

The Mitochondrion: A Promising Target for Kidney Disease
 , , , and
, , , and
Abstract
:1. Introduction
2. Mitochondria as a Key Regulator of Kidney Disease: The Pathophysiological Basis
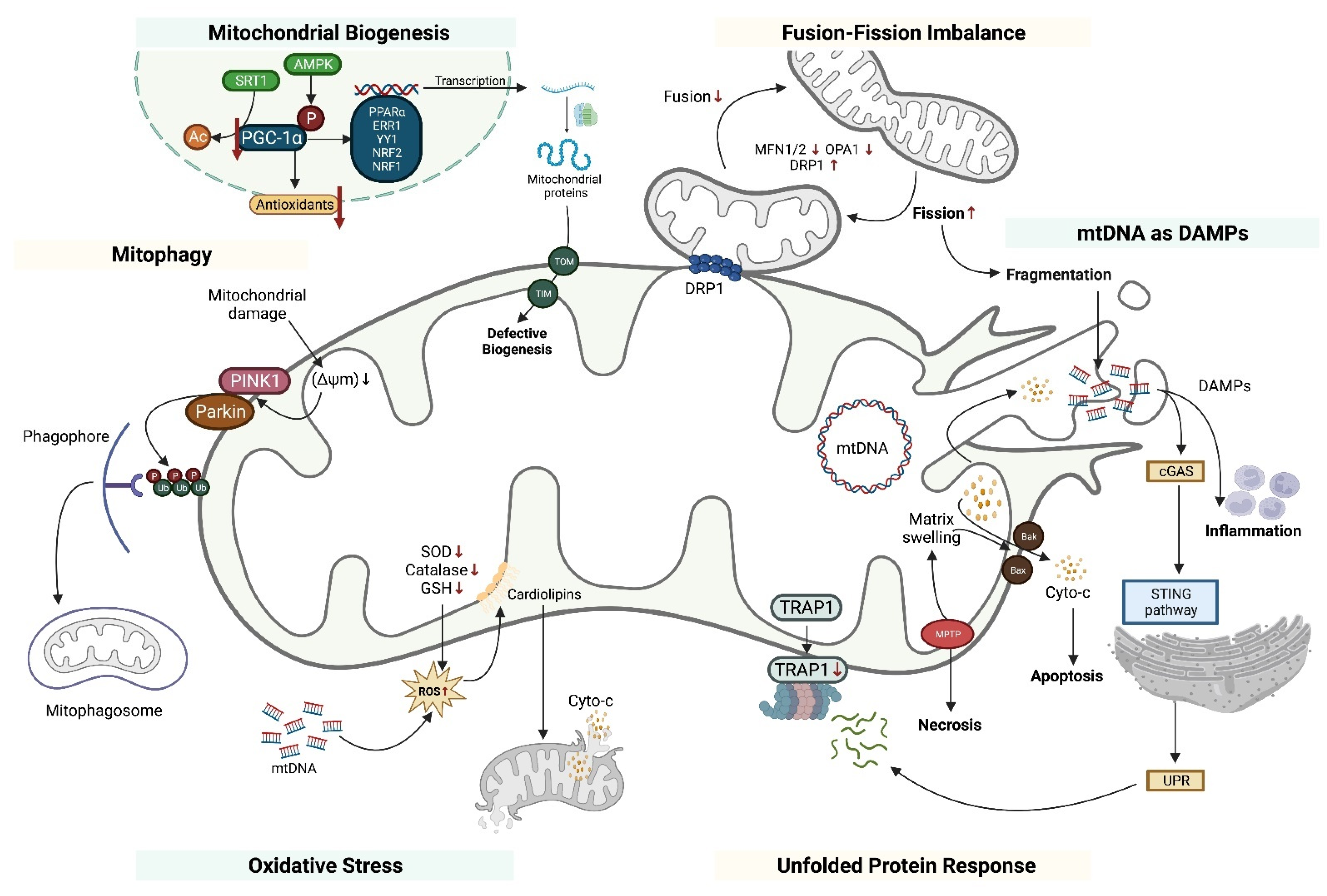
2.1. Alterations of Mitochondrial Biogenesis
2.1.1. PGC-1α and AKI
2.1.2. PGC-1α and Glomerular Diseases
2.2. Mitochondrial Fusion-Fission Imbalance
2.3. Mitochondrial DNA as DAMPs
2.4. Mitophagy
2.5. Oxidative Stress and Antioxidant Defense
2.6. Unfolded Protein Response
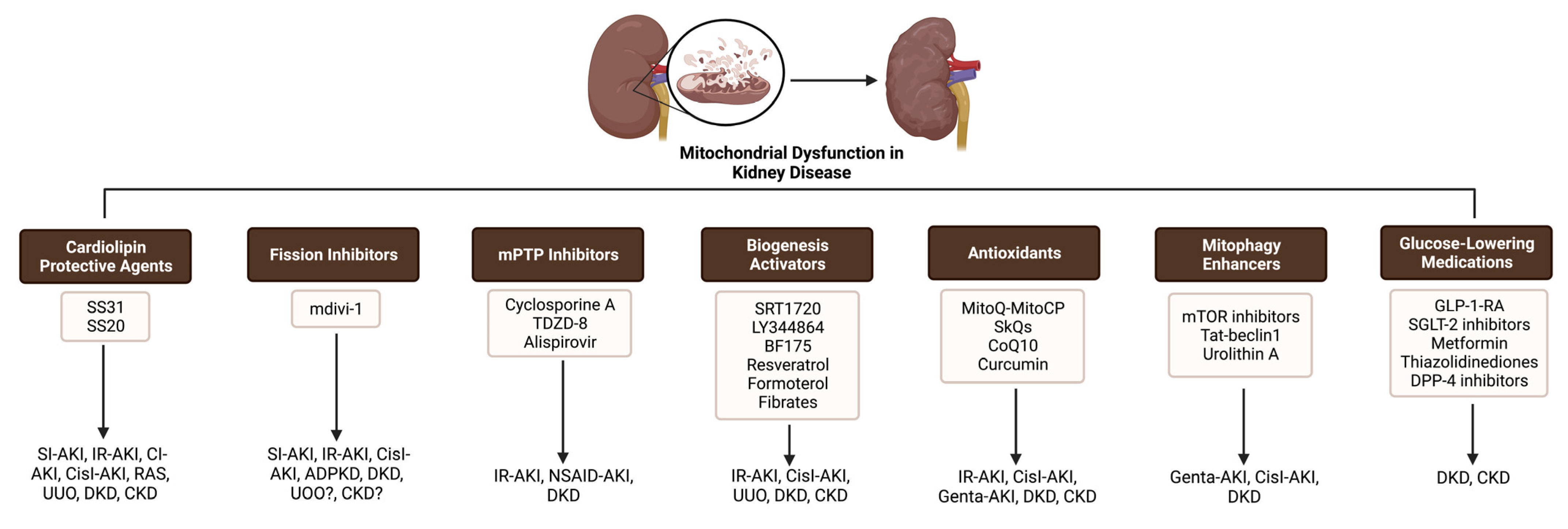
3. The Targeting of Mitochondria as a Therapeutic Approach in Kidney Disease
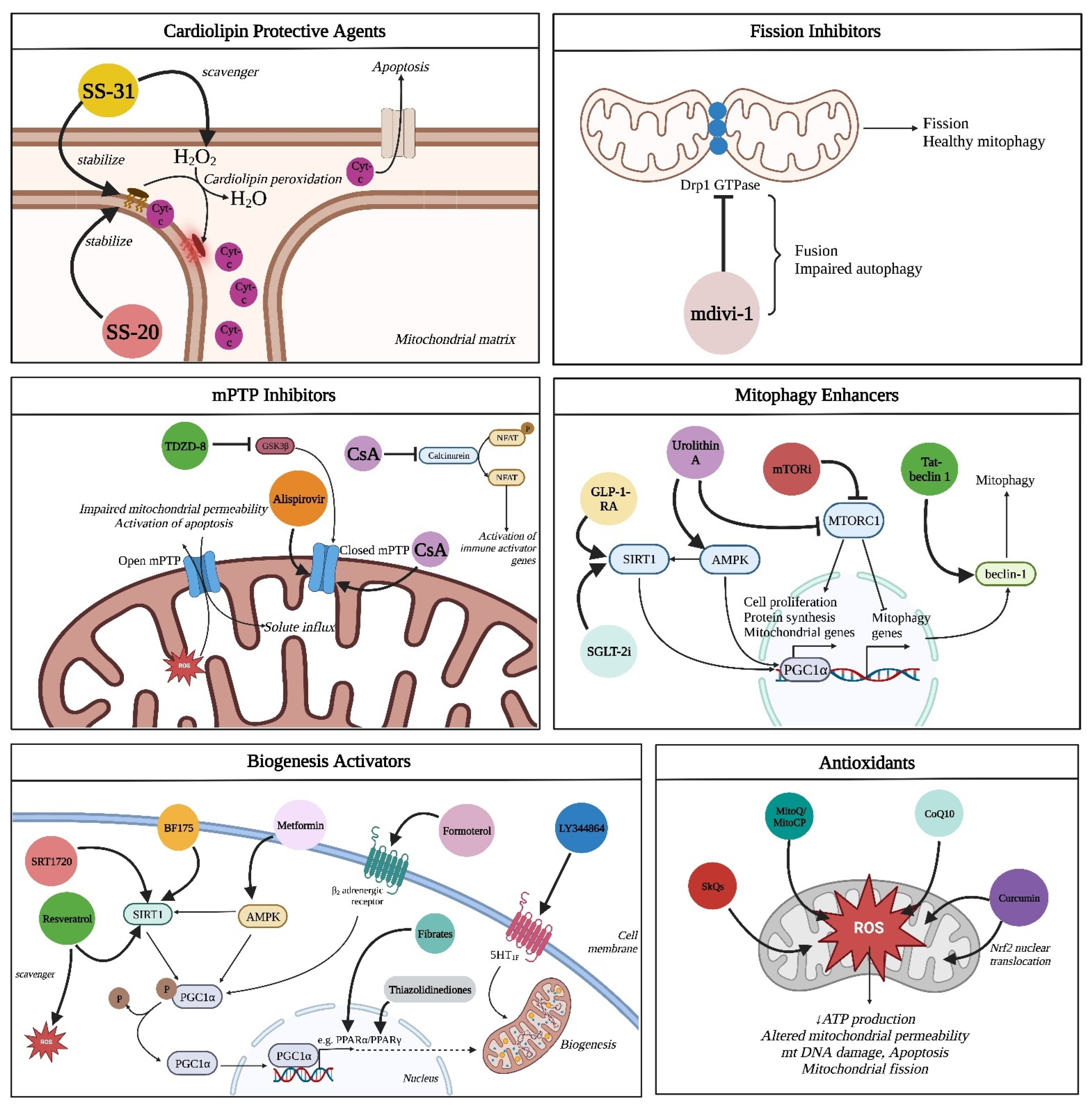
3.1. Cardiolipin Protective Agents
3.1.1. Szeto–Schiller Peptide 31 (SS-31)
3.1.2. Szeto–Schiller Peptide 20 (SS-20)
3.2. Fission Inhibitors
Mitochondrial Division Inhibitor-1
3.3. mPTP Inhibitors
3.3.1. Cyclosporine-A
3.3.2. TDZD-8
3.3.3. Alisporivir
3.4. Biogenesis Activators
3.4.1. SRT1720
3.4.2. LY344864
3.4.3. BF175
3.4.4. Resveratrol
3.4.5. Formoterol
3.4.6. Fibrates
3.5. Antioxidants
3.5.1. MitoQ and Mito-CP
3.5.2. SkQs
3.5.3. Ubiquinone (Coenzyme Q10, CoQ10)
3.5.4. Curcumin
3.6. Enhancement of Mitophagy and Autophagy
3.6.1. mTOR Inhibitors
3.6.2. Tat–Beclin 1
3.6.3. Urolithin A
3.6.4. Other Strategies
3.7. The Role of Mitochondria in the Treatment of Polycystic Kidney Disease
3.8. Glucose-Lowering Medications
3.8.1. GLP-1-RA
3.8.2. SGLT-2 Inhibitors
3.8.3. Other Glucose-Lowering Medications
4. Conclusions, Future Directions, and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Makris, K.; Spanou, L. Acute Kidney Injury: Definition, Pathophysiology and Clinical Phenotypes. Clin. Biochem. Rev. 2016, 37, 85–98. [Google Scholar]
- Lee, S.A.; Cozzi, M.; Bush, E.L.; Rabb, H. Distant Organ Dysfunction in Acute Kidney Injury: A Review. Am. J. Kidney Dis. 2018, 72, 846–856. [Google Scholar] [CrossRef]
- Zhang, X.; Agborbesong, E.; Li, X. The Role of Mitochondria in Acute Kidney Injury and Chronic Kidney Disease and Its Therapeutic Potential. Int. J. Mol. Sci. 2021, 22, 11253. [Google Scholar] [CrossRef]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef] [PubMed]
- Couser, W.G.; Remuzzi, G.; Mendis, S.; Tonelli, M. The contribution of chronic kidney disease to the global burden of major noncommunicable diseases. Kidney Int. 2011, 80, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Cai, J.; Yin, X.M.; Weinberg, J.M.; Venkatachalam, M.A.; Dong, Z. Mitochondrial quality control in kidney injury and repair. Nat. Rev. Nephrol. 2020, 17, 299–318. [Google Scholar] [CrossRef]
- Afsar, B.; Hornum, M.; Afsar, R.E.; Ertuglu, L.A.; Ortiz, A.; Covic, A.; van Raalte, D.H.; Cherney, D.Z.I.; Kanbay, M. Mitochondrion-driven nephroprotective mechanisms of novel glucose lowering medications. Mitochondrion 2021, 58, 72–82. [Google Scholar] [CrossRef]
- Nath, K.A.; Grande, J.P.; Croatt, A.J.; Likely, S.; Hebbel, R.P.; Enright, H. Intracellular targets in heme protein-induced renal injury. Kidney Int. 1998, 53, 100–111. [Google Scholar] [CrossRef]
- Funk, J.A.; Schnellmann, R.G. Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am. J. Physiol. Renal Physiol. 2012, 302, F853–F864. [Google Scholar] [CrossRef]
- Brooks, C.; Wei, Q.; Cho, S.-G.; Dong, Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J. Clin. Investig. 2009, 119, 1275–1285. [Google Scholar] [CrossRef]
- Perry, H.M.; Huang, L.; Wilson, R.J.; Bajwa, A.; Sesaki, H.; Yan, Z.; Rosin, D.L.; Kashatus, D.F.; Okusa, M.D. Dynamin-Related Protein 1 Deficiency Promotes Recovery from AKI. J. Am. Soc. Nephrol. 2018, 29, 194–206. [Google Scholar] [CrossRef]
- Szeto, H.H.; Liu, S.; Soong, Y.; Seshan, S.V.; Cohen-Gould, L.; Manichev, V.; Feldman, L.C.; Gustafsson, T. Mitochondria Protection after Acute Ischemia Prevents Prolonged Upregulation of IL-1β and IL-18 and Arrests CKD. J. Am. Soc. Nephrol. 2017, 28, 1437–1449. [Google Scholar] [CrossRef]
- Tran, M.; Tam, D.; Bardia, A.; Bhasin, M.; Rowe, G.C.; Kher, A.; Zsengeller, Z.K.; Akhavan-Sharif, M.R.; Khankin, E.V.; Saintgeniez, M.; et al. PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Investig. 2011, 121, 4003–4014. [Google Scholar] [CrossRef] [PubMed]
- Dominy, J.E.; Puigserver, P. Mitochondrial Biogenesis through Activation of Nuclear Signaling Proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a015008. [Google Scholar] [CrossRef]
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Martinez-Moreno, J.M.; Monsalve, M.; Ramos, A.M.; Sanchez-Nino, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. The Role of PGC-1α and Mitochondrial Biogenesis in Kidney Diseases. Biomolecules 2020, 10, 347. [Google Scholar] [CrossRef] [PubMed]
- Parekh, D.J.; Weinberg, J.M.; Ercole, B.; Torkko, K.C.; Hilton, W.; Bennett, M.; Devarajan, P.; Venkatachalam, M.A. Tolerance of the Human Kidney to Isolated Controlled Ischemia. J. Am. Soc. Nephrol. 2013, 24, 506–517. [Google Scholar] [CrossRef]
- Zager, R.A.; Johnson, A.C.; Hanson, S.Y. Renal tubular triglyercide accumulation following endotoxic, toxic, and ischemic injury. Kidney Int. 2005, 67, 111–121. [Google Scholar] [CrossRef]
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant Mechanisms in Renal Injury and Disease. Antioxid. Redox Signal. 2016, 25, 119–146. [Google Scholar] [CrossRef]
- Qin, N.; Cai, T.; Ke, Q.; Yuan, Q.; Luo, J.; Mao, X.; Jiang, L.; Cao, H.; Wen, P.; Zen, K.; et al. UCP2-dependent improvement of mitochondrial dynamics protects against acute kidney injury. J. Pathol. 2019, 247, 392–405. [Google Scholar] [CrossRef]
- Morigi, M.; Perico, L.; Rota, C.; Longaretti, L.; Conti, S.; Rottoli, D.; Novelli, R.; Remuzzi, G.; Benigni, A. Sirtuin 3–dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Investig. 2015, 125, 715–726. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Liu, J.; Zhou, F.; Wang, W.; Chen, N. PGC-1α ameliorates kidney fibrosis in mice with diabetic kidney disease through an antioxidative mechanism. Mol. Med. Rep. 2018, 17, 4490–4498. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-Y.; Kang, J.M.; Kim, D.-J.; Park, S.H.; Jeong, H.Y.; Lee, Y.H.; Kim, Y.G.; Yang, D.H.; Lee, S.H. PGC1α Activators Mitigate Diabetic Tubulopathy by Improving Mitochondrial Dynamics and Quality Control. J. Diabetes Res. 2017, 2017, 6483572. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.-X.; Wu, W.-H.; Qiu, H.-Y.; Bo, H.; Huang, S.-M. Amelioration of rhabdomyolysis-induced renal mitochondrial injury and apoptosis through suppression of Drp-1 translocation. J. Nephrol. 2013, 26, 1073–1082. [Google Scholar] [CrossRef]
- Fontecha-Barriuso, M.; Martín-Sánchez, D.; Martinez-Moreno, J.M.; Carrasco, S.; Ruiz-Andrés, O.; Monsalve, M.; Sanchez-Ramos, C.; Gómez, M.J.; Ruiz-Ortega, M.; Sánchez-Niño, M.D.; et al. PGC-1α deficiency causes spontaneous kidney inflammation and increases the severity of nephrotoxic AKI. J. Pathol. 2019, 249, 65–78. [Google Scholar] [CrossRef]
- Tran, M.T.; Zsengeller, Z.K.; Berg, A.H.; Khankin, E.V.; Bhasin, M.K.; Kim, W.; Clish, C.B.; Stillman, I.E.; Karumanchi, S.A.; Rhee, E.P.; et al. PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 2016, 531, 528–532. [Google Scholar] [CrossRef]
- Lynch, M.R.; Tran, M.T.; Ralto, K.M.; Zsengeller, Z.K.; Raman, V.; Bhasin, S.S.; Sun, N.; Chen, X.; Brown, D.; Rovira, I.I.; et al. TFEB-driven lysosomal biogenesis is pivotal for PGC1α-dependent renal stress resistance. JCI Insight 2019, 5. [Google Scholar] [CrossRef]
- Kim, D.H.; Jung, Y.J.; Lee, J.E.; Lee, A.S.; Kang, K.P.; Lee, S.; Park, S.K.; Han, M.K.; Lee, S.Y.; Ramkumar, K.M.; et al. SIRT1 activation by resveratrol ameliorates cisplatin-induced renal injury through deacetylation of p53. Am. J. Physiol. Renal Physiol. 2011, 301, F427–F435. [Google Scholar] [CrossRef]
