relation to Type 2 Diabetes Mellitus and Cataract Diseases. Bioinform. Biol. Insights 2017, 11, 1177932217728515. [Google Scholar] [CrossRef]
- Lalrohlui, F.; Thapa, S.; Ghatak, S.; Zohmingthanga, J.; Kumar, N.S. Mitochondrial complex I and V gene polymorphisms in type II diabetes mellitus among high risk Mizo-Mongoloid population, Northeast India. Genes Environ. 2016, 38, 5. [Google Scholar] [CrossRef] [PubMed]
- Lalrohlui, F.; Zohmingthanga, J.; Hruaii, V.; Kumar, N.S. Genomic profiling of mitochondrial DNA reveals novel complex gene mutations in familial type 2 diabetes mellitus individuals from Mizo ethnic population, Northeast India. Mitochondrion 2019, 51, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Carreno-Gago, L.; Gamez, J.; Cámara, Y.; de la Campa, E.A.; Aller-Alvarez, J.S.; Moncho, D.; Salvado, M.; Galan, A.; de la Cruz, X.; Pinós, T.; et al. Identification and characterization of the novel point mutation m.3634A>G in the mitochondrial MT-ND1 gene associated with LHON syndrome. Biochim. Biophys Acta Mol. Basis Dis. 2017, 1863, 182–187. [Google Scholar] [CrossRef]
- Baertling, F.; Sánchez-Caballero, L.; Brand, M.A.V.D.; Distelmaier, F.; Janssen, M.C.; Rodenburg, R.J.; Smeitink, J.A.; Nijtmans, L.G. A Heterozygous NDUFV1 Variant Aggravates Mitochondrial Complex I Deficiency in a Family with a Homoplasmic ND1 Variant. J. Pediatr. 2018, 196, 309–313.e3. [Google Scholar] [CrossRef]
- Smolina, N.; Khudiakov, A.; Knyazeva, A.; Zlotina, A.; Sukhareva, K.; Kondratov, K.; Gogvadze, V.; Zhivotovsky, B.; Sejersen, T.; Kostareva, A. Desmin mutations result in mitochondrial dysfunction regardless of their aggregation properties. Biochim. et Biophys. Acta (BBA)—Mol. Basis Dis. 2020, 1866, 165745. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, Y.; Li, F.; Liu, P.; Liu, Y.; Yang, C.; Chen, Z. The biochemical characterization of a missense mutation m.8914C>T in ATP6 gene associated with mitochondrial encephalomyopathy. Int. J. Dev. Neurosci. 2018, 71, 172–174. [Google Scholar] [CrossRef]
- Dafinca, R.; Barbagallo, P.; Farrimond, L.; Candalija, A.; Scaber, J.; Nida’a, A.A.; Talbot, K. Impairment of Mitochondrial Calcium Buffering Links Mutations in C9ORF72 and TARDBP in iPS-Derived Motor Neurons from Patients with ALS/FTD. Stem Cell Rep. 2020, 14, 892–908. [Google Scholar] [CrossRef]
- van Horssen, J.; van Schaik, P.; Witte, M. Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders? Neurosci Lett. 2019, 710, 132931. [Google Scholar] [CrossRef] [PubMed]
- Chu, Q.; Luo, X.; Zhan, X.; Ren, Y.; Pang, H. Female genetic distribution bias in mitochondrial genome observed in Parkinson’s Disease patients in northern China. Sci. Rep. 2015, 5, 17170. [Google Scholar] [CrossRef] [PubMed]
- Bi, R.; Zhang, W.; Yu, D.; Li, X.; Wang, H.Z.; Hu, Q.X.; Yao, Y.G.; Zhang, C.; Lu, W.; Ni, J.; et al. Mitochondrial DNA haplogroup B5 confers genetic susceptibility to Alzheimer’s disease in Han Chinese. Neurobiol. Aging 2015, 36, 1604.e7–1604.e16. [Google Scholar] [CrossRef] [PubMed]
- Ban, R.; Guo, J.-H.; Pu, C.-Q.; Shi, Q.; Liu, H.-X.; Zhang, Y.-T. A Novel Mutation of Mitochondrial T14709C Causes Myoclonic Epilepsy with Ragged Red Fibers Syndrome in a Chinese Patient. Chin. Med. J. 2018, 131, 1569–1574. [Google Scholar] [CrossRef]
- Henry, C.; Patel, N.; Shaffer, W.; Murphy, L.; Park, J.; Spieler, B. Mitochondrial Encephalomyopathy With Lactic Acidosis and Stroke-Like Episodes-MELAS Syndrome. Ochsner J. 2017, 17, 296–301. [Google Scholar]
- de Mello, A.H.; Costa, A.B.; Engel, J.D.G.; Rezin, G.T. Mitochondrial dysfunction in obesity. Life Sci. 2018, 192, 26–32. [Google Scholar]
