

Improve BBB Penetration and Cytotoxicity of Palbociclib in U87-MG Glioblastoma Cells Delivered by Dual Peptide Functionalized Nanoparticles
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis and Characterization of Peptide-Conjugated PLGA-PEG Copolymers
2.3. Preparation and Characterization of PBC Loaded NPs (PBC@NPs)
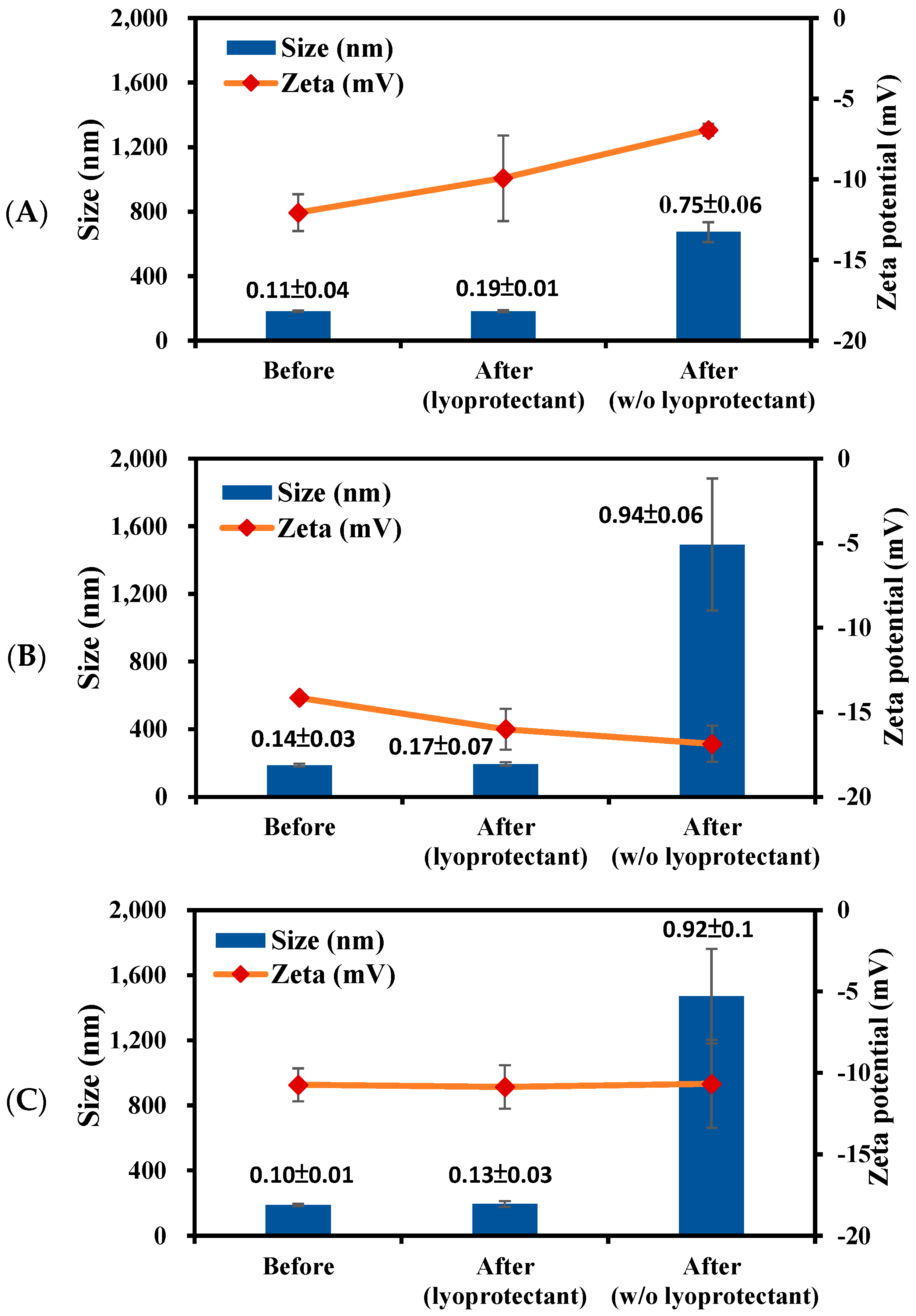
2.4. Stability Study
2.5. In Vitro Release Study
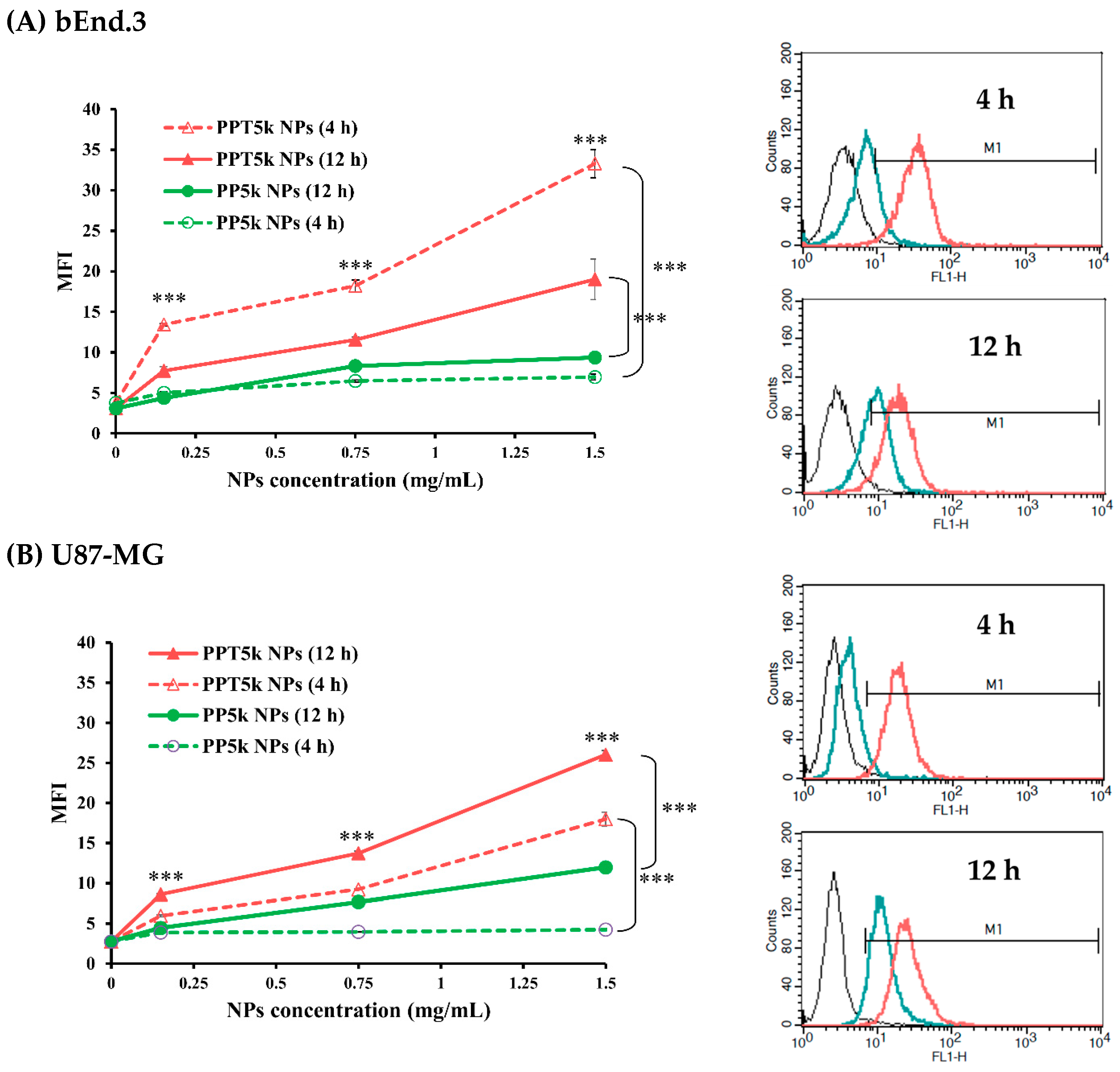
2.6. Cellular Uptake in a Co-Cultured BBB Model
2.7. In Vitro Transport of PBC@NPs in a bEnd.3 Monolayer
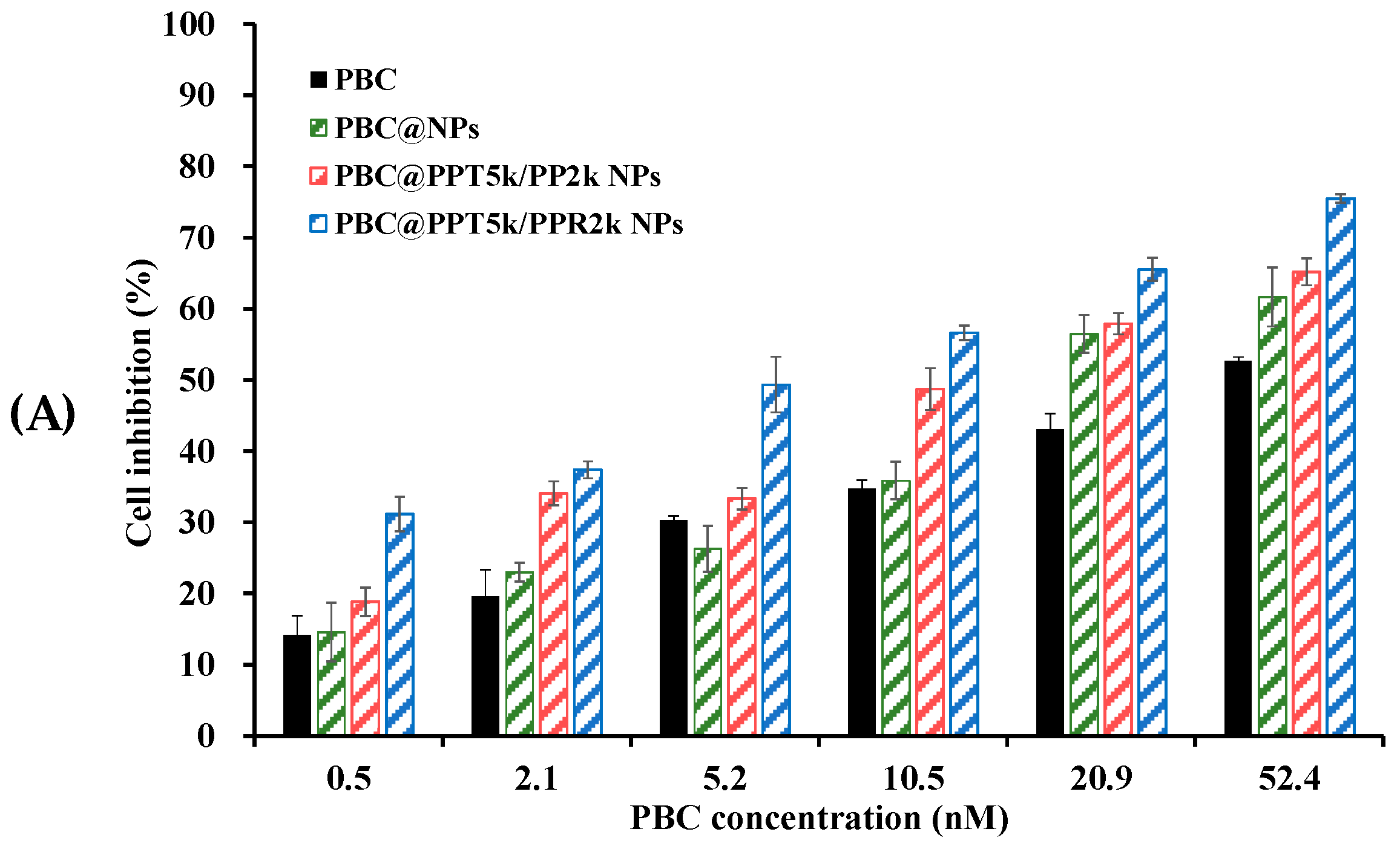
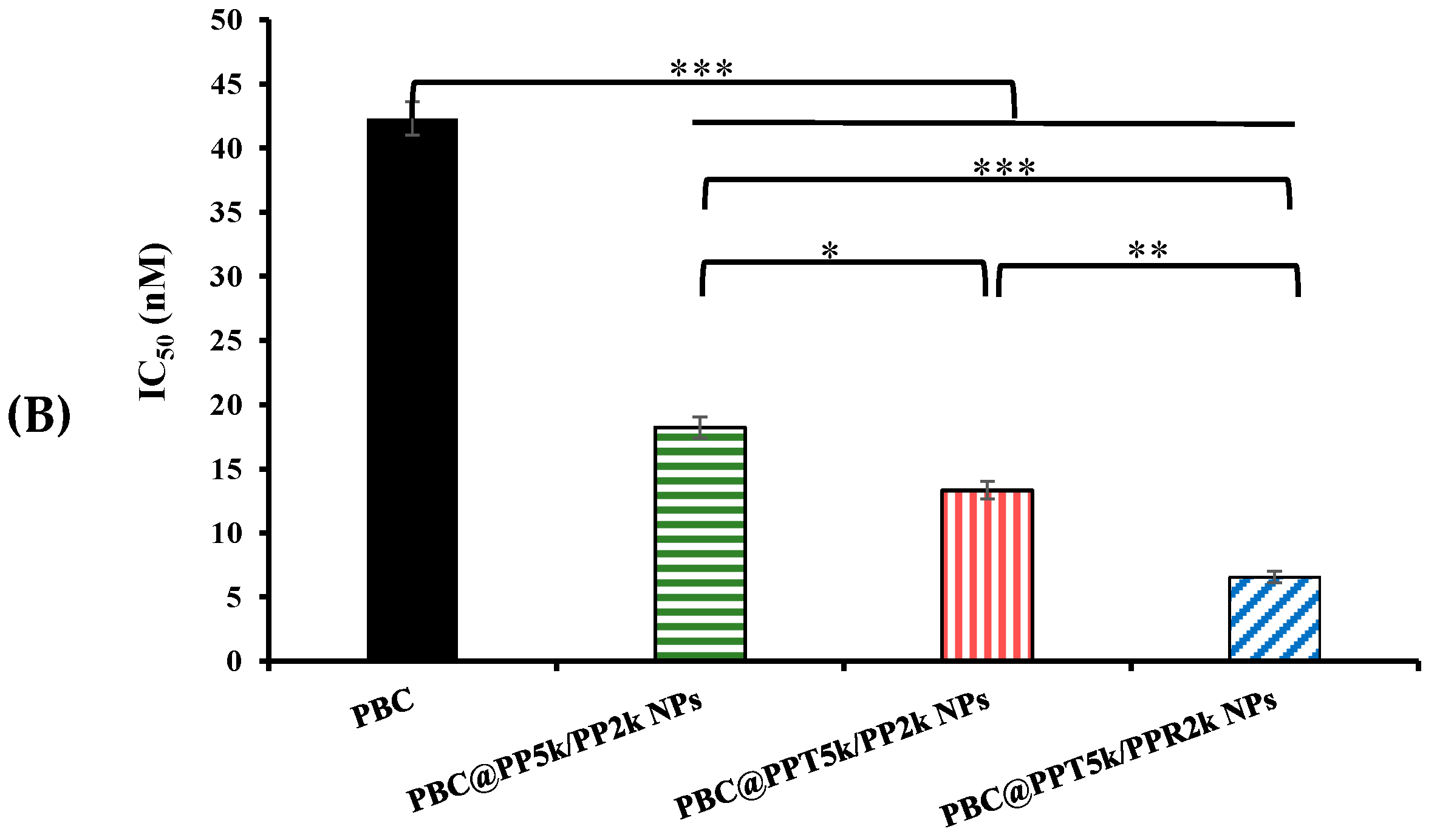
2.8. Cytotoxicity
2.9. Statistics
3. Results and Discussion
3.1. Characterization of Peptide-Conjugated PLGA-PEG Copolymers
3.2. Characterization of PBC Loaded Nanoparticles (PBC@NPs)
3.3. Stability of PBC@NPs
3.4. In Vitro Release Study
3.5. Cellular Uptake in a Co-Cultured BBB Model
3.6. Transport of PBC@NPs across bEnd.3 Cell Model
3.7. Cytotoxicity of PBC@NPs in U87-MG Cells
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Abbreviation | |
| PBC | Palbociclib |
| PBC@NP | Palbociclib loaded NPs |
| PP5k | PLGA-PEG5k copolymer (MW of PEG ~5000 Da) |
| PP2k | PLGA-PEG2k copolymer (MW of PEG ~2000 Da) |
| PPT5k | T7 peptide conjugated with PP5k |
| PPR2k | R9 peptide conjugated with PP2k |
