
SARS-CoV-2 Fusion Peptide Conjugated to a Tetravalent Dendrimer Selectively Inhibits Viral Infection
 , , , , , , , , and
, , , , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Peptide and Branched Amino Acid Core Synthesis and Characterization
2.3. Conjugation Reaction, Purification, and Characterization of the Peptide Dendrimer
2.4. Cells and Viruses
2.5. Cell Viability Assay
2.6. Antiviral Assays: HCoV-229E and SARS-CoV-2
2.6.1. Co-Treatment Assay
2.6.2. Cell Pre-Treatment Assay
2.6.3. Virucidal Assay
2.6.4. Post-Treatment Assay
2.7. Antiviral Assays: HCoV-OC43
2.8. Temperature-Shift Assays: SARS-CoV-2
2.9. Yield Reduction Assay
2.10. Statistical Analysis
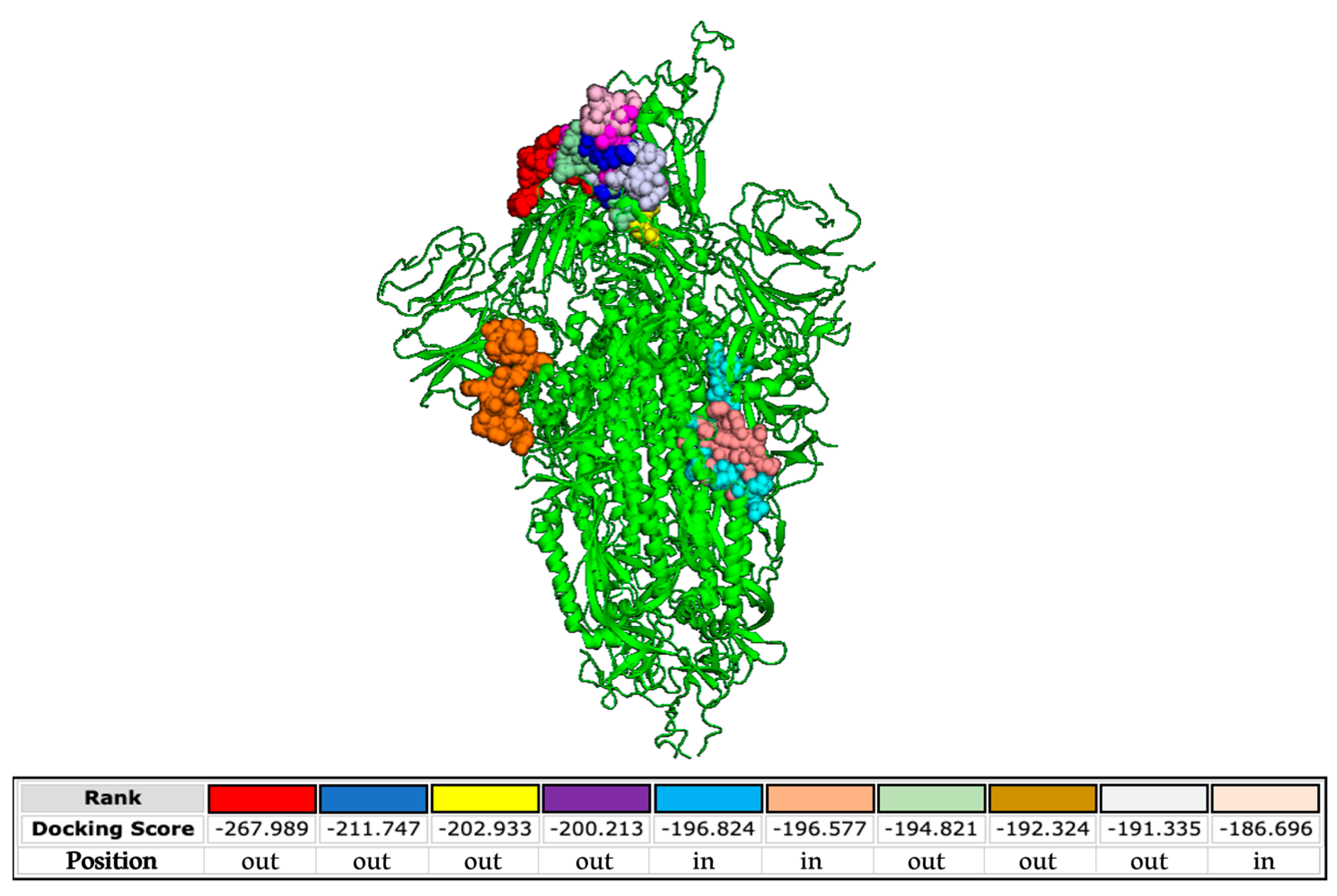
2.11. Molecular Docking
3. Results
3.1. Design and Preparation of Dendrimer R1
3.2. Cytotoxicity Analysis
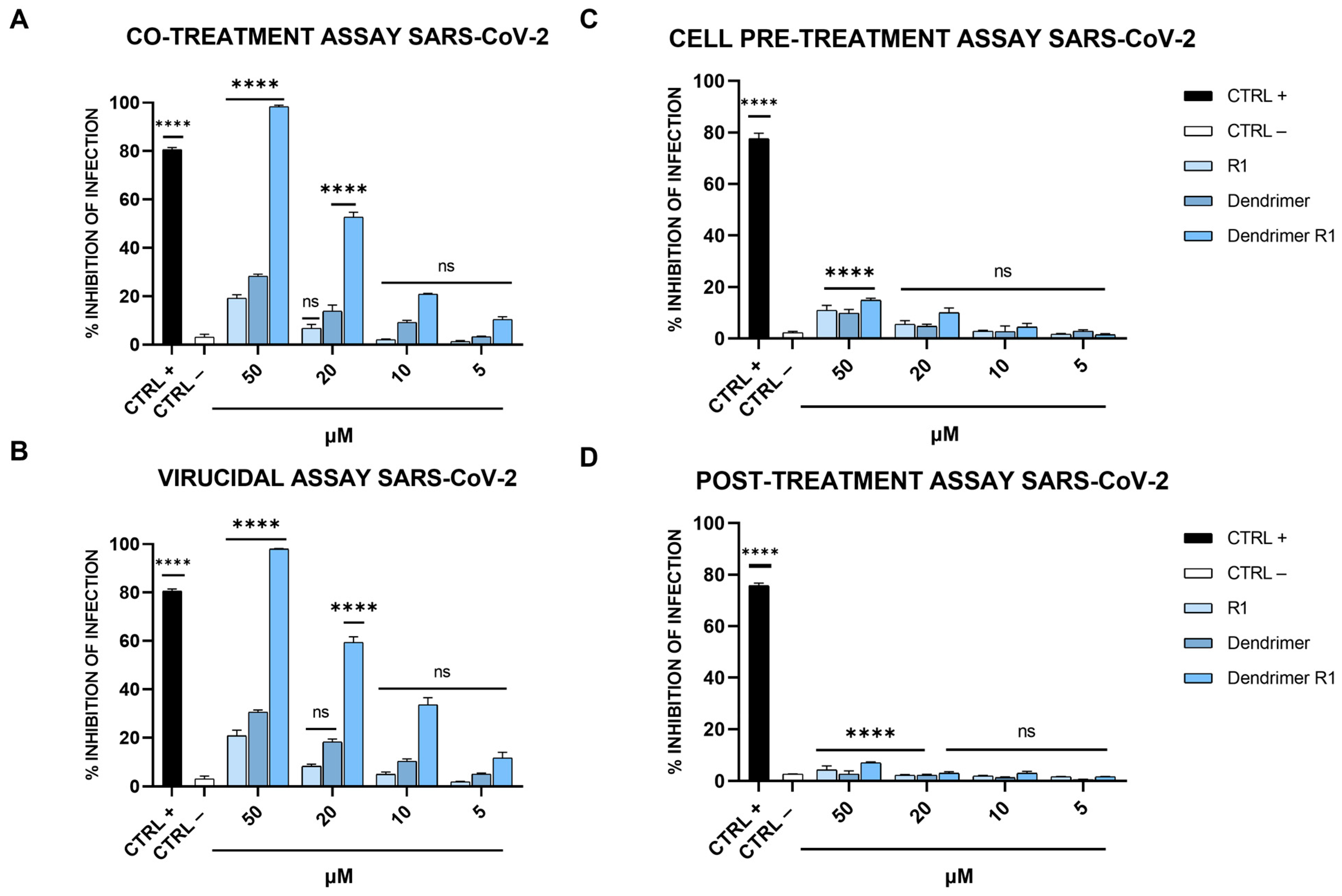
3.3. Antiviral Activity against Vero-76 Cells
3.4. Antiviral Activity on Calu-3 Cells
3.5. In Silico Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aboul-Fotouh, S.; Mahmoud, A.N.; Elnahas, E.M.; Habib, M.Z.; Abdelraouf, S.M. What are the current anti-COVID-19 drugs? From traditional to smart molecular mechanisms. Virol. J. 2023, 20, 241. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Recommends Highly Successful COVID-19 Therapy and Calls for Wide Geographical Distribution and Transparency from Originator. Available online: https://www.who.int/news/item/22-04-2022-who-recommends-highly-successful-covid-19-therapy-and-calls-for-wide-geographical-distribution-and-transparency-from-originator (accessed on 4 December 2023).
- FDA. FDA Approves First Oral Antiviral for Treatment of COVID-19 in Adults. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-oral-antiviral-treatment-covid-19-adults (accessed on 4 December 2023).
- FDA. Fact Sheet for Healthcare Providers: Emergency Use Authorization for Lagevrio™ (Molnupiravir) Capsules. Available online: https://www.fda.gov/media/155054/download (accessed on 4 December 2023).
- Hedskog, C.; Rodriguez, L.; Roychoudhury, P.; Huang, M.L.; Jerome, K.R.; Hao, L.; Ireton, R.C.; Li, J.; Perry, J.K.; Han, D.; et al. Viral Resistance Analyses from the Remdesivir Phase 3 Adaptive COVID-19 Treatment Trial-1 (ACTT-1). J. Infect. Dis. 2023, 228, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Parums, D.V. Rebound COVID-19 and Cessation of Antiviral Treatment for SARS-CoV-2 with Paxlovid and Molnupiravir. Med. Sci. Monit. 2022, 28, e938532. [Google Scholar] [CrossRef] [PubMed]
