
Computational Amendment of Parenteral In Situ Forming Particulates’ Characteristics: Design of Experiment and PBPK Physiological Modeling
 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Preparation of Drug-Loaded In Situ-Forming Particulate Formulations (IFPs)
2.2.2. Design of Experiment (DOE) and Construction of the 21.31 Full Factorial Experimental Design
2.2.3. Characterization of the Prepared IFPs
Particle Size and Polydispersity Index (PDI) Determination
In Vitro Drug Release Profile and Kinetic Modeling of the Prepared IFPs
2.2.4. Desirability Study
2.2.5. Investigation of the Effect of Further Variations in Formulation Factors on the Selected Formulation
Effect of Adding Some Structural Additives
Effect of Solvent Variation
2.2.6. Characterization of the Optimized Formulation
Morphological Study Using Transmission Electron Microscope (TEM)
Rheological Study
PBPK Physiological Modeling
- Construction of the PBPK Model
- PBPK Model Evaluation
2.2.7. Statistical Analysis of Data
3. Results and Discussion
3.1. Preparation of Drug-Loaded In Situ-Forming Particulate Formulations (IFPs)
3.2. Statistical Analysis of the 21.32 Full Factorial Experimental Design
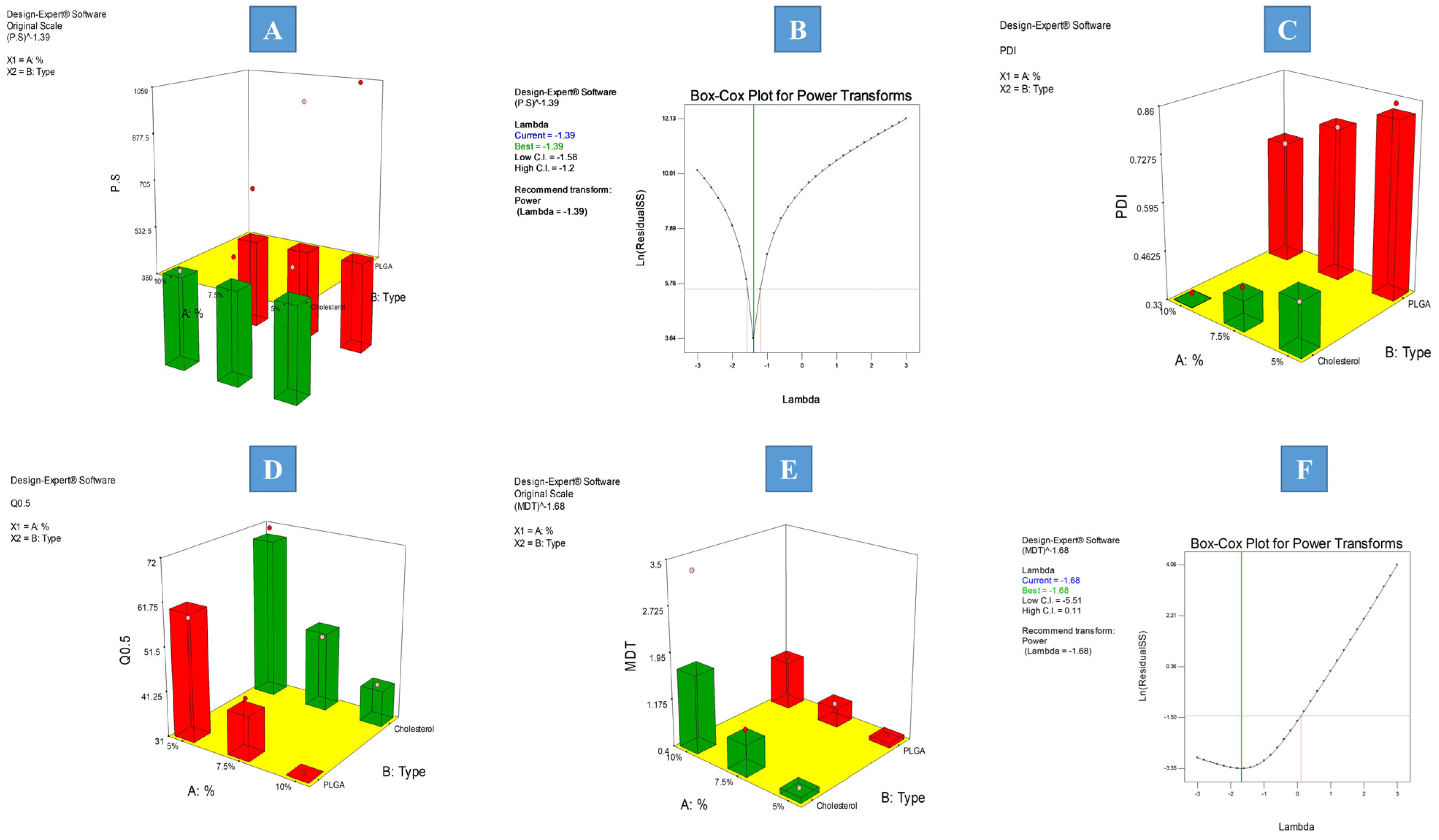
3.2.1. Effect of Formulation Factors on Average PS and PDI Values of the Prepared IFPs
3.2.2. Effect of Formulation Factors on In Vitro Release Parameters
Effect of Formulation Factors on Q0.5
Effect of Formulation Factors on the Mean Dissolution Time (MDT)
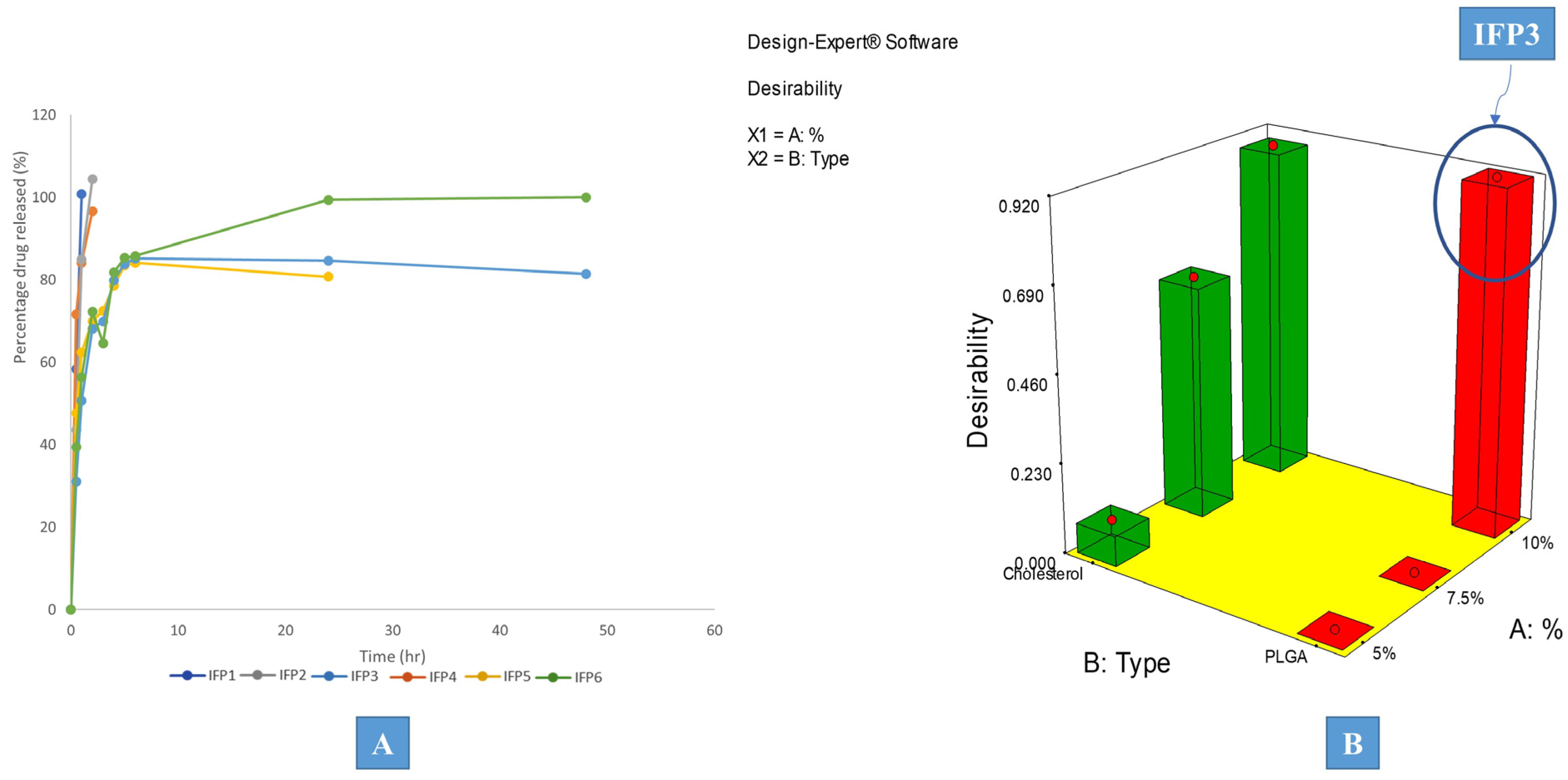
3.3. Desirability Study
3.4. Further Investigation of the Effect of Formulation Factors on the Selected Formulation
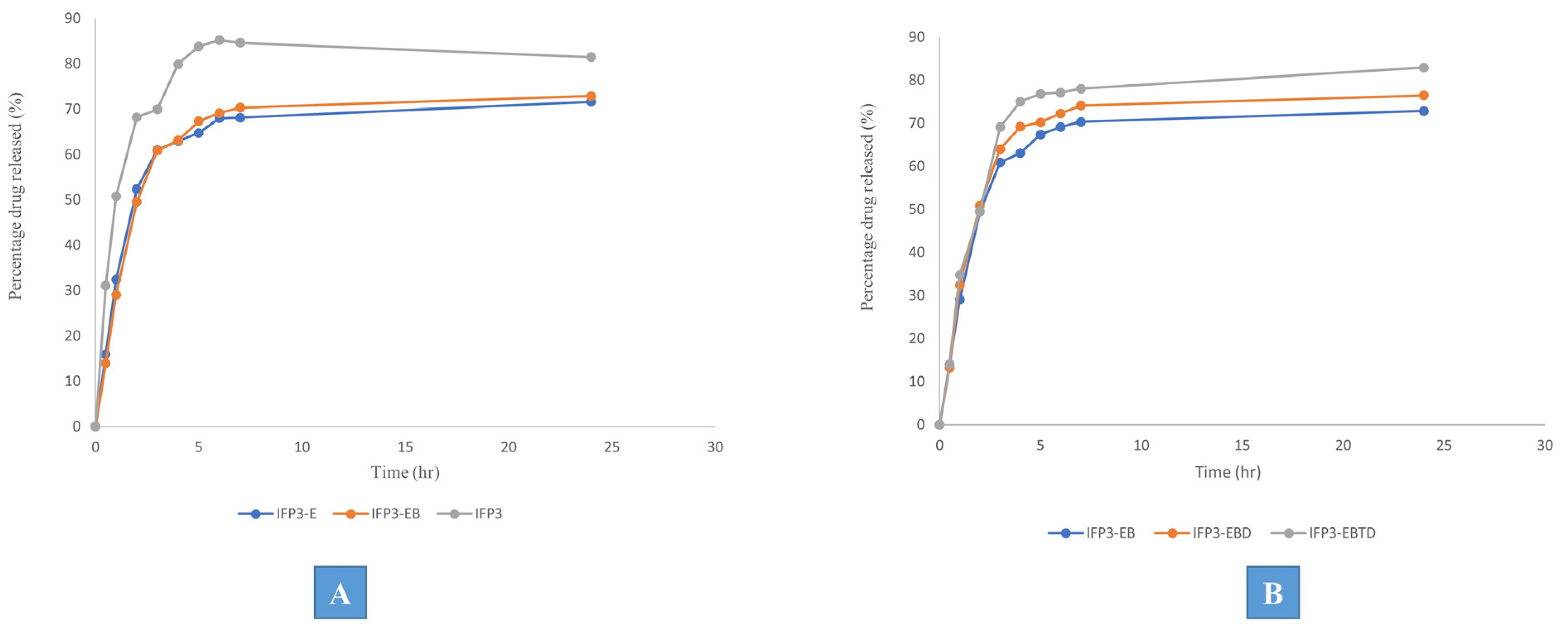
3.4.1. Effect of Some Structural Additives
3.4.2. Effect of Solvent Variation
3.5. Characterization of the Optimized Formulation
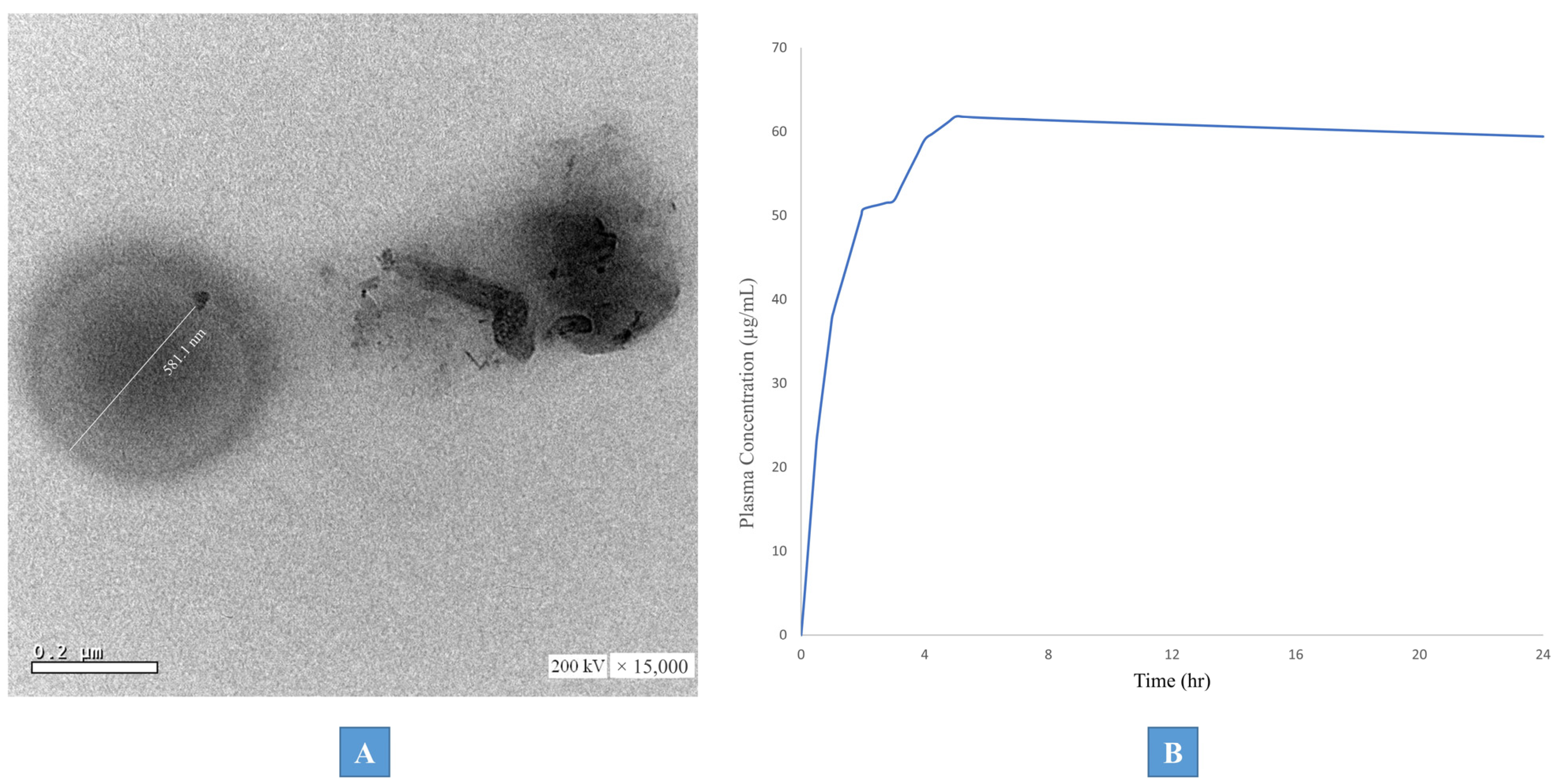
3.5.1. Transmission Electron Microscopy (TEM)
3.5.2. Rheological Study
3.5.3. PBPK Physiological Modeling
