
Small Extracellular Vesicles as a New Class of Medicines

Abstract
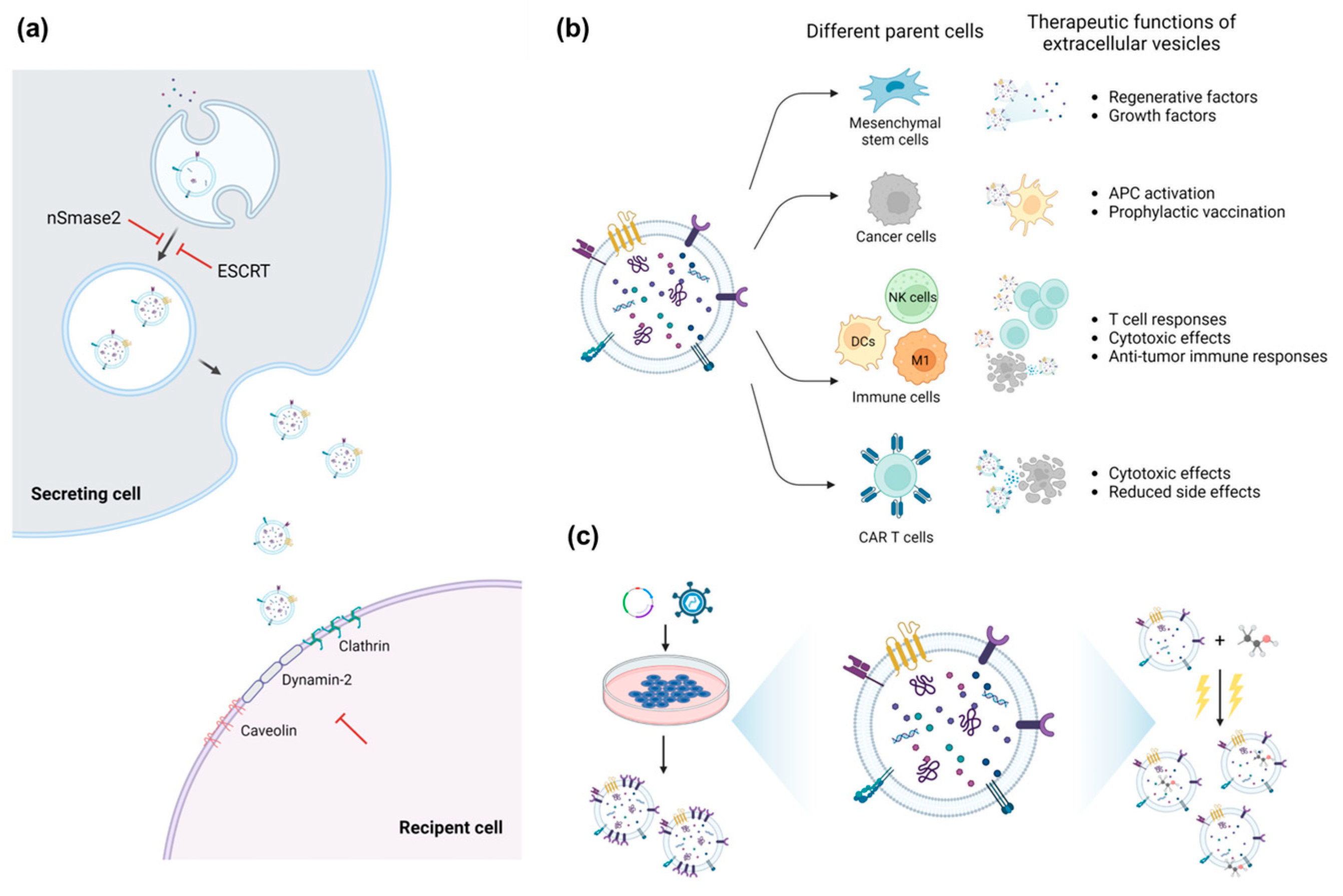
:1. Introduction
2. Types of sEV Therapeutics
2.1. Inhibition of the Release and Uptake of sEVs

{kind=link}
| Strategy | Reagent | Disease | Mode of Action | Treatment | Result | Reference |
|---|---|---|---|---|---|---|
| Exosome biogenesis and release inhibition | Manumycin A | C42B prostate cancer | ERK-dependent inhibition of hnRNP H1 | 250 nM treatment for 48 h | Decreased Ras activation by GTPγS | [11] |
| GW4869 | U937 acute myeloblastic leukemia | Enhance doxorubicin sensitivity on resistant cells by inhibition doxorubicin expulsionvia sEVs | 20 µM GW4869 + 0.5–2 uM PEGylated liposomal doxorubicin for 24 h | Enhanced cytotoxicity | [14] | |
| Tipifarnib | Metastatic renal cell carcinoma | Disrupt ESCRT-dependent and ESCRT-independent functional proteins (Alix, nSMase, and Rab27a) | 0.25–1 µM treatment for 48 h at 37 °C | Reduced sEV load in sunitinib-sensitive renal cell carcinomas | [12] | |
| 5-(N-ethyl-N-isopropyl) amiloride | A431 human epidermoid carcinoma | Disrupt Rac1 activation and assembly of actin | 50–100 µM pretreatment for 30 min | Inhibited clathrin-independent endocytosis and macropinocytosis of sEVs | [22] | |
| Imipramine | PC3 prostate cancer | Reduce macropinocytosis | 25 µM treatment | Reduced total EV release by 77% in PC3 | [23] | |
| 4T1 mammary carcinoma | 5 μM treatment for 1 h | Inhibited membrane ruffle formation | [24] | |||
| Calpeptin | Worms | Inhibit calpain | 80 μM treatment | Prevented the secretion of miRNAs from adult worms | [25] | |
| Exosome endocytosis inhibition | Dynasore | HUVEC | Interfere with the internalization of transferrin through the clathrin-dependent pathway | Pretreatment on HUVECs with 20 µM dynasore for 30 min at 37 °C | Prevented pancreatic cancer cell-derived sEVs | [16] |
| B cells | Pretreatment on uninfected B cells with 150 μM dynasor for 30 min at 37 °C | Prevented virus spread by inhibiting the uptake of Epstein–Barr virus-infected B cell-derived exosomes | [17] | |||
| Human bone marrow stromal cells | Pretreatment on bone marrow stromal cells with 50 μM dynasore for 30 min | Suppressed the effects of multiple myeloma cell-derived exosome uptake | [26] | |||
| Heparin | U-87 MG glioblastoma | Competitively inhibit cancer cell surface receptors depending on heparin sulfate proteoglycan coreceptors for the uptake of sEVs | Pretreatment on U-87 MG with 10 μg/mL heparin for 1 h | Reduced sEV uptake in U87 cells by 55% | [19] | |
| SW780 human bladder cancer cell line | Pretreatment on SW780 with 10 μg/mL heparin at 4 °C for 4 h | Inhibited receptors on recipient cells | [20] |
2.2. Naïve sEV Therapeutics
2.2.1. Stem Cell-Derived Naïve sEVs
2.2.2. Immune Cell-Derived Naïve sEVs
2.2.3. Other Cell-Derived Naïve sEVs
2.3. Engineered sEV Therapeutics
2.4. sEV Therapeutics in Clinical Trials
| Strategy | Source cell | sEV Purification and Engineering | Loaded Cargo Amount | Disease | Therapeutic Schedule | Result | Reference |
|---|---|---|---|---|---|---|---|
| Surface engineering of sEVs | Mouse spleen-derived CD4+ T cells | Ultracentrifugation and incubation with anti-VEGF | 10 anti VEGF molecules per sEV | Choroidal neovascularization | 10 μg sEVs, intravitreal injection per eyes | Suppressed angiogenesis and inflammation | [94] |
| Adipose-derived stem cells | Ultracentrifugation and primed with 100 ng/mL IFNγ overnight | N/A | Tendon injury | EV-laden collagen sheet containing 5–6 × 109 EVs applied around the injury site | Ameliorated the repair site’s inflammatory response, regenerated tendon matrix | [95] | |
| Human umbilical cord MSCs | Ultracentrifugation and transfection of IL-10-overexpressing vector plasmids | N/A | Autoimmune uveitis | 50 μg sEVs, intravenous injection on day 11 post-immunization | Modulated T cell immunity | [96] | |
| Human placental MSCs | Ultracentrifugation and expression of collagen-binding peptide using click chemistry | N/A | Ischemic disease | 1 × 1010 sEVs, intramuscular injection into four different sites around the hind limb ischemic region | Reduced inflammation and increased muscle regeneration | [97] | |
| Human umbilical vascular endothelial cells | Ultracentrifugation and incubation with anti-angiogenic peptide KV11-anchoring peptide CP05 | 83.1% EVs anchoring KV11 peptides | Retina neovascularization | 50 μg sEVs, retro-orbital injection on day 12 | Suppressed neovascularization | [98] | |
| Loading cargo into sEVs | Mouse bone marrow-derived macrophages | ExoQuick-TC™ Kit and incubation and sonication with Paclitaxel | 10 mg/mL Paclitaxel mixed with 1011 sEVs | Pulmonary metastases | 4 × 1011 particles of sEVs, systemic injection, three times on day 1, 4, and 7 | Suppressed neoplastic effect and increased survival period | [99] |
| Rat marrow stromal cells | ExoQuick-TC™ Kit and transfection of miR-67 or 146b plasmid | N/A | Brain tumor | 50 μg sEV, intratumoral injection on day 5 | Reduced glioma growth | [104] | |
| Human adipose-derived stem cells | Ultracentrifugation and loading of miR-21-5p into sEVs by electroporation | N/A | Diabetic wound | 5 μg/200 μL solution applied to the wound bed every 3 days for 15 days | Ameliorated diabetic wounds | [102] | |
| HEK293 | exoEasy Maxi Kit and incubation for loading of curcumin into sEVs | 1 μg curcumin in 2.04–2.46 × 109 sEVs | Acute lung injury | 15 μg sEVs, intratracheal instillation into the lungs | Reduced proinflammatory cytokines | [103] | |
| A549 | Centrifugation and transfection of TAT and TAR-miR-449a plasmids | N/A | Non-small-cell lung cancer | 2 mg/kg sEVs, intravenous injection | Suppressed tumor growth | [105] | |
| RAW 264.7 | ExoQuick-TC™ Kit and incubation, electroporation, and sonication with Paclitaxel | N/A | Lung metastasis | 1 × 107 particles, intravenous injection every other day, 7 times | Anticancer effect in Lewis lung carcinoma | [100] | |
| RAW 264.7 | Ultracentrifugation and incubation, electroporation, and sonication with catalase | 1376 ± 64.1 U of catalase activity in 4 × 1011 sEV/mL | Parkinson’s disease | 1.2 × 109 sEVs, injection into each nostril, 10 times every other day | Reduced microgliosis and protected neurons | [106] | |
| RAW 264.7 M1 macrophage | Ultracentrifugation and sonication with Paclitaxel | N/A | Breast cancer | 0.1 mg sEVs, intravenous injection every 3 days for 27 days | Anti-tumor effects | [66] | |