- Satsangi, S.; Gupta, N.; Kodan, P. Current and New Drugs for COVID-19 Treatment and Its Effects on the Liver. J. Clin. Transl. Hepatol. 2021, 9, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Singla, K.; Kumar, S.; Behl, A.; Puri, G.D. Remdesivir induced bradycardia and QT prolongation: A rare side effect of a ubiquitous drug of the COVID-19 era. J. Anaesthesiol. Clin. Pharmacol. 2022, 38 (Suppl. 1), S148–S149. [Google Scholar] [CrossRef] [PubMed]
- Al-Azzam, S.; Ding, Y.; Liu, J.; Pandya, P.; Ting, J.P.; Afshar, S. Peptides to combat viral infectious diseases. Peptides 2020, 134, 170402. [Google Scholar] [CrossRef]
- Chianese, A.; Zannella, C.; Monti, A.; Doti, N.; Sanna, G.; Manzin, A.; De Filippis, A.; Galdiero, M. Hylin-a1: A Pan-Inhibitor against Emerging and Re-Emerging Respiratory Viruses. Int. J. Mol. Sci. 2023, 24, 13888. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Bojarska, J.; Chai, T.T.; Elnagdy, S.; Kaczmarek, K.; Matsoukas, J.; New, R.; Parang, K.; Lopez, O.P.; Parhiz, H.; et al. A Global Review on Short Peptides: Frontiers and Perspectives. Molecules 2021, 26, 430. [Google Scholar] [CrossRef]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Chianese, A.; Zannella, C.; Monti, A.; De Filippis, A.; Doti, N.; Franci, G.; Galdiero, M. The Broad-Spectrum Antiviral Potential of the Amphibian Peptide AR-23. Int. J. Mol. Sci. 2022, 23, 883. [Google Scholar] [CrossRef]
- Zannella, C.; Chianese, A.; Palomba, L.; Marcocci, M.E.; Bellavita, R.; Merlino, F.; Grieco, P.; Folliero, V.; De Filippis, A.; Mangoni, M.; et al. Broad-Spectrum Antiviral Activity of the Amphibian Antimicrobial Peptide Temporin L and Its Analogs. Int. J. Mol. Sci. 2022, 23, 2060. [Google Scholar] [CrossRef] [PubMed]
- Nigro, E.; Colavita, I.; Sarnataro, D.; Scudiero, O.; Zambrano, G.; Granata, V.; Daniele, A.; Carotenuto, A.; Galdiero, S.; Folliero, V.; et al. An ancestral host defence peptide within human beta-defensin 3 recapitulates the antibacterial and antiviral activity of the full-length molecule. Sci. Rep. 2015, 5, 18450. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, L.; Stellato, M.I.; Oliva, R.; Falanga, A.; Galdiero, M.; Petraccone, L.; D’Errico, G.; De Santis, A.; Galdiero, S.; Del Vecchio, P. Antimicrobial peptides at work: Interaction of myxinidin and its mutant WMR with lipid bilayers mimicking the P. aeruginosa and E. coli membranes. Sci. Rep. 2017, 7, 44425. [Google Scholar] [CrossRef] [PubMed]