- Lempiäinen, J.; Finckenberg, P.; Levijoki, J.; Mervaala, E. AMPK activator AICAR ameliorates ischaemia reperfusion injury in the rat kidney. Br. J. Pharmacol. 2012, 166, 1905–1915. [Google Scholar] [CrossRef]
- Chander, V.; Chopra, K. Protective Effect of Nitric Oxide Pathway in Resveratrol Renal Ischemia-Reperfusion Injury in Rats. Arch. Med. Res. 2006, 37, 19–26. [Google Scholar] [CrossRef]
- Poyan Mehr, A.; Tran, M.T.; Ralto, K.M.; Leaf, D.E.; Washco, V.; Messmer, J.; Lerner, A.; Kher, A.; Kim, S.H.; Khoury, C.C.; et al. De novo NAD+ biosynthetic impairment in acute kidney injury in humans. Nat. Med. 2018, 24, 1351–1359. [Google Scholar] [CrossRef]
- Zhao, W.-Y.; Zhang, L.; Sui, M.-X.; Zhu, Y.-H.; Zeng, L. Protective effects of sirtuin 3 in a murine model of sepsis-induced acute kidney injury. Sci. Rep. 2016, 6, 33201. [Google Scholar] [CrossRef]
- Sharma, K.; Karl, B.; Mathew, A.V.; Gangoiti, J.A.; Wassel, C.L.; Saito, R.; Pu, M.; Sharma, S.; You, Y.-H.; Wang, L.; et al. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J. Am. Soc. Nephrol. 2013, 24, 1901–1912. [Google Scholar] [CrossRef]
- Zhang, T.; Chi, Y.; Ren, Y.; Du, C.; Shi, Y.; Li, Y. Resveratrol Reduces Oxidative Stress and Apoptosis in Podocytes via Sir2-Related Enzymes, Sirtuins1 (SIRT1)/Peroxisome Proliferator-Activated Receptor γ Co-Activator 1α (PGC-1α) Axis. Med. Sci. Monit. 2019, 25, 1220–1231. [Google Scholar] [CrossRef]
- Wu, L.; Wang, Q.; Guo, F.; Zhou, Y.; Ji, H.; Liu, F.; Ma, X.; Zhao, Y.; Qin, G. Activation of FoxO1/ PGC-1α prevents mitochondrial dysfunction and ameliorates mesangial cell injury in diabetic rats. Mol. Cell. Endocrinol. 2015, 413, 1–12. [Google Scholar] [CrossRef]
- Long, J.; Badal, S.S.; Ye, Z.; Wang, Y.; Ayanga, B.A.; Galvan, D.L.; Green, N.H.; Chang, B.H.; Overbeek, P.A.; Danesh, F.R. Long noncoding RNA Tug1 regulates mitochondrial bioenergetics in diabetic nephropathy. J. Clin. Investig. 2016, 126, 4205–4218. [Google Scholar] [CrossRef]
- Qi, W.; Keenan, H.A.; Li, Q.; Ishikado, A.; Kannt, A.; Sadowski, T.; Yorek, M.A.; Wu, I.H.; Lockhart, S.; Coppey, L.J.; et al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat. Med. 2017, 23, 753–762. [Google Scholar] [CrossRef]
- Zuo, Y.; Yang, H.-C.; Potthoff, S.A.; Najafian, B.; Kon, V.; Ma, L.-J.; Fogo, A.B. Protective effects of PPAR agonist in acute nephrotic syndrome. Nephrol. Dial. Transplant. 2012, 27, 174–181. [Google Scholar] [CrossRef]
- Yang, H.-C.; Ma, L.-J.; Ma, J.; Fogo, A.B. Peroxisome proliferator-activated receptor-gamma agonist is protective in podocyte injury-associated sclerosis. Kidney Int. 2006, 69, 1756–1764. [Google Scholar] [CrossRef]
- Yuan, S.; Liu, X.; Zhu, X.; Qu, Z.; Gong, Z.; Li, J.; Xiao, L.; Yang, Y.; Liu, H.; Sun, L.; et al. The Role of TLR4 on PGC-1α-Mediated Oxidative Stress in Tubular Cell in Diabetic Kidney Disease. Oxidative Med. Cell. Longev. 2018, 2018, 6296802. [Google Scholar] [CrossRef]
- Chen, K.-H.; Hsu, H.-H.; Lee, C.-C.; Yen, T.-H.; Ko, Y.-C.; Yang, C.-W.; Hung, C.-C. The AMPK Agonist AICAR Inhibits TGF-β1 Induced Activation of Kidney Myofibroblasts. PLoS ONE 2014, 9, e106554. [Google Scholar] [CrossRef] [Green Version]
- Makled, M.N.; El-Kashef, D.H. Saroglitazar attenuates renal fibrosis induced by unilateral ureteral obstruction via inhibiting TGF-β/Smad signaling pathway. Life Sci. 2020, 253, 117729. [Google Scholar] [CrossRef]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef]
- Lee, J.E.; Westrate, L.M.; Wu, H.; Page, C.; Voeltz, G.K. Multiple dynamin family members collaborate to drive mitochondrial division. Nature 2016, 540, 139–143. [Google Scholar] [CrossRef]
- Ban, T.; Ishihara, T.; Kohno, H.; Saita, S.; Ichimura, A.; Maenaka, K.; Oka, T.; Mihara, K.; Ishihara, N. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol. 2017, 19, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Ma, Z.; Zhu, J.; Zeng, M.; Liu, H.; Dong, Z. miR-214 represses mitofusin-2 to promote renal tubular apoptosis in ischemic acute kidney injury. Am. J. Physiol. Renal Physiol. 2020, 318, F878–F887. [Google Scholar] [CrossRef]
- Saikumar, P.; Dong, Z.; Patel, Y.; Hall, K.; Hopfer, U.; Weinberg, J.M.; Venkatachalam, M.A. Role of hypoxia-induced Bax translocation and cytochrome c release in reoxygenation injury. Oncogene 1998, 17, 3401–3415. [Google Scholar] [CrossRef]
- Mikhailov, V.; Mikhailova, M.; Degenhardt, K.; Venkatachalam, M.A.; White, E.; Saikumar, P. Association of Bax and Bak homo-oligomers in mitochondria. Bax requirement for Bak reorganization and cytochrome c release. J. Biol. Chem. 2003, 278, 5367–5376. [Google Scholar] [CrossRef]
- Tanaka, T.; Nangaku, M.; Miyata, T.; Inagi, R.; Ohse, T.; Ingelfinger, J.R.; Fujita, T. Blockade of Calcium Influx through L-Type Calcium Channels Attenuates Mitochondrial Injury and Apoptosis in Hypoxic Renal Tubular Cells. J. Am. Soc. Nephrol. 2004, 15, 2320–2333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Q.; Dong, G.; Chen, J.-K.; Ramesh, G.; Dong, Z. Bax and Bak have critical roles in ischemic acute kidney injury in global and proximal tubule–specific knockout mouse models. Kidney Int. 2013, 84, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Hu, Y.; Quirós, P.M.; Wei, Q.; López-Otín, C.; Dong, Z. OMA1 mediates OPA1 proteolysis and mitochondrial fragmentation in experimental models of ischemic kidney injury. Am. J. Physiol. Renal Physiol. 2014, 306, F1318–F1326. [Google Scholar] [CrossRef]
- Hu, Y.; Yang, C.; Amorim, T.; Maqbool, M.; Lin, J.; Li, C.; Fang, C.; Xue, L.; Kwart, A.; Fang, H.; et al. Cisplatin-Mediated Upregulation of APE2 Binding to MYH9 Provokes Mitochondrial Fragmentation and Acute Kidney Injury. Cancer Res. 2021, 81, 713–723. [Google Scholar] [CrossRef]
- Cassina, L.; Chiaravalli, M.; Boletta, A. Increased mitochondrial fragmentation in polycystic kidney disease acts as a modifier of disease progression. FASEB J. 2020, 34, 6493–6507. [Google Scholar] [CrossRef]
- Bhatia, D.; Capili, A.; Choi, M.E. Mitochondrial dysfunction in kidney injury, inflammation, and disease: Potential therapeutic approaches. Kidney Res. Clin. Pract. 2020, 39, 244–258. [Google Scholar] [CrossRef]
- Walter, F.; O’Brien, A.; Concannon, C.G.; Düssmann, H.; Prehn, J.H.M. ER stress signaling has an activating transcription factor 6α (ATF6)-dependent “off-switch”. J. Biol. Chem. 2018, 293, 18270–18284. [Google Scholar] [CrossRef]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef]
- Moretton, A.; Morel, F.; Macao, B.; Lachaume, P.; Ishak, L.; Lefebvre, M.; Garreau-Balandier, I.; Vernet, P.; Falkenberg, M.; Farge, G. Selective mitochondrial DNA degradation following double-strand breaks. PLoS ONE 2017, 12, e0176795. [Google Scholar] [CrossRef]
- Sun, Z.; Zhang, X.; Ito, K.; Li, Y.; Montgomery, R.A.; Tachibana, S.; Williams, G.M. Amelioration of oxidative mitochondrial DNA damage and deletion after renal ischemic injury by the KATP channel opener diazoxide. Am. J. Physiol. Renal Physiol. 2008, 294, F491–F498. [Google Scholar] [CrossRef]
- Tan, X.; Zhang, L.; Jiang, Y.; Yang, Y.; Zhang, W.; Li, Y.; Zhang, X. Postconditioning ameliorates mitochondrial DNA damage and deletion after renal ischemic injury. Nephrol. Dial. Transplant. 2013, 28, 2754–2765. [Google Scholar] [CrossRef] [PubMed]
- Bartz, R.R.; Fu, P.; Suliman, H.B.; Crowley, S.D.; MacGarvey, N.C.; Welty-Wolf, K.; Piantadosi, C.A. Staphylococcus aureus Sepsis Induces Early Renal Mitochondrial DNA Repair and Mitochondrial Biogenesis in Mice. PLoS ONE 2014, 9, e100912. [Google Scholar] [CrossRef] [PubMed]