| Mouse bone marrow-derived dendritic cells | Ultracentrifugation and sonication with Paclitaxel | 5 μg TGFB1 and 5 μg IL10 in 1 × 109 | Periodontitis | 200 µL sEVs, intravenous or palatal tissue local injection on day 3 | Prevented cytokines from proteolytic attack and alveolar bone loss | [101] |
| Applied EVs | Diseases | Dosing Schedules | Expected Results | Patients | Phase | Recruitment Status | Identifier |
|---|---|---|---|---|---|---|---|
| Amniotic MSC-sEV | Hair loss, alopecia | 1012 sEVs administered through an interval of 14 days over two months | Anticipation of growth factors contained in stem cell-sEV to improve hair loss | 20 | N/A | Recruiting | NCT05658094 |
| Placenta MSC-sEV | Complex anal fistula | N/A | Anticipation of anal fistula treatment effect using stem cell sEVs | 80 | Phase 1, Phase 2 | Recruiting | NCT05402748 |
| MSC-sEV | Cerebrovascular disorders | N/A | Evaluation of improvement in acute ischemic stroke patients receiving MSC-sEVs | 5 | Phase 1, Phase 2 | Unknown | NCT03384433 |
| Embryonic kidney T-REx™-293 cell-sEV | COVID-19 | 1010 sEVs/4 mL normal saline administered through inhalation, once a day for 5 days | Evaluation of the safety and efficacy of sEVs overexpressing CD24 | 155 | Phase 2 | Active, not recruiting | NCT04969172 |
| Adipose tissue stem cell-sEV | Periodontitis | N/A | Evaluation of regeneration effect | 10 | Early Phase 1 | Unknown | NCT04270006 |
| Plasma-sEV | Ulcer | N/A | Anticipation of skin wound-healing efficacy of plasma sEVs | 5 | Early Phase 1 | Unknown | NCT02565264 |
| sEV | Neuralgia | N/A | Evaluation of safety and efficacy of sEVs in patients with craniofacial neuralgia | 100 | N/A | Suspended | NCT04202783 |
| MSC-sEV | Myocardial infarction, myocardial ischemia, myocardial stunning | 100 μg/mL sEVs administered through intracoronary and intra-myocardial injection | Anticipation of improvement in patient outcomes from the detrimental effects of ischemia and reperfusion injury | 20 | Phase 1, Phase 2 | Recruiting | NCT05669144 |
| sEV | Refractory depression, anxiety disorders, neurodegenerative diseases | 2.1 × 107 allogenic sEVs/15 mL administered through intravenous injection | Evaluation of efficacy of sEVs in patients with neurodegenerative dementia | 300 | N/A | Suspended | NCT04202770 |
| MSC-sEV | SARS-CoV-2 infection | sEV administered through intravenous injection twice, in day 1 and day 7 of 2 weeks | Evaluation of efficacy of MSC-sEVs in reducing hyper-inflammation in patients with moderate COVID-19 | 60 | Phase 2, Phase 3 | Recruiting | NCT05216562 |
| MSC-sEV | Multiple organ failure | 180 mg sEV administered through intravenous injection once a day for 14 days | Evaluation of efficacy of MSC-sEVs for multiple organ dysfunction syndrome | 60 | N/A | Not yet recruiting | NCT04356300 |
| Mesenchymal progenitor cell-sEV | Drug-resistant | (8 or 16) × 103 sEVs/3 mL administered through inhalation 7 times, day 1 to 7 | Evaluation of efficacy of sEV treatment for pulmonary infection caused by carbapenem-resistant gram-negative bacilli | 60 | Phase 1, Phase 2 | Recruiting | NCT04544215 |
| Wharton jelly MSC-sEV | Retinitis pigmentosa | sEVs administered through single subtenon injection | Evaluation of efficacy of umbilical cord MSC-sEVs in the treatment of retinitis pigmentosa | 135 | Phase 2, Phase 3 | Recruiting | NCT05413148 |
| COVID-19-specific T cell-sEV | Corona virus infection pneumonia | 2.0 × 103 sEVs/3 mL administered once a day for 5 days | Evaluation of efficacy after targeted delivery of T cell sEVs by metered-dose inhaler | 60 | Phase 1 | Unknown | NCT04389385 |
| MSC-sEV | Psoriasis | 100 µg MSC sEV/g of ointment was dripped once a day for 20 days | Anticipation of psoriasis treatment efficacy using MSC-sEV ointment | 10 | Phase 1 | Completed | NCT05523011 |
| Adipose MSC-sEV | Corona virus | 2.0 × 108 sEVs/3 mL administered through aerosol inhalation 5 times for 5 days | Anticipation of safety and efficacy of stem cell sEVs in treatment of severely ill patients hospitalized with novel coronavirus pneumonia | 24 | Phase 1 | Completed | NCT04276987 |
| MSC-sEV | COVID-19, novel coronavirus pneumonia, acute respiratory distress syndrome | (2, 4, and 8 × 109 or 8, 4, and 8 × 109) sEVs/mL administered through injection every other day for 5 days or 8 × 109 sEV administered through injection every other day for 5 days | Anticipation of treatment effects of stem cell sEVs in treatments of patients with acute respiratory distress syndrome and novel coronavirus pneumonia | 55 | Phase 1, Phase 2 | Not yet recruiting | NCT04798716 |
| Neonatal stem cell-sEV | Neuralgia | N/A | Anticipation of treatment in neuralgia patients using neonatal sEVs | 100 | N/A | Suspended | NCT04202783 |
| MSC-sEV | Osteoarthritis | (3–5) × 1011 sEVs administered through single dose injection | Anticipation of knee arthritis treatment effect using stem cell sEVs | 10 | Phase 1 | Not yet recruiting | NCT05060107 |
| T-REx™-293 cell-sEV | SARS-CoV-2 | (1, 5, 10, 100) × 108 sEVs/2 mL saline) administered through QD inhalation device | Anticipation of therapeutic effect in moderate/severe COVID-19 patients with sEV-CD24 | 35 | Phase 1 | Recruiting | NCT04747574 |
| BM MSC-sEV | Familial hypercholesterolemia | (0.044, 0.088, 0.145, 0.220, 0.295, or 0.394 mg/kg) sEV administered through single dose injection | Anticipation of treatment of hypercholesterolemia patients with sEV-based LDLR mRNA nano platform | 30 | Phase 1 | Not yet recruiting | NCT05043181 |
| Synovial fluid MSC-sEV | Degenerative meniscal injury | 1 × 106 sEVs/kg administered through intra-articular injection | Anticipation of efficacy of synovial MSC-sEVs in patients with degenerative meniscal cartilage damage | 30 | Phase 2 | Recruiting | NCT05261360 |
| Cell free umbilical cord-blood-MSC-sEV | Diabetes mellitus type 1 | (1.22–1.51) × 106 sEVs/kg administered through intravenous injection | Anticipation of cord blood-sEVs to treat type 1 diabetes patients | 20 | Phase 2, Phase 3 | Unknown | NCT02138331 |
| Umbilical MSC-sEV | Dry eye | 10 ug/drop sEV administered through eye drops 4 times a day for 14 days | Anticipation of MSC-sEVs to alleviate dry eye symptoms in patients with chronic graft versus host diseases | 27 | Phase 1, Phase 2 | Recruiting | NCT04213248 |