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Yun, Y.H.; Lee, B.K.; Park, K. Controlled Drug Delivery: Historical perspective for the next generation. J. Control. Release 2015, 219, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, G.; Tiwari, R.; Sriwastawa, B.; Bhati, L.; Pandey, S.; Pandey, P.; Bannerjee, S.K. Drug delivery systems: An updated review. Int. J. Pharm. Investig. 2012, 2, 2–11. [Google Scholar] [CrossRef]
- Trenfield, S.J.; Basit, A.W. Modified Drug Release: Current Strategies and Novel Technologies for Oral Drug Delivery. In Nanotechnology for Oral Drug Delivery; Martins, J.P., Santos, H.A., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 177–197, Chapter 6. [Google Scholar]
- Rahnfeld, L.; Luciani, P. Injectable Lipid-Based Depot Formulations: Where Do We Stand? Pharmaceutics 2020, 12, 567. [Google Scholar] [CrossRef] [PubMed]
- Patrick, M.G.; Vladimir, R.M. Pharmacokinetic and Pharmacodynamic Properties of Drug Delivery Systems. J. Pharmacol. Exp. Ther. 2019, 370, 570. [Google Scholar]
- Quarterman, J.C.; Geary, S.M.; Salem, A.K. Evolution of drug-eluting biomedical implants for sustained drug delivery. Eur. J. Pharm. Biopharm. 2021, 159, 21–35. [Google Scholar] [CrossRef]
- Prasad, S.; Dangi, J.S. Targeting efficacy and anticancer activity of polymeric nanoparticles of SN-38 on colon cancer cell lines. Futur. J. Pharm. Sci. 2023, 9, 1–9. [Google Scholar] [CrossRef]
- Camargo, J.A.; Sapin, A.; Nouvel, C.; Daloz, D.; Leonard, M.; Bonneaux, F.; Six, J.L.; Maincent, P. Injectable PLA-based in situ forming implants for controlled release of Ivermectin a BCS Class II drug: Solvent selection based on physico-chemical characterization. Drug Dev. Ind. Pharm. 2013, 39, 146–155. [Google Scholar] [CrossRef]
- Śmiga-Matuszowicz, M.; Korytkowska-Wałach, A.; Nowak, B.; Pilawka, R.; Lesiak, M.; Sieroń, A.L. Poly(isosorbide succinate)-based in situ forming implants as potential systems for local drug delivery: Preliminary studies. Mater. Sci. Eng. C 2018, 91, 311–317. [Google Scholar] [CrossRef]