- Kim, H.; Komiyama, T.; Inomoto, C.; Kamiguchi, H.; Kajiwara, H.; Kobayashi, H.; Nakamura, N.; Terachi, T. Mutations in the Mitochondrial ND1 Gene Are Associated with Postoperative Prognosis of Localized Renal Cell Carcinoma. Int. J. Mol. Sci. 2016, 17, 2049. [Google Scholar] [CrossRef]
- Ganly, I.; Makarov, V.; Deraje, S.; Dong, Y.; Reznik, E.; Seshan, V.; Chan, T.A.; Nanjangud, G.; Eng, S.; Bose, P.; et al. Integrated Genomic Analysis of Hurthle Cell Cancer Reveals Oncogenic Drivers, Recurrent Mitochondrial Mutations, and Unique Chromosomal Landscapes. Cancer Cell 2018, 34, 256–270.e5. [Google Scholar] [CrossRef]
- Ganly, I.; McFadden, D.G. Short Review: Genomic Alterations in Hürthle Cell Carcinoma. Thyroid 2019, 29, 471–479. [Google Scholar] [CrossRef]
- Smith, A.L.M.; Whitehall, J.C.; Greaves, L.C. Mitochondrial DNA mutations in ageing and cancer. Mol. Oncol. 2022, 16, 3276–3294. [Google Scholar] [CrossRef]
- Muramatsu, H.; Honda, K.; Akanuma, S.; Ishizawa, F.; Umino, K.; Iwabuchi, Y.; Mochizuki, N.; Sugano, Y. Trial to search for mitochondrial DNA mutation associated with cancer detected by massively parallel sequencing. Forensic Sci. Int. Genet. Suppl. Ser. 2019, 7, 698–700. [Google Scholar] [CrossRef]
- Gasparre, G.; Porcelli, A.M.; Bonora, E.; Pennisi, L.F.; Toller, M.; Iommarini, L.; Ghelli, A.; Moretti, M.; Betts, C.M.; Martinelli, G.N.; et al. Disruptive mitochondrial DNA mutations in complex I subunits are markers of oncocytic phenotype in thyroid tumors. Proc. Natl. Acad. Sci. USA 2007, 104, 9001–9006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thapa, S.; Lalrohlui, F.; Ghatak, S.; Zohmingthanga, J.; Lallawmzuali, D.; Pautu, J.L.; Kumar, N.S. Mitochondrial complex I and V gene polymorphisms associated with breast cancer in mizo-mongloid population. Breast Cancer 2015, 23, 607–616. [Google Scholar] [CrossRef]
- Porcelli, A.M.; Ghelli, A.; Ceccarelli, C.; Lang, M.; Cenacchi, G.; Capristo, M.; Pennisi, L.F.; Morra, I.; Ciccarelli, E.; Melcarne, A.; et al. The genetic and metabolic signature of oncocytic transformation implicates HIF1α destabilization. Hum. Mol. Genet. 2009, 19, 1019–1032. [Google Scholar] [CrossRef] [PubMed]
- Lang, M.; Vocke, C.D.; Merino, M.J.; Schmidt, L.S.; Linehan, W.M. Mitochondrial DNA mutations distinguish bilateral multifocal renal oncocytomas from familial Birt–Hogg–Dubé tumors. Mod. Pathol. 2015, 28, 1458–1469. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G.; Baracca, A.; Carelli, V.; D’Aurelio, M.; Sgarbi, G.; Solaini, G. Bioenergetics of mitochondrial diseases associated with mtDNA mutations. Biochim. et Biophys. Acta (BBA)—Bioenerg. 2004, 1658, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Moslemi, A.-R.; Darin, N.; Tulinius, M.; Wiklund, L.-M.; Holme, E.; Oldfors, A. Progressive Encephalopathy and Complex I Deficiency Associated with Mutations in MTND1. Neuropediatrics 2008, 39, 24–28. [Google Scholar] [CrossRef]
- Ammar, M.; Tabebi, M.; Sfaihi, L.; Alila-Fersi, O.; Maalej, M.; Felhi, R.; Chabchoub, I.; Keskes, L.; Hachicha, M.; Fakhfakh, F.; et al. Mutational screening in patients with profound sensorineural hearing loss and neurodevelopmental delay: Description of a novel m.3861A > C mitochondrial mutation in the MT-ND1 gene. Biochem. Biophys. Res. Commun. 2016, 474, 702–708. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Z.; Khan, A.; Zheng, H.; Yuan, C.; Jiang, H. Advances in drug therapy for mitochondrial diseases. Ann. Transl. Med. 2020, 8, 17. [Google Scholar] [CrossRef]
- Weissig, V. Drug Development for the Therapy of Mitochondrial Diseases. Trends Mol. Med. 2019, 26, 40–57. [Google Scholar] [CrossRef]