- Maniccia-Bozzo, E.; Espiritu, M.B.; Singh, G. Differential effects of cisplatin on mouse hepatic and renal mitochrondrial DNA. Mol. Cell. Biochem. 1990, 94, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.C.; Coughlan, M.T. Mitochondrial dysfunction and mitophagy: The beginning and end to diabetic nephropathy? Br. J. Pharmacol. 2014, 171, 1917–1942. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Cooper, J.M.; Chau, K.-Y.; Rojo, M.; Schapira, A.H.; Taanman, J.-W. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 2010, 19, 4861–4870. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef]
- Tang, C.; Han, H.; Liu, Z.; Liu, Y.; Yin, L.; Cai, J.; He, L.; Liu, Y.; Chen, G.; Zhang, Z.; et al. Activation of BNIP3-mediated mitophagy protects against renal ischemia–reperfusion injury. Cell Death Dis. 2019, 10, 677. [Google Scholar] [CrossRef]
- Tang, C.; Han, H.; Yan, M.; Zhu, S.; Liu, J.; Liu, Z.; He, L.; Tan, J.; Liu, Y.; Liu, H.; et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy 2018, 14, 880–897. [Google Scholar] [CrossRef]
- Wang, J.; Zhu, P.; Li, R.; Ren, J.; Zhou, H. Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission. Redox Biol. 2020, 30, 101415. [Google Scholar] [CrossRef]
- Lin, Q.; Li, S.; Jiang, N.; Shao, X.; Zhang, M.; Jin, H.; Zhang, Z.; Shen, J.; Zhou, Y.; Zhou, W.; et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol. 2019, 26, 101254. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, C.; Cai, J.; Chen, G.; Zhang, D.; Zhang, Z.; Dong, Z. PINK1/Parkin-mediated mitophagy is activated in cisplatin nephrotoxicity to protect against kidney injury. Cell Death Dis. 2018, 9, 1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Dai, X.; Li, Y.; Li, G.; Lin, X.; Ai, C.; Cao, Y.; Li, T.; Lin, B. Role of Parkin-mediated mitophagy in the protective effect of polydatin in sepsis-induced acute kidney injury. J. Transl. Med. 2020, 18, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Xu, X.; Zhang, F.; Wang, M.; Xu, Y.; Tang, D.; Wang, J.; Qin, Y.; Liu, Y.; Tang, C.; et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017, 11, 297–311. [Google Scholar] [CrossRef]
- Zhan, M.; Usman, I.M.; Sun, L.; Kanwar, Y.S. Disruption of renal tubular mitochondrial quality control by myo-inositol oxygenase in diabetic kidney disease. J. Am. Soc. Nephrol. 2015, 26, 1304–1321. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Dai, H.; Yuan, J.; Chen, J.; Lin, L.; Zhang, W.; Wang, L.; Zhang, J.; Li, K.; He, Y. Optineurin-mediated mitophagy protects renal tubular epithelial cells against accelerated senescence in diabetic nephropathy. Cell Death Dis. 2018, 9, 105. [Google Scholar] [CrossRef]
- Li, W.; Du, M.; Wang, Q.; Ma, X.; Wu, L.; Guo, F.; Ji, H.; Huang, F.; Qin, G. FoxO1 Promotes Mitophagy in the Podocytes of Diabetic Male Mice via the PINK1/Parkin Pathway. Endocrinology 2017, 158, 2155–2167. [Google Scholar] [CrossRef]
- Sun, J.; Zhu, H.; Wang, X.; Gao, Q.; Li, Z.; Huang, H. CoQ10 ameliorates mitochondrial dysfunction in diabetic nephropathy through mitophagy. J. Endocrinol. 2019, 240, 445–465. [Google Scholar] [CrossRef]
- Li, S.; Lin, Q.; Shao, X.; Zhu, X.; Wu, J.; Wu, B.; Zhang, M.; Zhou, W.; Zhou, Y.; Jin, H.; et al. Drp1-regulated PARK2-dependent mitophagy protects against renal fibrosis in unilateral ureteral obstruction. Free. Radic. Biol. Med. 2020, 152, 632–649. [Google Scholar] [CrossRef]
- Bhatia, D.; Chung, K.-P.; Nakahira, K.; Patino, E.; Rice, M.C.; Torres, L.K.; Muthukumar, T.; Choi, A.M.; Akchurin, O.M.; Choi, M.E. Mitophagy-dependent macrophage reprogramming protects against kidney fibrosis. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Kim, J.; Seok, Y.M.; Jung, K.-J.; Park, K.M. Reactive oxygen species/oxidative stress contributes to progression of kidney fibrosis following transient ischemic injury in mice. Am. J. Physiol. Renal Physiol. 2009, 297, F461–F470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mapuskar, K.A.; Wen, H.; Holanda, D.G.; Rastogi, P.; Steinbach, E.; Han, R.; Coleman, M.C.; Attanasio, M.; Riley, D.P.; Spitz, D.R.; et al. Persistent increase in mitochondrial superoxide mediates cisplatin-induced chronic kidney disease. Redox Biol. 2019, 20, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Vasko, R. Peroxisomes and Kidney Injury. Antioxid. Redox Signal. 2016, 25, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K.; Wakino, S.; Yoshioka, K.; Tatematsu, S.; Hara, Y.; Minakuchi, H.; Sueyasu, K.; Washida, N.; Tokuyama, H.; Tzukerman, M.; et al. Kidney-specific Overexpression of Sirt1 Protects against Acute Kidney Injury by Retaining Peroxisome Function. J. Biol. Chem. 2010, 285, 13045–13056. [Google Scholar] [CrossRef]
- Szeto, H.H. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol. 2014, 171, 2029–2050. [Google Scholar] [CrossRef]
- Liu, D.; Jin, F.; Shu, G.; Xu, X.; Qi, J.; Kang, X.; Yu, H.; Lu, K.; Jiang, S.; Han, F.; et al. Enhanced efficiency of mitochondria-targeted peptide SS-31 for acute kidney injury by pH-responsive and AKI-kidney targeted nanopolyplexes. Biomaterials 2019, 211, 57–67. [Google Scholar] [CrossRef]
- Liu, S.; Soong, Y.; Seshan, S.V.; Szeto, H.H. Novel cardiolipin therapeutic protects endothelial mitochondria during renal ischemia and mitigates microvascular rarefaction, inflammation, and fibrosis. Am. J. Physiol. Renal Physiol. 2014, 306, F970–F980. [Google Scholar] [CrossRef]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Schumacker, P.T.; Chandel, N.S. Mitochondrial reactive oxygen species trigger hypoxia-inducible factor-dependent extension of the replicative life span during hypoxia. Mol. Cell. Biol. 2007, 27, 5737–5745. [Google Scholar] [CrossRef]
- Ding, W.; Guo, H.; Xu, C.; Wang, B.; Zhang, M.; Ding, F. Mitochondrial reactive oxygen species-mediated NLRP3 inflammasome activation contributes to aldosterone-induced renal tubular cells injury. Oncotarget 2016, 7, 17479–17491. [Google Scholar] [CrossRef]
- Jain, M.; Rivera, S.; Monclus, E.A.; Synenki, L.; Zirk, A.; Eisenbart, J.; Feghali-Bostwick, C.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial Reactive Oxygen Species Regulate Transforming Growth Factor-β Signaling. J. Biol. Chem. 2013, 288, 770–777. [Google Scholar] [CrossRef]
- Vazquez-Calvo, C.; Suhm, T.; Büttner, S.; Ott, M. The basic machineries for mitochondrial protein quality control. Mitochondrion 2020, 50, 121–131. [Google Scholar] [CrossRef]
- Saisawat, P.; Kohl, S.; Hilger, A.C.; Hwang, D.-Y.; Yung Gee, H.; Dworschak, G.C.; Tasic, V.; Pennimpede, T.; Natarajan, S.; Sperry, E.; et al. Whole-exome resequencing reveals recessive mutations in TRAP1 in individuals with CAKUT and VACTERL association. Kidney Int. 2014, 85, 1310–1317. [Google Scholar] [CrossRef]
- Stacchiotti, A.; Ricci, F.; Rezzani, R.; Li Volti, G.; Borsani, E.; Lavazza, A.; Bianchi, R.; Rodella, L.F. Tubular stress proteins and nitric oxide synthase expression in rat kidney exposed to mercuric chloride and melatonin. J. Histochem. Cytochem. 2006, 54, 1149–1157. [Google Scholar] [CrossRef]
- Hernádez-Pando, R.; Pedraza-Chaverri, J.; Orozco-Estévez, H.; Silva-Serna, P.; Moreno, I.; Rondán-Zárate, A.; Elinos, M.; Correa-Rotter, R.; Larriva-Sahd, J. Histological and subcellular distribution of 65 and 70 kD heat shock proteins in experimental nephrotoxic injury. Exp. Toxicol. Pathol. 1995, 47, 501–508. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, J.; DeHoog, R.J.; Pennathur, S.; Anderton, C.R.; Venkatachalam, M.A.; Alexandrov, T.; Eberlin, L.S.; Sharma, K. DESI-MSI and METASPACE indicates lipid abnormalities and altered mitochondrial membrane components in diabetic renal proximal tubules. Metabolomics 2020, 16, 11. [Google Scholar] [CrossRef]
- Kim, S.R.; Eirin, A.; Zhang, X.; Lerman, A.; Lerman, L.O. Mitochondrial Protection Partly Mitigates Kidney Cellular Senescence in Swine Atherosclerotic Renal Artery Stenosis. Cell. Physiol. Biochem. 2019, 52, 617–632. [Google Scholar] [CrossRef]
- Zhu, Y.; Luo, M.; Bai, X.; Li, J.; Nie, P.; Li, B.; Luo, P. SS-31, a Mitochondria-Targeting Peptide, Ameliorates Kidney Disease. Oxidative Med. Cell. Longev. 2022, 2022, 1295509. [Google Scholar] [CrossRef]