| PP5k/PP2k NP (peptide-free NPs) | NPs prepared by PP5k and PP2k (3:1 w/w) |
| PPT5k/PP2k NPs (T7 peptide-conjugated NPs) | NPs prepared by PPT5k and PP2k (3:1 w/w) |
| PPT5k/PPR2k NPs (T7/R9 dual peptide conjugated NPs) | NPs prepared by PPT5k and PPR2k (3:1 w/w) |
References
- Mair, M.J.; Geurts, M.; van den Bent, M.J.; Berghoff, A.S. A basic review on systemic treatment options in WHO grade II–III gliomas. Cancer Treat. Rev. 2021, 92, 102124. [Google Scholar] [CrossRef] [PubMed]
- Shah, F.H.; Salman, S.; Idrees, J.; Idrees, F.; Shah, S.T.A.; Khan, A.A.; Ahmad, B. Current progress of phytomedicine in glioblastoma therapy. Curr. Med. Sci. 2020, 40, 1067–1074. [Google Scholar] [CrossRef] [PubMed]
- Erthal, L.C.S.; Gobbo, O.L.; Ruiz-Hernandez, E. Biocompatible copolymer formulations to treat glioblastoma multiforme. Acta Biomater. 2021, 121, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Ro, J.; Andr, F.; Loi, S.; Verma, S.; Iwata, H.; Harbeck, N.; Loibl, S.; Bartlett, C.H.; Zhang, K.; et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 2015, 373, 209–219. [Google Scholar] [CrossRef]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef]
- Spring, L.; Bardia, A.; Modi, S. Targeting the cyclin D-cyclin-dependent kinase (CDK)4/6-retinoblastoma pathway with selective CDK 4/6 inhibitors in hormone receptor-positive breast cancer: Rationale, current status, and future directions. Discov. Med. 2016, 21, 65–74. [Google Scholar]