| Adipose-sEV | Wounds and injuries | N/A | Anticipation of sEVs to promote wound healing | 5 | N/A | Not yet recruiting | NCT05475418 |
| MSC-sEV | COVID-19 SARS-CoV-2 pneumonia | (0.5–2 × 1010) sEVs of the first or second type/3 mL administered through inhalation twice a day for 10 days | Anticipation of safety and efficacy of sEV inhalation method for COVID-19-associated pneumonia | 90 | Phase 2 | Unknown | NCT04602442 |
| sEV | COVID-19 | (1 or 10) × 109 sEVs administered once a day for 5 days | Anticipation of safety and efficacy assessment of CD24 overexpressing sEVs for patients with severe COVID-19 | 90 | Phase 2 | Recruiting | NCT04902183 |
| MSC-sEV | Acute respiratory distress syndrome | (2, 8, 16) × 108 sEVs administered through inhalation 7 times 1/4 MTD/day, or MTD/day at day 1 to 7 | Anticipation of treatment effects of MSC-sEVs in acute respiratory distress syndrome | 169 | Phase 1, Phase 2 | Unknown | NCT04602104 |
| MSC-sEV | Macular hole | 50 or 20 μg/10 μL sEV was dripped into vitreous cavity around macular holes | Evaluation of safety and efficacy evaluation of MSC-sEVs for promoting healing of large and refractory macular holes | 44 | Early Phase 1 | Active, not recruiting | NCT03437759 |
| Adipose MSC-sEV | Alzheimer’s disease | (5, 10, or 20 μg/1 mL) sEV administered through nasal drip twice a week for 12 weeks | Evaluation of safety and efficacy evaluation of allogeneic adipose MSC-sEVs in patients with Alzheimer’s disease | 9 | Phase 1, Phase 2 | Unknown | NCT04388982 |
| MSC-sEV | Dystrophic epidermolysis bullosa | N/A | Evaluation of the safety and efficacy of sEVs in the treatment of lesions in patients with epidermolysis bullosa | 10 | Phase 1, Phase 2 | Not yet recruiting | NCT04173650 |
| ASO-STAT6-sEV | Advanced hepatocellular carcinoma, gastric cancer metastatic to liver colorectal cancer metastatic to liver | N/A | Evaluation of treatment efficacy for advanced hepatocellular carcinoma and primary gastric cancer with ASO-STAT6 sEVs (CDK-004) | 30 | Phase 1 | Recruiting | NCT05375604 |
| sEV | Cutaneous T-cell lymphoma | N/A | Evaluation of safety, tolerability, pharmacokinetics, and pharmacodynamic effects of CDK-003 | 2 | Phase 1 | Terminated | NCT05156229 |
| BM MSC-sEV | COVID-19 acute respiratory distress syndrome | 1.2 × 1012 sEVs/85 mL, 8 × 1011 sEVs/90 mL administered through intravenous injection | Evaluation of safety and efficacy of acute respiratory distress syndrome treatment using DB-0018 SEV | 120 | Phase 2 | Completed | NCT04493242 |
| DC-sEV | Non-small-cell lung cancer | 1.3 × 1013 MHC class II molecules administered through injection, four doses at weekly intervals | Evaluation of the safety, feasibility, and efficacy of administering tumor antigen-loaded autologous dexosomes to patients with advanced non-small-cell lung cancer (NSCLC) | 13 | Phase 1 | Completed | |
| Turmeric-sEV | Colon cancer | 3.6 g curcumin-conjugated sEV tablets taken daily for 7 days | Evaluation of the ability of sEVs to deliver curcumin more effectively to normal colon tissues and colon tumors | 7 | Phase 1 | Active, not recruiting | NCT01294072 |
3. Issues to Overcome for Realization of sEV Therapeutics
3.1. Biodistribution
3.2. Large-Scale Manufacturing
3.3. Quality Control
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, G.; Choi, Y.; Kim, G.B.; Kim, S.; Kim, S.A.; Kim, I. Emerging Prospects of Exosomes for Cancer Treatment: From Conventional Therapy to Immunotherapy. Adv. Mater. 2020, 32, e2002440. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.F. Extracellular Vesicles and Neurodegenerative Diseases. J. Neurosci. 2019, 39, 9269–9273. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Zhang, Y.; Li, Y.; Luo, L.; Zhao, Y.; Yao, Y. Extracellular vesicles in cardiovascular diseases. Cell Death Discov. 2020, 6, 68. [Google Scholar] [CrossRef]
- Sahoo, S.; Adamiak, M.; Mathiyalagan, P.; Kenneweg, F.; Kafert-Kasting, S.; Thum, T. Therapeutic and Diagnostic Translation of Extracellular Vesicles in Cardiovascular Diseases: Roadmap to the Clinic. Circulation 2021, 143, 1426–1449. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Choi, Y.; Nam, G.-H.; Kim, I.-S. Functionalized exosome harboring bioactive molecules for cancer therapy. Cancer Lett. 2020, 489, 155–162. [Google Scholar] [CrossRef]
- Bai, S.; Wang, Z.; Wang, M.; Li, J.; Wei, Y.; Xu, R.; Du, J. Tumor-Derived Exosomes Modulate Primary Site Tumor Metastasis. Front. Cell Dev. Biol. 2022, 10, 752818. [Google Scholar] [CrossRef]
- Tian, W.; Liu, S.; Li, B. Potential Role of Exosomes in Cancer Metastasis. BioMed Res. Int. 2019, 2019, 4649705. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Dong, M.; Chen, Q. Tumor-Derived Exosomes and Their Role in Breast Cancer Metastasis. Int. J. Mol. Sci. 2022, 23, 13993. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell. Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef]
- Datta, A.; Kim, H.; Lal, M.; McGee, L.; Johnson, A.; Moustafa, A.A.; Jones, J.C.; Mondal, D.; Ferrer, M.; Abdel-Mageed, A.B. Manumycin A suppresses exosome biogenesis and secretion via targeted inhibition of Ras/Raf/ERK1/2 signaling and hnRNP H1 in castration-resistant prostate cancer cells. Cancer Lett. 2017, 408, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, J.W.; Kim, H.; Ahn, M.; Moustafa, A.A.; Zhou, H.; Barata, P.C.; Boulares, A.H.; Abdel-Mageed, A.B.; Krane, L.S. Combination of Tipifarnib and Sunitinib Overcomes Renal Cell Carcinoma Resistance to Tyrosine Kinase Inhibitors via Tumor-Derived Exosome and T Cell Modulation. Cancers 2022, 14, 903. [Google Scholar] [CrossRef]
- Kosaka, N.; Iguchi, H.; Hagiwara, K.; Yoshioka, Y.; Takeshita, F.; Ochiya, T. Neutral Sphingomyelinase 2 (nSMase2)-dependent Exosomal Transfer of Angiogenic MicroRNAs Regulate Cancer Cell Metastasis. J. Biol. Chem. 2013, 288, 10849–10859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hekmatirad, S.; Moloudizargari, M.; Moghadamnia, A.A.; Kazemi, S.; Mohammadnia-Afrouzi, M.; Baeeri, M.; Moradkhani, F.; Asghari, M.H. Inhibition of Exosome Release Sensitizes U937 Cells to PEGylated Liposomal Doxorubicin. Front. Immunol. 2021, 12, 692654. [Google Scholar] [CrossRef]