- Mizuguchi, Y.; Chen, J.; Seshan, S.V.; Poppas, D.P.; Szeto, H.H.; Felsen, D. A novel cell-permeable antioxidant peptide decreases renal tubular apoptosis and damage in unilateral ureteral obstruction. Am. J. Physiol. Renal Physiol. 2008, 295, F1545–F1553. [Google Scholar] [CrossRef]
- Liu, Z.-R.; Chen, S.-Q.; Zou, Y.-W.; Wu, X.-Y.; Li, H.-Y.; Wang, X.-Q.; Shi, Y.; Niu, H.-X. Hypochlorite modified albumins promote cell death in the tubule interstitium in rats via mitochondrial damage in obstructive nephropathy and the protective effects of antioxidant peptides. Free. Radic. Res. 2018, 52, 616–628. [Google Scholar] [CrossRef]
- Sun, M.; Ma, J.; Ye, J.; Fan, H.; Le, J.; Zhu, J. Protective effect of mitochondria-targeted antioxidant peptide SS-31 in sepsis-induced acute kidney injury. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 2021, 33, 1418–1422. [Google Scholar] [CrossRef]
- Wyss, J.-C.; Kumar, R.; Mikulic, J.; Schneider, M.; Mary, J.-L.; Aebi, J.D.; Juillerat-Jeanneret, L.; Golshayan, D. Differential Effects of the Mitochondria-Active Tetrapeptide SS-31 (D-Arg-dimethylTyr-Lys-Phe-NH2) and Its Peptidase-Targeted Prodrugs in Experimental Acute Kidney Injury. Front. Pharmacol. 2019, 10, 1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, S.; Zhang, G.; Hall, D.; Oates, P.J.; Maity, S.; Madesh, M.; Han, X.; Sharma, K. Restoring mitochondrial superoxide levels with elamipretide (MTP-131) protects db/db mice against progression of diabetic kidney disease. J. Biol. Chem. 2020, 295, 7249–7260. [Google Scholar] [CrossRef]
- Yang, S.-K.; Li, A.-M.; Han, Y.-C.; Peng, C.-H.; Song, N.; Yang, M.; Zhan, M.; Zeng, L.-F.; Song, P.-A.; Zhang, W.; et al. Mitochondria-Targeted Peptide SS31 Attenuates Renal Tubulointerstitial Injury via Inhibiting Mitochondrial Fission in Diabetic Mice. Oxidative Med. Cell. Longev. 2019, 2019, 2346580. [Google Scholar] [CrossRef]
- Wang, X.; Tang, D.; Zou, Y.; Wu, X.; Chen, Y.; Li, H.; Chen, S.; Shi, Y.; Niu, H. A mitochondrial-targeted peptide ameliorated podocyte apoptosis through a HOCl-alb-enhanced and mitochondria-dependent signalling pathway in diabetic rats and in vitro. J. Enzym. Inhib. Med. Chem. 2019, 34, 394–404. [Google Scholar] [CrossRef]
- Hou, Y.; Li, S.; Wu, M.; Wei, J.; Ren, Y.; Du, C.; Wu, H.; Han, C.; Duan, H.; Shi, Y. Mitochondria-targeted peptide SS-31 attenuates renal injury via an antioxidant effect in diabetic nephropathy. Am. J. Physiol. Renal Physiol. 2016, 310, F547–F559. [Google Scholar] [CrossRef] [PubMed]
- Eirin, A.; Ebrahimi, B.; Zhang, X.; Zhu, X.-Y.; Woollard, J.R.; He, Q.; Textor, S.C.; Lerman, A.; Lerman, L.O. Mitochondrial protection restores renal function in swine atherosclerotic renovascular disease. Cardiovasc. Res. 2014, 103, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-H.; Zhu, X.Y.; Eirin, A.; Nargesi, A.A.; Woollard, J.R.; Santelli, A.; Sun, I.O.; Textor, S.C.; Lerman, L.O. Early podocyte injury and elevated levels of urinary podocyte-derived extracellular vesicles in swine with metabolic syndrome: Role of podocyte mitochondria. Am. J. Physiol. Renal Physiol. 2019, 317, F12–F22. [Google Scholar] [CrossRef]
- Eirin, A.; Hedayat, A.F.; Ferguson, C.M.; Textor, S.C.; Lerman, A.; Lerman, L.O. Mitoprotection preserves the renal vasculature in porcine metabolic syndrome. Exp. Physiol. 2018, 103, 1020–1029. [Google Scholar] [CrossRef]
- Szeto, H.H.; Liu, S.; Soong, Y.; Alam, N.; Prusky, G.T.; Seshan, S.V. Protection of mitochondria prevents high-fat diet–induced glomerulopathy and proximal tubular injury. Kidney Int. 2016, 90, 997–1011. [Google Scholar] [CrossRef]
- Szeto, H.H.; Liu, S.; Soong, Y.; Wu, D.; Darrah, S.F.; Cheng, F.-Y.; Zhao, Z.; Ganger, M.; Tow, C.Y.; Seshan, S.V. Mitochondria-targeted peptide accelerates atp recovery and reduces ischemic kidney injury. J. Am. Soc. Nephrol. 2011, 22, 1041–1052. [Google Scholar] [CrossRef]
- Zhao, W.-Y.; Han, S.; Zhang, L.; Zhu, Y.-H.; Wang, L.-M.; Zeng, L. Mitochondria-targeted antioxidant peptide ss31 prevents hypoxia/reoxygenation-induced apoptosis by down-regulating p66Shc in renal tubular epithelial cells. Cell. Physiol. Biochem. 2013, 32, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Liu, Y.-J.; Liu, Z.-R.; Tang, D.-D.; Chen, X.-W.; Chen, Y.-H.; Zhou, R.-N.; Chen, S.-Q.; Niu, H.-X. Role of mitochondrial dysfunction in renal fibrosis promoted by hypochlorite-modified albumin in a remnant kidney model and protective effects of antioxidant peptide SS-31. Eur. J. Pharmacol. 2017, 804, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.-B.; Yang, S.-K.; Zhou, Q.-Y.; Pan, P.; Zhang, H.; Liu, F.; Xu, X.-Q. Mitochondria-targeted peptides prevent on contrast-induced acute kidney injury in the rats with hypercholesterolemia. Ren. Fail. 2013, 35, 1124–1129. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-K.; Han, Y.-C.; He, J.-R.; Yang, M.; Zhang, W.; Zhan, M.; Li, A.-M.; Li, L.; Song, N.; Liu, Y.-T.; et al. Mitochondria targeted peptide SS-31 prevent on cisplatin-induced acute kidney injury via regulating mitochondrial ROS-NLRP3 pathway. Biomed. Pharmacother. 2020, 130, 110521. [Google Scholar] [CrossRef] [PubMed]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Saad, A.; Herrmann, S.M.S.; Eirin, A.; Ferguson, C.M.; Glockner, J.F.; Bjarnason, H.; McKusick, M.A.; Misra, S.; Lerman, L.O.; Textor, S.C. Phase 2a Clinical Trial of Mitochondrial Protection (Elamipretide) During Stent Revascularization in Patients With Atherosclerotic Renal Artery Stenosis. Circ. Cardiovasc. Interv. 2017, 10. [Google Scholar] [CrossRef]
- Szeto, H.H.; Liu, S.; Soong, Y.; Birk, A.V. Improving mitochondrial bioenergetics under ischemic conditions increases warm ischemia tolerance in the kidney. Am. J. Physiol. Renal Physiol. 2015, 308, F11–F21. [Google Scholar] [CrossRef]
- Tagaya, M.; Kume, S.; Yasuda-Yamahara, M.; Kuwagata, S.; Yamahara, K.; Takeda, N.; Tanaka, Y.; Chin-Kanasaki, M.; Nakae, Y.; Yokoi, H.; et al. Inhibition of mitochondrial fission protects podocytes from albumin-induced cell damage in diabetic kidney disease. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166368. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Q.; Cai, J.; Wu, P.; Wang, D.; Shi, Y.; Huyan, T.; Su, J.; Li, X.; Wang, Q.; et al. Emodin prevents renal ischemia-reperfusion injury via suppression of CAMKII/DRP1-mediated mitochondrial fission. Eur. J. Pharmacol. 2022, 916, 174603. [Google Scholar] [CrossRef]
- Wang, J.; Zhu, P.; Toan, S.; Li, R.; Ren, J.; Zhou, H. Pum2-Mff axis fine-tunes mitochondrial quality control in acute ischemic kidney injury. Cell Biol. Toxicol. 2020, 36, 365–378. [Google Scholar] [CrossRef]
- Zhao, C.; Chen, Z.; Qi, J.; Duan, S.; Huang, Z.; Zhang, C.; Wu, L.; Zeng, M.; Zhang, B.; Wang, N.; et al. Drp1-dependent mitophagy protects against cisplatin-induced apoptosis of renal tubular epithelial cells by improving mitochondrial function. Oncotarget 2017, 8, 20988–21000. [Google Scholar] [CrossRef] [Green Version]
- Li, H.-B.; Zhang, X.-Z.; Sun, Y.; Zhou, Q.; Song, J.-N.; Hu, Z.-F.; Li, Y.; Wu, J.-N.; Guo, Y.; Zhang, Y.; et al. HO-1/PINK1 Regulated Mitochondrial Fusion/Fission to Inhibit Pyroptosis and Attenuate Septic Acute Kidney Injury. BioMed Res. Int. 2020, 2020, 2148706. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, M.; Xiong, L.; Fan, J.; Zhou, Y.; Li, H.; Peng, X.; Zhong, Z.; Wang, Y.; Huang, F.; et al. Drp1-mediated mitochondrial fission promotes renal fibroblast activation and fibrogenesis. Cell Death Dis. 2020, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Wang, S.-C.; Li, M.; Ma, X.-H.; Jia, X.-N.; Bu, Y.; Sun, L.; Yu, K.-J. An Inhibitor of DRP1 (Mdivi-1) Alleviates LPS-Induced Septic AKI by Inhibiting NLRP3 Inflammasome Activation. BioMed Res. Int. 2020, 2020, 2398420. [Google Scholar] [CrossRef]