- Michaud, K.; Solomon, D.A.; Oermann, E.; Kim, J.S.; Zhong, W.Z.; Prados, M.D.; Ozawa, T.; James, C.D.; Waldman, T. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 2010, 70, 3228–3238. [Google Scholar] [CrossRef]
- Schroder, L.B.; McDonald, K.L. CDK4/6 Inhibitor PD0332991 in glioblastoma treatment: Does it have a future? Front. Oncol. 2015, 5, 259. [Google Scholar] [CrossRef]
- Barton, K.L.; Misuraca, K.; Cordero, F.; Dobrikova, E.; Min, H.D.; Gromeier, M.; Kirsch, D.G.; Becher, O.J. PD-0332991, a CDK4/6 inhibitor, significantly prolongs survival in a genetically engineered mouse model of brainstem glioma. PLoS ONE 2013, 8, e77639. [Google Scholar] [CrossRef]
- de Gooijer, M.C.; Zhang, P.; Thota, N.; Mayayo-Peralta, I.; Buil, L.C.M.; Beijnen, J.H.; van Tellingen, O. P-glycoprotein and breast cancer resistance protein restrict the brain penetration of the CDK4/6 inhibitor Palbociclib. Investig. New Drugs 2015, 33, 1012–1019. [Google Scholar] [CrossRef]
- Bronner, S.M.; Merrick, K.A.; Murray, J.; Salphati, L.; Moffat, J.G.; Pang, J.; Sneeringer, C.J.; Dompe, N.; Cyr, P.; Purkey, H.; et al. Design of a brain-penetrant CDK4/6 inhibitor for glioblastoma. Bioorg. Med. Chem. Lett. 2019, 29, 2294–2301. [Google Scholar] [CrossRef] [PubMed]