- Haider, M.; Elsayed, I.; Ahmed, I.S.; Fares, A.R. In Situ-Forming Microparticles for Controlled Release of Rivastigmine: In Vitro Optimization and In Vivo Evaluation. Pharmaceuticals 2021, 14, 66. [Google Scholar] [CrossRef]
- Wang, X.; Burgess, D.J. Drug release from in situ forming implants and advances in release testing. Adv. Drug Deliv. Rev. 2021, 178, 113912. [Google Scholar] [CrossRef]
- Thakur, S.; Riyaz, B.; Patil, A.; Kaur, A.; Kapoor, B.; Mishra, V. Novel drug delivery systems for NSAIDs in management of rheumatoid arthritis: An overview. Biomed. Pharmacother. 2018, 106, 1011–1023. [Google Scholar] [CrossRef] [PubMed]
- Derry, S.; Massey, T.; Moore, R.A.; McQuay, H.J. Topical NSAIDS for chronic musculoskeletal pain in adults. Cochrane Database Syst. Rev. 2008. [Google Scholar] [CrossRef]
- Kasemiire, A.; Avohou, H.T.; De Bleye, C.; Sacre, P.-Y.; Dumont, E.; Hubert, P.; Ziemons, E. Design of experiments and design space approaches in the pharmaceutical bioprocess optimization. Eur. J. Pharm. Biopharm. 2021, 166, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Bowden, G.D.; Pichler, B.J.; Maurer, A. A Design of Experiments (DoE) Approach Accelerates the Optimization of Copper-Mediated 18F-Fluorination Reactions of Arylstannanes. Sci. Rep. 2019, 9, 11370. [Google Scholar] [CrossRef]
- Fairman, K.; Li, M.; Ning, B.; Lumen, A. Physiologically based pharmacokinetic (PBPK) modeling of RNAi therapeutics: Opportunities and challenges. Biochem. Pharmacol. 2021, 189, 114468. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Rowland, M.; Huang, S.-M. Best Practice in the Use of Physiologically Based Pharmacokinetic Modeling and Simulation to Address Clinical Pharmacology Regulatory Questions. Clin. Pharmacol. Ther. 2012, 92, 17–20. [Google Scholar] [CrossRef]
