
Effects of Lactate Transport Inhibition by AZD3965 in Muscle-Invasive Urothelial Bladder Cancer
 , , , , and
, , , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell lines and Conditions for Cell Culture
2.2. Protein Extraction and Western Blot
2.3. Immunofluorescence
2.4. Quantification of Extracellular Lactate Levels by Colorimetry
2.5. High-Performance Liquid Chromatography (HPLC)
2.6. Assesment of Mithocondrial Activity
2.7. Bioluminescence ATP Quantification Assay
2.8. Cell Viability Assay
2.9. Cell Proliferation Assay
2.10. Wound-Healing Assay
2.11. Cell Invasion Assay
2.12. Cell Cycle Analysis
2.13. Chick Chorioallantoic Membrane (CAM) Assay
2.14. Immunohistochemistry
2.15. Statistical Analysis
3. Results
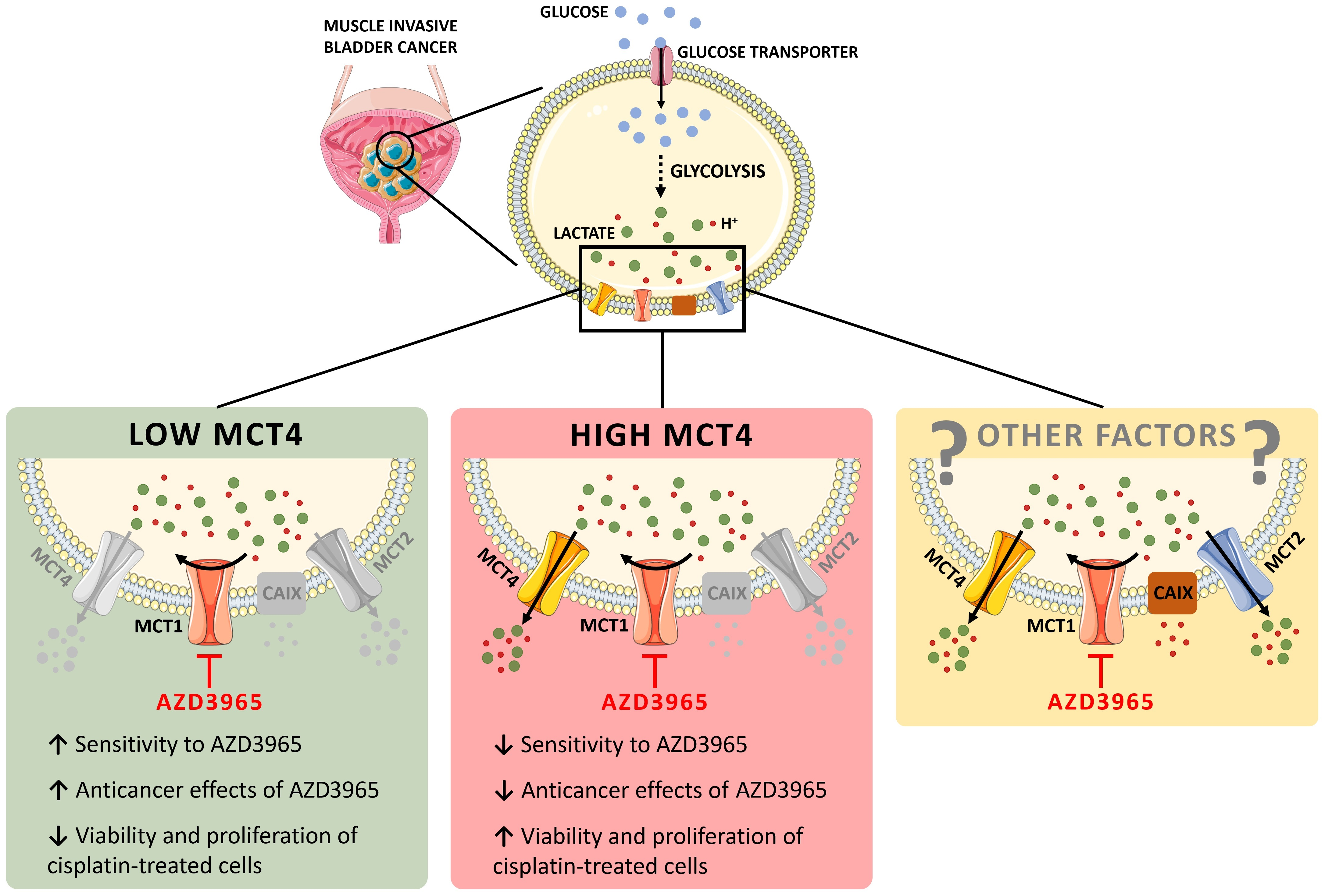
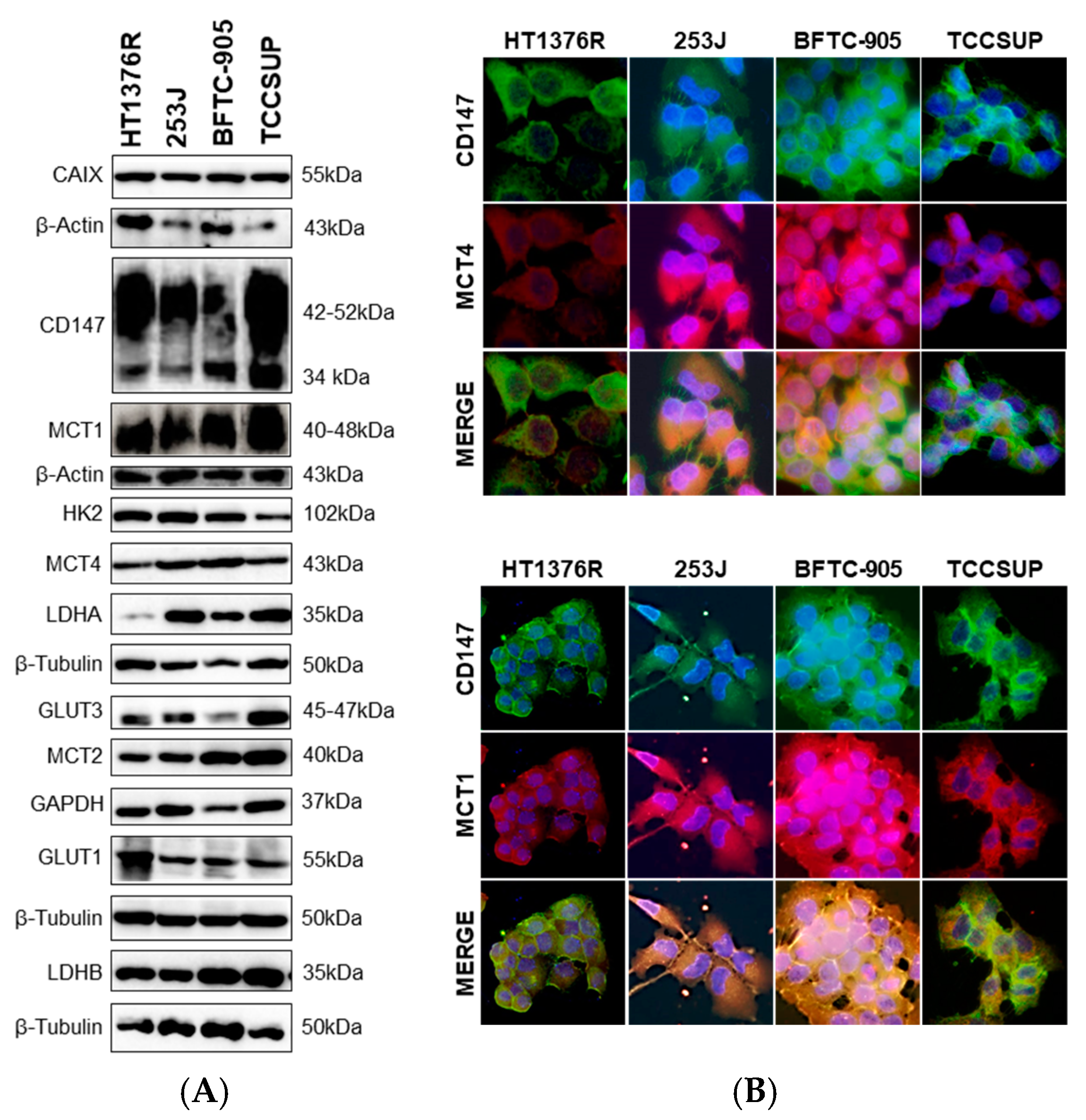
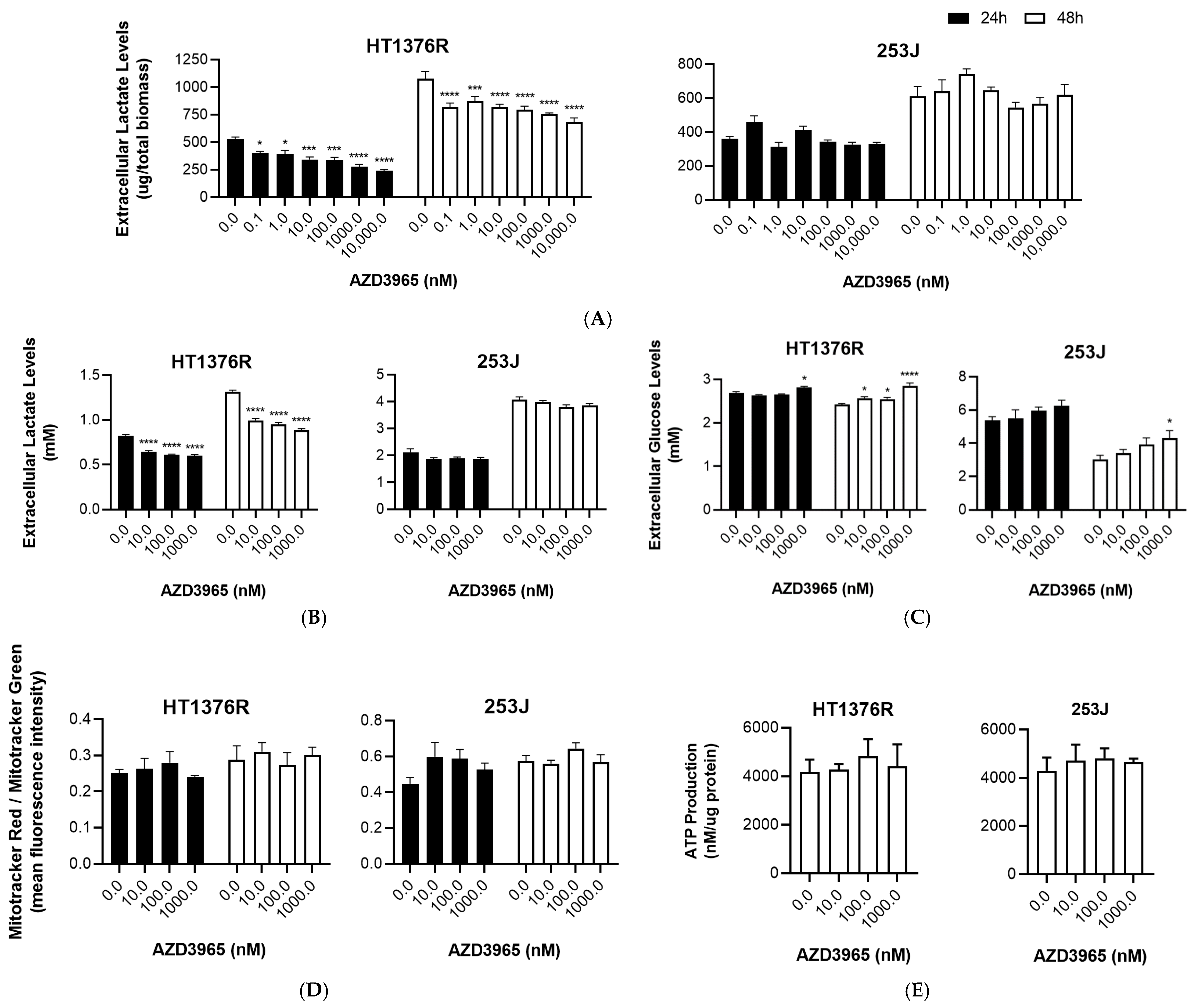
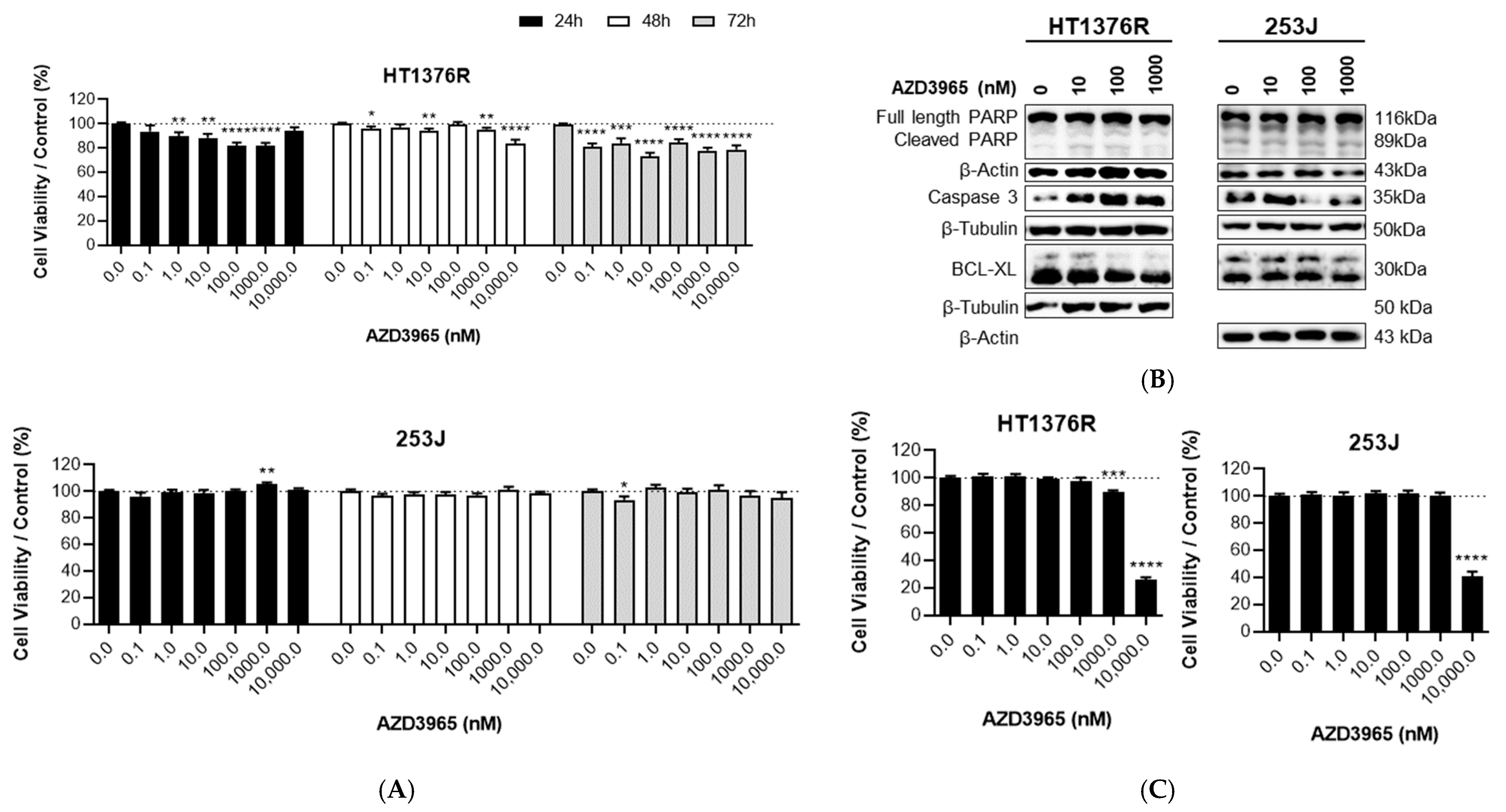
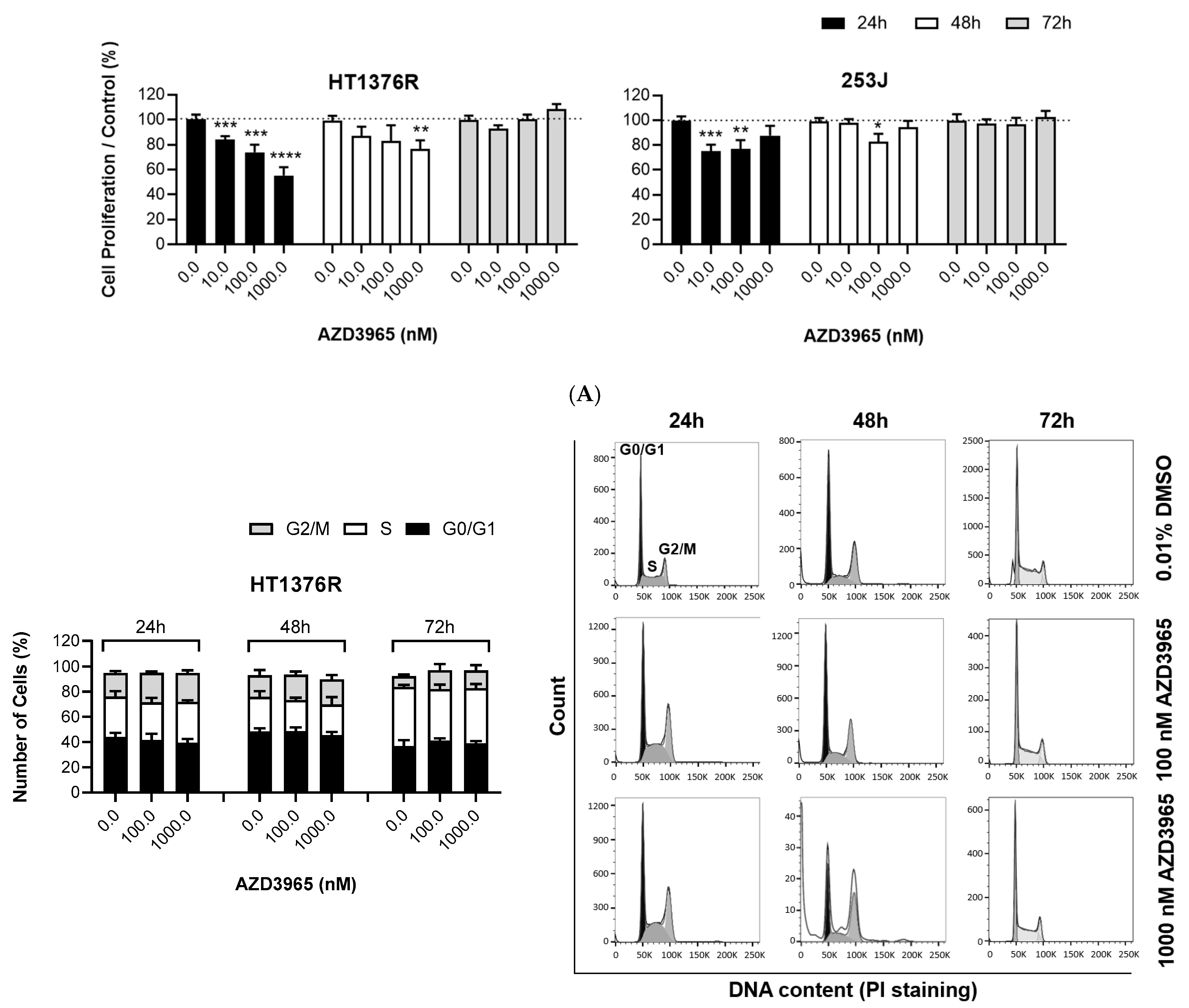
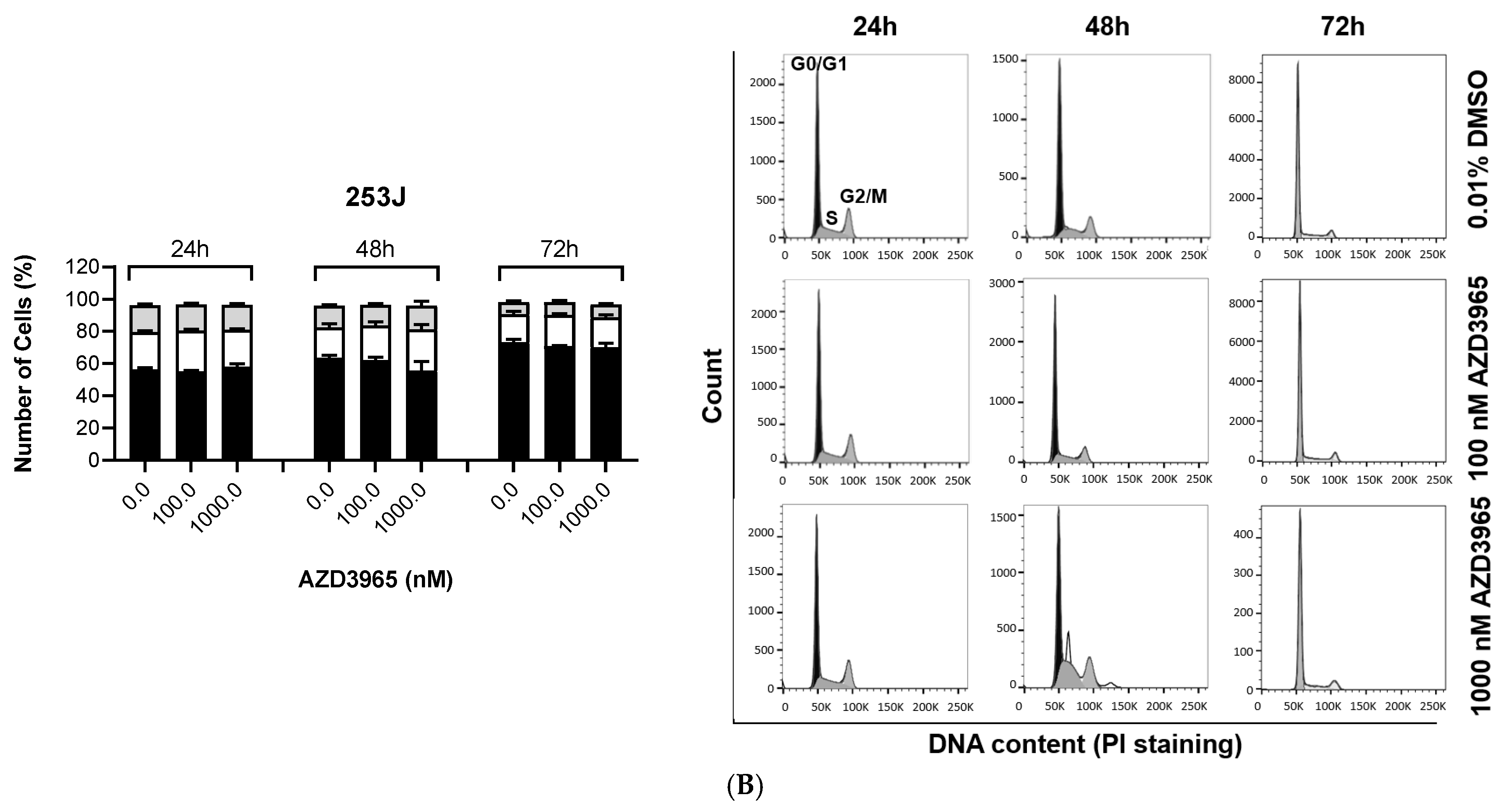
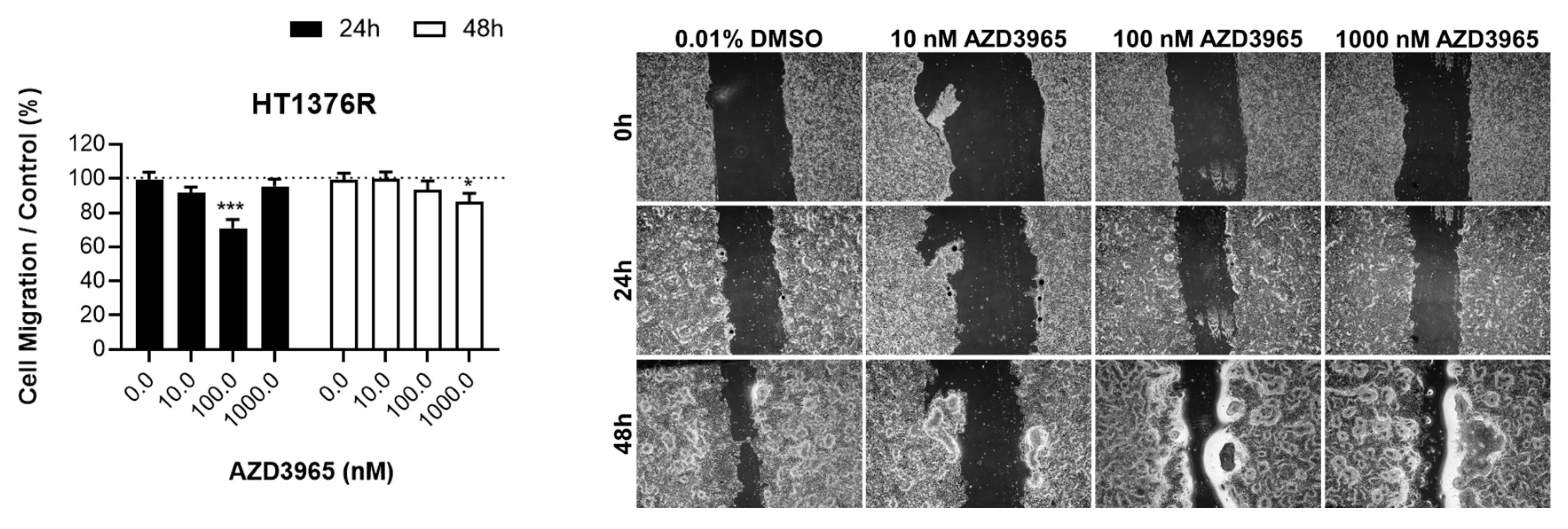
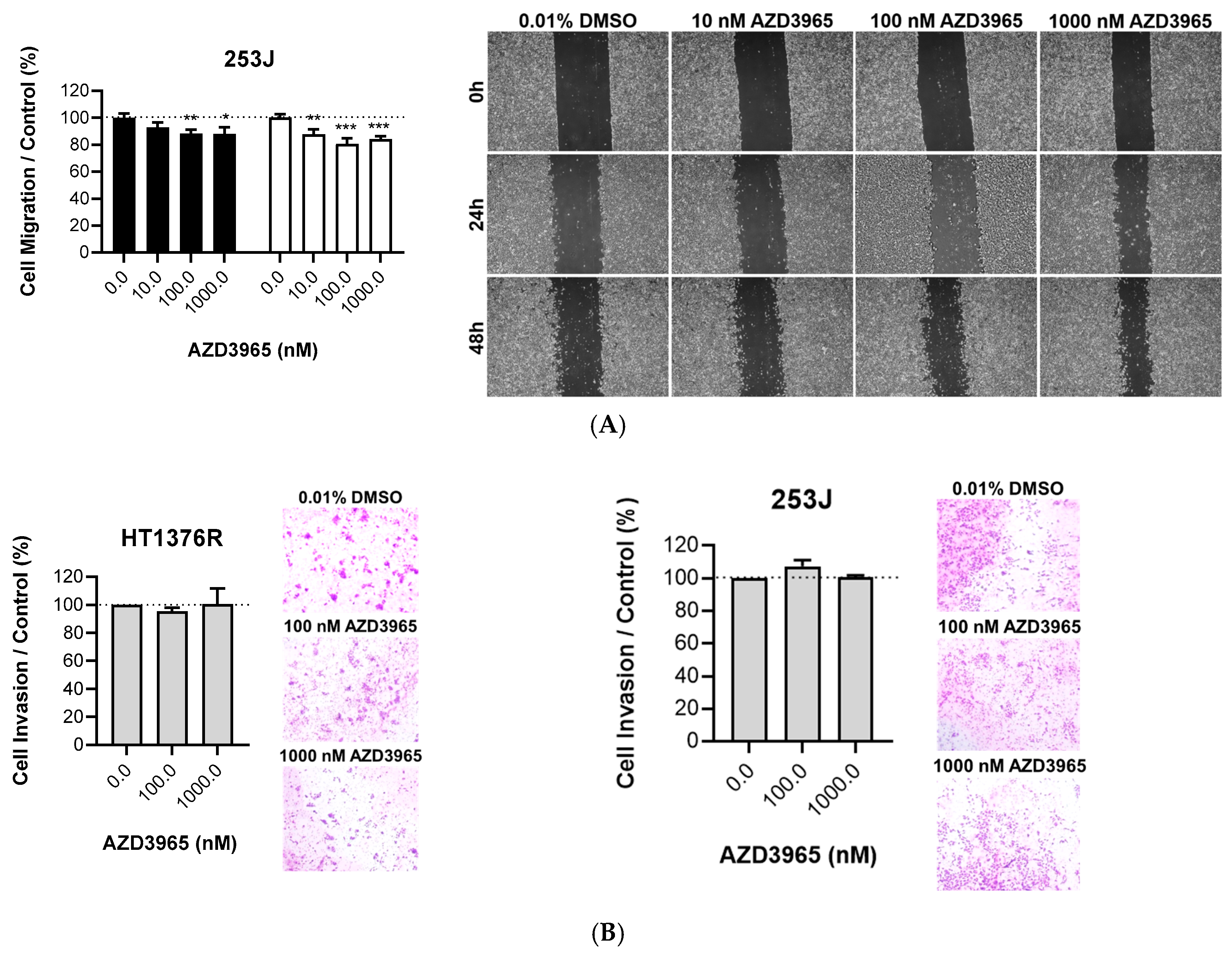
3.1. Impact of MCT1 Inhibition by AZD3965 in Muscle-Invasive UBC Cells
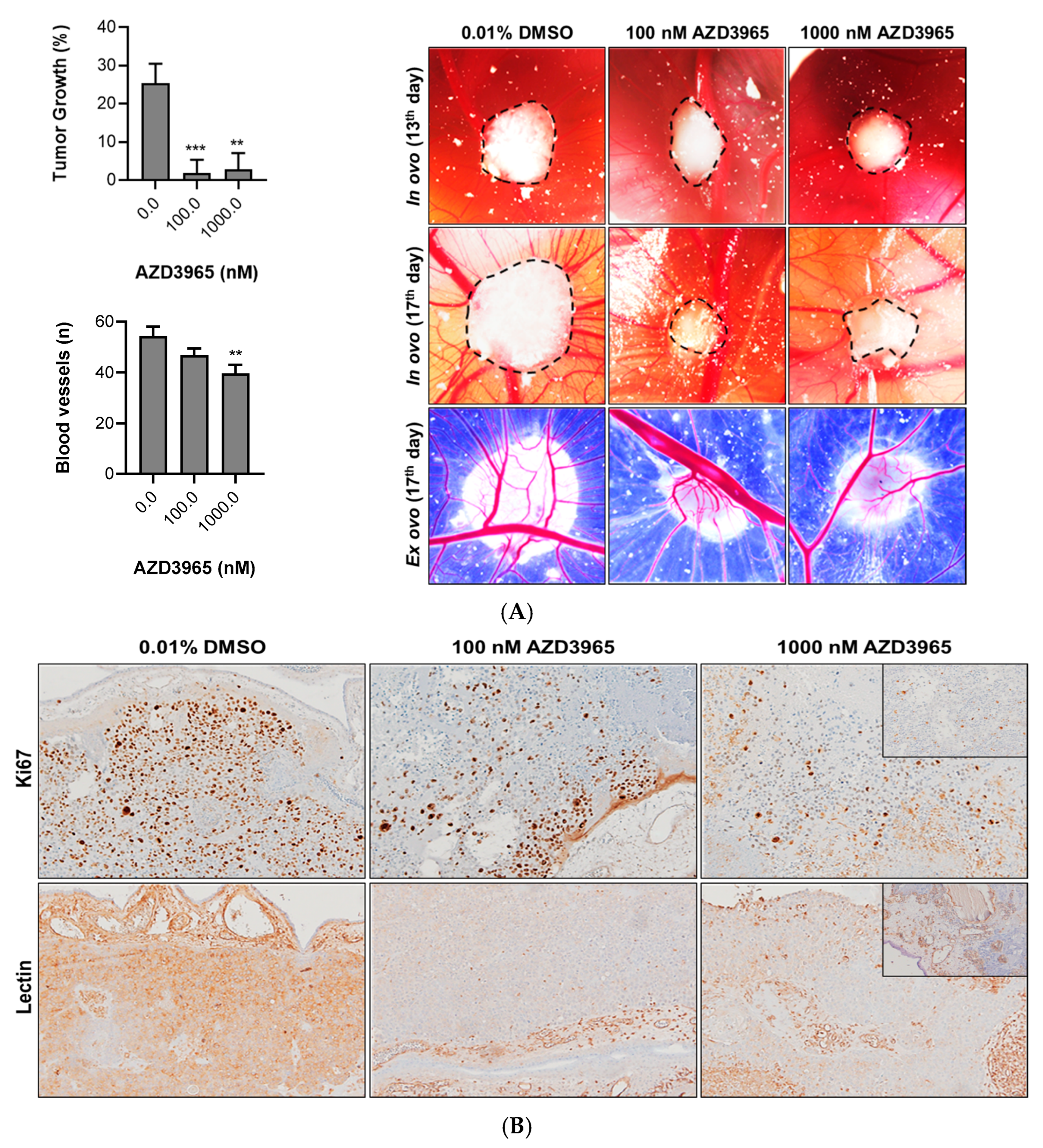
3.2. Impact of MCT1 Inhibition by AZD3965 in an “In Vivo” Muscle-Invasive UBC Model
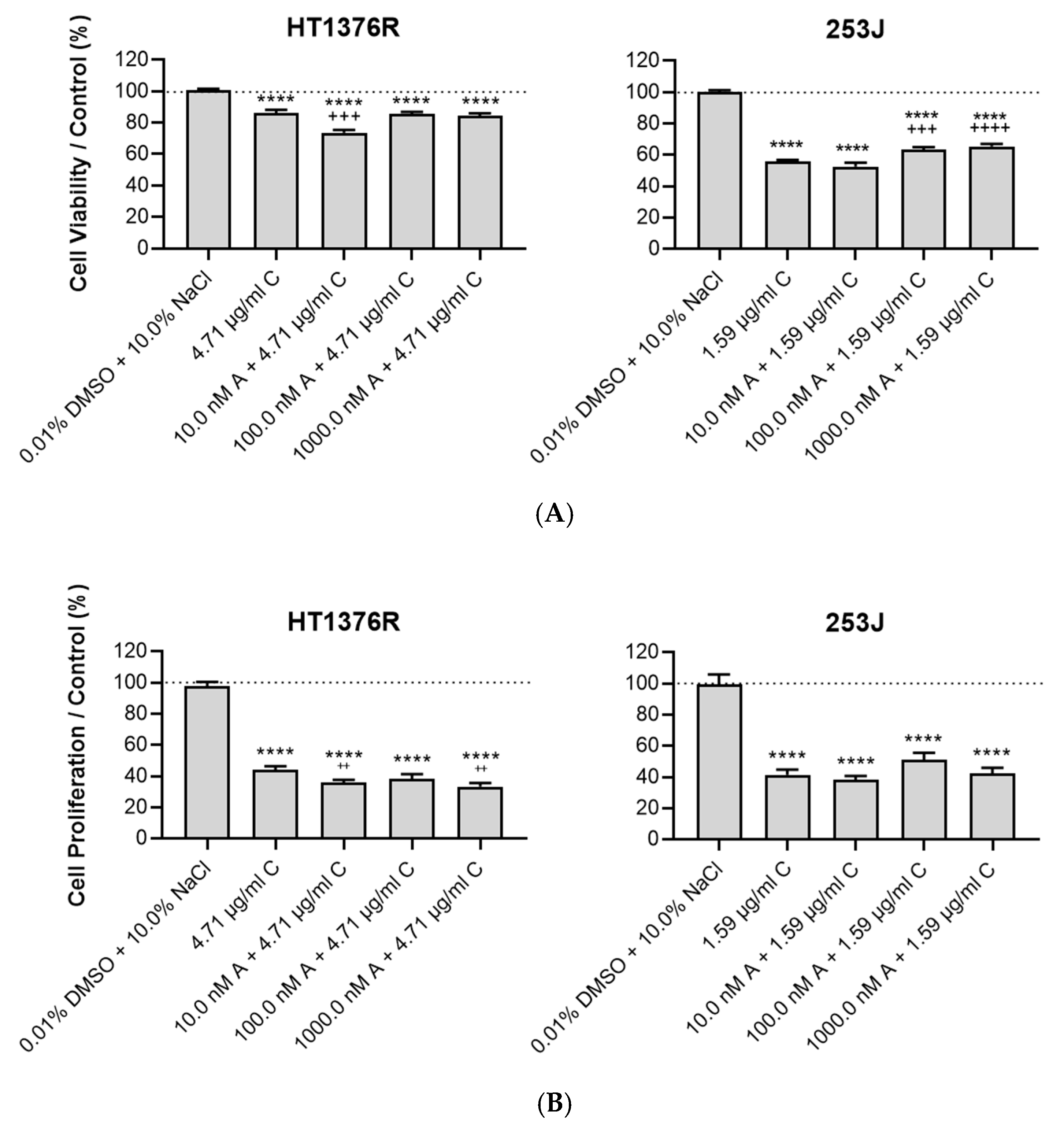
3.3. Impact of AZD3965 and Cisplatin Combined Therapy in Muscle-Invasive UBC Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Saginala, K.; Barsouk, A.; Aluru, J.S.; Rawla, P.; Padala, S.A.; Barsouk, A. Epidemiology of Bladder Cancer. Med. Sci. 2020, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Antoni, S.; Ferlay, J.; Soerjomataram, I.; Znaor, A.; Jemal, A.; Bray, F. Bladder Cancer Incidence and Mortality: A Global Overview and Recent Trends. Eur. Urol. 2017, 71, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Babjuk, M.; Burger, M.; Capoun, O.; Cohen, D.; Comperat, E.M.; Dominguez Escrig, J.L.; Gontero, P.; Liedberg, F.; Masson-Lecomte, A.; Mostafid, A.H.; et al. European Association of Urology Guidelines on Non-muscle-invasive Bladder Cancer (Ta, T1, and Carcinoma In Situ). Eur. Urol. 2022, 81, 75–94. [Google Scholar] [CrossRef] [PubMed]