- Tian, T.; Zhu, Y.-L.; Zhou, Y.-Y.; Liang, G.-F.; Wang, Y.-Y.; Hu, F.-H.; Xiao, Z.-D. Exosome Uptake through Clathrin-mediated Endocytosis and Macropinocytosis and Mediating miR-21 Delivery. J. Biol. Chem. 2014, 289, 22258–22267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiba, M.; Kubota, S.; Sato, K.; Monzen, S. Exosomes released from pancreatic cancer cells enhance angiogenic activities via dynamin-dependent endocytosis in endothelial cells in vitro. Sci. Rep. 2018, 8, 11972. [Google Scholar] [CrossRef] [Green Version]
- Nanbo, A.; Kawanishi, E.; Yoshida, R.; Yoshiyama, H. Exosomes Derived from Epstein-Barr Virus-Infected Cells Are Internalized via Caveola-Dependent Endocytosis and Promote Phenotypic Modulation in Target Cells. J. Virol. 2013, 87, 10334–10347. [Google Scholar] [CrossRef] [Green Version]
- Atai, N.A.; Balaj, L.; Van Veen, H.; Breakefield, X.O.; Jarzyna, P.A.; Van Noorden, C.J.F.; Skog, J.; Maguire, C.A. Heparin blocks transfer of extracellular vesicles between donor and recipient cells. J. Neurooncol. 2013, 115, 343–351. [Google Scholar] [CrossRef] [Green Version]
- Christianson, H.C.; Svensson, K.J.; van Kuppevelt, T.H.; Li, J.-P.; Belting, M. Cancer cell exosomes depend on cell-surface heparan sulfate proteoglycans for their internalization and functional activity. Proc. Natl. Acad. Sci. USA 2013, 110, 17380–17385. [Google Scholar] [CrossRef] [Green Version]
- Franzen, C.A.; Simms, P.E.; Van Huis, A.F.; Foreman, K.E.; Kuo, P.C.; Gupta, G.N. Characterization of Uptake and Internalization of Exosomes by Bladder Cancer Cells. BioMed Res. Int. 2014, 2014, 619829. [Google Scholar] [CrossRef] [PubMed]
- McKelvey, K.J.; Powell, K.L.; Ashton, A.W.; Morris, J.M.; McCracken, S.A. Exosomes: Mechanisms of Uptake. J. Circ. Biomark. 2015, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Verdera, H.C.; Gitz-Francois, J.J.; Schiffelers, R.M.; Vader, P. Cellular uptake of extracellular vesicles is mediated by clathrin-independent endocytosis and macropinocytosis. J. Control Release 2017, 266, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.Y.; Park, J.; Kim, Y.T.; Kim, M.J. Imipramine Inhibits Migration and Invasion in Metastatic Castration-Resistant Prostate Cancer PC-3 Cells via AKT-Mediated NF-κB Signaling Pathway. Molecules 2020, 25, 4619. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-P.; Singla, B.; Ghoshal, P.; Faulkner, J.L.; Cherian-Shaw, M.; O’Connor, P.M.; She, J.-X.; de Chantemele, E.J.B.; Csányi, G. Identification of novel macropinocytosis inhibitors using a rational screen of Food and Drug Administration-approved drugs. Br. J. Pharmacol. 2018, 175, 3640–3655. [Google Scholar] [CrossRef] [Green Version]
- Kumagai, T.; Shimogawara, R.; Ichimura, K.; Iwanaga, S. Calpain inhibitor suppresses both extracellular vesicle-mediated secretion of miRNAs and egg production from paired adults of Schistosoma japonicum. Parasitol. Int. 2022, 87, 102540. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Tu, C.; Zhang, J.; Wang, J. Inhibition of multiple myeloma-derived exosomes uptake suppresses the functional response in bone marrow stromal cell. Int. J. Oncol. 2019, 54, 1061–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimbrel, E.A.; Lanza, R. Next-generation stem cells—Ushering in a new era of cell-based therapies. Nat. Rev. Drug Discov. 2020, 19, 463–479. [Google Scholar] [CrossRef]
- Yagi, H.; Soto-Gutierrez, A.; Parekkadan, B.; Kitagawa, Y.; Tompkins, R.G.; Kobayashi, N.; Yarmush, M.L. Mesenchymal Stem Cells: Mechanisms of Immunomodulation and Homing. Cell Transplant. 2010, 19, 667–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikfarjam, S.; Rezaie, J.; Zolbanin, N.M.; Jafari, R. Mesenchymal stem cell derived-exosomes: A modern approach in translational medicine. J. Transl. Med. 2020, 18, 449. [Google Scholar] [CrossRef]
- Gowen, A.; Shahjin, F.; Chand, S.; Odegaard, K.E.; Yelamanchili, S.V. Mesenchymal Stem Cell-Derived Extracellular Vesicles: Challenges in Clinical Applications. Front. Cell Dev. Biol. 2020, 8, 149. [Google Scholar] [CrossRef]
- Lai, R.C.; Arslan, F.; Lee, M.M.; Sze, N.S.K.; Choo, A.; Chen, T.S.; Salto-Tellez, M.; Timmers, L.; Lee, C.N.; El Oakley, R.M.; et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010, 4, 214–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, J.-Y.; Wang, H.-W.; Chang, S.-J.; Liao, K.-H.; Lee, I.-H.; Lin, W.-S.; Wu, C.-H.; Lin, W.-Y.; Cheng, S.-M. Mesenchymal Stem Cells from Human Umbilical Cord Express Preferentially Secreted Factors Related to Neuroprotection, Neurogenesis, and Angiogenesis. PLoS ONE 2013, 8, e72604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Li, X.; Hu, J.; Chen, F.; Qiao, S.; Sun, X.; Gao, L.; Xie, J.; Xu, B. Mesenchymal stromal cell-derived exosomes attenuate myocardial ischaemia-reperfusion injury through miR-182-regulated macrophage polarization. Cardiovasc. Res. 2019, 115, 1205–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cone, A.S.; Yuan, X.; Sun, L.; Duke, L.C.; Vreones, M.P.; Carrier, A.N.; Kenyon, S.M.; Carver, S.R.; Benthem, S.D.; Stimmell, A.C.; et al. Mesenchymal stem cell-derived extracellular vesicles ameliorate Alzheimer’s disease-like phenotypes in a preclinical mouse model. Theranostics 2021, 11, 8129–8142. [Google Scholar] [CrossRef]
- Shen, Z.; Huang, W.; Liu, J.; Tian, J.; Wang, S.; Rui, K. Effects of Mesenchymal Stem Cell-Derived Exosomes on Autoimmune Diseases. Front. Immunol. 2021, 12, 749192. [Google Scholar] [CrossRef] [PubMed]
- Kholia, S.; Sanchez, M.B.H.; Cedrino, M.; Papadimitriou, E.; Tapparo, M.; Deregibus, M.C.; Brizzi, M.F.; Tetta, C.; Camussi, G. Human Liver Stem Cell-Derived Extracellular Vesicles Prevent Aristolochic Acid-Induced Kidney Fibrosis. Front. Immunol. 2018, 9, 1639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.-Z.; Liu, Y.-M.; Niu, X.; Yin, J.-Y.; Hu, B.; Guo, S.-C.; Fan, Y.; Wang, Y.; Wang, N.-S. Exosomes secreted by human urine-derived stem cells could prevent kidney complications from type I diabetes in rats. Stem Cell Res. Ther. 2016, 7, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, S.; Miura, Y.; Fujishiro, A.; Shindo, T.; Shimazu, Y.; Hirai, H.; Tahara, H.; Takaori-Kondo, A.; Ichinohe, T.; Maekawa, T. Graft-Versus-Host Disease Amelioration by Human Bone Marrow Mesenchymal Stromal/Stem Cell-Derived Extracellular Vesicles Is Associated with Peripheral Preservation of Naive T Cell Populations. Stem Cells 2017, 36, 434–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, J.-H.; Li, Q.; Bhang, D.-H.; Song, W.-J.; Youn, H.-Y. TNF-α and INF-γ primed canine stem cell-derived extracellular vesicles alleviate experimental murine colitis. Sci. Rep. 2020, 10, 2115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruno, S.; Collino, F.; Deregibus, M.C.; Grange, C.; Tetta, C.; Camussi, G. Microvesicles Derived from Human Bone Marrow Mesenchymal Stem Cells Inhibit Tumor Growth. Stem Cells Dev. 2013, 22, 758–771. [Google Scholar] [CrossRef]