- Marcocci, M.E.; Amatore, D.; Villa, S.; Casciaro, B.; Aimola, P.; Franci, G.; Grieco, P.; Galdiero, M.; Palamara, A.T.; Mangoni, M.L.; et al. The Amphibian Antimicrobial Peptide Temporin B Inhibits In Vitro Herpes Simplex Virus 1 Infection. Antimicrob. Agents Chemother. 2018, 62, e02367-17. [Google Scholar] [CrossRef] [PubMed]
- Chianese, A.; Iovane, V.; Zannella, C.; Capasso, C.; Nastri, B.M.; Monti, A.; Doti, N.; Montagnaro, S.; Pagnini, U.; Iovane, G.; et al. Synthetic Frog-Derived-like Peptides: A New Weapon against Emerging and Potential Zoonotic Viruses. Viruses 2023, 15, 1804. [Google Scholar] [CrossRef] [PubMed]
- Chianese, A.; Zannella, C.; Palma, F.; Di Clemente, L.; Monti, A.; Doti, N.; De Filippis, A.; Galdiero, M. Melittin-Related Peptides Interfere with Sandfly Fever Naples Virus Infection by Interacting with Heparan Sulphate. Microorganisms 2023, 11, 2446. [Google Scholar] [CrossRef] [PubMed]
- Zannella, C.; Chianese, A.; Greco, G.; Santella, B.; Squillaci, G.; Monti, A.; Doti, N.; Sanna, G.; Manzin, A.; Morana, A.; et al. Design of Three Residues Peptides against SARS-CoV-2 Infection. Viruses 2022, 14, 2103. [Google Scholar] [CrossRef]
- Doti, N.; Mardirossian, M.; Sandomenico, A.; Ruvo, M.; Caporale, A. Recent Applications of Retro-Inverso Peptides. Int. J. Mol. Sci. 2021, 22, 8677. [Google Scholar] [CrossRef]
- Mali, A.; Franci, G.; Zannella, C.; Chianese, A.; Anthiya, S.; Lopez-Estevez, A.M.; Monti, A.; De Filippis, A.; Doti, N.; Alonso, M.J.; et al. Antiviral Peptides Delivered by Chitosan-Based Nanoparticles to Neutralize SARS-CoV-2 and HCoV-OC43. Pharmaceutics 2023, 15, 1621. [Google Scholar] [CrossRef]
- Chis, A.A.; Dobrea, C.; Morgovan, C.; Arseniu, A.M.; Rus, L.L.; Butuca, A.; Juncan, A.M.; Totan, M.; Vonica-Tincu, A.L.; Cormos, G.; et al. Applications and Limitations of Dendrimers in Biomedicine. Molecules 2020, 25, 3982. [Google Scholar] [CrossRef]
- Folliero, V.; Zannella, C.; Chianese, A.; Stelitano, D.; Ambrosino, A.; De Filippis, A.; Galdiero, M.; Franci, G.; Galdiero, M. Application of Dendrimers for Treating Parasitic Diseases. Pharmaceutics 2021, 13, 343. [Google Scholar] [CrossRef] [PubMed]