- Zhao, P.; Zhang, L.; Grillo, J.A.; Liu, Q.; Bullock, J.M.; Moon, Y.J.; Song, P.; Brar, S.S.; Madabushi, R.; Wu, T.C.; et al. Applications of Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulation During Regulatory Review. Clin. Pharmacol. Ther. 2011, 89, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Deepika, D.; Kumar, V. The Role of “Physiologically Based Pharmacokinetic Model (PBPK)” New Approach Methodology (NAM) in Pharmaceuticals and Environmental Chemical Risk Assessment. Int. J. Environ. Res. Public Health 2023, 20, 3473. [Google Scholar] [CrossRef]
- Alshawwa, S.Z.; Kassem, A.A.; Farid, R.M.; Mostafa, S.K.; Labib, G.S. Nanocarrier Drug Delivery Systems: Characterization, Limitations, Future Perspectives and Implementation of Artificial Intelligence. Pharmaceutics 2022, 14, 883. [Google Scholar] [CrossRef] [PubMed]
- Sager, J.E.; Yu, J.; Ragueneau-Majlessi, I.; Isoherranen, N. Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulation Approaches: A Systematic Review of Published Models, Applications, and Model Verification. Drug Metab. Dispos. 2015, 43, 1823. [Google Scholar] [CrossRef]
- Smits, A.; De Cock, P.; Vermeulen, A.; Allegaert, K. Physiologically based pharmacokinetic (PBPK) modeling and simulation in neonatal drug development: How clinicians can contribute. Expert Opin. Drug Metab. Toxicol. 2019, 15, 25–34. [Google Scholar] [CrossRef]
- Ammar, H.O.; Ibrahim, M.; Mahmoud, A.A.; Shamma, R.N.; El Hoffy, N.M. Non-ionic Surfactant Based In Situ Forming Vesicles as Controlled Parenteral Delivery Systems. AAPS PharmSciTech 2018, 19, 1001–1010. [Google Scholar] [CrossRef]
- Antony, J. (Ed.) 1—Introduction to Industrial Experimentation. In Design of Experiments for Engineers and Scientists, 2nd ed.; Elsevier: Oxford, UK, 2014; pp. 1–6. [Google Scholar]
- Muralidharan, K.; Romero, M.; Wüthrich, K. Factorial Designs, Model Selection, and (Incorrect) Inference in Randomized Experiments. Rev. Econ. Stat. 2023, 1–44. [Google Scholar] [CrossRef]