- Mathew, B.; Ravindran, S.; Liu, X.; Torres, L.; Chennakesavalu, M.; Huang, C.-C.; Feng, L.; Zelka, R.; Lopez, J.; Sharma, M.; et al. Mesenchymal stem cell-derived extracellular vesicles and retinal ischemia-reperfusion. Biomaterials 2019, 197, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, J.; Liu, Z.; Gong, Y.; Zheng, Z. Human umbilical cord mesenchymal stem cell-derived exosomal miR-27b attenuates subretinal fibrosis via suppressing epithelial–mesenchymal transition by targeting HOXC6. Stem Cell Res. Ther. 2021, 12, 42. [Google Scholar] [CrossRef] [PubMed]
- He, G.-H.; Zhang, W.; Ma, Y.-X.; Yang, J.; Chen, L.; Song, J.; Chen, S. Mesenchymal stem cells-derived exosomes ameliorate blue light stimulation in retinal pigment epithelium cells and retinal laser injury by VEGF-dependent mechanism. Int. J. Ophthalmol. 2018, 11, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Joshi, B.S.; Youssef, S.A.; Bron, R.; de Bruin, A.; Kampinga, H.H.; Zuhorn, I.S. DNAJB6b-enriched small extracellular vesicles decrease polyglutamine aggregation in in vitro and in vivo models of Huntington disease. Iscience 2021, 24, 103282. [Google Scholar] [CrossRef]
- Chen, Y.-A.; Lu, C.-H.; Ke, C.-C.; Chiu, S.-J.; Jeng, F.-S.; Chang, C.-W.; Yang, B.-H.; Liu, R.-S. Mesenchymal Stem Cell-Derived Exosomes Ameliorate Alzheimer’s Disease Pathology and Improve Cognitive Deficits. Biomedicines 2021, 9, 594. [Google Scholar] [CrossRef]
- Ding, M.; Shen, Y.; Wang, P.; Xie, Z.; Xu, S.; Zhu, Z.; Wang, Y.; Lyu, Y.; Wang, D.; Xu, L.; et al. Exosomes Isolated From Human Umbilical Cord Mesenchymal Stem Cells Alleviate Neuroinflammation and Reduce Amyloid-Beta Deposition by Modulating Microglial Activation in Alzheimer’s Disease. Neurochem. Res. 2018, 43, 2165–2177. [Google Scholar] [CrossRef]
- Cui, G.-H.; Guo, H.-D.; Li, H.; Zhai, Y.; Gong, Z.-B.; Wu, J.; Liu, J.-S.; Dong, Y.-R.; Hou, S.-X. RVG-modified exosomes derived from mesenchymal stem cells rescue memory deficits by regulating inflammatory responses in a mouse model of Alzheimer’s disease. Immun. Ageing 2019, 16, 10. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.-S.; Jia, J.; Wang, Z. Mesenchymal Stem Cell-Derived Extracellular Vesicles Suppresses iNOS Expression and Ameliorates Neural Impairment in Alzheimer’s Disease Mice. J. Alzheimer’s Dis. 2018, 61, 1005–1013. [Google Scholar] [CrossRef]
- Cui, G.-H.; Wu, J.; Mou, F.-F.; Xie, W.-H.; Wang, F.-B.; Wang, Q.-L.; Fang, J.; Xu, Y.-W.; Dong, Y.-R.; Liu, J.-R.; et al. Exosomes derived from hypoxia-preconditioned mesenchymal stromal cells ameliorate cognitive decline by rescuing synaptic dysfunction and regulating inflammatory responses in APP/PS1 mice. FASEB J. 2018, 32, 654–668. [Google Scholar] [CrossRef] [Green Version]
- Elia, C.A.; Tamborini, M.; Rasile, M.; Desiato, G.; Marchetti, S.; Swuec, P.; Mazzitelli, S.; Clemente, F.; Anselmo, A.; Matteoli, M.; et al. Intracerebral Injection of Extracellular Vesicles from Mesenchymal Stem Cells Exerts Reduced Aβ Plaque Burden in Early Stages of a Preclinical Model of Alzheimer’s Disease. Cells 2019, 8, 1059. [Google Scholar] [CrossRef]
- Yang, L.; Zhai, Y.; Hao, Y.; Zhu, Z.; Cheng, G. The Regulatory Functionality of Exosomes Derived from hUMSCs in 3D Culture for Alzheimer’s Disease Therapy. Small 2019, 16, e1906273. [Google Scholar] [CrossRef] [PubMed]
- Wolfers, J.; Lozier, A.; Raposo, G.; Regnault, A.; Théry, C.; Masurier, C.; Flament, C.; Pouzieux, S.; Faure, F.; Tursz, T.; et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat. Med. 2001, 7, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Droste, M.; Thakur, B.K.; Eliceiri, B.P. Tumor-Derived Extracellular Vesicles and the Immune System—Lessons From Immune-Competent Mouse-Tumor Models. Front. Immunol. 2020, 11, 606859. [Google Scholar] [CrossRef] [PubMed]
- Santos, P.; Almeida, F. Exosome-Based Vaccines: History, Current State, and Clinical Trials. Front. Immunol. 2021, 12, 711565. [Google Scholar] [CrossRef]
- Stefanius, K.; Servage, K.A.; Santos, M.D.S.; Gray, H.F.; Toombs, J.E.; Chimalapati, S.; Kim, M.S.; Malladi, V.S.; Brekken, R.A.; Orth, K. Human pancreatic cancer cell exosomes, but not human normal cell exosomes, act as an initiator in cell transformation. eLife 2019, 8, e40226. [Google Scholar] [CrossRef]
- Kucharzewska, P.; Christianson, H.C.; Welch, J.E.; Svensson, K.J.; Fredlund, E.; Ringnér, M.; Mörgelin, M.; Bourseau-Guilmain, E.; Bengzon, J.; Belting, M. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc. Natl. Acad. Sci. USA 2013, 110, 7312–7317. [Google Scholar] [CrossRef] [Green Version]
- Capello, M.; Vykoukal, J.V.; Katayama, H.; Bantis, L.E.; Wang, H.; Kundnani, D.L.; Aguilar-Bonavides, C.; Aguilar, M.; Tripathi, S.C.; Dhillon, D.S.; et al. Exosomes harbor B cell targets in pancreatic adenocarcinoma and exert decoy function against complement-mediated cytotoxicity. Nat. Commun. 2019, 10, 254. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Mark, M.T.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Romagnoli, G.G.; Zelante, B.B.; Toniolo, P.A.; Migliori, I.K.; Barbuto, J.A.M. Dendritic Cell-Derived Exosomes may be a Tool for Cancer Immunotherapy by Converting Tumor Cells into Immunogenic Targets. Front. Immunol. 2015, 5, 692. [Google Scholar] [CrossRef] [Green Version]
- Markov, O.; Oshchepkova, A.; Mironova, N. Immunotherapy Based on Dendritic Cell-Targeted/-Derived Extracellular Vesicles—A Novel Strategy for Enhancement of the Anti-tumor Immune Response. Front. Pharmacol. 2019, 10, 1152. [Google Scholar] [CrossRef]
- Wahlund, C.J.E.; Güclüler, G.; Hiltbrunner, S.; Veerman, R.E.; Näslund, T.I.; Gabrielsson, S. Exosomes from antigen-pulsed dendritic cells induce stronger antigen-specific immune responses than microvesicles in vivo. Sci. Rep. 2017, 7, 17095. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.; Miao, Y.; Wang, X.; Huang, X.; Dai, J. Recent progress of dendritic cell-derived exosomes (Dex) as an anti-cancer nanovaccine. Biomed. Pharmacother. 2022, 152, 113250. [Google Scholar] [CrossRef]
