
Self-Penetrating Oligonucleotide Derivatives: Features of Self-Assembly and Interactions with Serum and Intracellular Proteins
 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Buffer Composition
2.3. Oligonucleotide Synthesis
2.4. Duplex Thermal Denaturation Experiments
2.5. Critical Aggregation Concentration (CAC) Determination by Nile Red Encapsulation Assay
2.6. Characterization of Self-Assembled Micellar Structures by Dynamic Light Scattering (DLS)
2.7. Electrophoretic Mobility Shift Assay (EMSA)
2.8. Determination of Kd by Fluorescence Spectroscopy
2.9. 5′-dRp-DNA Obtaining
2.10. Affinity Modification of Ku Antigen, PARP1, and DNA Polymerase β by 5′-dRp DNAs
2.11. Cell Lines
2.12. Cellular Accumulation Assay
3. Results and Discussion
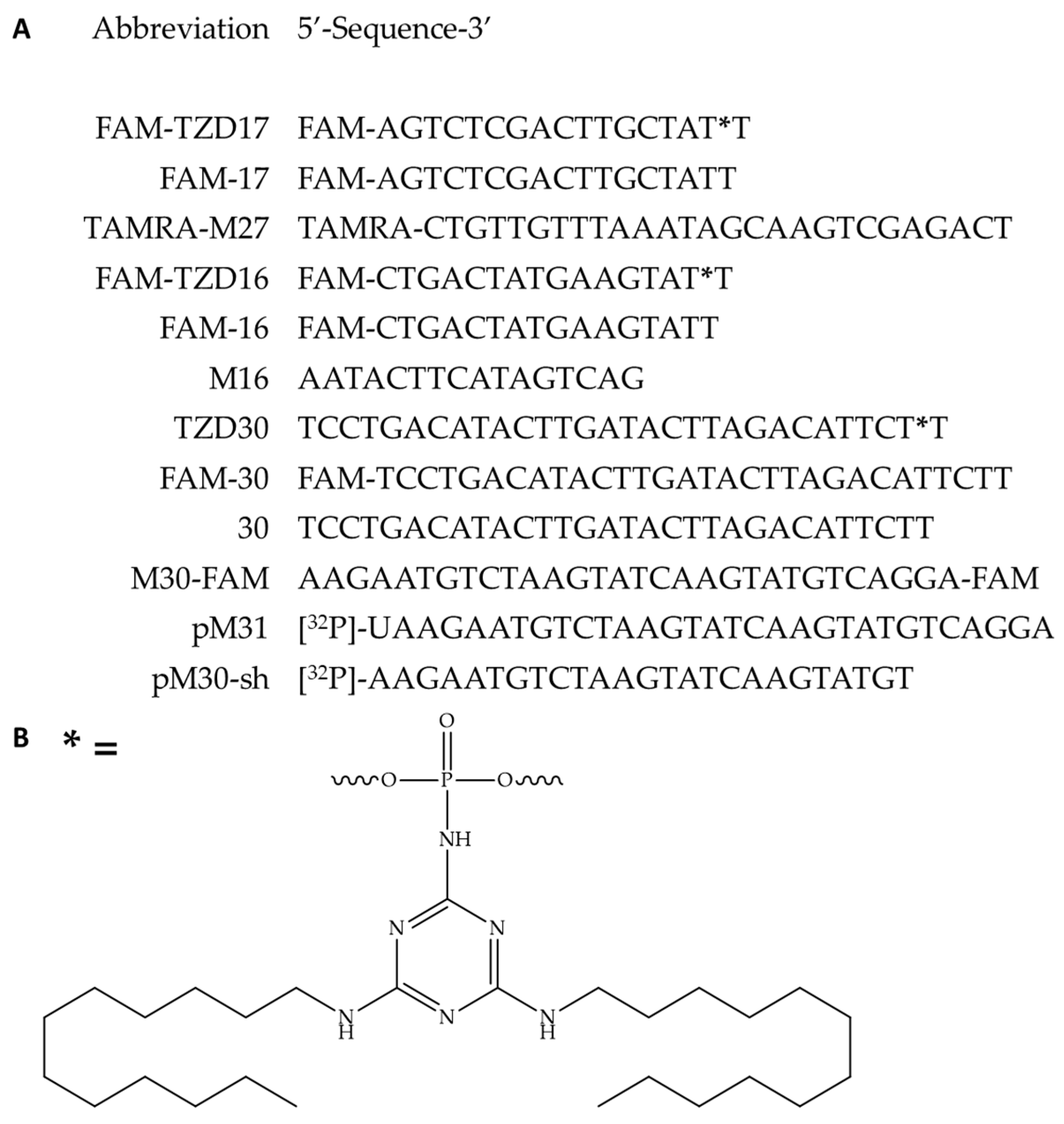
3.1. Oligonucleotides and Their Derivatives Used in This Study
3.2. Hybridization Properties of Modified Oligonucleotides
3.3. CAC Determination by Nile Red Encapsulation Assay
3.4. The Size Assessment of Oligonucleotide Micelle-like Structures and Their Complexes with HSA by DLS
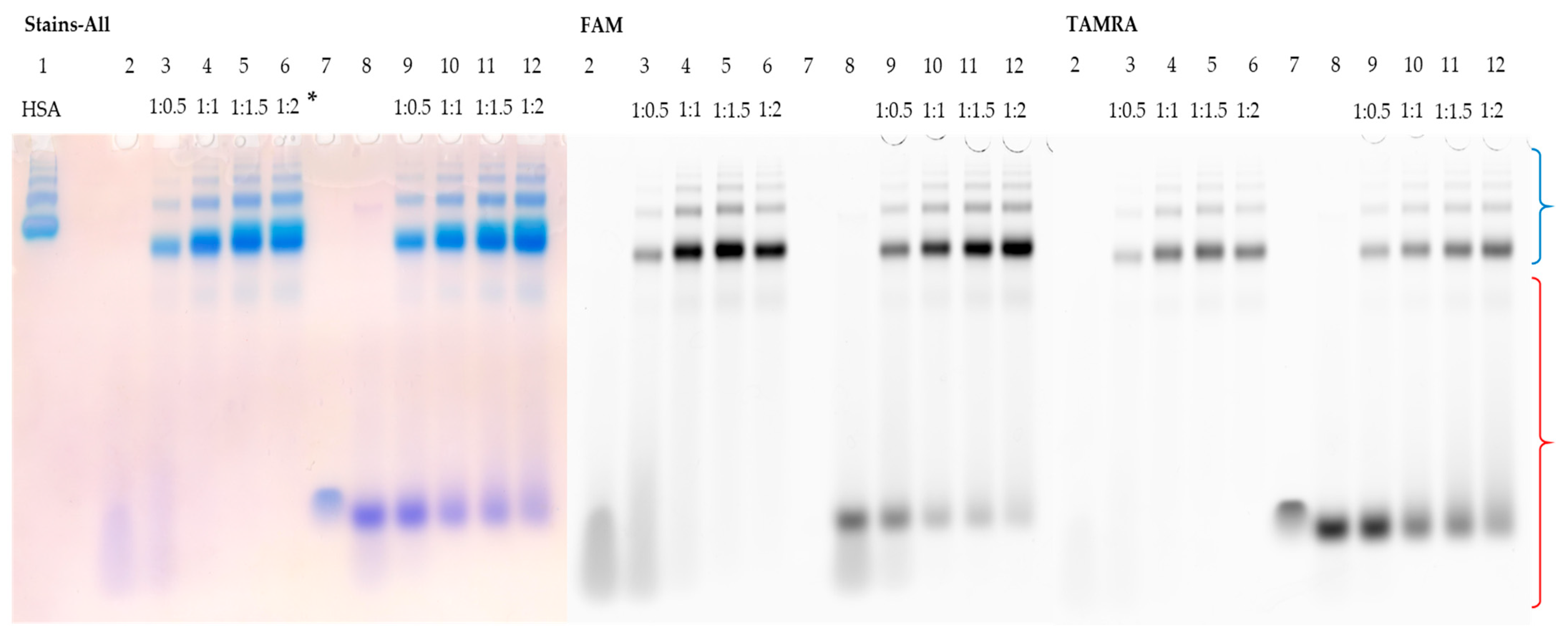
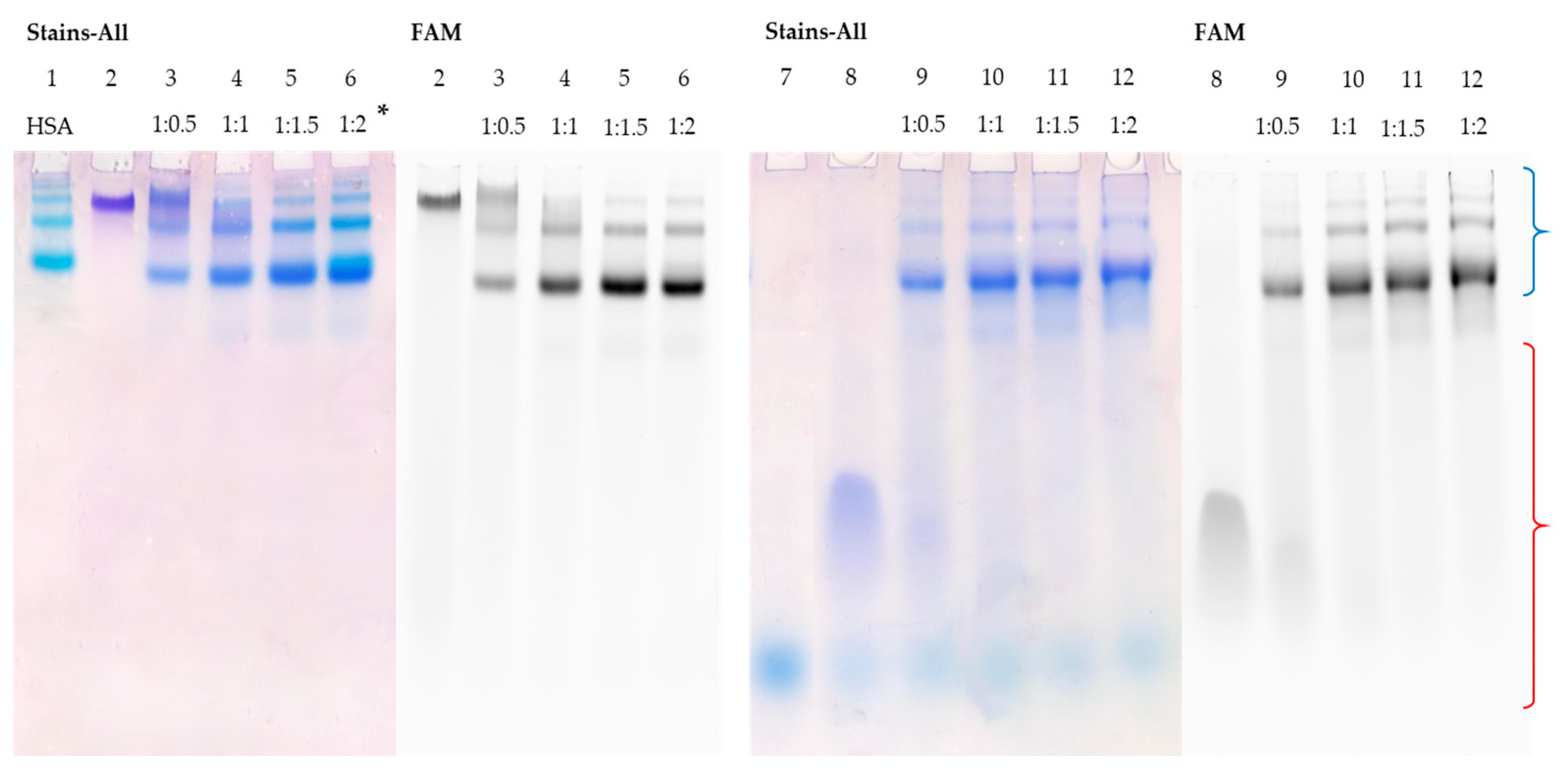
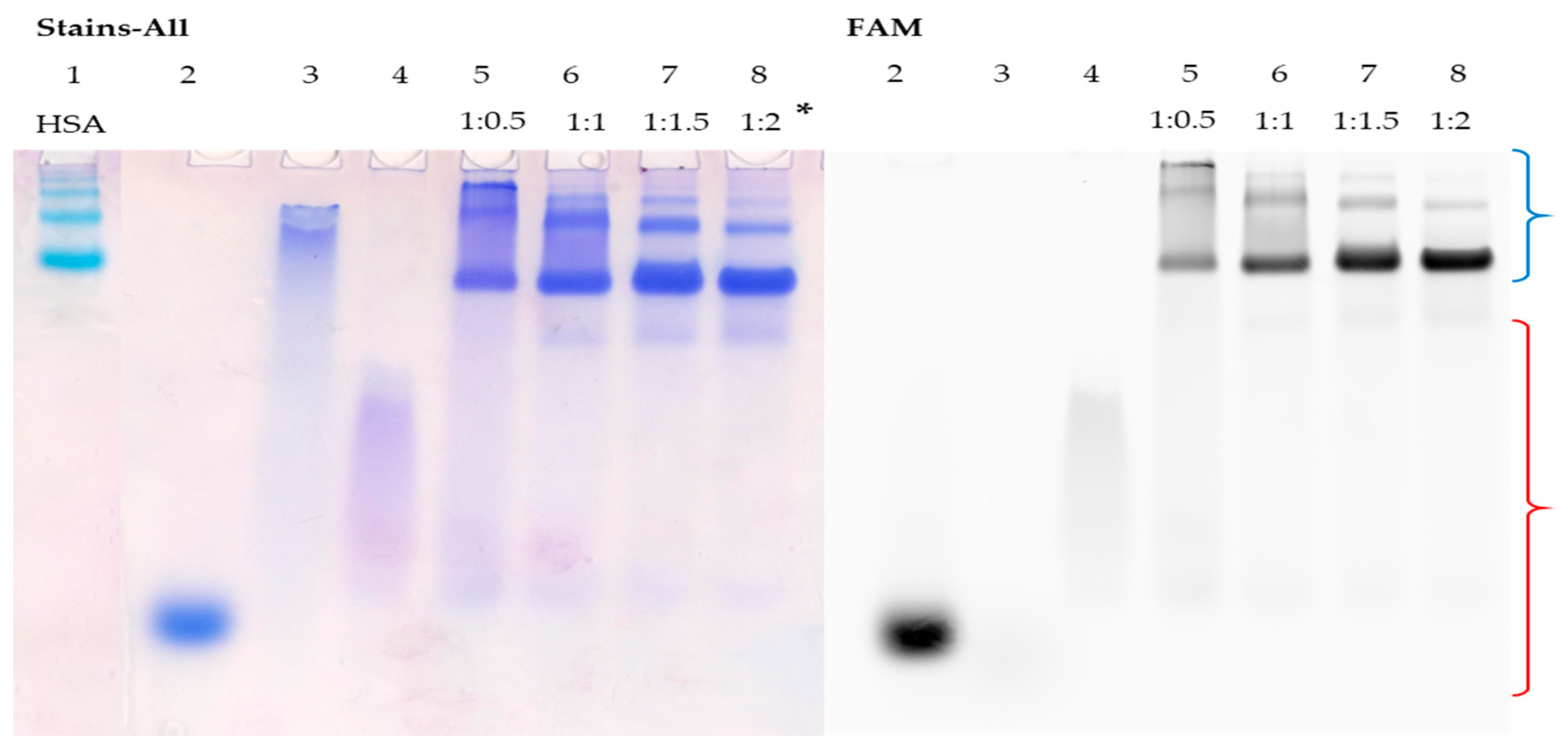
3.5. Detection of HSA–Oligonucleotide Complexes by EMSA

3.6. Kd Determination of HSA–Oligonucleotide Complexes by Fluorescence Titration Experiment
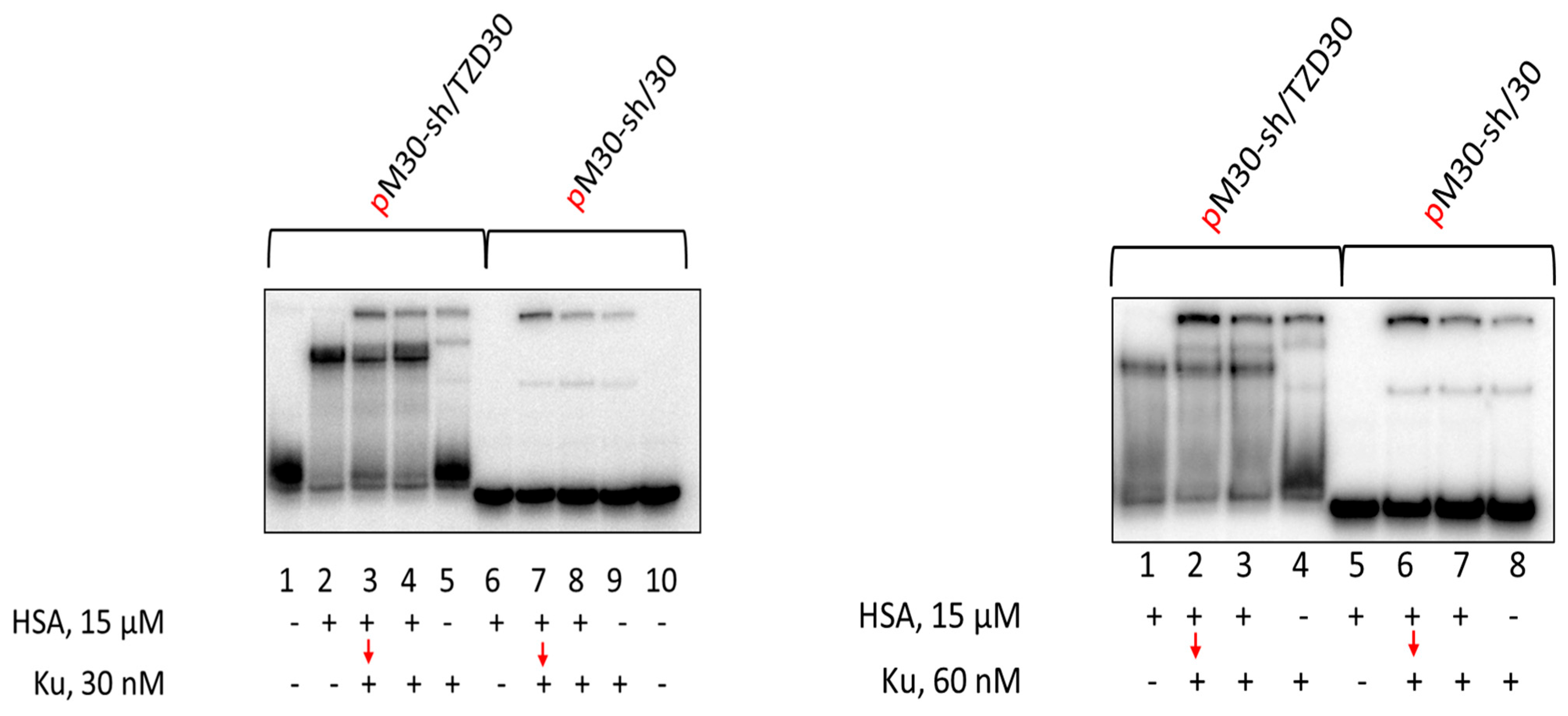
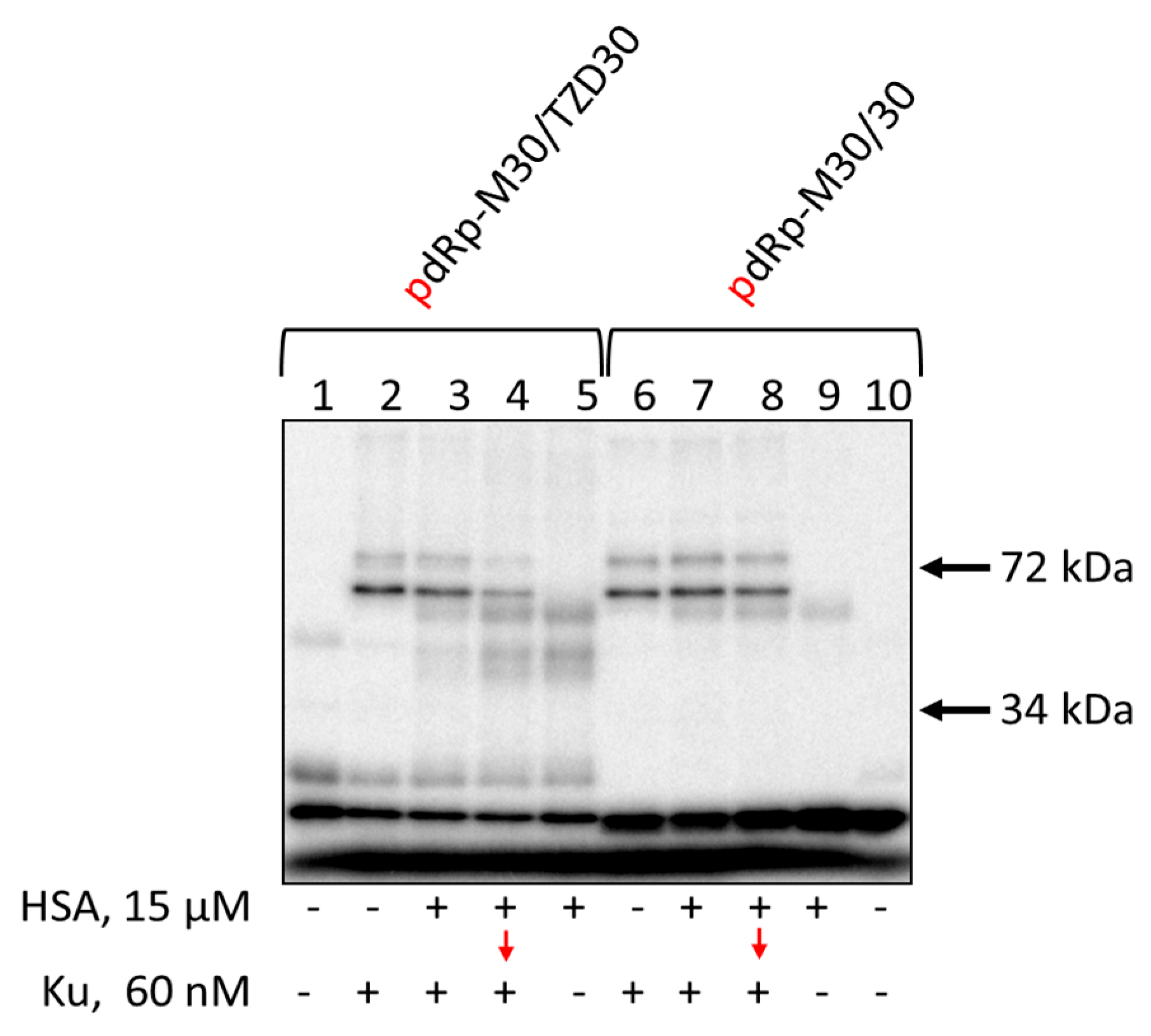
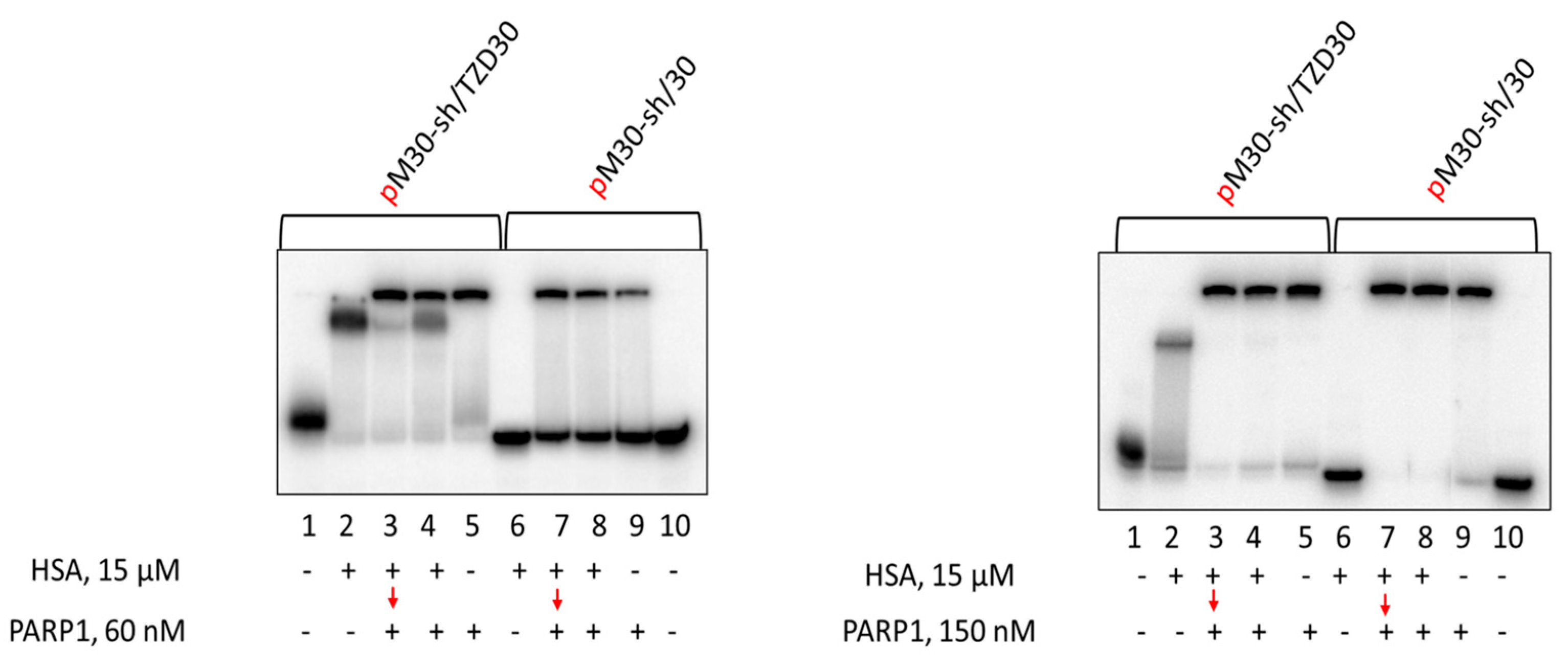
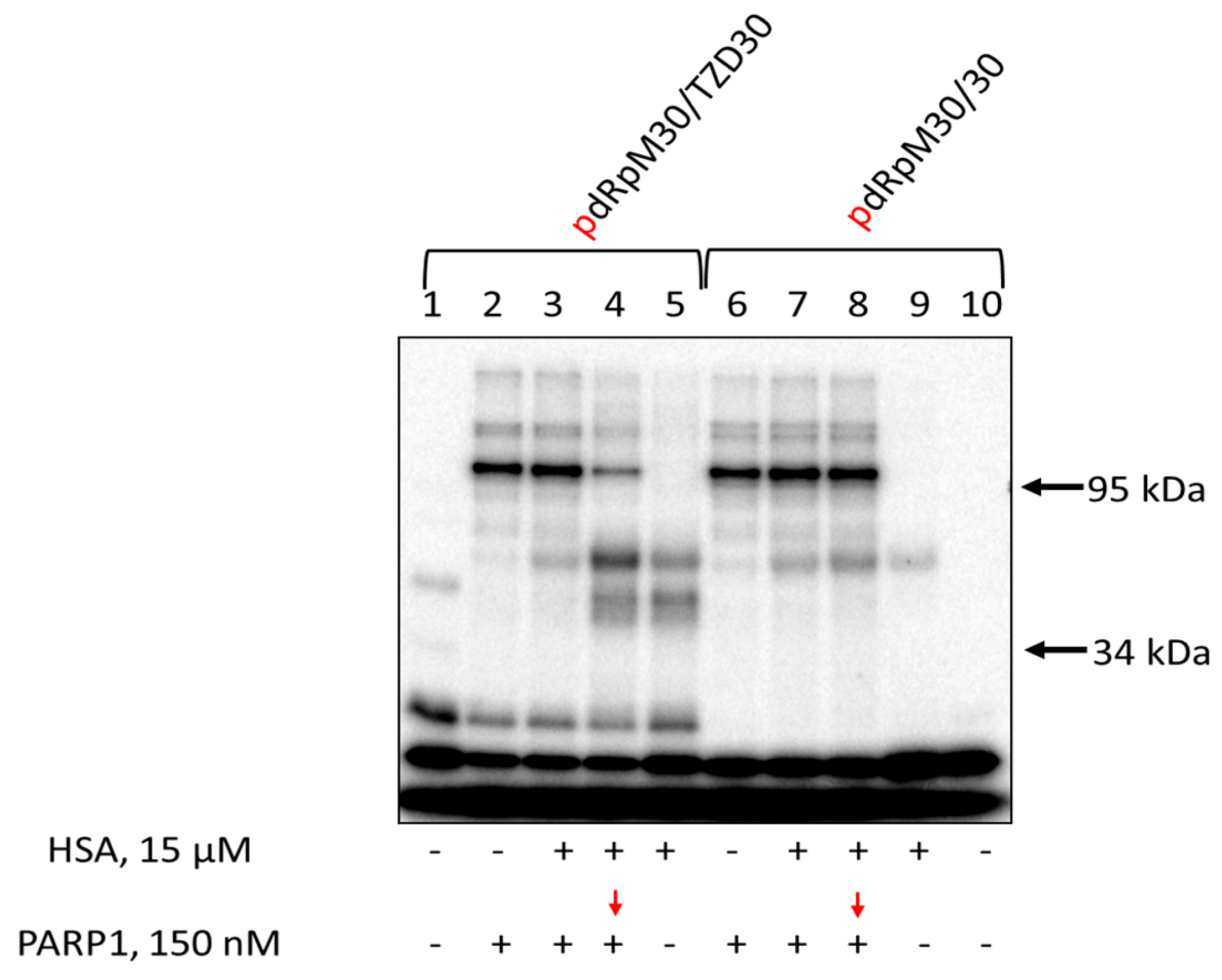
3.7. Affinity Modification of Ku Antigen, PARP1 and DNA Polymerase β by [32P]5′-dRp-DNAs
3.8. Studying the Mutual Influence of PARP1 and HSA or Ku Antigen and HSA upon Interaction with DNA Duplexes by EMSA
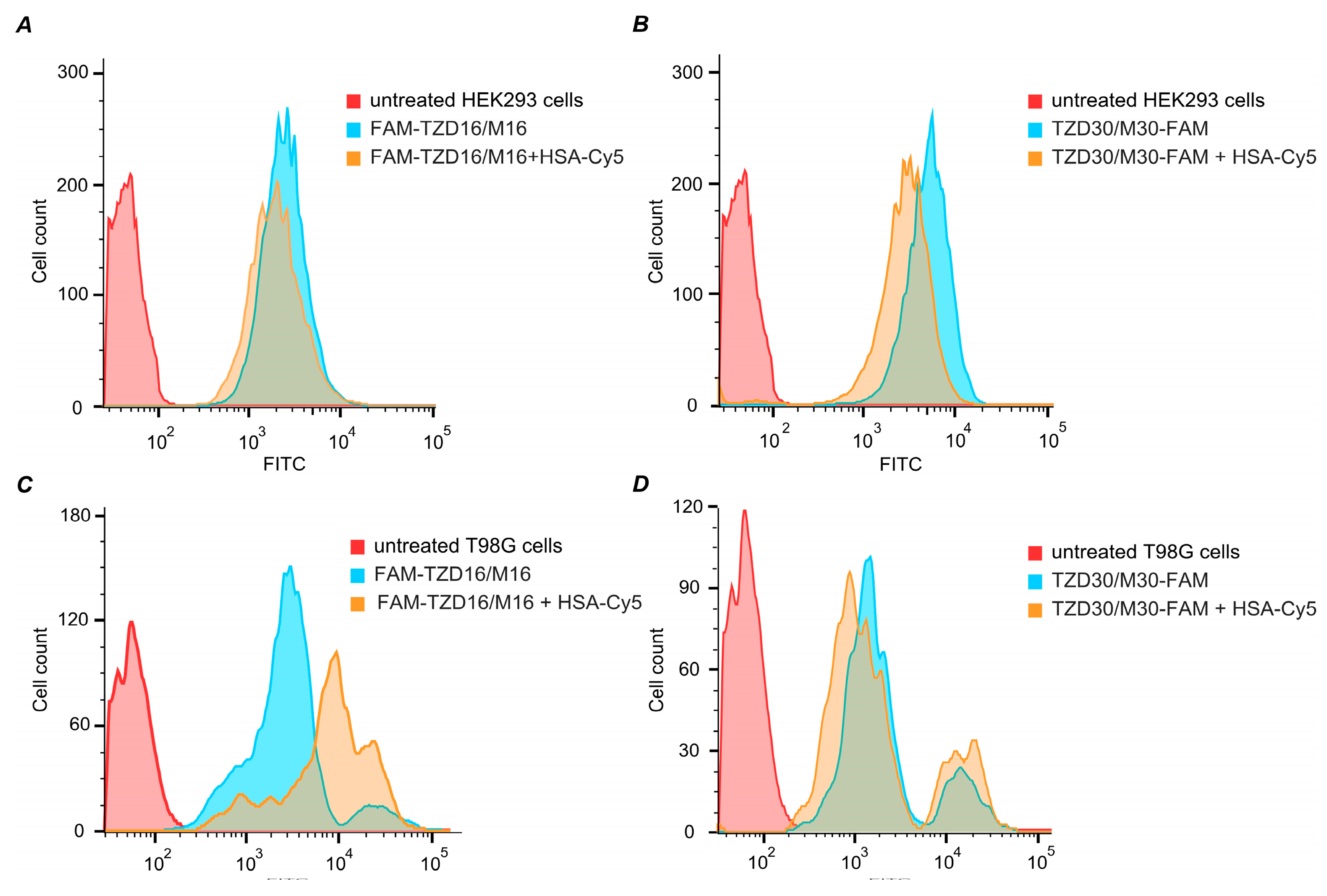
3.9. Intracellular Accumulation of DNA Duplexes Bound within Complexes with HSA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smith, C.I.E.; Zain, R. Therapeutic Oligonucleotides: State of the Art. Annu. Rev. Pharmacol. Toxicol. 2018, 14, 605–630. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Witzigmann, D.; Thomson, S.B.; Chen, S.; Leavitt, B.R.; Cullis, P.R.; van der Meel, R. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 2021, 16, 630–643. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, C.; Wood, M.J.A. Antisense oligonucleotides: The next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 2017, 14, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Juskowiak, B. Nucleic acid-based fluorescent probes and their analytical potential. Anal. Bioanal. Chem. 2010, 399, 3157–3176. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Pumm, A.; Maffeo, C.; Kohler, F.; Feigl, E.; Zhao, W.; Verschueren, D.; Golestanian, R.; Aksimentiev, A.; Dietz, H.; et al. A DNA turbine powered by a transmembrane potential across a nanopore. Nat. Nanotechnol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Bao, C.J.; Duan, J.L.; Xie, Y.; Feng, X.P.; Cui, W.; Chen, S.Y.; Li, P.S.; Liu, Y.X.; Wang, J.L.; Wang, G.L.; et al. Bioorthogonal Engineered Virus-Like Nanoparticles for Efficient Gene Therapy. Nano-Micro Lett. 2023, 15, 197. [Google Scholar] [CrossRef] [PubMed]
- Mejía-Méndez, J.L.; Vazquez-Duhalt, R.; Hernández, L.R.; Sánchez-Arreola, E.; Bach, H. Virus-like Particles: Fundamentals and Biomedical Applications. Int. J. Mol. Sci. 2022, 23, 8579. [Google Scholar] [CrossRef] [PubMed]