- Audisio, A.; Buttigliero, C.; Delcuratolo, M.D.; Parlagreco, E.; Audisio, M.; Ungaro, A.; Di Stefano, R.F.; Di Prima, L.; Turco, F.; Tucci, M. New Perspectives in the Medical Treatment of Non-Muscle-Invasive Bladder Cancer: Immune Checkpoint Inhibitors and Beyond. Cells 2022, 11, 357. [Google Scholar] [CrossRef]
- Sanli, O.; Dobruch, J.; Knowles, M.A.; Burger, M.; Alemozaffar, M.; Nielsen, M.E.; Lotan, Y. Bladder cancer. Nat. Rev. Dis. Primers 2017, 3, 17022. [Google Scholar] [CrossRef]
- Witjes, J.A.; Bruins, H.M.; Cathomas, R.; Comperat, E.M.; Cowan, N.C.; Gakis, G.; Hernandez, V.; Linares Espinos, E.; Lorch, A.; Neuzillet, Y.; et al. European Association of Urology Guidelines on Muscle-invasive and Metastatic Bladder Cancer: Summary of the 2020 Guidelines. Eur. Urol. 2021, 79, 82–104. [Google Scholar] [CrossRef]
- Patel, V.G.; Oh, W.K.; Galsky, M.D. Treatment of muscle-invasive and advanced bladder cancer in 2020. CA Cancer J. Clin. 2020, 70, 404–423. [Google Scholar] [CrossRef]
- Ripoll, J.; Ramos, M.; Montano, J.; Pons, J.; Ameijide, A.; Franch, P. Cancer-specific survival by stage of bladder cancer and factors collected by Mallorca Cancer Registry associated to survival. BMC Cancer 2021, 21, 676. [Google Scholar] [CrossRef]
- Bellmunt, J.; Powles, T.; Henriksson, R.; Steinberg, G.D.; Batyrbekova, N.; Schain, F.; Fleming, S.; Shalaby, W.; Siefker-Radtke, A.O. Clinical outcomes and economic burden for bladder cancer patients: An analysis from a Swedish cancer registry. J. Clin. Oncol. 2020, 38, 5026. [Google Scholar] [CrossRef]
- Barone, B.; Calogero, A.; Scafuri, L.; Ferro, M.; Lucarelli, G.; Di Zazzo, E.; Sicignano, E.; Falcone, A.; Romano, L.; De Luca, L.; et al. Immune Checkpoint Inhibitors as a Neoadjuvant/Adjuvant Treatment of Muscle-Invasive Bladder Cancer: A Systematic Review. Cancers 2022, 14, 2545. [Google Scholar] [CrossRef] [PubMed]
- Ascione, C.M.; Napolitano, F.; Esposito, D.; Servetto, A.; Belli, S.; Santaniello, A.; Scagliarini, S.; Crocetto, F.; Bianco, R.; Formisano, L. Role of FGFR3 in bladder cancer: Treatment landscape and future challenges. Cancer Treat. Rev. 2023, 115, 102530. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.M.; Gupta, S.; Kitchlu, A.; Meraz-Munoz, A.; North, S.A.; Alimohamed, N.S.; Blais, N.; Sridhar, S.S. Defining cisplatin eligibility in patients with muscle-invasive bladder cancer. Nat. Rev. Urol. 2021, 18, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Afonso, J.; Santos, L.L.; Miranda-Goncalves, V.; Morais, A.; Amaro, T.; Longatto-Filho, A.; Baltazar, F. CD147 and MCT1-potential partners in bladder cancer aggressiveness and cisplatin resistance. Mol. Carcinog. 2015, 54, 1451–1466. [Google Scholar] [CrossRef]