- Solmonson, A.D.; DeBerardinis, R.J. Lipoic acid metabolism and mitochondrial redox regulation. J. Biol. Chem. 2018, 293, 7522–7530. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.; The Mitochondrial Medicine Society; Saneto, R.; Falk, M.J.; Anselm, I.; Cohen, B.H.; Haas, R. A modern approach to the treatment of mitochondrial disease. Curr. Treat. Options Neurol. 2009, 11, 414–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, M.C.; Macdonald, J.R.; Mahoney, D.J.; Parise, G.; Beal, M.F.; Tarnopolsky, M.A. Beneficial effects of creatine, CoQ10, and lipoic acid in mitochondrial disorders. Muscle Nerve 2007, 35, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Avula, S.; Parikh, S.; Demarest, S.; Kurz, J.; Gropman, A. Treatment of Mitochondrial Disorders. Curr. Treat. Options Neurol. 2014, 16, 292. [Google Scholar] [CrossRef] [PubMed]
- Komura, K.; Hobbiebrunken, E.; Wilichowski, E.K.; A Hanefeld, F. Effectiveness of creatine monohydrate in mitochondrial encephalomyopathies. Pediatr. Neurol. 2003, 28, 53–58. [Google Scholar] [CrossRef]
- Tarnopolsky, M.A. Creatine as a therapeutic strategy for myopathies. Amino Acids 2011, 40, 1397–1407. [Google Scholar] [CrossRef]
- Schlame, M.; Ren, M. The role of cardiolipin in the structural organization of mitochondrial membranes. Biochim. et Biophys. Acta (BBA)—Biomembr. 2009, 1788, 2080–2083. [Google Scholar] [CrossRef]
- Szeto, H.H. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol. 2014, 171, 2029–2050. [Google Scholar] [CrossRef]
- Zhao, K.; Zhao, G.-M.; Wu, D.; Soong, Y.; Birk, A.V.; Schiller, P.W.; Szeto, H.H. Cell-permeable Peptide Antioxidants Targeted to Inner Mitochondrial Membrane inhibit Mitochondrial Swelling, Oxidative Cell Death, and Reperfusion Injury. J. Biol. Chem. 2004, 279, 34682–34690. [Google Scholar] [CrossRef]
- Birk, A.V.; Chao, W.M.; Bracken, C.; Warren, J.D.; Szeto, H.H. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br. J. Pharmacol. 2014, 171, 2017–2028. [Google Scholar] [CrossRef]
- Rudolph, G.; Dimitriadis, K.; Büchner, B.; Heck, S.; Al-Tamami, J.; Seidensticker, F.; Rummey, C.; Leinonen, M.; Meier, T.; Klopstock, T. Effects of Idebenone on Color Vision in Patients With Leber Hereditary Optic Neuropathy. J. Neuro-Ophthalmol. 2013, 33, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Trivedi, S.; Pandey, T.; Ranjan, S.; Trivedi, M.; Pandey, R. Wedelolactone Mitigates Parkinsonism Via Alleviating Oxidative Stress and Mitochondrial Dysfunction Through NRF2/SKN-1. Mol. Neurobiol. 2021, 58, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Kavelaars, A.; Dougherty, P.M.; Heijnen, C.J. Beyond symptomatic relief for chemotherapy-induced peripheral neuropathy: Targeting the source. Cancer 2018, 124, 2289–2298. [Google Scholar] [CrossRef] [PubMed]
- Chokchaiwong, S.; Kuo, Y.T.; Lin, S.H.; Hsu, Y.C.; Hsu, S.P.; Liu, Y.T.; Kao, S.H. Coenzyme Q10 serves to couple mitochondrial oxidative phosphorylation and fatty acid beta-oxidation, and attenuates NLRP3 inflammasome activation. Free. Radic. Res. 2018, 52, 1445–1455. [Google Scholar] [CrossRef]
- Athanasopoulos, T.; Munye, M.M.; Yáñez-Muñoz, R.J. Nonintegrating Gene Therapy Vectors. Hematol. Clin. N. Am. 2017, 31, 753–770. [Google Scholar] [CrossRef]
- Gruntman, A.M.; Flotte, T.R. The rapidly evolving state of gene therapy. FASEB J. 2018, 32, 1733–1740. [Google Scholar] [CrossRef]
- Faria, R.; Paul, M.; Biswas, S.; Vivès, E.; Boisguérin, P.; Sousa, Â.; Costa, D. Peptides vs. Polymers: Searching for the Most Efficient Delivery System for Mitochondrial Gene Therapy. Pharmaceutics 2022, 14, 757. [Google Scholar] [CrossRef]
- Coutinho, E.; Batista, C.; Sousa, F.; Queiroz, J.; Costa, D. Mitochondrial Gene Therapy: Advances in Mitochondrial Gene Cloning, Plasmid Production, and Nanosystems Targeted to Mitochondria. Mol. Pharm. 2017, 14, 626–638. [Google Scholar] [CrossRef]
- FC Lopes, A. Mitochondrial metabolism and DNA methylation: A review of the interaction between two genomes. Clin. Epigenetics 2020, 12, 182. [Google Scholar] [CrossRef]