- Jahns, H.; Degaonkar, R.; Podbevsek, P.; Gupta, S.; Bisbe, A.; Aluri, K.; Szeto, J.; Kumar, P.; LeBlanc, S.; Racie, T.; et al. Small circular interfering RNAs (sciRNAs) as a potent therapeutic platform for gene-silencing. Nucleic Acids Res. 2021, 49, 10250–10264. [Google Scholar] [CrossRef]
- Chubarov, A.S.; Oscorbin, I.P.; Novikova, L.M.; Filipenko, M.L.; Lomzov, A.A.; Pyshnyi, D.V. Allele-Specific PCR for PIK3CA Mutation Detection Using Phosphoryl Guanidine Modified Primers. Diagnostics 2023, 13, 250. [Google Scholar] [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Berthault, N.; Bergam, P.; Pereira, F.; Girard, P.M.; Dutreix, M. Inhibition of DNA Repair by Inappropriate Activation of ATM, PARP, and DNA-PK with the Drug Agonist AsiDNA. Cells 2022, 11, 2149. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Mladenova, V.; Stuschke, M.; Iliakis, G. New Facets of DNA Double Strand Break Repair: Radiation Dose as Key Determinant of HR versus c-NHEJ Engagement. Int. J. Mol. Sci. 2023, 24, 14956. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.M.; Myung, K. Crosstalk between different DNA repair pathways for DNA double strand break repairs. Mutat. Res. Toxicol. Environ. Mutagen. 2022, 873, 503438. [Google Scholar] [CrossRef]
- Fell, V.L.; Schild-Poulter, C. The Ku heterodimer: Function in DNA repair and beyond. Mutat. Res. Mutat. Res. 2015, 763, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Caracciolo, D.; Riillo, C.; Di Martino, M.T.; Tagliaferri, P.; Tassone, P. Alternative Non-Homologous End-Joining: Error-Prone DNA Repair as Cancer’s Achilles’ Heel. Cancers 2021, 13, 1392. [Google Scholar] [CrossRef] [PubMed]
- Lavrik, O.I. PARPs’ impact on base excision DNA repair. DNA Repair 2020, 93, 102911. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.; Bauman, J.; Kang, H.; Ming, X. Biological barriers to therapy with antisense and siRNA oligonucleotides. Mol. Pharm. 2009, 6, 686–695. [Google Scholar] [CrossRef]
- Merki, E.; Graham, M.J.; Mullick, A.E.; Miller, E.R.; Crooke, R.M.; Pitas, R.E.; Witztum, J.L.; Tsimikas, S. Antisense Oligonucleotide Directed to Human Apolipoprotein B-100 Reduces Lipoprotein(a) Levels and Oxidized Phospholipids on Human Apolipoprotein B-100 Particles in Lipoprotein(a) Transgenic Mice. Circulation 2008, 118, 743–753. [Google Scholar] [CrossRef]
- Wurster, C.D.; Ludolph, A.C. Nusinersen for spinal muscular atrophy. Ther. Adv. Neurol. Disord. 2018, 11, 175628561875445. [Google Scholar] [CrossRef]
- Gales, L. Tegsedi (Inotersen): An Antisense Oligonucleotide Approved for the Treatment of Adult Patients with Hereditary Transthyretin Amyloidosis. Pharmaceuticals 2019, 12, 78. [Google Scholar] [CrossRef]
- Heo, Y.A. Golodirsen: First Approval. Drugs 2020, 80, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, E.; Cortesi, R.; Preti, L.; Rimessi, P.; Sguizzato, M.; Bovolenta, M.; Perrone, D. Antisense Oligonucleotides Conjugated with Lipophilic Compounds: Synthesis and In Vitro Evaluation of Exon Skipping in Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2022, 23, 4270. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.; Ngo, N.M.; Medhi, R.; Srinoi, P.; Liu, T.; Rittikulsittichai, S.; Lee, T.R. Multifunctional Iron Oxide Magnetic Nanoparticles for Biomedical Applications: A Review. Materials 2022, 15, 503. [Google Scholar] [CrossRef] [PubMed]