- Falanga, A.; Del Genio, V.; Kaufman, E.A.; Zannella, C.; Franci, G.; Weck, M.; Galdiero, S. Engineering of Janus-Like Dendrimers with Peptides Derived from Glycoproteins of Herpes Simplex Virus Type 1: Toward a Versatile and Novel Antiviral Platform. Int. J. Mol. Sci. 2021, 22, 6488. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Ferreiro, M.; Abelairas, A.; Criado, A.; Gómez, I.J.; Mosquera, J. Dendrimers: Exploring Their Wide Structural Variety and Applications. Polymers 2023, 15, 4369. [Google Scholar] [CrossRef] [PubMed]
- Mirakabad, F.S.T.; Khoramgah, M.S.; Keshavarz, F.K.; Tabarzad, M.; Ranjbari, J. Peptide dendrimers as valuable biomaterials in medical sciences. Life Sci. 2019, 233, 116754. [Google Scholar] [CrossRef]
- Scorciapino, M.A.; Pirri, G.; Vargiu, A.V.; Ruggerone, P.; Giuliani, A.; Casu, M.; Buerck, J.; Wadhwani, P.; Ulrich, A.S.; Rinaldi, A.C. A novel dendrimeric peptide with antimicrobial properties: Structure-function analysis of SB056. Biophys. J. 2012, 102, 1039–1048. [Google Scholar] [CrossRef]
- Cardoso, P.; Glossop, H.; Meikle, T.G.; Aburto-Medina, A.; Conn, C.E.; Sarojini, V.; Valery, C. Molecular engineering of antimicrobial peptides: Microbial targets, peptide motifs and translation opportunities. Biophys. Rev. 2021, 13, 35–69. [Google Scholar] [CrossRef]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef]
- Scorciapino, M.A.; Serra, I.; Manzo, G.; Rinaldi, A.C. Antimicrobial Dendrimeric Peptides: Structure, Activity and New Therapeutic Applications. Int. J. Mol. Sci. 2017, 18, 542. [Google Scholar] [CrossRef]
- Jeong, W.J.; Bu, J.; Mickel, P.; Han, Y.; Rawding, P.A.; Wang, J.; Kang, H.; Hong, H.; Kral, P.; Hong, S. Dendrimer-Peptide Conjugates for Effective Blockade of the Interactions between SARS-CoV-2 Spike Protein and Human ACE2 Receptor. Biomacromolecules 2023, 24, 141–149. [Google Scholar] [CrossRef]
- Joshi, V.G.; Dighe, V.D.; Thakuria, D.; Malik, Y.S.; Kumar, S. Multiple antigenic peptide (MAP): A synthetic peptide dendrimer for diagnostic, antiviral and vaccine strategies for emerging and re-emerging viral diseases. Indian. J. Virol. 2013, 24, 312–320. [Google Scholar] [CrossRef]
- Francis, M.J.; Hastings, G.Z.; Brown, F.; McDermed, J.; Lu, Y.A.; Tam, J.P. Immunological evaluation of the multiple antigen peptide (MAP) system using the major immunogenic site of foot-and-mouth disease virus. Immunology 1991, 73, 249–254. [Google Scholar] [PubMed]