- McManus, M.J.; Picard, M.; Chen, H.-W.; De Haas, H.J.; Potluri, P.; Leipzig, J.; Towheed, A.; Angelin, A.; Sengupta, P.; Morrow, R.M.; et al. Mitochondrial DNA Variation Dictates Expressivity and Progression of Nuclear DNA Mutations Causing Cardiomyopathy. Cell Metab. 2018, 29, 78–90.e5. [Google Scholar] [CrossRef]
- Reyes, A.; Melchionda, L.; Nasca, A.; Carrara, F.; Lamantea, E.; Zanolini, A.; Lamperti, C.; Fang, M.; Zhang, J.; Ronchi, D.; et al. RNASEH1 Mutations Impair mtDNA Replication and Cause Adult-Onset Mitochondrial Encephalomyopathy. Am. J. Hum. Genet. 2015, 97, 186–193. [Google Scholar] [CrossRef]
- Du, X.; Wang, J.; Zhou, Q.; Zhang, L.; Wang, S.; Zhang, Z.; Yao, C. Advanced physical techniques for gene delivery based on membrane perforation. Drug Deliv. 2018, 25, 1516–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrero, M.J.; Sendra, L.; Miguel, A.; Aliño, S.F. Physical Methods of Gene Delivery. Saf. Effic. Gene-Based Ther. 2017, 113–135. [Google Scholar] [CrossRef]
- Schwarz, D.; Kollo, M.; Bosch, C.; Feinauer, C.; Whiteley, I.; Margrie, T.W.; Cutforth, T.; Schaefer, A.T. Architecture of a mammalian glomerular domain revealed by novel volume electroporation using nanoengineered microelectrodes. Nat. Commun. 2018, 9, 183. [Google Scholar] [CrossRef] [PubMed]
- Batabyal, S.; Kim, Y.-T.; Mohanty, S. Ultrafast laser-assisted spatially targeted optoporation into cortical axons and retinal cells in the eye. J. Biomed. Opt. 2017, 22, 060504. [Google Scholar] [CrossRef]
- Nomikou, N.; Feichtinger, G.; Saha, S.; Nuernberger, S.; Heimel, P.; Redl, H.; McHale, A. Ultrasound-responsive gene-activated matrices for osteogenic gene therapy using matrix-assisted sonoporation. J. Tissue Eng. Regen. Med. 2017, 12, e250–e260. [Google Scholar] [CrossRef]
- Polyakova, T.; Zablotskii, V.; Dejneka, A. Cell Membrane Pore Formation and Change in Ion Channel Activity in High-Gradient Magnetic Fields. IEEE Magn. Lett. 2017, 8, 1507805. [Google Scholar] [CrossRef]
- Bonnefoy, N.; Fox, T.D. Directed Alteration of Saccharomyces cerevisiae Mitochondrial DNA by Biolistic Transformation and Homologous Recombination. In Mitochondria; Humana Press: Totowa, NJ, USA, 2007; pp. 372, 153–166. [Google Scholar] [CrossRef]
- Yasuzaki, Y.; Yamada, Y.; Ishikawa, T.; Harashima, H. Validation of Mitochondrial Gene Delivery in Liver and Skeletal Muscle via Hydrodynamic Injection Using an Artificial Mitochondrial Reporter DNA Vector. Mol. Pharm. 2015, 12, 4311–4320. [Google Scholar] [CrossRef]
- Jang, Y.-H.; Lim, K.-I. Recent Advances in Mitochondria-Targeted Gene Delivery. Molecules 2018, 23, 2316. [Google Scholar] [CrossRef]
- Horten, P.; Colina-Tenorio, L.; Rampelt, H. Biogenesis of Mitochondrial Metabolite Carriers. Biomolecules 2020, 10, 1008. [Google Scholar] [CrossRef]
- Bykov, Y.S.; Rapaport, D.; Herrmann, J.M.; Schuldiner, M. Cytosolic Events in the Biogenesis of Mitochondrial Proteins. Trends Biochem. Sci. 2020, 45, 650–667. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.; Joo, C.; Kim, G.-Y.; Ko, K.S.; Huh, K.M.; Han, J.; Choi, J.S. Cationic Oligopeptide-Functionalized Mitochondria Targeting Sequence Show Mitochondria Targeting and Anticancer Activity. Macromol. Res. 2019, 27, 1071–1080. [Google Scholar] [CrossRef]
- Faria, R.; Vivés, E.; Boisguerin, P.; Sousa, A.; Costa, D. Development of Peptide-Based Nanoparticles for Mitochondrial Plasmid DNA Delivery. Polymers 2021, 13, 1836. [Google Scholar] [CrossRef] [PubMed]
- Bayda, S.; Adeel, M.; Tuccinardi, T.; Cordani, M.; Rizzolio, F. The History of Nanoscience and Nanotechnology: From Chemical–Physical Applications to Nanomedicine. Molecules 2020, 25, 112. [Google Scholar] [CrossRef]