- Chernikov, I.V.; Gladkikh, D.V.; Meschaninova, M.I.; Ven’yaminova, A.G.; Zenkova, M.A.; Vlassov, V.V.; Chernolovskaya, E.L. Cholesterol-Containing Nuclease-Resistant siRNA Accumulates in Tumors in a Carrier-free Mode and Silences MDR1 Gene. Mol. Ther. Nucleic Acids 2017, 6, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Prakash, T.P.; Mullick, A.E.; Lee, R.G.; Yu, J.; Yeh, S.T.; Low, A.; Chappell, A.E.; Østergaard, M.E.; Murray, S.; Gaus, H.J.; et al. Fatty acid conjugation enhances potency of antisense oligonucleotides in muscle. Nucleic Acids Res. 2019, 47, 6029. [Google Scholar] [CrossRef] [PubMed]
- Benizri, S.; Gissot, A.; Martin, A.; Vialet, B.; Grinstaff, M.W.; Barthélémy, P. Bioconjugated Oligonucleotides: Recent Developments and Therapeutic Applications. Bioconjug. Chem. 2019, 30, 366–383. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.M.; Nair, J.K.; Janas, M.M.; Anglero-Rodriguez, Y.I.; Dang, L.T.H.; Peng, H.; Theile, C.S.; Castellanos-Rizaldos, E.; Brown, C.; Foster, D.; et al. Expanding RNAi therapeutics to extrahepatic tissues with lipophilic conjugates. Nat. Biotechnol. 2022, 40, 1500–1508. [Google Scholar] [CrossRef]
- Biscans, A.; Caiazzi, J.; McHugh, N.; Hariharan, V.; Muhuri, M.; Khvorova, A. Docosanoic acid conjugation to siRNA enables functional and safe delivery to skeletal and cardiac muscles. Mol. Ther. 2021, 29, 1382–1394. [Google Scholar] [CrossRef]
- Wada, S.; Yasuhara, H.; Wada, F.; Sawamura, M.; Waki, R.; Yamamoto, T.; Harada-Shiba, M.; Obika, S. Evaluation of the effects of chemically different linkers on hepatic accumulations, cell tropism and gene silencing ability of cholesterol-conjugated antisense oligonucleotides. J. Control. Release 2016, 226, 57–65. [Google Scholar] [CrossRef]
- Nishina, T.; Numata, J.; Nishina, K.; Yoshida-Tanaka, K.; Nitta, K.; Piao, W.; Iwata, R.; Ito, S.; Kuwahara, H.; Wada, T.; et al. Chimeric antisense oligonucleotide conjugated to α-Tocopherol. Mol. Ther. Nucleic Acids 2015, 4, e220. [Google Scholar] [CrossRef]
- Raouane, M.; Desmaele, D.; Gilbert-Sirieix, M.; Gueutin, C.; Zouhiri, F.; Bourgaux, C.; Lepeltier, E.; Gref, R.; Ben Salah, R.; Clayman, G.; et al. Synthesis, characterization, and in vivo delivery of siRNA-squalene nanoparticles targeting fusion oncogene in papillary thyroid carcinoma. J. Med. Chem. 2011, 54, 4067–4076. [Google Scholar] [CrossRef] [PubMed]
- Biscans, A.; Coles, A.; Haraszti, R.; Echeverria, D.; Hassler, M.; Osborn, M.; Khvorova, A. Diverse lipid conjugates for functional extra-hepatic siRNA delivery in vivo. Nucleic Acids Res. 2019, 47, 1082–1096. [Google Scholar] [CrossRef] [PubMed]
- Markov, O.V.; Filatov, A.V.; Kupryushkin, M.S.; Chernikov, I.V.; Patutina, O.A.; Strunov, A.A.; Chernolovskaya, E.L.; Vlassov, V.V.; Pyshnyi, D.V.; Zenkova, M.A. Transport Oligonucleotides—A Novel System for Intracellular Delivery of Antisense Therapeutics. Molecules 2020, 25, 3663. [Google Scholar] [CrossRef] [PubMed]
- Karaki, S.; Benizri, S.; Mejías, R.; Baylot, V.; Branger, N.; Nguyen, T.; Vialet, B.; Oumzil, K.; Barthélémy, P.; Rocchi, P. Lipid-oligonucleotide conjugates improve cellular uptake and efficiency of TCTP-antisense in castration-resistant prostate cancer. J. Control. Release 2017, 258, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Asahi, W.; Kurihara, R.; Takeyama, K.; Umehara, Y.; Kimura, Y.; Kondo, T.; Tanabe, K. Aggregate Formation of BODIPY-Tethered Oligonucleotides That Led to Efficient Intracellular Penetration and Gene Regulation. ACS Appl. Bio Mater. 2019, 2, 4456–4463. [Google Scholar] [CrossRef] [PubMed]