- Afonso, J.; Santos, L.L.; Morais, A.; Amaro, T.; Longatto-Filho, A.; Baltazar, F. Metabolic coupling in urothelial bladder cancer compartments and its correlation to tumor aggressiveness. Cell Cycle 2016, 15, 368–380. [Google Scholar] [CrossRef]
- Drayton, R.M.; Catto, J.W. Molecular mechanisms of cisplatin resistance in bladder cancer. Expert. Rev. Anticancer Ther. 2012, 12, 271–281. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Pascale, R.M.; Calvisi, D.F.; Simile, M.M.; Feo, C.F.; Feo, F. The Warburg Effect 97 Years after Its Discovery. Cancers 2020, 12, 2819. [Google Scholar] [CrossRef]
- Kocianova, E.; Piatrikova, V.; Golias, T. Revisiting the Warburg Effect with Focus on Lactate. Cancers 2022, 14, 6028. [Google Scholar] [CrossRef]
- Afonso, J.; Santos, L.L.; Longatto-Filho, A.; Baltazar, F. Competitive glucose metabolism as a target to boost bladder cancer immunotherapy. Nat. Rev. Urol. 2020, 17, 77–106. [Google Scholar] [CrossRef]
- Singh, M.; Afonso, J.; Sharma, D.; Gupta, R.; Kumar, V.; Rani, R.; Baltazar, F.; Kumar, V. Targeting monocarboxylate transporters (MCTs) in cancer: How close are we to the clinics? Semin. Cancer Biol. 2023, 90, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.; Cerqueira, M.C.; Rosa, B.; Sobral, C.; Pinto-Ribeiro, F.; Costa, M.F.; Baltazar, F.; Afonso, J. Prognostic Value of Monocarboxylate Transporter 1 Overexpression in Cancer: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 5141. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zhang, Y.; Dong, D.; Wang, F.; Ma, X.; Guan, F.; Sun, L. MCT1 regulates aggressive and metabolic phenotypes in bladder cancer. J. Cancer 2018, 9, 2492–2501. [Google Scholar] [CrossRef] [PubMed]
- Halford, S.E.R.; Walter, H.; McKay, P.; Townsend, W.; Linton, K.; Heinzmann, K.; Dragoni, I.; Brotherton, L.; Veal, G.; Siskos, A.; et al. Phase I expansion study of the first-in-class monocarboxylate transporter 1 (MCT1) inhibitor AZD3965 in patients with diffuse large B-cell lymphoma (DLBCL) and Burkitt lymphoma (BL). J. Clin. Oncol. 2021, 39, 3115. [Google Scholar] [CrossRef]