- Oberoi, R.K.; Parrish, K.E.; Sio, T.T.; Mittapalli, R.K.; Elmquist, W.F.; Sarkaria, J.N. Strategies to improve delivery of anticancer drugs across the blood-brain barrier to treat glioblastoma. Neuro. Oncol. 2016, 18, 27–36. [Google Scholar] [CrossRef]
- Mehata, A.K.; Singh, V.; Vikas, S.N.; Mandal, A.; Dash, D.; Koch, B.; Muthu, M.S. Chitosan-g-estrone nanoparticles of palbociclib vanished hypoxic breast tumor after targeted delivery: Development and ultrasound/photoacoustic imaging. ACS Appl. Mater. Interfaces 2023, 15, 34343–34359. [Google Scholar] [CrossRef] [PubMed]
- Persha, H.E.; Kato, S.; De, P.; Adashek, J.J.; Sicklick, J.K.; Subbiah, V.; Kurzrock, R. Osteosarcoma with cell-cycle and fibroblast growth factor genomic alterations: Case report of molecular tumor board combination strategy resulting in long-term exceptional response. J. Hematol. Oncol. 2022, 15, 119. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Kim, B.J.; Singh, N.P.; Lai, H.; Sasaki, T. Synthesis and anti-cancer activity of covalent conjugates of artemisinin and a transferrin-receptor targeting peptide. Cancer Lett. 2009, 274, 33–39. [Google Scholar] [CrossRef]
- Bi, Y.; Liu, L.; Lu, Y.; Sun, T.; Shen, C.; Chen, X.; Chen, Q.; An, S.; He, X.; Ruan, C.; et al. T7 peptide-functionalized PEG-PLGA micelles loaded with carmustine for targeting therapy of glioma. ACS Appl. Mater. Interfaces 2016, 8, 27465–27473. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zhang, M.; Zeng, F.; Jin, H.; Xu, Q.; Huang, Y. Dual-targeting magnetic PLGA nanoparticles for codelivery of paclitaxel and curcumin for brain tumor therapy. ACS Appl Mater. Interfaces 2016, 8, 32159–32169. [Google Scholar] [CrossRef]
- Han, L.; Huang, R.; Liu, S.; Huang, S.; Jiang, C. Peptide-conjugated PAMAM for targeted doxorubicin delivery to transferrin receptor overexpressed tumors. Mol. Pharm. 2010, 7, 2156–2165. [Google Scholar] [CrossRef]
- Lee, J.H.; Engler, J.A.; Collawn, J.F.; Moore, B.A. Receptor mediated uptake of peptides that bind the human transferrin receptor. Eur. J. Biochem. 2001, 268, 2004–2012. [Google Scholar] [CrossRef]
- Wang, X.; Mao, W.; Wang, Z.; Li, X.; Xiong, Y.; Lu, H.; Wang, X.; Yin, H.; Cao, X.; Xin, H. Enhanced anti-brain metastasis from non-small cell lung cancer of osimertinib and doxorubicin co-delivery targeted nanocarrier. Inter. J. Nanomed. 2020, 15, 5491–5501. [Google Scholar] [CrossRef]
- Wang, H.; Chen, W.; Wu, G.; Kong, J.; Yuan, S.; Chen, L. A magnetic T7 peptide&AS1411 aptamer-modified microemulsion for triple glioma-targeted delivery of shikonin and docetaxel. J. Pharm. Sci. 2021, 110, 2946–2954. [Google Scholar] [PubMed]