- Craig, K.; Abrams, M.; Amiji, M. Recent preclinical and clinical advances in oligonucleotide conjugates. Expert Opin. Drug Deliv. 2018, 15, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Osborn, M.F.; Coles, A.H.; Biscans, A.; Haraszti, R.A.; Roux, L.; Davis, S.; Ly, S.; Echeverria, D.; Hassler, M.R.; Godinho, B.M.D.C.; et al. Hydrophobicity drives the systemic distribution of lipid-conjugated siRNAs via lipid transport pathways. Nucleic Acids Res. 2019, 47, 1070–1081. [Google Scholar] [CrossRef]
- Sarett, S.M.; Werfel, T.A.; Lee, L.; Jackson, M.A.; Kilchrist, K.V.; Brantley-Sieders, D.; Duvall, C.L. Lipophilic siRNA targets albumin in situ and promotes bioavailability, tumor penetration, and carrier-free gene silencing. Proc. Natl. Acad. Sci. USA 2017, 114, E6490–E6497. [Google Scholar] [CrossRef]
- Chappell, A.E.; Gaus, H.J.; Berdeja, A.; Gupta, R.; Jo, M.; Prakash, T.P.; Oestergaard, M.; Swayze, E.E.; Seth, P.P. Mechanisms of palmitic acid-conjugated antisense oligonucleotide distribution in mice. Nucleic Acids Res. 2020, 48, 4382–4395. [Google Scholar] [CrossRef]
- Lacroix, A.; Fakih, H.H.; Sleiman, H.F. Detailed cellular assessment of albumin-bound oligonucleotides: Increased stability and lower non-specific cell uptake. J. Control. Release 2020, 324, 34–46. [Google Scholar] [CrossRef]
- Fanali, G.; di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: From bench to bedside. Mol. Aspects Med. 2012, 33, 209–290. [Google Scholar] [CrossRef]
- Zeeshan, F.; Madheswaran, T.; Panneerselvam, J.; Taliyan, R.; Kesharwani, P. Human Serum Albumin as Multifunctional Nanocarrier for Cancer Therapy. J. Pharm. Sci. 2021, 110, 3111–3117. [Google Scholar] [CrossRef] [PubMed]
- Varanko, A.; Saha, S.; Chilkoti, A. Recent trends in protein and peptide-based biomaterials for advanced drug delivery. Adv. Drug Deliv. Rev. 2020, 156, 133–187. [Google Scholar] [CrossRef]
- Prajapati, R.; Somoza, Á. Albumin nanostructures for nucleic acid delivery in cancer: Current trend, emerging issues, and possible solutions. Cancers 2021, 13, 3454. [Google Scholar] [CrossRef] [PubMed]
- Chubarov, A.S. Serum Albumin for Magnetic Nanoparticles Coating. Magnetochemistry 2022, 8, 13. [Google Scholar] [CrossRef]
- Hu, H.; Quintana, J.; Weissleder, R.; Parangi, S.; Miller, M. Deciphering albumin-directed drug delivery by imaging. Adv. Drug Deliv. Rev. 2022, 185, 114237. [Google Scholar] [CrossRef] [PubMed]
- De Simone, G.; Di Masi, A.; Ascenzi, P. Serum albumin: A multifaced enzyme. Int. J. Mol. Sci. 2021, 22, 10086. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Xue, X.; Yu, P.; Ni, Y.; Chen, F. Evans Blue Dye: A Revisit of Its Applications in Biomedicine. Contrast Media Mol. Imaging 2018, 2018, 7628037. [Google Scholar] [CrossRef]
- Lau, J.; Jacobson, O.; Niu, G.; Lin, K.S.; Bénard, F.; Chen, X. Bench to Bedside: Albumin Binders for Improved Cancer Radioligand Therapies. Bioconjug. Chem. 2019, 30, 487–502. [Google Scholar] [CrossRef]
- Tian, R.; Jacobson, O.; Niu, G.; Kiesewetter, D.O.; Wang, Z.; Zhu, G.; Ma, Y.; Liu, G.; Chen, X. Evans blue attachment enhances somatostatin receptor subtype-2 imaging and radiotherapy. Theranostics 2018, 8, 735–745. [Google Scholar] [CrossRef]
- Kunde, S.S.; Wairkar, S. Targeted delivery of albumin nanoparticles for breast cancer: A review. Colloids Surf. B Biointerfaces 2022, 213, 112422. [Google Scholar] [CrossRef] [PubMed]