- Halford, S.; Veal, G.J.; Wedge, S.R.; Payne, G.S.; Bacon, C.M.; Sloan, P.; Dragoni, I.; Heinzmann, K.; Potter, S.; Salisbury, B.M.; et al. A Phase I Dose-escalation Study of AZD3965, an Oral Monocarboxylate Transporter 1 Inhibitor, in Patients with Advanced Cancer. Clin. Cancer Res. 2023, 29, 1429–1439. [Google Scholar] [CrossRef]
- Bola, B.M.; Chadwick, A.L.; Michopoulos, F.; Blount, K.G.; Telfer, B.A.; Williams, K.J.; Smith, P.D.; Critchlow, S.E.; Stratford, I.J. Inhibition of monocarboxylate transporter-1 (MCT1) by AZD3965 enhances radiosensitivity by reducing lactate transport. Mol. Cancer Ther. 2014, 13, 2805–2816. [Google Scholar] [CrossRef] [PubMed]
- Polanski, R.; Hodgkinson, C.L.; Fusi, A.; Nonaka, D.; Priest, L.; Kelly, P.; Trapani, F.; Bishop, P.W.; White, A.; Critchlow, S.E.; et al. Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin. Cancer Res. 2014, 20, 926–937. [Google Scholar] [CrossRef]
- Silva, A.; Antunes, B.; Batista, A.; Pinto-Ribeiro, F.; Baltazar, F.; Afonso, J. In Vivo Anticancer Activity of AZD3965: A Systematic Review. Molecules 2021, 27, 181. [Google Scholar] [CrossRef]
- Benyahia, Z.; Blackman, M.; Hamelin, L.; Zampieri, L.X.; Capeloa, T.; Bedin, M.L.; Vazeille, T.; Schakman, O.; Sonveaux, P. In Vitro and In Vivo Characterization of MCT1 Inhibitor AZD3965 Confirms Preclinical Safety Compatible with Breast Cancer Treatment. Cancers 2021, 13, 569. [Google Scholar] [CrossRef]
- Michaelis, M.; Wass, M.N.; Cinatl, J. Drug-adapted cancer cell lines as preclinical models of acquired resistance. Cancer Drug Resist. 2019, 2, 447–456. [Google Scholar] [CrossRef]
- Michaelis, M.; Rothweiler, F.; Barth, S.; Cinatl, J.; van Rikxoort, M.; Loschmann, N.; Voges, Y.; Breitling, R.; von Deimling, A.; Rodel, F.; et al. Adaptation of cancer cells from different entities to the MDM2 inhibitor nutlin-3 results in the emergence of p53-mutated multi-drug-resistant cancer cells. Cell Death Dis. 2011, 2, e243. [Google Scholar] [CrossRef] [PubMed]
- Afonso, J.; Goncalves, C.; Costa, M.; Ferreira, D.; Santos, L.; Longatto-Filho, A.; Baltazar, F. Glucose Metabolism Reprogramming in Bladder Cancer: Hexokinase 2 (HK2) as Prognostic Biomarker and Target for Bladder Cancer Therapy. Cancers 2023, 15, 982. [Google Scholar] [CrossRef] [PubMed]