- Yu, M.; Yuan, W.; Li, D.; Schwendeman, A.; Schwendeman, S.P. Predicting drug release kinetics from nanocarriers inside dialysis bags. J. Control. Release 2019, 315, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.C.; Kumar, K.S.; Ramalingam, M. Planejamento e características de liberação de comprimidos de liberação controlada de cloridrato de metformina. Rev. Bras. Cienc. Farm. 2008, 44, 477–483. [Google Scholar] [CrossRef]
- Onnainty, R.; Granero, G. Chitosan-based nanocomposites: Promising materials for drug delivery applications. In Biomedical Applications of Nanoparticles; Grumezescu, A.M., Ed.; William Andrew Publishing: New York, NY, USA, 2019; pp. 375–407, Chapter 14. [Google Scholar]
- Buhr, E.; Senftleben, N.; Klein, T.; Bergmann, D.; Gnieser, D.; Frase, C.G.; Bosse, H. Characterization of nanoparticles by scanning electron microscopy in transmission mode. Meas. Sci. Technol. 2009, 20, 084025. [Google Scholar] [CrossRef]
- Blasco-Martinez, A.B.; Mateo-Orobia, A.; Blasco-Alberto, J.; Pablo-Julvez, L. Rheological Behavior Patterns in Artificial Tears. Optom. Vis. Sci. 2022, 99, 455–462. [Google Scholar] [CrossRef]
- Thelen, K.; Coboeken, K.; Willmann, S.; Burghaus, R.; Dressman, J.B.; Lippert, J. Evolution of a detailed physiological model to simulate the gastrointestinal transit and absorption process in humans, Part 1: Oral solutions. J. Pharm. Sci. 2011, 100, 5324–5345. [Google Scholar] [CrossRef] [PubMed]
- Ishizaki, T.; Nomura, T.; Abe, T. Pharmacokinetics of piroxicam, a new nonsteroidal anti-inflammatory agent, under fasting and postprandial states in man. J. Pharmacokinet. Biopharm. 1979, 7, 369–381. [Google Scholar] [CrossRef]
- Schmitt, W. General approach for the calculation of tissue to plasma partition coefficients. Toxicol. In Vitro 2008, 22, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Hindmarsh, A.C.; Brown, P.N.; Grant, K.E.; Lee, S.L.; Serban, R.; Shumaker, D.E.; Woodward, C.S. SUNDIALS: Suite of nonlinear and differential/algebraic equation solvers. ACM Trans. Math. Softw. 2005, 31, 363–396. [Google Scholar] [CrossRef]