- Lugini, L.; Cecchetti, S.; Huber, V.; Luciani, F.; Macchia, G.; Spadaro, F.; Paris, L.; Abalsamo, L.; Colone, M.; Molinari, A.; et al. Immune surveillance properties of human NK cell-derived exosomes. J. Immunol. 2012, 189, 2833–2842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Kalimuthu, S.; Gangadaran, P.; Oh, J.M.; Lee, H.W.; Baek, S.H.; Jeong, S.Y.; Lee, S.-W.; Lee, J.; Ahn, B.-C. Exosomes derived from natural killer cells exert therapeutic effect in melanoma. Theranostics 2017, 7, 2732–2745. [Google Scholar] [CrossRef] [PubMed]
- Shoae-Hassani, A.; Hamidieh, A.A.; Behfar, M.; Mohseni, R.; Mortazavi-Tabatabaei, S.A.; Asgharzadeh, S. NK Cell–derived Exosomes From NK Cells Previously Exposed to Neuroblastoma Cells Augment the Antitumor Activity of Cytokine-activated NK Cells. J. Immunother. 2017, 40, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wang, H.; Huang, Q.; Peng, C.; Yao, L.; Chen, H.; Qiu, Z.; Wu, Y.; Wang, L.; Chen, W. Exosomes from M1-Polarized Macrophages Enhance Paclitaxel Antitumor Activity by Activating Macrophages-Mediated Inflammation. Theranostics 2019, 9, 1714–1727. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wang, Y.; Huang, L. Exosomes from M1-Polarized Macrophages Potentiate the Cancer Vaccine by Creating a Pro-inflammatory Microenvironment in the Lymph Node. Mol. Ther. 2017, 25, 1665–1675. [Google Scholar] [CrossRef] [Green Version]
- Aharon, A.; Horn, G.; Bar-Lev, T.H.; Yohay, M.E.Z.; Waks, M.T.; Levin, M.; Unger, N.D.; Avivi, I.; Levin, A.G. Extracellular Vesicles Derived from Chimeric Antigen Receptor-T Cells: A Potential Therapy for Cancer. Hum. Gene Ther. 2021, 32, 1224–1241. [Google Scholar] [CrossRef]
- Fu, W.; Lei, C.; Liu, S.; Cui, Y.; Wang, C.; Qian, K.; Li, T.; Shen, Y.; Fan, X.; Lin, F.; et al. CAR exosomes derived from effector CAR-T cells have potent antitumour effects and low toxicity. Nat. Commun. 2019, 10, 4355. [Google Scholar] [CrossRef] [Green Version]
- Zitvogel, L.; Regnault, A.; Lozier, A.; Wolfers, J.; Flament, C.; Tenza, D.; Ricciardi-Castagnoli, P.; Raposo, G.; Amigorena, S. Eradication of established murine tumors using a novel cell-free vaccine: Dendritic cell derived exosomes. Nat. Med. 1998, 4, 594–600. [Google Scholar] [CrossRef]
- Qiu, Y.; Yang, Y.; Yang, R.; Liu, C.; Hsu, J.-M.; Jiang, Z.; Sun, L.; Wei, Y.; Li, C.-W.; Yu, D.; et al. Activated T cell-derived exosomal PD-1 attenuates PD-L1-induced immune dysfunction in triple-negative breast cancer. Oncogene 2021, 40, 4992–5001. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Hu, W.; Chen, H.; Shou, X.; Ye, T.; Xu, Y. Cocktail Strategy Based on NK Cell-Derived Exosomes and Their Biomimetic Nanoparticles for Dual Tumor Therapy. Cancers 2019, 11, 1560. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Cao, X.; Cai, H.; Feng, P.; Chen, X.; Zhu, Y.; Yang, Y.; An, W.; Yang, Y.; Jie, J. The exosomes derived from CAR-T cell efficiently target mesothelin and reduce triple-negative breast cancer growth. Cell. Immunol. 2020, 360, 104262. [Google Scholar] [CrossRef] [PubMed]
- Gurbel, P.A.; Jeong, Y.-H.; Navarese, E.P.; Tantry, U.S. Platelet-Mediated Thrombosis: From Bench to Bedside. Circ. Res. 2016, 118, 1380–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, S.-C.; Guo, S.-C.; Zhang, C.-Q. Platelet-derived Extracellular Vesicles: An Emerging Therapeutic Approach. Int. J. Biol. Sci. 2017, 13, 828–834. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.-C.; Tao, S.-C.; Yin, W.-J.; Qi, X.; Yuan, T.; Zhang, C.-Q. Exosomes derived from platelet-rich plasma promote the re-epithelization of chronic cutaneous wounds via activation of YAP in a diabetic rat model. Theranostics 2017, 7, 81–96. [Google Scholar] [CrossRef] [Green Version]
- Hayon, Y.; Dashevsky, O.; Shai, E.; Brill, A.; Varon, D.; Leker, R.R. Platelet Microparticles Induce Angiogenesis and Neurogenesis after Cerebral Ischemia. Curr. Neurovascular Res. 2012, 9, 185–192. [Google Scholar] [CrossRef]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [Green Version]
- Thangaraju, K.; Neerukonda, S.N.; Katneni, U.; Buehler, P.W. Extracellular Vesicles from Red Blood Cells and Their Evolving Roles in Health, Coagulopathy and Therapy. Int. J. Mol. Sci. 2020, 22, 153. [Google Scholar] [CrossRef]
- Zhang, G.; Huang, X.; Xiu, H.; Sun, Y.; Chen, J.; Cheng, G.; Song, Z.; Peng, Y.; Shen, Y.; Wang, J.; et al. Extracellular vesicles: Natural liver-accumulating drug delivery vehicles for the treatment of liver diseases. J. Extracell. Vesicles 2020, 10, e12030. [Google Scholar] [CrossRef]
- Chiangjong, W.; Netsirisawan, P.; Hongeng, S.; Chutipongtanate, S. Red Blood Cell Extracellular Vesicle-Based Drug Delivery: Challenges and Opportunities. Front. Med. 2021, 8, 761362. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.; Nam, G.-H.; Hong, Y.; Kim, Y.K.; Kim, D.-H.; Yang, Y.; Kim, I.-S. Comparison of exosomes and ferritin protein nanocages for the delivery of membrane protein therapeutics. J. Control Release 2018, 279, 326–335. [Google Scholar] [CrossRef]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Kim, G.B.; Nam, G.-H.; Hong, Y.; Woo, J.; Cho, Y.; Kwon, I.C.; Yang, Y.; Kim, I.-S. Xenogenization of tumor cells by fusogenic exosomes in tumor microenvironment ignites and propagates antitumor immunity. Sci. Adv. 2020, 6, eaaz2083. [Google Scholar] [CrossRef]
- Koh, E.; Lee, E.J.; Nam, G.-H.; Hong, Y.; Cho, E.; Yang, Y.; Kim, I.-S. Exosome-SIRPα, a CD47 blockade increases cancer cell phagocytosis. Biomaterials 2017, 121, 121–129. [Google Scholar] [CrossRef]
- Conceição, M.; Forcina, L.; Wiklander, O.P.; Gupta, D.; Nordin, J.Z.; Vrellaku, B.; McClorey, G.; Mäger, I.; Görgens, A.; Lundin, P.; et al. Engineered extracellular vesicle decoy receptor-mediated modulation of the IL6 trans-signalling pathway in muscle. Biomaterials 2020, 266, 120435. [Google Scholar] [CrossRef] [PubMed]
- Dooley, K.; McConnell, R.E.; Xu, K.; Lewis, N.D.; Haupt, S.; Youniss, M.R.; Martin, S.; Sia, C.L.; McCoy, C.; Moniz, R.J.; et al. A versatile platform for generating engineered extracellular vesicles with defined therapeutic properties. Mol. Ther. 2021, 29, 1729–1743. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; McKay, P.F.; Samnuan, K.; Najer, A.; Blakney, A.K.; Che, J.; O’Driscoll, G.; Cihova, M.; Stevens, M.M.; Shattock, R.J. Presentation of antigen on extracellular vesicles using transmembrane domains from viral glycoproteins for enhanced immunogenicity. J. Extracell. Vesicles 2022, 11, e12199. [Google Scholar] [CrossRef] [PubMed]