- Afonso, J.; Pinto, T.; Simoes-Sousa, S.; Schmitt, F.; Longatto-Filho, A.; Pinheiro, C.; Marques, H.; Baltazar, F. Clinical significance of metabolism-related biomarkers in non-Hodgkin lymphoma—MCT1 as potential target in diffuse large B cell lymphoma. Cell. Oncol. 2019, 42, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Morris, M.E. In Vitro and In Vivo Efficacy of AZD3965 and Alpha-Cyano-4-Hydroxycinnamic Acid in the Murine 4T1 Breast Tumor Model. AAPS J. 2020, 22, 84. [Google Scholar] [CrossRef] [PubMed]
- Barbato, A.; Giallongo, C.; Giallongo, S.; Romano, A.; Scandura, G.; Concetta, S.; Zuppelli, T.; Lolicato, M.; Lazzarino, G.; Parrinello, N.; et al. Lactate trafficking inhibition restores sensitivity to proteasome inhibitors and orchestrates immuno-microenvironment in multiple myeloma. Cell Prolif. 2023, 56, e13388. [Google Scholar] [CrossRef]
- Curtis, N.J.; Mooney, L.; Hopcroft, L.; Michopoulos, F.; Whalley, N.; Zhong, H.; Murray, C.; Logie, A.; Revill, M.; Byth, K.F.; et al. Pre-clinical pharmacology of AZD3965, a selective inhibitor of MCT1: DLBCL, NHL and Burkitt’s lymphoma anti-tumor activity. Oncotarget 2017, 8, 69219–69236. [Google Scholar] [CrossRef] [PubMed]
- Braga, M.; Kaliszczak, M.; Carroll, L.; Schug, Z.T.; Heinzmann, K.; Baxan, N.; Benito, A.; Valbuena, G.N.; Stribbling, S.; Beckley, A.; et al. Tracing Nutrient Flux Following Monocarboxylate Transporter-1 Inhibition with AZD3965. Cancers 2020, 12, 1703. [Google Scholar] [CrossRef]
- Noble, R.A.; Bell, N.; Blair, H.; Sikka, A.; Thomas, H.; Phillips, N.; Nakjang, S.; Miwa, S.; Crossland, R.; Rand, V.; et al. Inhibition of monocarboxyate transporter 1 by AZD3965 as a novel therapeutic approach for diffuse large B-cell lymphoma and Burkitt lymphoma. Haematologica 2017, 102, 1247–1257. [Google Scholar] [CrossRef]
- Beloueche-Babari, M.; Wantuch, S.; Casals Galobart, T.; Koniordou, M.; Parkes, H.G.; Arunan, V.; Chung, Y.L.; Eykyn, T.R.; Smith, P.D.; Leach, M.O. MCT1 Inhibitor AZD3965 Increases Mitochondrial Metabolism, Facilitating Combination Therapy and Noninvasive Magnetic Resonance Spectroscopy. Cancer Res. 2017, 77, 5913–5924. [Google Scholar] [CrossRef]