- Calvo, A.M.; Santos, G.M.; Dionísio, T.J.; Marques, M.P.; Brozoski, D.T.; Lanchote, V.L.; Fernandes, M.H.R.; Faria, F.A.C.; Santos, C.F. Quantification of piroxicam and 5′-hydroxypiroxicam in human plasma and saliva using liquid chromatography–tandem mass spectrometry following oral administration. J. Pharm. Biomed. Anal. 2016, 120, 212–220. [Google Scholar] [CrossRef]
- Cho, C.; Kang, P.; Park, H.-J.; Ko, E.; Mu, C.Y.; Lee, Y.J.; Choi, C.-I.; Kim, H.S.; Jang, C.-G.; Bae, J.; et al. Physiologically based pharmacokinetic (PBPK) modeling of piroxicam with regard to CYP2C9 genetic polymorphism. Arch. Pharm. Res. 2022, 45, 352–366. [Google Scholar] [CrossRef]
- Porras, M.; Solans, C.; González, C.; Gutiérrez, J. Properties of water-in-oil (W/O) nano-emulsions prepared by a low-energy emulsification method. Colloids Surf. A Physicochem. Eng. Asp. 2008, 324, 181–188. [Google Scholar] [CrossRef]
- Saberi, A.H.; Fang, Y.; McClements, D.J. Fabrication of vitamin E-enriched nanoemulsions: Factors affecting particle size using spontaneous emulsification. J. Colloid Interface Sci. 2013, 391, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Goindi, S. Development and Optimization of Itraconazole-Loaded Solid Lipid Nanoparticles for Topical Administration Using High Shear Homogenization Process by Design of Experiments: In Vitro, Ex Vivo and In Vivo Evaluation. AAPS PharmSciTech 2021, 22, 248. [Google Scholar] [CrossRef]
- Ammar, H.O.; Ibrahim, M.; Mahmoud, A.A.; Shamma, R.N.; El Hoffy, N.M. Polymer-Free Injectable In Situ Forming Nanovesicles as a New Platform for Controlled Parenteral Drug Delivery Systems. J. Pharm. Innov. 2022, 17, 391–398. [Google Scholar] [CrossRef]
- Saharan, P.; Bahmani, K. Preparation, Optimization and In vitro Evaluation of Glipizide Nanoparticles Integrated with Eudragit RS-100. Pharm. Nanotechnol. 2019, 7, 72–85. [Google Scholar] [CrossRef]