- Donalisio, M.; Rusnati, M.; Civra, A.; Bugatti, A.; Allemand, D.; Pirri, G.; Giuliani, A.; Landolfo, S.; Lembo, D. Identification of a dendrimeric heparan sulfate-binding peptide that inhibits infectivity of genital types of human papillomaviruses. Antimicrob. Agents Chemother. 2010, 54, 4290–4299. [Google Scholar] [CrossRef] [PubMed]
- Luganini, A.; Giuliani, A.; Pirri, G.; Pizzuto, L.; Landolfo, S.; Gribaudo, G. Peptide-derivatized dendrimers inhibit human cytomegalovirus infection by blocking virus binding to cell surface heparan sulfate. Antiviral Res. 2010, 85, 532–540. [Google Scholar] [CrossRef]
- Luganini, A.; Nicoletto, S.F.; Pizzuto, L.; Pirri, G.; Giuliani, A.; Landolfo, S.; Gribaudo, G. Inhibition of herpes simplex virus type 1 and type 2 infections by peptide-derivatized dendrimers. Antimicrob. Agents Chemother. 2011, 55, 3231–3239. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Yan, L.; Xu, W.; Agrawal, A.S.; Algaissi, A.; Tseng, C.K.; Wang, Q.; Du, L.; Tan, W.; Wilson, I.A.; et al. A pan-coronavirus fusion inhibitor targeting the HR1 domain of human coronavirus spike. Sci. Adv. 2019, 5, eaav4580. [Google Scholar] [CrossRef]
- Eggink, D.; Bontjer, I.; de Taeye, S.W.; Langedijk, J.P.M.; Berkhout, B.; Sanders, R.W. HIV-1 anchor inhibitors and membrane fusion inhibitors target distinct but overlapping steps in virus entry. J. Biol. Chem. 2019, 294, 5736–5746. [Google Scholar] [CrossRef]
- Liu, S.; Xiao, G.; Chen, Y.; He, Y.; Niu, J.; Escalante, C.R.; Xiong, H.; Farmar, J.; Debnath, A.K.; Tien, P.; et al. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: Implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet 2004, 363, 938–947. [Google Scholar] [CrossRef]
- Xing, L.; Xu, X.; Xu, W.; Liu, Z.; Shen, X.; Zhou, J.; Xu, L.; Pu, J.; Yang, C.; Huang, Y.; et al. A Five-Helix-Based SARS-CoV-2 Fusion Inhibitor Targeting Heptad Repeat 2 Domain against SARS-CoV-2 and Its Variants of Concern. Viruses 2022, 14, 597. [Google Scholar] [CrossRef]
- Watanabe, M.; Hashimoto, K.; Abe, Y.; Kodama, E.N.; Nabika, R.; Oishi, S.; Ohara, S.; Sato, M.; Kawasaki, Y.; Fujii, N.; et al. A Novel Peptide Derived from the Fusion Protein Heptad Repeat Inhibits Replication of Subacute Sclerosing Panencephalitis Virus In Vitro and In Vivo. PLoS ONE 2016, 11, e0162823. [Google Scholar] [CrossRef]