- Boulares, A.H.; Yakovlev, A.G.; Ivanova, V.; Stoica, B.A.; Wang, G.; Iyer, S.; Smulson, M. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J. Biol. Chem. 1999, 274, 22932–22940. [Google Scholar] [CrossRef]
- Grasa, L.; Chueca, E.; Arechavaleta, S.; Garcia-Gonzalez, M.A.; Saenz, M.A.; Valero, A.; Hordnler, C.; Lanas, A.; Piazuelo, E. Antitumor effects of lactate transport inhibition on esophageal adenocarcinoma cells. J. Physiol. Biochem. 2023, 79, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.Y.; de Wang, G.; Liu, P.F.; Cao, Y.W.; Wang, Y.H.; Yang, X.C.; Hu, C.X.; Sun, L.J.; Niu, H.T. Targeting of MCT1 and PFKFB3 influences cell proliferation and apoptosis in bladder cancer by altering the tumor microenvironment. Oncol. Rep. 2016, 36, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Saulle, E.; Spinello, I.; Quaranta, M.T.; Pasquini, L.; Pelosi, E.; Iorio, E.; Castelli, G.; Chirico, M.; Pisanu, M.E.; Ottone, T.; et al. Targeting Lactate Metabolism by Inhibiting MCT1 or MCT4 Impairs Leukemic Cell Proliferation, Induces Two Different Related Death-Pathways and Increases Chemotherapeutic Sensitivity of Acute Myeloid Leukemia Cells. Front. Oncol. 2020, 10, 621458. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.S.; Graham, N.A.; Gu, W.; Espindola Camacho, C.; Mah, V.; Maresh, E.L.; Alavi, M.; Bagryanova, L.; Krotee, P.A.L.; Gardner, B.K.; et al. MCT1 Modulates Cancer Cell Pyruvate Export and Growth of Tumors that Co-express MCT1 and MCT4. Cell Rep. 2016, 14, 1590–1601. [Google Scholar] [CrossRef] [PubMed]
- Pivovarova, A.I.; MacGregor, G.G. Glucose-dependent growth arrest of leukemia cells by MCT1 inhibition: Feeding Warburg’s sweet tooth and blocking acid export as an anticancer strategy. Biomed. Pharmacother. 2018, 98, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Wittekind, C.; Neid, M. Cancer invasion and metastasis. Oncology 2005, 69 (Suppl. S1), 14–16. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.C.; Nohr-Nielsen, A.; Zeeberg, K.; Reshkin, S.J.; Hoffmann, E.K.; Novak, I.; Pedersen, S.F. Monocarboxylate Transporters MCT1 and MCT4 Regulate Migration and Invasion of Pancreatic Ductal Adenocarcinoma Cells. Pancreas 2016, 45, 1036–1047. [Google Scholar] [CrossRef]