- Zhu, Y.; Kuang, L.; Wu, Y.; Deng, H.; She, H.; Zhou, Y.; Zhang, J.; Liu, L.; Li, T. Protective Effects of Inhibition of Mitochondrial Fission on Organ Function After Sepsis. Front. Pharmacol. 2021, 12, 712489. [Google Scholar] [CrossRef]
- Sumida, M.; Doi, K.; Ogasawara, E.; Yamashita, T.; Hamasaki, Y.; Kariya, T.; Takimoto, E.; Yahagi, N.; Nangaku, M.; Noiri, E. Regulation of Mitochondrial Dynamics by Dynamin-Related Protein-1 in Acute Cardiorenal Syndrome. J. Am. Soc. Nephrol. 2015, 26, 2378–2387. [Google Scholar] [CrossRef]
- Ayanga, B.A.; Badal, S.S.; Wang, Y.; Galvan, D.L.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Dynamin–Related Protein 1 Deficiency Improves Mitochondrial Fitness and Protects against Progression of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2016, 27, 2733–2747. [Google Scholar] [CrossRef]
- Giorgio, V.; Soriano, M.E.; Basso, E.; Bisetto, E.; Lippe, G.; Forte, M.A.; Bernardi, P. Cyclophilin D in mitochondrial pathophysiology. Biochim. Biophys. Acta 2010, 1797, 1113–1118. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Woodfield, K.-Y.; Connern, C.P. Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase. J. Biol. Chem. 1997, 272, 3346–3354. [Google Scholar] [CrossRef]
- Leung, A.W.; Varanyuwatana, P.; Halestrap, A.P. The mitochondrial phosphate carrier interacts with cyclophilin d and may play a key role in the permeability transition. J. Biol. Chem. 2008, 283, 26312–26323. [Google Scholar] [CrossRef]
- Shanmughapriya, S.; Rajan, S.; Hoffman, N.E.; Higgins, A.M.; Tomar, D.; Nemani, N.; Hines, K.J.; Smith, D.J.; Eguchi, A.; Vallem, S.; et al. SPG7 Is an Essential and Conserved Component of the Mitochondrial Permeability Transition Pore. Mol. Cell 2015, 60, 47–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zou, Y.; Cheng-Jing, Y.; Xiang-Heng, L.; Wang, X.P.; Yu, X.J.; Li, G.S.; Wang, J. Pioglitazone alleviates cisplatin nephrotoxicity by suppressing mitochondria-mediated apoptosis via SIRT1/p53 signalling. J. Cell. Mol. Med. 2020, 24, 11718–11728. [Google Scholar] [CrossRef]
- Hu, C.; Zhou, G.; Liu, K.; Yin, W.; Zhou, L.; Wang, J.; Chen, L.; Zuo, S.; Xie, Y.; Zuo, X. CaMKII as a key regulator of contrast-induced nephropathy through mPTP opening in HK-2 cells. Cell. Signal. 2020, 75, 109734. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, L.; Zhao, J.; Guo, X.; Luo, Y.; Hu, W.; Zhao, T. Tumor Necrosis Factor Receptor-Associated Protein 1 Protects against Mitochondrial Injury by Preventing High Glucose-Induced mPTP Opening in Diabetes. Oxidative Med. Cell. Longev. 2020, 2020, 6431517. [Google Scholar] [CrossRef]
- Wang, Z.-K.; Zhou, X.-L.; Song, X.-B.; Zhuang, D.-M.; Wang, Z.-Y.; Yang, D.-B.; Wang, L. Alleviation of Lead-Induced Apoptosis by Puerarin via Inhibiting Mitochondrial Permeability Transition Pore Opening in Primary Cultures of Rat Proximal Tubular Cells. Biol. Trace Elem. Res. 2016, 174, 166–176. [Google Scholar] [CrossRef]
- Garbaisz, D.; Turoczi, Z.; Aranyi, P.; Fulop, A.; Rosero, O.; Hermesz, E.; Ferencz, A.; Lotz, G.; Harsanyi, L.; Szijarto, A. Attenuation of skeletal muscle and renal injury to the lower limb following ischemia-reperfusion using mptp inhibitor NIM-811. PLoS ONE 2014, 9, e101067. [Google Scholar] [CrossRef]
- de Arriba, G.; Calvino, M.; Benito, S.; Parra, T. Cyclosporine A-induced apoptosis in renal tubular cells is related to oxidative damage and mitochondrial fission. Toxicol. Lett. 2013, 218, 30–38. [Google Scholar] [CrossRef]
- Guan, N.; Ren, Y.-L.; Liu, X.-Y.; Zhang, Y.; Pei, P.; Zhu, S.-N.; Fan, Q. Protective role of cyclosporine A and minocycline on mitochondrial disequilibrium-related podocyte injury and proteinuria occurrence induced by adriamycin. Nephrol. Dial. Transplant. 2015, 30, 957–969. [Google Scholar] [CrossRef]
- Singh, D.; Chander, V.; Chopra, K. Cyclosporine protects against ischemia/reperfusion injury in rat kidneys. Toxicology 2005, 207, 339–347. [Google Scholar] [CrossRef]
- Wang, Z.; Bao, H.; Ge, Y.; Zhuang, S.; Peng, A.; Gong, R. Pharmacological targeting of GSK3β confers protection against podocytopathy and proteinuria by desensitizing mitochondrial permeability transition. Br. J. Pharmacol. 2015, 172, 895–909. [Google Scholar] [CrossRef]
- Singh, S.P.; Tao, S.; Fields, T.A.; Webb, S.; Harris, R.C.; Rao, R. Glycogen synthase kinase-3 inhibition attenuates fibroblast activation and development of fibrosis following renal ischemia/reperfusion in mice. Dis. Model. Mech. 2015, 8, 931–940. [Google Scholar] [CrossRef] [Green Version]
- Bao, H.; Ge, Y.; Zhuang, S.; Dworkin, L.D.; Liu, Z.; Gong, R. Inhibition of glycogen synthase kinase-3β prevents NSAID-induced acute kidney injury. Kidney Int. 2012, 81, 662–673. [Google Scholar] [CrossRef]
- Lindblom, R.S.J.; Higgins, G.C.; Nguyen, T.-V.; Arnstein, M.; Henstridge, D.C.; Granata, C.; Snelson, M.; Thallas-Bonke, V.; Cooper, M.E.; Forbes, J.M.; et al. Delineating a role for the mitochondrial permeability transition pore in diabetic kidney disease by targeting cyclophilin D. Clin. Sci. 2020, 134, 239–259. [Google Scholar] [CrossRef]
- Ren, Y.; Du, C.; Shi, Y.; Wei, J.; Wu, H.; Cui, H. The Sirt1 activator, SRT1720, attenuates renal fibrosis by inhibiting CTGF and oxidative stress. Int. J. Mol. Med. 2017, 39, 1317–1324. [Google Scholar] [CrossRef]
- Ponnusamy, M.; Zhuang, M.A.; Zhou, X.; Tolbert, E.; Bayliss, G.; Zhao, T.C.; Zhuang, S. Activation of Sirtuin-1 Promotes Renal Fibroblast Activation and Aggravates Renal Fibrogenesis. J. Pharmacol. Exp. Ther. 2015, 354, 142–151. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Jo, J.; Kim, K.; An, H.-J.; Gwon, M.-G.; Gu, H.; Kim, H.-J.; Yang, A.Y.; Kim, S.-W.; Jeon, E.J.; et al. Pharmacological Activation of Sirt1 Ameliorates Cisplatin-Induced Acute Kidney Injury by Suppressing Apoptosis, Oxidative Stress, and Inflammation in Mice. Antioxidants 2019, 8, 322. [Google Scholar] [CrossRef]
- Khader, A.; Yang, W.-L.; Kuncewitch, M.; Jacob, A.; Prince, J.M.; Asirvatham, J.R.; Nicastro, J.; Coppa, G.F.; Wang, P. Sirtuin 1 activation stimulates mitochondrial biogenesis and attenuates renal injury after ischemia-reperfusion. Transplantation 2014, 98, 148–156. [Google Scholar] [CrossRef]
- Funk, J.A.; Schnellmann, R.G. Accelerated recovery of renal mitochondrial and tubule homeostasis with SIRT1/PGC-1α activation following ischemia–reperfusion injury. Toxicol. Appl. Pharmacol. 2013, 273, 345–354. [Google Scholar] [CrossRef]
- Gibbs, W.S.; Garrett, S.M.; Beeson, C.C.; Schnellmann, R.G. Identification of dual mechanisms mediating 5-hydroxytryptamine receptor 1F-induced mitochondrial biogenesis. Am. J. Physiol. Renal Physiol. 2018, 314, F260–F268. [Google Scholar] [CrossRef]
- Garrett, S.M.; Whitaker, R.M.; Beeson, C.C.; Schnellmann, R.G. Agonism of the 5-hydroxytryptamine 1F receptor promotes mitochondrial biogenesis and recovery from acute kidney injury. J. Pharmacol. Exp. Ther. 2014, 350, 257–264. [Google Scholar] [CrossRef]
- Hong, Q.; Zhang, L.; Das, B.; Li, Z.; Liu, B.; Cai, G.; Chen, X.; Chuang, P.Y.; He, J.C.; Lee, K. Increased podocyte Sirtuin-1 function attenuates diabetic kidney injury. Kidney Int. 2018, 93, 1330–1343. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, R.; Zhang, W.; Hyink, D.P.; Das, G.C.; Das, B.; Li, Z.; Wang, A.; Yuan, W.; Klotman, P.E.; et al. Role of SIRT1 in HIV-associated kidney disease. Am. J. Physiol. Renal Physiol. 2020, 319, F335–F344. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Kume, S.; Imaizumi, N.; Koya, D. Resveratrol improves oxidative stress and protects against diabetic nephropathy through normalization of Mn-SOD dysfunction in AMPK/SIRT1-independent pathway. Diabetes 2011, 60, 634–643. [Google Scholar] [CrossRef] [PubMed]