- Yacoub, A.S.; Ammar, H.O.; Ibrahim, M.; Mansour, S.M.; El Hoffy, N.M. Artificial intelligence-assisted development of in situ forming nanoparticles for arthritis therapy via intra-articular delivery. Drug Deliv. 2022, 29, 1423–1436. [Google Scholar] [CrossRef]
- Galvao, J.; Davis, B.; Tilley, M.; Normando, E.; Duchen, M.R.; Cordeiro, M.F. Unexpected low-dose toxicity of the universal solvent DMSO. FASEB J. 2014, 28, 1317–1330. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Sharma, R.; Jain, D.K. Nanotechnology Based Approaches for Enhancing Oral Bioavailability of Poorly Water Soluble Antihypertensive Drugs. Scientifica 2016, 2016, 8525679. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulae | Independent Factors | Dependent Factors * | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Percentage of Particulate Forming Agent | Type of Particulate Forming Agent | Particle Size Mean ± SD (nm) | PDI Mean ± SD (nm) | Zeta Potential Mean ± SD (mV) | Q0.5 ± SD (h) | K ± SD (h−1) | T25% ± SD (h) | T50% ± SD (h) | T90% ± SD (h) | MDT ± SD (h) | |
| IFP1 | 5% | PLGA | 1043 ± 98.26 | 0.854 ± 0.064 | −11.6 ± 0.81 | 58.4 ± 3.31 | 2.20 ± 0.14 | 0.13 ± 0.016 | 0.32 ± 0.017 | 1.51 ±0.12 | 0.46 ± 0.03 |
| IFP2 | 7.5% | PLGA | 951 ± 87.10 | 0.751 ± 0.041 | −11.4 ± 0.92 | 43.65 ± 2.34 | 1.53 ± 0.12 | 0.19 ± 0.013 | 0.45 ± 0.020 | 3.60 ± 0.42 | 0.68 ± 0.04 |
| IFP3 | 10% | PLGA | 569 ± 34.21 | 0.663 ± 0.036 | −18.4 ± 1.56 | 31.10 ± 2.80 | 0.50 ± 0.06 | 0.58 ± 0.041 | 1.40 ± 0.160 | 4.66 ± 0.51 | 1.24 ± 0.14 |
| IFP4 | 5% | Cholesterol | 470.5 ± 23.89 | 0.452 ± 0.035 | −12.6 ± 0.76 | 71.65 ± 5.42 | 2.27 ± 0.19 | 0.13 ± 0.018 | 0.31 ± 0.024 | 4.17 ± 0.36 | 0.48 ± 0.06 |
| IFP5 | 7.5% | Cholesterol | 462 ± 39.74 | 0.426 ± 0.024 | −12.5 ± 1.10 | 47.70 ± 3.90 | 0.64 ± 0.08 | 0.45 ± 0.032 | 1.10 ± 0.250 | 1.05 ± 0.16 | 1.04 ± 0.09 |
| IFP6 | 10% | Cholesterol | 363.5 ± 21.67 | 0.347 ± 0.017 | −12.8 ± 1.07 | 39.45 ± 1.23 | 0.55 ± 0.02 | 0.52 ± 0.047 | 1.26 ± 0.19 | 1.02 ± 0.22 | 1.32 ± 0.28 |
| Model Parameters | PDI Mean (nm) | Particle Size Mean (nm) | Q0.5 (h) | MDT (h) |