- Payen, V.L.; Hsu, M.Y.; Radecke, K.S.; Wyart, E.; Vazeille, T.; Bouzin, C.; Porporato, P.E.; Sonveaux, P. Monocarboxylate Transporter MCT1 Promotes Tumor Metastasis Independently of Its Activity as a Lactate Transporter. Cancer Res. 2017, 77, 5591–5601. [Google Scholar] [CrossRef]
- Sheng, G.; Gao, Y.; Wu, H.; Liu, Y.; Yang, Y. Functional heterogeneity of MCT1 and MCT4 in metabolic reprogramming affects osteosarcoma growth and metastasis. J. Orthop. Surg. Res. 2023, 18, 131. [Google Scholar] [CrossRef]
- Liao, C.G.; Liang, X.H.; Ke, Y.; Yao, L.; Liu, M.; Liu, Z.K.; He, L.; Guo, Y.X.; Bian, H.; Chen, Z.N.; et al. Active demethylation upregulates CD147 expression promoting non-small cell lung cancer invasion and metastasis. Oncogene 2022, 41, 1780–1794. [Google Scholar] [CrossRef]
- Voigt, H.; Vetter-Kauczok, C.S.; Schrama, D.; Hofmann, U.B.; Becker, J.C.; Houben, R. CD147 impacts angiogenesis and metastasis formation. Cancer Investig. 2009, 27, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Yan, Y.; Guo, J.; Bai, C. CD147 promotes breast cancer invasion and metastasis by inducing EMT and upregulating MMPs via the MAPK/ERK signaling pathway—PREPRINT. Res. Sq. 2023. [Google Scholar] [CrossRef]
- Beloueche-Babari, M.; Casals Galobart, T.; Delgado-Goni, T.; Wantuch, S.; Parkes, H.G.; Tandy, D.; Harker, J.A.; Leach, M.O. Monocarboxylate transporter 1 blockade with AZD3965 inhibits lipid biosynthesis and increases tumour immune cell infiltration. Br. J. Cancer 2020, 122, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Dalton, K.M.; Lochmann, T.L.; Floros, K.V.; Calbert, M.L.; Kurupi, R.; Stein, G.T.; McClanaghan, J.; Murchie, E.; Egan, R.K.; Greninger, P.; et al. Catastrophic ATP loss underlies a metabolic combination therapy tailored for MYCN-amplified neuroblastoma. Proc. Natl. Acad. Sci. USA 2021, 118, e2009620118. [Google Scholar] [CrossRef] [PubMed]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef]
- Byun, J.K. Tumor lactic acid: A potential target for cancer therapy. Arch. Pharm. Res. 2023, 46, 90–110. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Wilson, M.C. The monocarboxylate transporter family--role and regulation. IUBMB Life 2012, 64, 109–119. [Google Scholar] [CrossRef]