- Dinesen, A.; Winther, A.; Wall, A.; Märcher, A.; Palmfeldt, J.; Chudasama, V.; Wengel, J.; Gothelf, K.V.; Baker, J.R.; Howard, K.A. Albumin Biomolecular Drug Designs Stabilized through Improved Thiol Conjugation and a Modular Locked Nucleic Acid Functionalized Assembly. Bioconjug. Chem. 2022, 33, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Kragh-Hansen, U. Human Serum Albumin: A Multifunctional Protein. In Albumine in Medicine; Springer: Singapore, 2016; pp. 1–24. ISBN 978-981-10-2115-2. [Google Scholar]
- Wei, D.; Zhang, X. Biosynthesis, bioactivity, biotoxicity and applications of antimicrobial peptides for human health. Biosaf. Health 2022, 4, 118–134. [Google Scholar] [CrossRef]
- Song, M.; Liu, G.; Liu, Y.; Cheng, Z.; Lin, H.; Liu, J.; Wu, Z.; Xue, J.; Hong, W.; Huang, M.; et al. Using porphyrins as albumin-binding molecules to enhance antitumor efficacies and reduce systemic toxicities of antimicrobial peptides. Eur. J. Med. Chem. 2021, 217, 113382. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhao, P.; Wang, Y.; Wu, A.; Rao, Y. Roles of Albumin-binding Proteins in Cancer Progression and Biomimetic Targeted Drug Delivery. ChemBioChem 2018, 56, 1796–1805. [Google Scholar] [CrossRef]
- Hoogenboezem, E.N.; Duvall, C.L. Harnessing Albumin as a Carrier for Cancer Therapies. Adv Drug Deliv Rev. 2018, 130, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Ishima, Y.; Maruyama, T.; Otagiri, M.; Chuang, V.T.G.; Ishida, T. The New Delivery Strategy of Albumin Carrier Utilizing the Interaction with Albumin Receptors. Chem. Pharm. Bull 2022, 70, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Kragh-Hansen, U. Albumin in Medicine; Springer: Singapore, 2016; ISBN 978-981-10-2115-2. [Google Scholar]
- Sockolosky, J.T.; Szoka, F.C. The neonatal Fc receptor, FcRn, as a target for drug delivery and therapy. Adv. Drug Deliv. Rev. 2015, 91, 109–124. [Google Scholar] [CrossRef]
- Dobrynin, S.; Kutseikin, S.; Morozov, D.; Krumkacheva, O.; Spitsyna, A.; Gatilov, Y.; Silnikov, V.; Angelovski, G.; Bowman, M.K.; Kirilyuk, I.; et al. Human Serum Albumin Labelled with Sterically-Hindered Nitroxides as Potential MRI Contrast Agents. Molecules 2020, 25, 1709. [Google Scholar] [CrossRef]
- Wang, S.Y.; Hu, H.Z.; Qing, X.C.; Zhang, Z.C.; Shao, Z.W. Recent advances of drug delivery nanocarriers in osteosarcoma treatment. J. Cancer 2020, 11, 69–82. [Google Scholar] [CrossRef]
- Yi, X.; Zhou, H.; Zhang, Z.; Xiong, S.; Yang, K. X-rays-optimized delivery of radiolabeled albumin for cancer theranostics. Biomaterials 2020, 233, 119764. [Google Scholar] [CrossRef] [PubMed]
- Hornok, V. Serum Albumin Nanoparticles: Problems and Prospects. Polymers 2021, 13, 3759. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, A.S.; Dovydenko, I.S.; Kupryushkin, M.S.; Grigor’eva, A.E.; Pyshnaya, I.A.; Pyshnyi, D.V. Amphiphilic “like-a-brush” oligonucleotide conjugates with three dodecyl chains: Self-assembly features of novel scaffold compounds for nucleic acids delivery. Nanomaterials 2020, 10, 1948. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, A.S.; Ilyushchenko, V.V.; Kupryushkin, M.S.; Zharkov, T.D.; Dyudeeva, E.S.; Bauer, I.A.; Chubarov, A.S.; Pyshnyi, D.V.; Pyshnaya, I.A. Complexes and Supramolecular Associates of Dodecyl-Containing Oligonucleotides with Serum Albumin. Biochemistry 2023, 88, 1165–1180. [Google Scholar] [CrossRef] [PubMed]