- Kinnear, C.; Moore, T.L.; Rodriguez-Lorenzo, L.; Rothen-Rutishauser, B.; Petri-Fink, A. Form Follows Function: Nanoparticle Shape and Its Implications for Nanomedicine. Chem. Rev. 2017, 117, 11476–11521. [Google Scholar] [CrossRef]
- Weissig, V.; Pettinger, T.K.; Murdock, N. Nanopharmaceuticals (part 1): Products on the market. Int. J. Nanomed. 2014, 9, 4357–4373. [Google Scholar] [CrossRef]
- Yuan, Y.; Gu, Z.; Yao, C.; Luo, D.; Yang, D. Nucleic Acid–Based Functional Nanomaterials as Advanced Cancer Therapeutics. Small 2019, 15, e1900172. [Google Scholar] [CrossRef]
- Cordani, M.; Somoza, Á. Targeting autophagy using metallic nanoparticles: A promising strategy for cancer treatment. Cell Mol. Life Sci. 2018, 76, 1215–1242. [Google Scholar] [CrossRef]
- Indoria, S.; Singh, V.; Hsieh, M.-F. Recent advances in theranostic polymeric nanoparticles for cancer treatment: A review. Int. J. Pharm. 2020, 582, 119314. [Google Scholar] [CrossRef]
- Zheng, F.; Xiong, W.; Sun, S.; Zhang, P.; Zhu, J.J. Recent advances in drug release monitoring. Nanophotonics 2019, 8, 391–413. [Google Scholar] [CrossRef]
- Khair, S.Z.N.M.; Radzak, S.M.A.; Yusoff, A.A.M. The Uprising of Mitochondrial DNA Biomarker in Cancer. Dis. Markers 2021, 2021, 7675269. [Google Scholar] [CrossRef]
- Trecarichi, A.; Duggett, N.A.; Granat, L.; Lo, S.; Malik, A.N.; Zuliani-Álvarez, L.; Flatters, S.J.L. Preclinical evidence for mitochondrial DNA as a potential blood biomarker for chemotherapy-induced peripheral neuropathy. PLoS ONE 2022, 17, e0262544. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Qi, R.; Zhang, L.; Yu, Y.; Hou, J.; Gu, Y.; Song, D.; Wang, X. Potential biomarkers and targets of mitochondrial dynamics. Clin. Transl. Med. 2021, 11, e529. [Google Scholar] [CrossRef]
- Tian, H.; Chen, J.; Chen, X. Nanoparticles for gene delivery. Small 2013, 9, 2034–2044. [Google Scholar] [CrossRef]
- Seeman, N.C.; Sleiman, H.F. DNA nanotechnology. Nat. Rev. Mater. 2017, 3, 17068. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, Q.; Xia, X.; Li, X.; Ruan, W.; Zheng, M.; Zou, Y.; Shi, B. Polymeric Nanoparticles for Mitochondria Targeting Mediated Robust Cancer Therapy. Front. Bioeng. Biotechnol. 2021, 9, 755727. [Google Scholar] [CrossRef]
- Allemailem, K.S.; Almatroudi, A.; Alsahli, M.A.; Aljaghwani, A.; El-Kady, A.M.; Rahmani, A.H.; Khan, A.A. Novel Strategies for Disrupting Cancer-Cell Functions with Mitochondria-Targeted Antitumor Drug-Loaded Nanoformulations. Int. J. Nanomed. 2021, 16, 3907–3936. [Google Scholar]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef]
- Guzman-Villanueva, D.; Mendiola, M.R.; Nguyen, H.X.; Yambao, F.; Yu, N.; Weissig, V. Conjugation of Triphenylphosphonium Cation to Hydrophobic Moieties to Prepare Mitochondria-Targeting Nanocarriers. Basic Protoc. 2019, 2000, 183–189. [Google Scholar] [CrossRef]
- Marrache, S.; Tundup, S.; Harn, D.A.; Dhar, S. Ex Vivo Programming of Dendritic Cells by Mitochondria-Targeted Nanoparticles to Produce Interferon-Gamma for Cancer Immunotherapy. ACS Nano 2013, 7, 7392–7402. [Google Scholar] [CrossRef]
- Bae, Y.; Jung, M.K.; Lee, S.; Song, S.J.; Mun, J.Y.; Green, E.S.; Han, J.; Ko, K.S.; Choi, J.S. Dequalinium-based functional nanosomes show increased mitochondria targeting and anticancer effect. Eur. J. Pharm. Biopharm. 2018, 124, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Mallick, S.; Song, S.J.; Bae, Y.; Choi, J.S. Self-assembled nanoparticles composed of glycol chitosan-dequalinium for mitochondria-targeted drug delivery. Int. J. Biol. Macromol. 2019, 132, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Faria, R.; Albuquerque, T.; Neves, A.R.; Bhatt, H.; Biswas, S.; Cardoso, A.M.; de Lima, M.C.P.; Jurado, A.S.; Costa, D. Physicochemical characterization and targeting performance of triphenylphosphonium nano-polyplexes. J. Mol. Liq. 2020, 316, 113873. [Google Scholar] [CrossRef]