- Palsamy, P.; Subramanian, S. Resveratrol protects diabetic kidney by attenuating hyperglycemia-mediated oxidative stress and renal inflammatory cytokines via Nrf2–Keap1 signaling. Biochim. Biophys. Acta 2011, 1812, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guan, Y.; Karamercan, M.A.; Ye, L.; Bhatti, T.; Becker, L.B.; Baur, J.A.; Sims, C.A. Resveratrol Rescues Kidney Mitochondrial Function Following Hemorrhagic Shock. Shock 2015, 44, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Sattarinezhad, A.; Roozbeh, J.; Shirazi Yeganeh, B.; Omrani, G.R.; Shams, M. Resveratrol reduces albuminuria in diabetic nephropathy: A randomized double-blind placebo-controlled clinical trial. Diabetes Metab. 2019, 45, 53–59. [Google Scholar] [CrossRef]
- Arif, E.; Solanki, A.K.; Srivastava, P.; Rahman, B.; Fitzgibbon, W.R.; Deng, P.; Budisavljevic, M.N.; Baicu, C.F.; Zile, M.R.; Megyesi, J.; et al. Mitochondrial biogenesis induced by the β2-adrenergic receptor agonist formoterol accelerates podocyte recovery from glomerular injury. Kidney Int. 2019, 96, 656–673. [Google Scholar] [CrossRef]
- Cleveland, K.H.; Brosius, F.C., 3rd; Schnellmann, R.G. Regulation of mitochondrial dynamics and energetics in the diabetic renal proximal tubule by the β2-adrenergic receptor agonist formoterol. Am. J. Physiol. Renal Physiol. 2020, 319, F773–F779. [Google Scholar] [CrossRef]
- Cameron, R.B.; Gibbs, W.S.; Miller, S.R.; Dupre, T.V.; Megyesi, J.; Beeson, C.C.; Schnellmann, R.G. Proximal Tubule β2-Adrenergic Receptor Mediates Formoterol-Induced Recovery of Mitochondrial and Renal Function after Ischemia-Reperfusion Injury. J. Pharmacol. Exp. Ther. 2019, 369, 173–180. [Google Scholar] [CrossRef]
- Jesinkey, S.R.; Funk, J.A.; Stallons, L.J.; Wills, L.P.; Megyesi, J.K.; Beeson, C.C.; Schnellmann, R.G. Formoterol restores mitochondrial and renal function after ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2014, 25, 1157–1162. [Google Scholar] [CrossRef]
- Negishi, K.; Noiri, E.; Maeda, R.; Portilla, D.; Sugaya, T.; Fujita, T. Renal L-type fatty acid-binding protein mediates the bezafibrate reduction of cisplatin-induced acute kidney injury. Kidney Int. 2008, 73, 1374–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadler, K.; Goldberg, I.J.; Susztak, K. The evolving understanding of the contribution of lipid metabolism to diabetic kidney disease. Curr. Diabetes Rep. 2015, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Dare, A.J.; Bolton, E.A.; Pettigrew, G.J.; Bradley, J.A.; Saeb-Parsy, K.; Murphy, M.P. Protection against renal ischemia–reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ. Redox Biol. 2015, 5, 163–168. [Google Scholar] [CrossRef]
- Chacko, B.K.; Reily, C.; Srivastava, A.; Johnson, M.S.; Ye, Y.; Ulasova, E.; Agarwal, A.; Zinn, K.R.; Murphy, M.P.; Kalyanaraman, B.; et al. Prevention of diabetic nephropathy in Ins2+/−AkitaJ mice by the mitochondria-targeted therapy MitoQ. Biochem. J. 2010, 432, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Horváth, B.; Zsengellér, Z.; Zielonka, J.; Tanchian, G.; Holovac, E.; Kechrid, M.; Patel, V.; Stillman, I.E.; Parikh, S.M.; et al. Mitochondrial-targeted antioxidants represent a promising approach for prevention of cisplatin-induced nephropathy. Free. Radic. Biol. Med. 2012, 52, 497–506. [Google Scholar] [CrossRef]
- Plotnikov, E.Y.; Chupyrkina, A.A.; Jankauskas, S.S.; Pevzner, I.B.; Silachev, D.N.; Skulachev, V.P.; Zorov, D.B. Mechanisms of nephroprotective effect of mitochondria-targeted antioxidants under rhabdomyolysis and ischemia/reperfusion. Biochim. Biophys. Acta 2011, 1812, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Sourris, K.C.; Harcourt, B.E.; Tang, P.H.; Morley, A.L.; Huynh, K.; Penfold, S.A.; Coughlan, M.T.; Cooper, M.E.; Nguyen, T.-V.; Ritchie, R.H.; et al. Ubiquinone (coenzyme Q10) prevents renal mitochondrial dysfunction in an experimental model of type 2 diabetes. Free. Radic. Biol. Med. 2012, 52, 716–723. [Google Scholar] [CrossRef]
- Lu, M.; Yin, N.; Liu, W.; Cui, X.; Chen, S.; Wang, E. Curcumin Ameliorates Diabetic Nephropathy by Suppressing NLRP3 Inflammasome Signaling. BioMed Res. Int. 2017, 2017, 1516985. [Google Scholar] [CrossRef]
- Soetikno, V.; Sari, F.R.; Lakshmanan, A.P.; Arumugam, S.; Harima, M.; Suzuki, K.; Kawachi, H.; Watanabe, K. Curcumin alleviates oxidative stress, inflammation, and renal fibrosis in remnant kidney through the Nrf2-keap1 pathway. Mol. Nutr. Food Res. 2013, 57, 1649–1659. [Google Scholar] [CrossRef]
- Antonenko, Y.N.; Avetisyan, A.V.; Bakeeva, L.E.; Chernyak, B.V.; Chertkov, V.A.; Domnina, L.V.; Ivanova, O.Y.; Izyumov, D.S.; Khailova, L.S.; Klishin, S.S.; et al. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 1. Cationic plastoquinone derivatives: Synthesis and in vitro studies. Biochemistry 2008, 73, 1273–1287. [Google Scholar] [CrossRef]
- Kitada, M.; Xu, J.; Ogura, Y.; Monno, I.; Koya, D. Manganese Superoxide Dismutase Dysfunction and the Pathogenesis of Kidney Disease. Front. Physiol. 2020, 11, 755. [Google Scholar] [CrossRef]
- Shabalina, I.G.; Vyssokikh, M.Y.; Gibanova, N.; Csikasz, R.I.; Edgar, D.; Hallden-Waldemarson, A.; Rozhdestvenskaya, Z.; Bakeeva, L.E.; Vays, V.B.; Pustovidko, A.V.; et al. Improved health-span and lifespan in mtDNA mutator mice treated with the mitochondrially targeted antioxidant SkQ1. Aging 2017, 9, 315–339. [Google Scholar] [CrossRef]
- Jankauskas, S.S.; Plotnikov, E.Y.; Morosanova, M.A.; Pevzner, I.B.; Zorova, L.D.; Skulachev, V.; Zorov, D.B. Mitochondria-targeted antioxidant SkQR1 ameliorates gentamycin-induced renal failure and hearing loss. Biochemistry 2012, 77, 666–670. [Google Scholar] [CrossRef]
- Petrov, A.; Perekhvatova, N.; Skulachev, M.; Stein, L.; Ousler, G. SkQ1 Ophthalmic Solution for Dry Eye Treatment: Results of a Phase 2 Safety and Efficacy Clinical Study in the Environment and During Challenge in the Controlled Adverse Environment Model. Adv. Ther. 2016, 33, 96–115. [Google Scholar] [CrossRef]
- Yeung, C.K.; Billings, F.T.t.; Claessens, A.J.; Roshanravan, B.; Linke, L.; Sundell, M.B.; Ahmad, S.; Shao, B.; Shen, D.D.; Ikizler, T.A.; et al. Coenzyme Q10 dose-escalation study in hemodialysis patients: Safety, tolerability, and effect on oxidative stress. BMC Nephrol. 2015, 16, 183. [Google Scholar] [CrossRef]
- Tapia, E.; Zatarain-Barrón, Z.L.; Hernández-Pando, R.; Zarco-Márquez, G.; Molina-Jijón, E.; Cristóbal-García, M.; Santamaría, J.; Pedraza-Chaverri, J. Curcumin reverses glomerular hemodynamic alterations and oxidant stress in 5/6 nephrectomized rats. Phytomedicine 2013, 20, 359–366. [Google Scholar] [CrossRef]
- Hassanzadeh, K.; Buccarello, L.; Dragotto, J.; Mohammadi, A.; Corbo, M.; Feligioni, M. Obstacles against the Marketing of Curcumin as a Drug. Int. J. Mol. Sci. 2020, 21, 6619. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Functional role of mitochondrial reactive oxygen species in physiology. Free Radic. Biol. Med. 2016, 100, 81–85. [Google Scholar] [CrossRef]
- Bhatia, D.; Choi, M.E. The Emerging Role of Mitophagy in Kidney Diseases. J. Life Sci. 2019, 1, 13–22. [Google Scholar] [CrossRef]
- Wang, Y.; Cai, J.; Tang, C.; Dong, Z. Mitophagy in Acute Kidney Injury and Kidney Repair. Cells 2020, 9, 338. [Google Scholar] [CrossRef]
- Cui, J.; Bai, X.-Y.; Sun, X.; Cai, G.; Hong, Q.; Ding, R.; Chen, X. Rapamycin protects against gentamicin-induced acute kidney injury via autophagy in mini-pig models. Sci. Rep. 2015, 5, 11256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.; Wei, Q.; Dong, G.; Komatsu, M.; Su, Y.; Dong, Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012, 82, 1271–1283. [Google Scholar] [CrossRef] [PubMed]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; Nicolet-Dit-Félix, A.A.; Williams, E.G.; Jha, P.; Lo Sasso, G.; Huzard, D.; et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016, 22, 879–888. [Google Scholar] [CrossRef]