- Kupryushkin, M.S.; Zharkov, T.D.; Ilina, E.S.; Markov, O.V.; Kochetkova, A.S.; Akhmetova, M.M.; Lomzov, A.A.; Pyshnyi, D.V.; Lavrik, O.I.; Khodyreva, S.N. Triazinylamidophosphate Oligonucleotides: Synthesis and Study of Their Interaction with Cells and DNA-Binding Proteins. Russ. J. Bioorg. Chem. 2021, 47, 719–733. [Google Scholar] [CrossRef]
- Chubarov, A.; Spitsyna, A.; Krumkacheva, O.; Mitin, D.; Suvorov, D.; Tormyshev, V.; Fedin, M.; Bowman, M.K.; Bagryanskaya, E. Reversible Dimerization of Human Serum Albumin. Molecules 2021, 26, 108. [Google Scholar] [CrossRef] [PubMed]
- Chubarov, A.S.; Zakharova, O.D.; Koval, O.A.; Romaschenko, A.V.; Akulov, A.E.; Zavjalov, E.L.; Razumov, I.A.; Koptyug, I.V.; Knorre, D.G.; Godovikova, T.S. Design of protein homocystamides with enhanced tumor uptake properties for 19F magnetic resonance imaging. Bioorg. Med. Chem. 2015, 23, 6943–6954. [Google Scholar] [CrossRef] [PubMed]
- Ilina, E.S.; Lavrik, O.I.; Khodyreva, S.N. Ku antigen interacts with abasic sites. Biochim. Biophys. Acta 2008, 1784, 1777–1785. [Google Scholar] [CrossRef]
- Malke, H.J. SAMBROCK, E.F. FRITSCH and T. MANIATIS, Molecular Cloning, A Laboratory Manual (Second Edition), Volumes 1, 2 and 3. 1625 S., zahlreiche Abb. und Tab Cold Spring Harbor 1989. Cold Spring Harbor Laboratory Press. $ 115.00. ISBN: 0-87969-309-6. J. Basic Microbiol. 1990, 30, 623. [Google Scholar] [CrossRef]
- Kosova, A.A.; Khodyreva, S.N.; Lavrik, O.I. Ku antigen displays the AP lyase activity on a certain type of duplex DNA. Biochim. Biophys. Acta-Proteins Proteom. 2016, 1864, 1244–1252. [Google Scholar] [CrossRef]
- Amé, J.C.; Kalisch, T.; Dantzer, F.; Schreiber, V. Purification of Recombinant Poly(ADP-Ribose) Polymerases. Methods Mol. Biol. 2011, 780, 135–152. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Widen, S.G.; Williams, K.R.; Kedar, P.; Karpel, R.L.; Wilson, S.H. Studies of the domain structure of mammalian DNA polymerase β: Identification of a discrete template binding domain. J. Biol. Chem. 1990, 265, 2124–2131. [Google Scholar] [CrossRef] [PubMed]
- Biade, S.; Sobol, R.W.; Wilson, S.H.; Matsumoto, Y. Impairment of proliferating cell nuclear antigen-dependent apurinic/apyrimidinic site repair on linear DNA. J. Biol. Chem. 1998, 273, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Khodyreva, S.N.; Prasad, R.; Ilina, E.S.; Sukhanova, M.V.; Kutuzov, M.M.; Liu, Y.; Hou, E.W.; Wilson, S.H.; Lavrik, O.I. Apurinic/apyrimidinic (AP) site recognition by the 5′-dRP/AP lyase in poly(ADP-ribose) polymerase-1 (PARP-1). Proc. Natl. Acad. Sci. USA 2010, 107, 22090–22095. [Google Scholar] [CrossRef] [PubMed]
- Sabbarwal, S.; Dubey, A.K.; Pandey, M.; Kumar, M. Synthesis of biocompatible, BSA capped fluorescent CaCO3 pre-nucleation nanoclusters for cell imaging applications. J. Mater. Chem. B 2020, 8, 5729–5744. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Roy, S.; Monfregola, L.; Shang, S.; Shoemaker, R.; Caruthers, M.H. Oxidative substitution of boranephosphonate diesters as a route to post-synthetically modified DNA. J. Am. Chem. Soc. 2015, 137, 3253–3264. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Caruthers, M. Synthesis of DNA/RNA and Their Analogs via Phosphoramidite and H-Phosphonate Chemistries. Molecules 2013, 18, 14268–14284. [Google Scholar] [CrossRef]