- Wang, W.; De Feo, C.J.; Zhuang, M.; Vassell, R.; Weiss, C.D. Selection with a peptide fusion inhibitor corresponding to the first heptad repeat of HIV-1 gp41 identifies two genetic pathways conferring cross-resistance to peptide fusion inhibitors corresponding to the first and second heptad repeats (HR1 and HR2) of gp41. J. Virol. 2011, 85, 12929–12938. [Google Scholar]
- Liu, I.J.; Kao, C.L.; Hsieh, S.C.; Wey, M.T.; Kan, L.S.; Wang, W.K. Identification of a minimal peptide derived from heptad repeat (HR) 2 of spike protein of SARS-CoV and combination of HR1-derived peptides as fusion inhibitors. Antivir. Res. 2009, 81, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Monsó, M.; Kowalczyk, W.; Andreu, D.; de la Torre, B.G. Reverse thioether ligation route to multimeric peptide antigens. Org. Biomol. Chem. 2012, 10, 3116–3121. [Google Scholar] [CrossRef] [PubMed]
- Acosta, G.A.; del Fresno, M.; Paradis-Bas, M.; Rigau-DeLlobet, M.; Côté, S.; Royo, M.; Albericio, F. Solid-phase peptide synthesis using acetonitrile as a solvent in combination with PEG-based resins. J. Pept. Sci. 2009, 15, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Caporale, A.; Doti, N.; Monti, A.; Sandomenico, A.; Ruvo, M. Automatic procedures for the synthesis of difficult peptides using oxyma as activating reagent: A comparative study on the use of bases and on different deprotection and agitation conditions. Peptides 2018, 102, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Tarallo, R.; Carberry, T.P.; Falanga, A.; Vitiello, M.; Galdiero, S.; Galdiero, M.; Weck, M. Dendrimers functionalized with membrane-interacting peptides for viral inhibition. Int. J. Nanomed. 2013, 8, 521–534. [Google Scholar]
- Franci, G.; Falanga, A.; Zannella, C.; Folliero, V.; Martora, F.; Galdiero, M.; Galdiero, S.; Morelli, G.; Galdiero, M. Infectivity inhibition by overlapping synthetic peptides derived from the gH/gL heterodimer of herpes simplex virus type 1. J. Pept. Sci. 2017, 23, 311–319. [Google Scholar] [CrossRef]
- Basile, A.; Zannella, C.; De Marco, M.; Sanna, G.; Franci, G.; Galdiero, M.; Manzin, A.; De Laurenzi, V.; Chetta, M.; Rosati, A.; et al. Spike-mediated viral membrane fusion is inhibited by a specific anti-IFITM2 monoclonal antibody. Antivir. Res. 2023, 211, 105546. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.F.; Xu, W.; Liu, S.W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Magazine, N.; Zhang, T.; Wu, Y.; McGee, M.C.; Veggiani, G.; Huang, W. Mutations and Evolution of the SARS-CoV-2 Spike Protein. Viruses 2022, 14, 640. [Google Scholar] [CrossRef]