- Nikzamir, M.; Hanifehpour, Y.; Akbarzadeh, A.; Panahi, Y. Applications of Dendrimers in Nanomedicine and Drug Delivery: A Review. J. Inorg. Organomet. Polym. Mater. 2021, 31, 2246–2261. [Google Scholar] [CrossRef]
- Surekha, B.; Kommana, N.S.; Dubey, S.K.; Kumar, A.P.; Shukla, R.; Kesharwani, P. PAMAM dendrimer as a talented multifunctional biomimetic nanocarrier for cancer diagnosis and therapy. Colloids Surfaces B Biointerfaces 2021, 204, 111837. [Google Scholar] [CrossRef]
- Oddone, N.; Lecot, N.; Fernández, M.; Rodriguez-Haralambides, A.; Cabral, P.; Cerecetto, H.; Benech, J.C. In vitro and in vivo uptake studies of PAMAM G4.5 dendrimers in breast cancer. J. Nanobiotechnol. 2016, 14, 45. [Google Scholar] [CrossRef]
- Wang, X.; Shao, N.; Zhang, Q.; Cheng, Y. Mitochondrial targeting dendrimer allows efficient and safe gene delivery. J. Mater. Chem. B 2013, 2, 2546–2553. [Google Scholar] [CrossRef]
- Biswas, S.; Dodwadkar, N.S.; Piroyan, A.; Torchilin, V.P. Surface conjugation of triphenylphosphonium to target poly(amidoamine) dendrimers to mitochondria. Biomaterials 2012, 33, 4773–4782. [Google Scholar] [CrossRef]
- Dolatabadi, J.E.N.; Valizadeh, H.; Hamishehkar, H. Solid Lipid Nanoparticles as Efficient Drug and Gene Delivery Systems: Recent Breakthroughs. Adv. Pharm. Bull. 2015, 5, 151–159. [Google Scholar] [CrossRef]
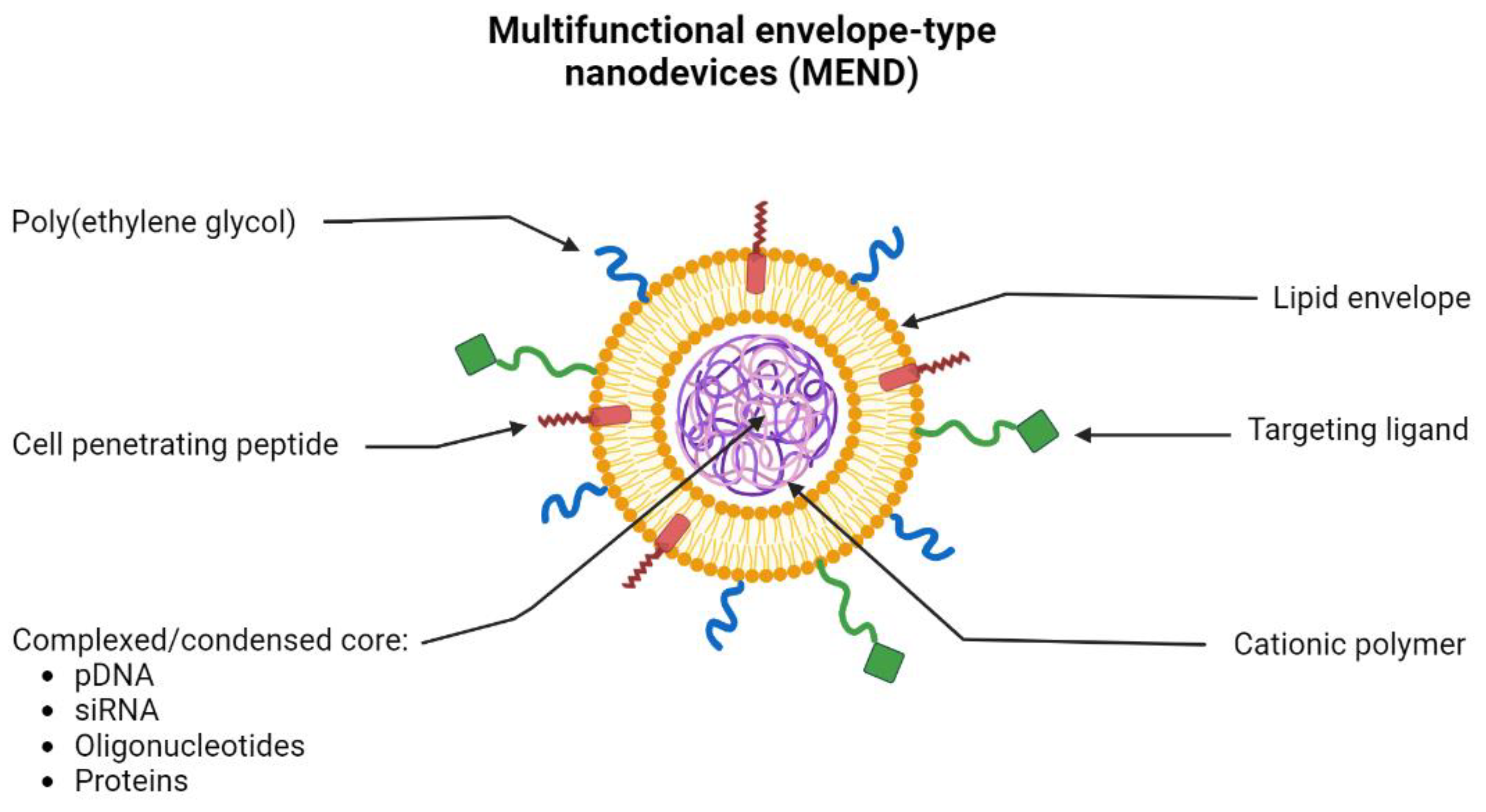
- Yamada, Y.; Akita, H.; Harashima, H. Multifunctional Envelope-Type Nano Device (MEND) for Organelle Targeting Via a Stepwise Membrane Fusion Process. Methods Enzymol. 2012, 509, 301–326. [Google Scholar] [CrossRef]
- Khalil, I.; Kogure, K.; Futaki, S.; Hama, S.; Akita, H.; Ueno, M.; Kishida, H.; Kudoh, M.; Mishina, Y.; Kataoka, K.; et al. Octaarginine-modified multifunctional envelope-type nanoparticles for gene delivery. Gene Ther. 2007, 14, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Furukawa, R.; Yasuzaki, Y.; Harashima, H. Dual Function MITO-Porter, a Nano Carrier Integrating Both Efficient Cytoplasmic Delivery and Mitochondrial Macromolecule Delivery. Mol. Ther. 2011, 19, 1449–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akita, H.; Kudo, A.; Minoura, A.; Yamaguti, M.; Khalil, I.A.; Moriguchi, R.; Masuda, T.; Danev, R.; Nagayama, K.; Kogure, K.; et al. Multi-layered nanoparticles for penetrating the endosome and nuclear membrane via a step-wise membrane fusion process. Biomaterials 2009, 30, 2940–2949. [Google Scholar] [CrossRef]
- Yamada, Y.; Fujishita, N.; Harashima, H. A nanocarrier for the mitochondrial delivery of nucleic acids to cardiomyocytes. Nucleosides Nucleotides Nucleic Acids 2019, 39, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.; Jung, M.K.; Song, S.J.; Green, E.S.; Lee, S.; Park, H.-S.; Jeong, S.H.; Han, J.; Mun, J.Y.; Ko, K.S.; et al. Functional nanosome for enhanced mitochondria-targeted gene delivery and expression. Mitochondrion 2017, 37, 27–40. [Google Scholar] [CrossRef]
- D’Souza, G.G.; Boddapati, S.V.; Weissig, V. Mitochondrial leader sequence-plasmid DNA conjugates delivered into mammalian cells by DQAsomes co-localize with mitochondria. Mitochondrion 2005, 5, 352–358. [Google Scholar] [CrossRef]
- Yamada, Y.; Akita, H.; Kamiya, H.; Kogure, K.; Yamamoto, T.; Shinohara, Y.; Yamashita, K.; Kobayashi, H.; Kikuchi, H.; Harashima, H. MITO-Porter: A liposome-based carrier system for delivery of macromolecules into mitochondria via membrane fusion. Biochim. et Biophys. Acta (BBA)—Biomembr. 2008, 1778, 423–432. [Google Scholar] [CrossRef]