|---|---|---|---|---|
| Model Type | Main Effects | Main Effects | Main Effects | Main Effects |
| R2 | 0.9891 | 0.9999 | 0.9797 | 0.9868 |
| Adjusted R2 | 0.9728 | 0.9997 | 0.9492 | 0.9670 |
| Predicted R2 | 0.9022 | 0.9990 | 0.8171 | 0.8811 |
| Adequate Precision | 18.163 | 187.885 | 14.411 | 16.929 |
| Final Equation in Terms of Coded Factors | PDI = +0.58 +0.071 * A [1] +6.333 × 10−3 * A [2] −0.17 * B | (P.S)^−1.39 = +1.585 × 10−4 −3.019 × 10−5 * A [1] −2.335 × 10−5 * A [2] +6.371 × 10−5 * B | Q0.5 = +48.66 +16.37 * A [1] −2.98 * A [2] +4.28 * B | (MDT)^−1.68 = +1.80 +1.77 * A [1] −0.38 * A [2] −0.29 * B |
| Reference | AUC0-24 (μg·h/mL) | Cmax (μg/mL) | Tmax (h) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Observed | Predicted | Fold Error * | Observed | Predicted | Fold Error * | Observed | Predicted | Fold Error * | |
| Calvo et al. [36] | 78.7 | 45.4 | 0.6 | 2.28 | 2 | 0.9 | 4 | 5.25 | 1.3 |
| AUC0-24 (μg·h/mL) | Cmax (μg/mL) | Tmax (h) |
|---|---|---|
| 1383.03 | 61.79 | 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Hoffy, N.M.; Yacoub, A.S.; Ghoneim, A.M.; Ibrahim, M.; Ammar, H.O.; Eissa, N. Computational Amendment of Parenteral In Situ Forming Particulates’ Characteristics: Design of Experiment and PBPK Physiological Modeling. Pharmaceutics 2023, 15, 2513. https://doi.org/10.3390/pharmaceutics15102513
El Hoffy NM, Yacoub AS, Ghoneim AM, Ibrahim M, Ammar HO, Eissa N. Computational Amendment of Parenteral In Situ Forming Particulates’ Characteristics: Design of Experiment and PBPK Physiological Modeling. Pharmaceutics. 2023; 15(10):2513. https://doi.org/10.3390/pharmaceutics15102513
Chicago/Turabian StyleEl Hoffy, Nada M., Ahmed S. Yacoub, Amira M. Ghoneim, Magdy Ibrahim, Hussein O. Ammar, and Nermin Eissa. 2023. "Computational Amendment of Parenteral In Situ Forming Particulates’ Characteristics: Design of Experiment and PBPK Physiological Modeling" Pharmaceutics 15, no. 10: 2513. https://doi.org/10.3390/pharmaceutics15102513