- Ou, X.; Zheng, W.; Shan, Y.; Mu, Z.; Dominguez, S.R.; Holmes, K.V.; Qian, Z. Identification of the Fusion Peptide-Containing Region in Betacoronavirus Spike Glycoproteins. J. Virol. 2016, 90, 5586–5600. [Google Scholar] [CrossRef]
- Zhu, J.; Xiao, G.; Xu, Y.; Yuan, F.; Zheng, C.; Liu, Y.; Yan, H.; Cole, D.K.; Bell, J.I.; Rao, Z.; et al. Following the rule: Formation of the 6-helix bundle of the fusion core from severe acute respiratory syndrome coronavirus spike protein and identification of potent peptide inhibitors. Biochem. Biophys. Res. Commun. 2004, 319, 283–288. [Google Scholar] [CrossRef]
- Yuan, K.; Yi, L.; Chen, J.; Qu, X.; Qing, T.; Rao, X.; Jiang, P.; Hu, J.; Xiong, Z.; Nie, Y.; et al. Suppression of SARS-CoV entry by peptides corresponding to heptad regions on spike glycoprotein. Biochem. Biophys. Res. Commun. 2004, 319, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Outlaw, V.K.; Bovier, F.T.; Mears, M.C.; Cajimat, M.N.; Zhu, Y.; Lin, M.J.; Addetia, A.; Lieberman, N.A.P.; Peddu, V.; Xie, X.; et al. Inhibition of Coronavirus Entry In Vitro and Ex Vivo by a Lipid-Conjugated Peptide Derived from the SARS-CoV-2 Spike Glycoprotein HRC Domain. mBio 2020, 11, e01935-20. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Duzgunes, N.; Fernandez-Fuentes, N.; Konopka, K. Inhibition of Viral Membrane Fusion by Peptides and Approaches to Peptide Design. Pathogens 2021, 10, 1599. [Google Scholar] [CrossRef] [PubMed]
- Konopka, K.; Pretzer, E.; Duzgunes, N. Differential effects of a hydrophobic tripeptide on human immunodeficiency virus type 1 (HIV-1)-induced syncytium formation and viral infectivity. Biochem. Biophys. Res. Commun. 1995, 208, 75–81. [Google Scholar] [CrossRef]
- Owens, R.J.; Tanner, C.C.; Mulligan, M.J.; Srinivas, R.V.; Compans, R.W. Oligopeptide inhibitors of HIV-induced syncytium formation. AIDS Res. Hum. Retroviruses 1990, 6, 1289–1296. [Google Scholar] [CrossRef]
- Silburn, K.A.; McPhee, D.A.; Maerz, A.L.; Poumbourios, P.; Whittaker, R.G.; Kirkpatrick, A.; Reilly, W.G.; Manthey, M.K.; Curtain, C.C. Efficacy of fusion peptide homologs in blocking cell lysis and HIV-induced fusion. AIDS Res. Hum. Retroviruses 1998, 14, 385–392. [Google Scholar] [CrossRef]