- Jayatunga, D.P.W.; Hone, E.; Khaira, H.; Lunelli, T.; Singh, H.; Guillemin, G.J.; Fernando, B.; Garg, M.L.; Verdile, G.; Martins, R.N. Therapeutic Potential of Mitophagy-Inducing Microflora Metabolite, Urolithin A for Alzheimer’s Disease. Nutrients 2021, 13, 3744. [Google Scholar] [CrossRef]
- Zou, D.; Ganugula, R.; Arora, M.; Nabity, M.B.; Sheikh-Hamad, D.; Kumar, M. Oral delivery of nanoparticle urolithin A normalizes cellular stress and improves survival in mouse model of cisplatin-induced AKI. Am. J. Physiol. Renal Physiol. 2019, 317, F1255–F1264. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; D’Amico, D.; Shankland, E.; Bhayana, S.; Garcia, J.M.; Aebischer, P.; Rinsch, C.; Singh, A.; Marcinek, D.J. Effect of Urolithin A Supplementation on Muscle Endurance and Mitochondrial Health in Older Adults: A Randomized Clinical Trial. JAMA Netw. Open 2022, 5, e2144279. [Google Scholar] [CrossRef]
- Guo, J.; Wang, R.; Liu, D. Bone Marrow-Derived Mesenchymal Stem Cells Ameliorate Sepsis-Induced Acute Kidney Injury by Promoting Mitophagy of Renal Tubular Epithelial Cells via the SIRT1/Parkin Axis. Front. Endocrinol. 2021, 12, 639165. [Google Scholar] [CrossRef]
- Tang, H.; Yang, M.; Liu, Y.; Zhu, X.; Liu, S.; Liu, H.; Sun, L.; Song, P. Melatonin alleviates renal injury by activating mitophagy in diabetic nephropathy. Front. Endocrinol. 2022, 13, 889729. [Google Scholar] [CrossRef]
- Livingston, M.J.; Ding, H.-F.; Huang, S.; Hill, J.A.; Yin, X.-M.; Dong, Z. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy 2016, 12, 976–998. [Google Scholar] [CrossRef]
- Li, H.; Peng, X.; Wang, Y.; Cao, S.; Xiong, L.; Fan, J.; Wang, Y.; Zhuang, S.; Yu, X.; Mao, H. Atg5-mediated autophagy deficiency in proximal tubules promotes cell cycle G2/M arrest and renal fibrosis. Autophagy 2016, 12, 1472–1486. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Tsakiridis, E.; Steinberg, G.R.; Pei, Y. Targeting AMP-activated protein kinase (AMPK) for treatment of autosomal dominant polycystic kidney disease. Cell. Signal. 2020, 73, 109704. [Google Scholar] [CrossRef]
- Padovano, V.; Podrini, C.; Boletta, A.; Caplan, M.J. Metabolism and mitochondria in polycystic kidney disease research and therapy. Nat. Rev. Nephrol. 2018, 14, 678–687. [Google Scholar] [CrossRef]
- Shillingford, J.M.; Murcia, N.S.; Larson, C.H.; Low, S.H.; Hedgepeth, R.; Brown, N.; Flask, C.A.; Novick, A.C.; Goldfarb, D.A.; Kramer-Zucker, A.; et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5466–5471. [Google Scholar] [CrossRef]
- Shillingford, J.M.; Piontek, K.B.; Germino, G.G.; Weimbs, T. Rapamycin Ameliorates PKD Resulting from Conditional Inactivation of Pkd1. J. Am. Soc. Nephrol. 2010, 21, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Serra, A.L.; Poster, D.; Kistler, A.D.; Krauer, F.; Raina, S.; Young, J.; Rentsch, K.M.; Spanaus, K.S.; Senn, O.; Kristanto, P.; et al. Sirolimus and Kidney Growth in Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 2010, 363, 820–829. [Google Scholar] [CrossRef]
- Walz, G.; Budde, K.; Mannaa, M.; Nürnberger, J.; Wanner, C.; Sommerer, C.; Kunzendorf, U.; Banas, B.; Hörl, W.H.; Obermüller, N.; et al. Everolimus in Patients with Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 2010, 363, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Hawley, S.A.; Ford, R.J.; Smith, B.K.; Gowans, G.J.; Mancini, S.J.; Pitt, R.D.; Day, E.A.; Salt, I.P.; Steinberg, G.R.; Hardie, D.G. The Na+/Glucose Cotransporter Inhibitor Canagliflozin Activates AMPK by Inhibiting Mitochondrial Function and Increasing Cellular AMP Levels. Diabetes 2016, 65, 2784–2794. [Google Scholar] [CrossRef]
- Zhang, G.; Darshi, M.; Sharma, K. The Warburg Effect in Diabetic Kidney Disease. Semin. Nephrol. 2018, 38, 111–120. [Google Scholar] [CrossRef]
- Wang, C.; Li, L.; Liu, S.; Liao, G.; Li, L.; Chen, Y.; Cheng, J.; Lu, Y.; Liu, J. GLP-1 receptor agonist ameliorates obesity-induced chronic kidney injury via restoring renal metabolism homeostasis. PLoS ONE 2018, 13, e0193473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kume, S.; Kitada, M.; Kanasaki, K.; Maegawa, H.; Koya, D. Anti-aging molecule, Sirt1: A novel therapeutic target for diabetic nephropathy. Arch. Pharmacal Res. 2013, 36, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Kume, S.; Uzu, T.; Horiike, K.; Chin-Kanasaki, M.; Isshiki, K.; Araki, S.; Sugimoto, T.; Haneda, M.; Kashiwagi, A.; Koya, D. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J. Clin. Investig. 2010, 120, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Huang, Q. Glucagon-like peptide-1 protects mouse podocytes against high glucose-induced apoptosis, and suppresses reactive oxygen species production and proinflammatory cytokine secretion, through sirtuin 1 activation in vitro. Mol. Med. Rep. 2018, 18, 1789–1797. [Google Scholar] [CrossRef]
- Yang, S.; Lin, C.; Zhuo, X.; Wang, J.; Rao, S.; Xu, W.; Cheng, Y.; Yang, L. Glucagon-like peptide-1 alleviates diabetic kidney disease through activation of autophagy by regulating AMP-activated protein kinase-mammalian target of rapamycin pathway. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E1019–E1030. [Google Scholar] [CrossRef]
- Germano, J.F.; Huang, C.; Sin, J.; Song, Y.; Tucker, K.C.; Taylor, D.J.R.; Saadaeijahromi, H.; Stotland, A.; Piplani, H.; Gottlieb, R.A.; et al. Intermittent Use of a Short-Course Glucagon-like Peptide-1 Receptor Agonist Therapy Limits Adverse Cardiac Remodeling via Parkin-dependent Mitochondrial Turnover. Sci. Rep. 2020, 10, 8284. [Google Scholar] [CrossRef]
- Packer, M. Role of Deranged Energy Deprivation Signaling in the Pathogenesis of Cardiac and Renal Disease in States of Perceived Nutrient Overabundance. Circulation 2020, 141, 2095–2105. [Google Scholar] [CrossRef]
- Nilsson, L.M.; Zhang, L.; Bondar, A.; Svensson, D.; Wernerson, A.; Brismar, H.; Scott, L.; Aperia, A. Prompt apoptotic response to high glucose in SGLT-expressing renal cells. Am. J. Physiol. Renal Physiol. 2019, 316, F1078–F1089. [Google Scholar] [CrossRef]
- Hatanaka, T.; Ogawa, D.; Tachibana, H.; Eguchi, J.; Inoue, T.; Yamada, H.; Takei, K.; Makino, H.; Wada, J. Inhibition of SGLT2 alleviates diabetic nephropathy by suppressing high glucose-induced oxidative stress in type 1 diabetic mice. Pharmacol. Res. Perspect. 2016, 4, e00239. [Google Scholar] [CrossRef]
- Cioce, M.; Pulito, C.; Strano, S.; Blandino, G.; Fazio, V.M. Metformin: Metabolic Rewiring Faces Tumor Heterogeneity. Cells 2020, 9, 2439. [Google Scholar] [CrossRef]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.-M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Zhang, M.; Torres, G.; Wu, S.; Ouyang, C.; Xie, Z.; Zou, M.-H. Metformin Suppresses Diabetes-Accelerated Atherosclerosis via the Inhibition of Drp1-Mediated Mitochondrial Fission. Diabetes 2017, 66, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Hondares, E.; Mora, O.; Yubero, P.; de la Concepción, M.R.; Iglesias, R.; Giralt, M.; Villarroya, F. Thiazolidinediones and rexinoids induce peroxisome proliferator-activated receptor-coactivator (PGC)-1α gene transcription: An autoregulatory loop controls PGC-1α expression in adipocytes via peroxisome proliferator-activated receptor-γ coactivation. Endocrinology 2006, 147, 2829–2838. [Google Scholar] [CrossRef]
- Sarafidis, P.A.; Stafylas, P.C.; Georgianos, P.I.; Saratzis, A.N.; Lasaridis, A.N. Effect of thiazolidinediones on albuminuria and proteinuria in diabetes: A meta-analysis. Am. J. Kidney Dis. 2010, 55, 835–847. [Google Scholar] [CrossRef]