- Schoenmaker, L.; Witzigmann, D.; Kulkarni, J.A.; Verbeke, R.; Kersten, G.; Jiskoot, W.; Crommelin, D.J. mRNA-lipid nanoparticle COVID-19 vaccines: Structure and stability. Int. J. Pharm. 2021, 601, 120586. [Google Scholar] [CrossRef] [PubMed]
- Sahay, G.; Querbes, W.; Alabi, C.; Eltoukhy, A.; Sarkar, S.; Zurenko, C.; Karagiannis, E.; Love, K.; Chen, D.; Zoncu, R.; et al. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat. Biotechnol. 2013, 31, 653–658. [Google Scholar] [CrossRef]
- Bonsergent, E.; Grisard, E.; Buchrieser, J.; Schwartz, O.; Théry, C.; Lavieu, G. Quantitative characterization of extracellular vesicle uptake and content delivery within mammalian cells. Nat. Commun. 2021, 12, 1864. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Qiao, Y.; Li, X.; Chen, J.; Ding, J.; Bai, L.; Shen, F.; Shi, B.; Liu, J.; Peng, L.; et al. Exosomes Exploit the Virus Entry Machinery and Pathway To Transmit Alpha Interferon-Induced Antiviral Activity. J. Virol. 2018, 92, e01578-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Duan, L.; Lu, J.; Xia, J. Engineering exosomes for targeted drug delivery. Theranostics 2021, 11, 3183–3195. [Google Scholar] [CrossRef]
- Tian, Y.; Zhang, F.; Qiu, Y.; Wang, S.; Li, F.; Zhao, J.; Pan, C.; Tao, Y.; Di Yu, D.; Wei, W. Reduction of choroidal neovascularization via cleavable VEGF antibodies conjugated to exosomes derived from regulatory T cells. Nat. Biomed. Eng. 2021, 5, 968–982. [Google Scholar] [CrossRef]
- Shen, H.; Yoneda, S.; Abu-Amer, Y.; Guilak, F.; Gelberman, R.H. Stem cell-derived extracellular vesicles attenuate the early inflammatory response after tendon injury and repair. J. Orthop. Res. 2019, 38, 117–127. [Google Scholar] [CrossRef]
- Li, Y.; Ren, X.; Zhang, Z.; Duan, Y.; Li, H.; Chen, S.; Shao, H.; Li, X.; Zhang, X. Effect of small extracellular vesicles derived from IL-10-overexpressing mesenchymal stem cells on experimental autoimmune uveitis. Stem Cell Res. Ther. 2022, 13, 100. [Google Scholar] [CrossRef]
- Hao, D.; Lu, L.; Song, H.; Duan, Y.; Chen, J.; Carney, R.; Li, J.J.; Zhou, P.; Nolta, J.; Lam, K.S.; et al. Engineered extracellular vesicles with high collagen-binding affinity present superior in situ retention and therapeutic efficacy in tissue repair. Theranostics 2022, 12, 6021–6037. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Lei, Y.; Yu, Z.; Wang, T.; Liu, Y.; Han, G.; Zhang, X.; Li, Y.; Song, Y.; Xu, H.; et al. Exosome-mediated delivery of an anti-angiogenic peptide inhibits pathological retinal angiogenesis. Theranostics 2021, 11, 5107–5126. [Google Scholar] [CrossRef]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Yuan, D.; Deygen, I.; Klyachko, N.L.; Kabanov, A.V.; Batrakova, E.V. Engineering macrophage-derived exosomes for targeted paclitaxel delivery to pulmonary metastases: In vitro and in vivo evaluations. Nanomedicine 2018, 14, 195–204. [Google Scholar] [CrossRef]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Mahajan, V.; Deygen, I.; Klyachko, N.L.; Inskoe, E.; Piroyan, A.; Sokolsky, M.; Okolie, O.; et al. Development of exosome-encapsulated paclitaxel to overcome mdr in cancer cells. Nanomedicine 2016, 12, 655–664. [Google Scholar] [CrossRef]
- Elashiry, M.; Elashiry, M.M.; Elsayed, R.; Rajendran, M.; Auersvald, C.; Zeitoun, R.; Rashid, M.H.; Ara, R.; Meghil, M.M.; Liu, Y.; et al. Dendritic cell derived exosomes loaded with immunoregulatory cargo reprogram local immune responses and inhibit degenerative bone disease in vivo. J. Extracell. Vesicles 2020, 9, 1795362. [Google Scholar] [CrossRef]
- Lv, Q.; Deng, J.; Chen, Y.; Wang, Y.; Liu, B.; Liu, J. Engineered Human Adipose Stem-Cell-Derived Exosomes Loaded with miR-21-5p to Promote Diabetic Cutaneous Wound Healing. Mol. Pharm. 2020, 17, 1723–1733. [Google Scholar] [CrossRef]
- Kim, G.; Lee, Y.; Ha, J.; Han, S.; Lee, M. Engineering exosomes for pulmonary delivery of peptides and drugs to inflammatory lung cells by inhalation. J. Control Release 2021, 330, 684–695. [Google Scholar] [CrossRef]
- Katakowski, M.; Buller, B.; Zheng, X.; Lu, Y.; Rogers, T.; Osobamiro, O.; Shu, W.; Jiang, F.; Chopp, M. Exosomes from marrow stromal cells expressing miR-146b inhibit glioma growth. Cancer Lett. 2013, 335, 201–204. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Xu, M.; Wang, Z.; Yang, M. Engineered exosomes loaded with miR-449a selectively inhibit the growth of homologous non-small cell lung cancer. Cancer Cell Int. 2021, 21, 485. [Google Scholar] [CrossRef]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Control Release 2015, 207, 18–30. [Google Scholar] [CrossRef] [Green Version]
- Skotland, T.; Iversen, T.G.; Llorente, A.; Sandvig, K. Biodistribution, pharmacokinetics and excretion studies of intravenously injected nanoparticles and extracellular vesicles: Possibilities and challenges. Adv. Drug Deliv. Rev. 2022, 186, 114326. [Google Scholar] [CrossRef]
- Kang, M.; Jordan, V.; Blenkiron, C.; Chamley, L.W. Biodistribution of extracellular vesicles following administration into animals: A systematic review. J. Extracell. Vesicles 2021, 10, e12085. [Google Scholar] [CrossRef]
- Shimomura, T.; Seino, R.; Umezaki, K.; Shimoda, A.; Ezoe, T.; Ishiyama, M.; Akiyoshi, K. New Lipophilic Fluorescent Dyes for Labeling Extracellular Vesicles: Characterization and Monitoring of Cellular Uptake. Bioconjug. Chem. 2021, 32, 680–684. [Google Scholar] [CrossRef]
- Goh, W.J.; Zou, S.; Ong, W.Y.; Torta, F.; Alexandra, A.F.; Schiffelers, R.M.; Storm, G.; Wang, J.-W.; Czarny, B.; Pastorin, G. Bioinspired Cell-Derived Nanovesicles versus Exosomes as Drug Delivery Systems: A Cost-Effective Alternative. Sci. Rep. 2017, 7, 14322. [Google Scholar] [CrossRef]
- Khan, A.A.; de Rosales, R.T.M. Radiolabelling of Extracellular Vesicles for PET and SPECT imaging. Nanotheranostics 2021, 5, 256–274. [Google Scholar] [CrossRef]
- Joshi, B.; Ortiz, D.; Zuhorn, I. Converting extracellular vesicles into nanomedicine: Loading and unloading of cargo. Mater. Today Nano 2021, 16, 100148. [Google Scholar] [CrossRef]
- An, H.J.; Cho, H.-K.; Song, D.H.; Kee, C. Quantitative analysis of exosomes in the aqueous humor of Korean patients with pseudoexfoliation glaucoma. Sci. Rep. 2022, 12, 12875. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Y.; Gao, X.; Yuan, Y.; Zhao, J.; Zhou, S.; Wang, H.; Wang, L.; Xu, G.; Li, X.; et al. Plasma-Derived Exosomal ALIX as a Novel Biomarker for Diagnosis and Classification of Pancreatic Cancer. Front. Oncol. 2021, 11, 628346. [Google Scholar] [CrossRef]