- Jiang, X.; Jia, Q.; Lu, L.; Yu, F.; Zheng, J.; Shi, W.; Cai, L.; Jiang, S.; Liu, K. A novel bispecific peptide HIV-1 fusion inhibitor targeting the N-terminal heptad repeat and fusion peptide domains in gp41. Amino Acids 2016, 48, 2867–2873. [Google Scholar] [CrossRef]
- Ghosh, J.K.; Shai, Y. A peptide derived from a conserved domain of Sendai virus fusion protein inhibits virus-cell fusion. A plausible mode of action. J. Biol. Chem. 1998, 273, 7252–7259. [Google Scholar] [CrossRef]
- Wu, W.; Lin, D.; Shen, X.; Li, F.; Fang, Y.; Li, K.; Xun, T.; Yang, G.; Yang, J.; Liu, S.; et al. New influenza A virus Entry Inhibitors Derived from the Viral Fusion Peptides. PLoS ONE 2015, 10, e0138426. [Google Scholar] [CrossRef] [PubMed]
- Hatano, K.; Matsubara, T.; Muramatsu, Y.; Ezure, M.; Koyama, T.; Matsuoka, K.; Kuriyama, R.; Kori, H.; Sato, T. Synthesis and influenza virus inhibitory activities of carbosilane dendrimers peripherally functionalized with hemagglutinin-binding Peptide. J. Med. Chem. 2014, 57, 8332–8339. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; He, J. Multivalent peptide dendrimers inhibit the fusion of viral-cellular membranes and the cellular NF-kappaB signaling pathway. Eur. J. Med. Chem. 2022, 230, 114140. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zannella, C.; Chianese, A.; Monti, A.; Giugliano, R.; Morone, M.V.; Secci, F.; Sanna, G.; Manzin, A.; De Filippis, A.; Doti, N.; et al. SARS-CoV-2 Fusion Peptide Conjugated to a Tetravalent Dendrimer Selectively Inhibits Viral Infection. Pharmaceutics 2023, 15, 2791. https://doi.org/10.3390/pharmaceutics15122791
Zannella C, Chianese A, Monti A, Giugliano R, Morone MV, Secci F, Sanna G, Manzin A, De Filippis A, Doti N, et al. SARS-CoV-2 Fusion Peptide Conjugated to a Tetravalent Dendrimer Selectively Inhibits Viral Infection. Pharmaceutics. 2023; 15(12):2791. https://doi.org/10.3390/pharmaceutics15122791
Chicago/Turabian StyleZannella, Carla, Annalisa Chianese, Alessandra Monti, Rosa Giugliano, Maria Vittoria Morone, Francesco Secci, Giuseppina Sanna, Aldo Manzin, Anna De Filippis, Nunzianna Doti, and et al. 2023. "SARS-CoV-2 Fusion Peptide Conjugated to a Tetravalent Dendrimer Selectively Inhibits Viral Infection" Pharmaceutics 15, no. 12: 2791. https://doi.org/10.3390/pharmaceutics15122791