- Yamada, Y.; Harashima, H. Mitochondrial drug delivery systems for macromolecule and their therapeutic application to mitochondrial diseases. Adv. Drug Deliv. Rev. 2008, 60, 1439–1462. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, E.; Maruyama, M.; Abe, J.; Sudo, A.; Takeda, A.; Takada, S.; Yokota, T.; Kinugawa, S.; Harashima, H.; Yamada, Y. Validation of Gene Therapy for Mutant Mitochondria by Delivering Mitochondrial RNA Using a MITO-Porter. Mol. Ther.—Nucleic Acids 2020, 20, 687–698. [Google Scholar] [CrossRef]
- Kawamura, E.; Hibino, M.; Harashima, H.; Yamada, Y. Targeted mitochondrial delivery of antisense RNA-containing nanoparticles by a MITO-Porter for safe and efficient mitochondrial gene silencing. Mitochondrion 2019, 49, 178–188. [Google Scholar] [CrossRef]
- Ishikawa, T.; Somiya, K.; Munechika, R.; Harashima, H.; Yamada, Y. Mitochondrial transgene expression via an artificial mitochondrial DNA vector in cells from a patient with a mitochondrial disease. J. Control Release 2018, 274, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Wolfram, R.K.; Heller, L.; Csuk, R. Targeting mitochondria: Esters of rhodamine B with triterpenoids are mitocanic triggers of apoptosis. Eur. J. Med. Chem. 2018, 152, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.; Sousa, F.; Queiroz, J.; Costa, D. Rhodamine based plasmid DNA nanoparticles for mitochondrial gene therapy. Colloids Surf. B Biointerfaces 2014, 121, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Salvado, R.; Sousa, F.; Queiroz, J.; Costa, D. Development of mitochondrial targeting plasmid DNA nanoparticles: Characterization and in vitro studies. Colloids Surf. A Physicochem. Eng. Asp. 2015, 480, 287–295. [Google Scholar] [CrossRef]
- Costa, D.; Costa, C.; Caldeira, M.; Cortes, L.; Queiroz, J.A.; Cruz, C. Targeting of Cellular Organelles by Fluorescent Plasmid DNA Nanoparticles. Biomacromolecules 2017, 18, 2928–2936. [Google Scholar] [CrossRef]
- Huang, S.; Shao, K.; Liu, Y.; Kuang, Y.; Li, J.; An, S.; Guo, Y.; Ma, H.; Jiang, C. Tumor-Targeting and Microenvironment-Responsive Smart Nanoparticles for Combination Therapy of Antiangiogenesis and Apoptosis. ACS Nano 2013, 7, 2860–2871. [Google Scholar] [CrossRef]
- Huang, R.; Ke, W.; Liu, Y.; Jiang, C.; Pei, Y. The use of lactoferrin as a ligand for targeting the polyamidoamine-based gene delivery system to the brain. Biomaterials 2008, 29, 238–246. [Google Scholar] [CrossRef]
- Han, L.; Guo, Y.; Ma, H.; He, X.; Kuang, Y.; Zhang, N.; Lim, E.; Zhou, W.; Jiang, C. Acid Active Receptor-Specific Peptide Ligand for In Vivo Tumor-Targeted Delivery. Small 2013, 9, 3647–3658. [Google Scholar] [CrossRef]
- Chen, H.; Tian, J.; Liu, D.; He, W.; Guo, Z. Dual aptamer modified dendrigraft poly-l-lysine nanoparticles for overcoming multi-drug resistance through mitochondrial targeting. J. Mater. Chem. B 2016, 5, 972–979. [Google Scholar] [CrossRef]
- Fan, X.-X.; Xu, M.-Z.; Leung, E.L.-H.; Jun, C.; Yuan, Z.; Liu, L. ROS-Responsive Berberine Polymeric Micelles Effectively Suppressed the Inflammation of Rheumatoid Arthritis by Targeting Mitochondria. Nano-Micro Lett. 2020, 12, 76. [Google Scholar] [CrossRef]
- Zhou, W.; Yu, H.; Zhang, L.-J.; Wu, B.; Wang, C.-X.; Wang, Q.; Deng, K.; Zhuo, R.-X.; Huang, S.-W. Redox-triggered activation of nanocarriers for mitochondria-targeting cancer chemotherapy. Nanoscale 2017, 9, 17044–17053. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xia, P.; Pan, S.; Qi, Z.; Fu, C.; Yu, Z.; Kong, W.; Chang, Y.; Wang, K.; Wu, D.; et al. The Advances of Ceria Nanoparticles for Biomedical Applications in Orthopaedics. Int. J. Nanomed. 2020, 15, 7199–7214. [Google Scholar] [CrossRef]