| Source Cell | sEV Purification | Disease | Therapeutic Schedule | Result | Reference |
|---|---|---|---|---|---|
| Mouse bone marrow MSCs | Ultracentrifugation | Acute myocardial infarction | 50 μg MSC-sEVs in 25 μL PBS, intramyocardial injection | Changed M1 macrophages to M2 by delivery of miR-182 | [33] |
| Human liver stem cells | Ultracentrifugation, purification by iodixanol | Chronic kidney disease | 1 × 1010 particles/mL MSC-sEVs, intravenous injection weekly for 4 weeks | Anti-fibrosis and improvement in kidney function | [36] |
| Human bone marrow MSCs | Filtration, Total Exosome Isolation Reagent | GVHD | 2 × 10⁶ particles/kg MSC-sEVs in 200 μL saline, intravenous injection | Immunomodulatory effects on T cells by delivering miRNA | [38] |
| Rat urine stem cells | Ultracentrifugation, purification by 30% sucrose/D2O cushion | Diabetic nephropathy | 100 μg MSC-sEVs in 200 μL of PBS, intravenous injection weekly for 4 weeks | Ameliorated kidney impairment | [37] |
| Canine adipose tissue MSCs | Ultracentrifugation | Inflammatory bowel disease | 100 µg MSC-sEVs from either naïve or primed cASCs, in 200 µL PBS, intraperitoneally injected at days 1, 3, and 5 | Increased the immune modulatory effect | [39] |
| Human bone marrow MSCs | Ultracentrifugation | Tumor | First, 100 μg MSC-sEVs; followed by 50 μg MSC-sEVs, in a volume of 20 mL of PBS, intravenous injection weekly for 4 weeks | Inhibited tumor growth | [40] |
| Human bone marrow MSCs | 100 kDa ultra-filtration, Exo Quick-TCTM Kit | Retinal ischemia | 4 μL of 1 × 109 particles/mL MSC-sEVs, intravitreous humor injection | Neuroprotection and regeneration | [41] |
| Human umbilical cord MSCs | Ultracentrifugation | Eye subretinal fibrosis | 2 μL MSC-sEVs, intravitreous tumor injection | Ameliorated subretinal fibrosis by delivering miR-27b | [42] |
| Human umbilical cord blood MSCs | Ultracentrifugation | Choroidal neovascularization | 1 μg, 2 μg, 3 μg of 50 μg/Ml MSC-sEVs, intravitreal humor injection | Ameliorated RPE cells and retina via downregulation of VEGF-A | [43] |
| Mouse neural stem cells | Ultracentrifugation | Huntington’s disease | 10 μL of 5 mg/mL MSC-sEVs, injection into area between the first and second lumbar vertebrae, twice after a 7-day interval each | Reduced mutant HTT aggregation in the brain | [44] |
| Wharton’s jelly | Ultracentrifugation, Exo-Prep kit | Alzheimer’s disease | 50 µg MSC-sEVs, intravenous injection, weekly for 4 weeks | Downregulated HDAC4, improved AD pathology | [45] |
| Human bone marrow MSCs | Centrifugation, purification by PEG solution | Alzheimer’s disease | 2 × 109 MSC-sEVs in 5 µL, intranasal injection every 4 days for 4 months | Improved in cognitive tests | [34] |
| Human umbilical cord blood MSCs | Exo Quick-TCTM kit | Alzheimer’s disease | 30 μg MSC-sEVs, intravenous injection, every 2 weeks, four times | Reduced neuroinflammation and Aβ deposition | [46] |
| Murine bone marrow MSCs | Ultracentrifugation | Alzheimer’s disease | 5 × 1011 MSC-sEVs, intravenous injection, monthly for 4 months | Lessened plaque deposition, restored inflammatory cytokine levels | [47] |
| Human bone marrow MSCs | Ultracentrifugation | Alzheimer’s disease | 100 μg MSC-sEVs, intracerebroventricular injection, once every 2 days for 2 weeks | Reduced iNOS expression, relieved synaptic impairment, and long-term potentiation | [48] |
| Mouse bone marrow-derived MSCs | ExoQuick | Alzheimer’s disease | 150 μg MSC-sEVs, intravenous injection, biweekly for 4 months | Recovered learning and memory capabilities and synaptic dysfunction | [49] |
| Mouse bone marrow MSCs | Ultracentrifugation | Alzheimer’s disease | 22.4 μg MSC-sEVs, single intracerebral injection | Ameliorated Aβ burden and dystrophic neurites | [50] |
| Human umbilical cord MSCs | Ultracentrifugation | Alzheimer’s disease | 2 mg/mL intracerebroventricular injection | Reduced Aβ generation and oxidative stress, prevented microglia activity | [51] |
| Human umbilical cord MSCs | Centrifugation | Alzheimer’s disease | 5 × 105 MSC-sEVs, intravenous injection, at weeks 2 and 3 | Improved neurogenesis and neuroinflammation properties | [51] |
| Source Cell | sEV Purification | Disease | Therapeutic Schedule | Result | Reference |
|---|---|---|---|---|---|
| Murine bone marrow-derived DCs | Ultracentrifugation | Mastocytoma, mammary adenocarcinoma | 3–5 μg sEVs, single intradermal injection | Suppressed tumor growth, primed tumor-specific cytotoxic T cells | [70] |
| NK-92MI human NK cells | Ultracentrifugation | Melanoma | 20 μg sEVs, intertumoral injection for two days | Suppressed tumor growth | [64] |
| Peripheral blood mononuclear cell-derived T cells | Ultracentrifugation | Triple-negative breast cancer | 240 μg sEVs, intraperitoneal injection, every 3 days for 27 days | Decreased tumor cell-induced T cell dysfunction | [71] |
| NK cells | Ultracentrifugation | SK-N-SH neuroblastoma | Treated with sEVs, intraperitoneal injection, repeated three times with a 7-day interval until death | Increased survival time | [65] |
| NK cells | Ultracentrifugation | Anti-tumor effect | 100 µg sEV injection | Suppression of tumor growth | [72] |
| CAR-T cells | Ultracentrifugation | MDA-MB-231, HCC827, SK-BR-3 | 25–125 µg sEV injection, every week for 40 days | Suppression of dose-dependent tumor growth | [69] |
| CAR-T cells | Ultracentrifugation | Breast cancer | 100–500 μg sEVs, intravenous injection on days 3, 6, 9, 12, and 15 | Inhibited tumor growth, low toxicity | [73] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, I.; Choi, Y.; Shin, D.-U.; Kwon, M.; Kim, S.; Jung, H.; Nam, G.-H.; Kwon, M. Small Extracellular Vesicles as a New Class of Medicines. Pharmaceutics 2023, 15, 325. https://doi.org/10.3390/pharmaceutics15020325
Lee I, Choi Y, Shin D-U, Kwon M, Kim S, Jung H, Nam G-H, Kwon M. Small Extracellular Vesicles as a New Class of Medicines. Pharmaceutics. 2023; 15(2):325. https://doi.org/10.3390/pharmaceutics15020325
Chicago/Turabian StyleLee, Inkyu, Yoonjeong Choi, Dong-U Shin, Minjeong Kwon, Seohyun Kim, Hanul Jung, Gi-Hoon Nam, and Minsu Kwon. 2023. "Small Extracellular Vesicles as a New Class of Medicines" Pharmaceutics 15, no. 2: 325. https://doi.org/10.3390/pharmaceutics15020325