- Quanz, M.; Bender, E.; Kopitz, C.; Grunewald, S.; Schlicker, A.; Schwede, W.; Eheim, A.; Toschi, L.; Neuhaus, R.; Richter, C.; et al. Preclinical Efficacy of the Novel Monocarboxylate Transporter 1 Inhibitor BAY-8002 and Associated Markers of Resistance. Mol. Cancer Ther. 2018, 17, 2285–2296. [Google Scholar] [CrossRef]
- Payen, V.L.; Mina, E.; Van Hee, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate transporters in cancer. Mol. Metab. 2020, 33, 48–66. [Google Scholar] [CrossRef]
- Xiang, A.P.; Chen, X.N.; Xu, P.F.; Shao, S.H.; Shen, Y.F. Expression and prognostic value of carbonic anhydrase IX (CA-IX) in bladder urothelial carcinoma. BMC Urol. 2022, 22, 120. [Google Scholar] [CrossRef]
- Klatte, T.; Seligson, D.B.; Rao, J.Y.; Yu, H.; de Martino, M.; Kawaoka, K.; Wong, S.G.; Belldegrun, A.S.; Pantuck, A.J. Carbonic anhydrase IX in bladder cancer: A diagnostic, prognostic, and therapeutic molecular marker. Cancer 2009, 115, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
- Ames, S.; Andring, J.T.; McKenna, R.; Becker, H.M. CAIX forms a transport metabolon with monocarboxylate transporters in human breast cancer cells. Oncogene 2020, 39, 1710–1723. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Simon, M.C. Cancer Cells Don’t Live Alone: Metabolic Communication within Tumor Microenvironments. Dev. Cell 2020, 54, 183–195. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, A.; Félix, A.; Cerqueira, M.; Gonçalves, C.S.; Sampaio-Marques, B.; Longatto-Filho, A.; Baltazar, F.; Afonso, J. Effects of Lactate Transport Inhibition by AZD3965 in Muscle-Invasive Urothelial Bladder Cancer. Pharmaceutics 2023, 15, 2688. https://doi.org/10.3390/pharmaceutics15122688
Silva A, Félix A, Cerqueira M, Gonçalves CS, Sampaio-Marques B, Longatto-Filho A, Baltazar F, Afonso J. Effects of Lactate Transport Inhibition by AZD3965 in Muscle-Invasive Urothelial Bladder Cancer. Pharmaceutics. 2023; 15(12):2688. https://doi.org/10.3390/pharmaceutics15122688
Chicago/Turabian StyleSilva, Ana, Ana Félix, Mónica Cerqueira, Céline S. Gonçalves, Belém Sampaio-Marques, Adhemar Longatto-Filho, Fátima Baltazar, and Julieta Afonso. 2023. "Effects of Lactate Transport Inhibition by AZD3965 in Muscle-Invasive Urothelial Bladder Cancer" Pharmaceutics 15, no. 12: 2688. https://doi.org/10.3390/pharmaceutics15122688