- Yu, H.; Jin, F.; Liu, D.; Shu, G.; Wang, X.; Qi, J.; Sun, M.; Yang, P.; Jiang, S.; Ying, X.; et al. ROS-responsive nano-drug delivery system combining mitochondria-targeting ceria nanoparticles with atorvastatin for acute kidney injury. Theranostics 2020, 10, 2342–2357. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, E.; Yamada, Y.; Harashima, H. Mitochondrial targeting functional peptides as potential devices for the mitochondrial delivery of a DF-MITO-Porter. Mitochondrion 2013, 13, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Nakamura, K.; Abe, J.; Hyodo, M.; Haga, S.; Ozaki, M.; Harashima, H. Mitochondrial delivery of Coenzyme Q10 via systemic administration using a MITO-Porter prevents ischemia/reperfusion injury in the mouse liver. J. Control Release 2015, 213, 86–95. [Google Scholar] [CrossRef]
- Sharma, A.; Liaw, K.; Sharma, R.; Zhang, Z.; Kannan, S.; Kannan, R.M. Targeting Mitochondrial Dysfunction and Oxidative Stress in Activated Microglia using Dendrimer-Based Therapeutics. Theranostics 2018, 8, 5529–5547. [Google Scholar] [CrossRef]
- Li, J.; Liu, Y.; Li, H.; Shi, W.; Bi, X.; Qiu, Q.; Qian, H.; Zhang, B. pH-Sensitive micelles with mitochondria-targeted and aggregation-induced emission characterization: Synthesis, cytotoxicity and biological applications. Biomater. Sci. 2018, 6, 2998–3008. [Google Scholar]
- Yang, P.; Sheng, D.; Guo, Q.; Wang, P.; Xu, S.; Qian, K.; Li, Y.; Cheng, Y.; Wang, L.; Zhang, Q.; et al. Neuronal mitochondria-targeted micelles relieving oxidative stress for delayed progression of Alzheimer’s disease. Biomaterials 2020, 238, 119844. [Google Scholar]
- Arafa, K.K.; Hamzawy, M.A.; Mousa, S.A.; El-Sherbiny, I.M. Mitochondria-targeted alginate/triphenylphosphonium-grafted-chitosan for treatment of hepatocellular carcinoma. RSC Adv. 2022, 12, 21690–21703. [Google Scholar] [CrossRef]
- Kulkarni, C.A.; Fink, B.D.; Gibbs, B.E.; Chheda, P.R.; Wu, M.; Sivitz, W.I.; Kerns, R.J. A Novel Triphenylphosphonium Carrier to Target Mitochondria without Uncoupling Oxidative Phosphorylation. J. Med. Chem. 2021, 64, 662–676. [Google Scholar] [CrossRef]
- Battogtokh, G.; Cho, Y.-Y.; Lee, J.Y.; Lee, H.S.; Kang, H.C. Mitochondrial-Targeting Anticancer Agent Conjugates and Nanocarrier Systems for Cancer Treatment. Front. Pharmacol. 2018, 9, 922. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutated Mitochondrial Gene | Associated Disease | Reference |
|---|---|---|
| ND1 | Type II diabetes | [44] |
| Leber’s hereditary optic neuropathy (LHON) | [46] | |
| Leigh’s syndrome | [66] | |
| Progressive cardiomyopathy | [67] | |
| Neurodevelopmental delay | [68] | |
| Sensorineural hearing loss | ||
| Renal cancer | [57] | |
| Thyroid carcinoma | [62] | |
| Oncocytic pituitary adenoma | [64] | |
| Breast carcinoma | [65] | |
| ND3 | Type II diabetes | [45] |
| Parkinson’s disease | [52] | |
| ND5 | Invasive ductal breast carcinoma | [62] |
| ATP6 | Type II diabetes | [43] |
| Mitochondrial encephalomyopathies | [49] | |
| Alzheimer’s disease | [53] | |
| Hurthle cell thyroid carcinoma | [58] | |
| Breast cancer | [63] | |
| Desmin | Desmin myopathies | [48] |
| Skeletal myopathy | ||
| C9orf72/TARDBP | Frontotemporal dementia | [50] |
| Amyotrophic lateral sclerosis | ||
| COXIII | Invasive lobular breast carcinoma | [62] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faria, R.; Boisguérin, P.; Sousa, Â.; Costa, D. Delivery Systems for Mitochondrial Gene Therapy: A Review. Pharmaceutics 2023, 15, 572. https://doi.org/10.3390/pharmaceutics15020572
Faria R, Boisguérin P, Sousa Â, Costa D. Delivery Systems for Mitochondrial Gene Therapy: A Review. Pharmaceutics. 2023; 15(2):572. https://doi.org/10.3390/pharmaceutics15020572
Chicago/Turabian StyleFaria, Rúben, Prisca Boisguérin, Ângela Sousa, and Diana Costa. 2023. "Delivery Systems for Mitochondrial Gene Therapy: A Review" Pharmaceutics 15, no. 2: 572. https://doi.org/10.3390/pharmaceutics15020572