- Sun, L.; Yuan, Q.; Xu, T.; Yao, L.; Feng, J.; Ma, J.; Wang, L.; Lu, C.; Wang, D. Pioglitazone Improves Mitochondrial Function in the Remnant Kidney and Protects against Renal Fibrosis in 5/6 Nephrectomized Rats. Front. Pharmacol. 2017, 8, 545. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Xi, X.; Zhang, S.; Zou, C.; Kuang, R.; Ye, Z.; Huang, Y.; Hu, H. Pioglitazone Protects Against Renal Ischemia-Reperfusion Injury via the AMP-Activated Protein Kinase-Regulated Autophagy Pathway. Front. Pharmacol. 2018, 9, 851. [Google Scholar] [CrossRef]
- Morrison, M.C.; Yakala, G.K.; Liang, W.; Wielinga, P.Y.; Salic, K.; van Koppen, A.; Tomar, T.; Kleemann, R.; Heeringa, P.; Kooistra, T. Protective effect of rosiglitazone on kidney function in high-fat challenged human-CRP transgenic mice: A possible role for adiponectin and miR-21? Sci. Rep. 2017, 7, 2915. [Google Scholar] [CrossRef]
- Yu, H.; Jin, F.; Liu, D.; Shu, G.; Wang, X.; Qi, J.; Sun, M.; Yang, P.; Jiang, S.; Ying, X.; et al. ROS-responsive nano-drug delivery system combining mitochondria-targeting ceria nanoparticles with atorvastatin for acute kidney injury. Theranostics 2020, 10, 2342–2357. [Google Scholar] [CrossRef]
- Hou, Y.; Shi, Y.; Han, B.; Liu, X.; Qiao, X.; Qi, Y.; Wang, L. The antioxidant peptide SS31 prevents oxidative stress, downregulates CD36 and improves renal function in diabetic nephropathy. Nephrol. Dial. Transplant. 2018, 33, 1908–1918. [Google Scholar] [CrossRef]
- Emani, S.M.; Piekarski, B.L.; Harrild, D.; del Nido, P.J.; McCully, J.D. Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 2017, 154, 286–289. [Google Scholar] [CrossRef]
- Kubat, G.B.; Ozler, M.; Ulger, O.; Ekinci, O.; Atalay, O.; Celik, E.; Safali, M.; Budak, M.T. The effects of mesenchymal stem cell mitochondrial transplantation on doxorubicin-mediated nephrotoxicity in rats. J. Biochem. Mol. Toxicol. 2021, 35, e22612. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Drug Name | MECHANISM of Action | Animal Models | Clinical Trials on Kidney Disease Identifier Number | Kidney Diseases Studied on Humans |
|---|---|---|---|---|
| Cardiolipin Protective Agents | ||||
| SS-31 | Binds cardiolipins, prevents their peroxidation, and maintains mitochondrial membrane structure, ROS scavenger | Unilateral ureteral obstruction (98, 99), sepsis-induced acute kidney injury (100), diabetic kidney disease (102–105), renovascular disease (106), metabolic syndrome (107–109), ischemia-reperfusion acute kidney injury (110, 111), contrast-induced nephropathy (113), cisplatin-induced acute kidney injury (114), chronic kidney disease (13, 87, 112) | NCT01755858, NCT02436447 | NCT02436447: patients with renal impairment NCT01755858: percutaneous transluminal renal angioplasty |
| SS-20 | Binds cardiolipin and maintains mitochondrial membrane structure, but does not scavenger ROS and inhibit inner mitochondrial membrane transition | Warm ischemia kidney models and ischemia-induced fibrosis (117) | N/A | N/A |
| Fission inhibitors | ||||
| mdivi-1 | Selectively inhibits DRP1 GTPase and favors mitochondrial fusion | Septic acute kidney injury (124, 125), cisplatin-induced acute kidney injury (11), ischemia reperfusion-induced acute kidney injury (11, 126), autosomal dominant polycystic kidney disease (55), chronic kidney disease (123), diabetic kidney disease (127), unilateral renal obstruction (78, 123) | N/A | N/A |
| mPTP Inhibitors | ||||
| TDZD-8 | Inhibits glycogen synthase kinase 3b and prevents mPTP channel opening | Adriamycin-induced kidney damage (140), ischemia-reperfusion acute kidney injury (141), NSAID-induced acute kidney injury (142) | N/A | N/A |
| Alisporivir | Analogue of cyclosporine A. Binds cyclophilin and inhibits mPTP channels | Diabetic kidney disease (143) | NCT01975337 | NCT01975337: hemodialysis patients |
| Biogenesis Activators | ||||
| SRT1720 | Activates Sirtuin1 and mitochondrial biogenesis | Unilateral ureteral obstruction (144), chronic kidney disease (144, 145), cisplatin-induced acute kidney injury (146), ischemia-reperfusion acute kidney injury (147, 148) | N/A | N/A |
| LY344864 | Selectively activates 5-HT1F receptor and activates mitochondrial biogenesis | Ischemia-reperfusion acute kidney injury (150) | N/A | N/A |
| BF175 | Selectively activates Sirtuin1 and mitochondrial biogenesis | Diabetic kidney disease (151), HIV kidney disease (152) | N/A | N/A |
| Resveratrol | Activates mitochondrial biogenesis and ROS scavenger | Diabetic kidney disease (153, 154), ischemia-reperfusion acute kidney injury (155) | NCT02433925, NCT03352895, NCT03597568, NCT03815786, NCT02704494, NCT03946176 | NCT02433925, NCT03352895, NCT03597568, NCT03815786: chronic kidney disease, NCT02704494: diabetic kidney disease, NCT03946176: hemodialysis patients |
| Antioxidants | ||||
| MitoQ | ROS scavenger and antioxidant which concentrates at the matrix in a mitochondrial membrane potential-dependent manner | Diabetic kidney disease (73, 164), cisplatin-induced acute kidney injury (165) | NCT02364648, NCT03960073 | NCT02364648: chronic kidney disease NCT03960073: HFpEF patients with and without chronic kidney disease |
| SkQ | Accumulates in mitochondria by penetrating planar phospholipid membranes | Mitochondrial DNA mutator mice (172), ischemia/reperfusion injury (166), gentamycin-induced nephrotoxicity (173) | N/A | N/A |
| Ubiquinone (coenzyme Q10, CoQ10) | Normalizes ATP production, attenuates mitochondria reactive oxidative species and decreases mitochondrial membrane potential | Diabetic kidney disease (167) | NCT04445779, NCT05422534, NCT03579693, NCT04972552, NCT00307996, NCT00908297, NCT05266794, NCT05214885, NCT01408680 | NCT04445779: acute kidney injury/failure NCT05422534: end stage renal disease NCT03579693: chronic kidney disease NCT04972552: kidney transplantation NCT00307996, NCT00908297, NCT05266794, NCT01408680 hemodialysis patients NCT05214885: clear cell renal cell carcinoma |
| Curcumin | Acts as an antioxidant and is responsible for altering mitochondrial dynamics and bioenergetics | 5/6 nephrectomy rats (169, 176), diabetic kidney disease (168) | NCT02369549, NCT03475017, NCT04413266, NCT03223883, NCT03935958, NCT03262363, NCT02494141, NCT04132648, NCT01285375, NCT01831193, NCT01225094, NCT05183737, NCT03019848, NCT04890704, NCT01906840 | NCT02369549, NCT03475017, NCT03223883, NCT03262363, NCT04132648, NCT05183737: chronic kidney disease NCT04413266: peritoneal dialysis NCT03935958, NCT01285375: kidney transplantation NCT02494141: autosomal dominant polycystic kidney, NCT01831193, NCT03019848: proteinuria NCT01225094, NCT04890704: acute kidney injury NCT01906840: end stage renal disease |
| Enhancement of Mitophagy and Autophagy | ||||
| Tat–beclin 1 | Inducer of autophagy, derived from a region of the autophagy protein, beclin 1 | Mice infected with chikungunya or West Nile virus (183) | N/A | N/A |
| Urolithin A | A gut microbial metabolite of ellagic acid and related compounds was reported to induce mitophagy | Cisplatin-induced AKI (186), two different mouse models of age-related decline in muscle function, as well as young rats and Caenorhabditis elegans (184) | N/A | N/A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanriover, C.; Copur, S.; Ucku, D.; Cakir, A.B.; Hasbal, N.B.; Soler, M.J.; Kanbay, M. The Mitochondrion: A Promising Target for Kidney Disease. Pharmaceutics 2023, 15, 570. https://doi.org/10.3390/pharmaceutics15020570
Tanriover C, Copur S, Ucku D, Cakir AB, Hasbal NB, Soler MJ, Kanbay M. The Mitochondrion: A Promising Target for Kidney Disease. Pharmaceutics. 2023; 15(2):570. https://doi.org/10.3390/pharmaceutics15020570
Chicago/Turabian StyleTanriover, Cem, Sidar Copur, Duygu Ucku, Ahmet B. Cakir, Nuri B. Hasbal, Maria Jose Soler, and Mehmet Kanbay. 2023. "The Mitochondrion: A Promising Target for Kidney Disease" Pharmaceutics 15, no. 2: 570. https://doi.org/10.3390/pharmaceutics15020570