- Takahashi, Y.; Kakuta, K.; Namioka, Y.; Igarashi, A.; Sakamoto, T.; Iwata Hara, R.; Sato, K.; Wada, T. Synthesis of P-Modified DNA from Boranophosphate DNA as a Precursor via Acyl Phosphite Intermediates. J. Org. Chem. 2023, 88, 10617–10631. [Google Scholar] [CrossRef]
- Vlaho, D.; Fakhoury, J.F.; Damha, M.J. Structural Studies and Gene Silencing Activity of siRNAs Containing Cationic Phosphoramidate Linkages. Nucleic Acid Ther. 2018, 28, 34–43. [Google Scholar] [CrossRef]
- Fokina, A.; Wang, M.; Ilyina, A.; Klabenkova, K.; Burakova, E.; Chelobanov, B.; Stetsenko, D. Analysis of new charge-neutral DNA/RNA analogues phosphoryl guanidine oligonucleotides (PGO) by gel electrophoresis. Anal. Biochem. 2018, 555, 9–11. [Google Scholar] [CrossRef]
- Greenspan, P.; Mayer, E.P.; Fowler, S.D. Nile red: A selective fluorescent stain for intracellular lipid droplets. J. Cell Biol. 1985, 100, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Pokholenko, O.; Gissot, A.; Vialet, B.; Bathany, K.; Thiéry, A.; Barthélémy, P. Lipid oligonucleotide conjugates as responsive nanomaterials for drug delivery. J. Mater. Chem. B 2013, 1, 5329–5334. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhu, Z.; Kang, H.; Wu, Y.; Sefan, K.; Tan, W. DNA-Based Micelles: Synthesis, Micellar Properties and Size-Dependent Cell Permeability. Chem.—A Eur. J. 2010, 16, 3791–3797. [Google Scholar] [CrossRef] [PubMed]
- Trinh, T.; Chidchob, P.; Bazzi, H.S.; Sleiman, H.F. DNA micelles as nanoreactors: Efficient DNA functionalization with hydrophobic organic molecules. Chem. Commun. 2016, 52, 10914–10917. [Google Scholar] [CrossRef] [PubMed]
- Kauss, T.; Arpin, C.; Bientz, L.; Vinh Nguyen, P.; Vialet, B.; Benizri, S.; Barthélémy, P. Lipid oligonucleotides as a new strategy for tackling the antibiotic resistance. Sci. Rep. 2020, 10, 1054. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Liu, X.; Bai, H.; Wang, R.; Tan, J.; Peng, X.; Tan, W. Engineering Stability-Tunable DNA Micelles Using Photocontrollable Dissociation of an Intermolecular G-Quadruplex. ACS Nano 2017, 11, 12087–12093. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; Kangs, S.-H.; Gryaznod, S.M.; Dedionision, L.; Heidenreichs, O.; Sullivanll, S.; Xu, X.; Nerenberg, M.I. Effect of Phosphorothioate Modification of Oligodeoxynucleotides on Specific Protein Binding. J. Biol. Chem. 1994, 269, 26801–26805. [Google Scholar] [CrossRef] [PubMed]
- Gaus, H.J.; Gupta, R.; Chappell, A.E.; Østergaard, M.E.; Swayze, E.E.; Seth, P.P. Characterization of the interactions of chemically-modified therapeutic nucleic acids with plasma proteins using a fluorescence polarization assay. Nucleic Acids Res. 2019, 47, 1110–1122. [Google Scholar] [CrossRef]
- Lacroix, A.; Edwardson, T.G.W.; Hancock, M.A.; Dore, M.D.; Sleiman, H.F. Development of DNA Nanostructures for High-Affinity Binding to Human Serum Albumin. J. Am. Chem. Soc. 2017, 139, 7355–7362. [Google Scholar] [CrossRef]
- Jin, C.; Zhang, H.; Zou, J.; Liu, Y.; Zhang, L.; Li, F.; Wang, R.; Xuan, W.; Ye, M.; Tan, W. Floxuridine Homomeric Oligonucleotides “Hitchhike” with Albumin In Situ for Cancer Chemotherapy. Angew. Chemie 2018, 130, 9132–9135. [Google Scholar] [CrossRef]
- Khodyreva, S.; Lavrik, O. Non-canonical interaction of DNA repair proteins with intact and cleaved AP sites. DNA Repair 2020, 90, 102847. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Strande, N.; Burkhalter, M.D.; Strom, C.; Havener, J.M.; Hasty, P.; Ramsden, D.A. Ku is a 5′-dRP/AP lyase that excises nucleotide damage near broken ends. Nature 2010, 464, 1214–1217. [Google Scholar] [CrossRef] [PubMed]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342, 249. [Google Scholar] [CrossRef]
- Ilina, E.S.; Lavrik, O.I.; Khodyreva, S.N. 5′-Deoxyribose Phosphate Lyase Activity of Apurinic/Apyrimidinic Endonuclease 1. Mol. Biol. 2021, 55, 269–276. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Strande, N.; Roberts, S.A.; Oh, S.; Hendrickson, E.A.; Ramsden, D.A. Specificity of the dRP/AP lyase of ku promotes nonhomologous end joining (NHEJ) fidelity at damaged ends. J. Biol. Chem. 2012, 287, 13686–13693. [Google Scholar] [CrossRef]
- Strande, N.T.; Carvajal-Garcia, J.; Hallett, R.A.; Waters, C.A.; Roberts, S.A.; Strom, C.; Kuhlman, B.; Ramsden, D.A. Requirements for 5′dRP/AP lyase activity in Ku. Nucleic Acids Res. 2014, 42, 11136. [Google Scholar] [CrossRef]
- Griffith, A.J.; Blier, P.R.; Mimori, T.; Hardin, J.A. Ku polypeptides synthesized in vitro assemble into complexes which recognize ends of double-stranded DNA. J. Biol. Chem. 1992, 267, 331–338. [Google Scholar] [CrossRef]
- Zharkov, T.D.; Mironova, E.M.; Markov, O.V.; Zhukov, S.A.; Khodyreva, S.N.; Kupryushkin, M.S. Fork- and Comb-like Lipophilic Structures: Different Chemical Approaches to the Synthesis of Oligonucleotides with Multiple Dodecyl Residues. Int. J. Mol. Sci. 2023, 24, 14637. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Parameter | |
|---|---|---|
| Tm, °C | ΔG0, kcal × mol−1 | |
| FAM-16/M-16 | 51.9 ± 0.1 | (−1.34 ± 0.01) × 104 |
| FAM-TZD16/M-16 | 48.7 ± 0.3 | (−1.15 ± 0.03) × 104 |
| FAM-17/TAMRA-M27 | 60.9 ± 0.2 | (−1.89 ± 0.09) × 104 |
| FAM-TZD17/TAMRA-M27 | 59.9 ± 0.1 | (−1.57 ± 0.03) × 104 |
| FAM-30/M30-FAM | 64.6 ± 0.5 | (−1.91 ± 0.05) × 104 |
| TZD30/M30-FAM | 65.3 ± 0.5 | (−2.1 ± 0.2) × 104 |
| Sample | Dh, nm | PDI |
|---|---|---|
| FAM-17 | 0.71 ± 0.13 | 0.249 |
| FAM-TZD16 | 11.9 ± 3.4 | 0.303 |
| FAM-TZD17 | 8.6 ± 2.5 | 0.265 |
| 11.6 ± 3.1 | 0.531 | |
| TZD30 | 10.4 ± 3.0 | 0.190 |
| 14.1 ± 4.0 | 0.245 | |
| TZD30 (T = 40 °C) | 14.8 ± 4.0 | 0.195 |
| HSA | 6.0 ± 1.3 | 0.284 |
| HSA/FAM-TZD16 | 6.7 ± 2.0 | 0.288 |
| HSA/FAM-TZD17 | 7.4 ± 2.0 | 0.423 |
| HSA/TZD30 | 7.0 ± 2.1 | 0.226 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bauer, I.; Ilina, E.; Zharkov, T.; Grigorieva, E.; Chinak, O.; Kupryushkin, M.; Golyshev, V.; Mitin, D.; Chubarov, A.; Khodyreva, S.; et al. Self-Penetrating Oligonucleotide Derivatives: Features of Self-Assembly and Interactions with Serum and Intracellular Proteins. Pharmaceutics 2023, 15, 2779. https://doi.org/10.3390/pharmaceutics15122779
Bauer I, Ilina E, Zharkov T, Grigorieva E, Chinak O, Kupryushkin M, Golyshev V, Mitin D, Chubarov A, Khodyreva S, et al. Self-Penetrating Oligonucleotide Derivatives: Features of Self-Assembly and Interactions with Serum and Intracellular Proteins. Pharmaceutics. 2023; 15(12):2779. https://doi.org/10.3390/pharmaceutics15122779
Chicago/Turabian StyleBauer, Irina, Ekaterina Ilina, Timofey Zharkov, Evgeniya Grigorieva, Olga Chinak, Maxim Kupryushkin, Victor Golyshev, Dmitry Mitin, Alexey Chubarov, Svetlana Khodyreva, and et al. 2023. "Self-Penetrating Oligonucleotide Derivatives: Features of Self-Assembly and Interactions with Serum and Intracellular Proteins" Pharmaceutics 15, no. 12: 2779. https://doi.org/10.3390/pharmaceutics15122779