- Li, C.; Guan, N.; Liu, F. T7 peptide-decorated exosome-based nanocarrier system for delivery of Galectin-9 siRNA to stimulate macrophage repolarization in glioblastoma. J. Neurooncol. 2023, 162, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Zhang, R.; Wang, Y.; Zhang, X.; Yang, Y.; Zhao, B.; Yang, L. A simple self-assembly nanomicelle based on brain tumor-targeting peptide-mediated siRNA delivery for glioma immunotherapy via intranasal administration. Acta Biomater. 2023, 155, 521–537. [Google Scholar] [CrossRef] [PubMed]
- Stiltner, J.; McCandless, K.; Zahid, M. Cell-penetrating peptides: Applications in tumor diagnosis and therapeutics. Pharmaceutics 2021, 13, 890. [Google Scholar] [CrossRef] [PubMed]
- Chiarpotti, M.V.; Longo, G.S.; Del Pópolo, M.G. Nanoparticles modified with cell penetrating peptides: Assessing adsorption on membranes containing acidic lipids. Colloids Surf. B Biointerfaces 2021, 197, 111373. [Google Scholar] [CrossRef]
- McErlean, E.M.; Ziminska, M.; McCrudden, C.M.; McBride, J.W.; Loughran, S.P.; Cole, G.; Mulholland, E.J.; Kett, V.; Buckley, N.E.; Robson, T.; et al. Rational design and characterisation of a linear cell penetrating peptide for non-viral gene delivery. J. Control. Release 2021, 330, 1288–1299. [Google Scholar] [CrossRef]
- Sadeghian, I.; Heidari, R.; Raee, M.J.; Negahdaripour, M. Cell-penetrating peptide-mediated delivery of therapeutic peptides/proteins to manage the diseases involving oxidative stress, inflammatory response and apoptosis. J. Pharm. Pharmacol. 2022, 74, 1085–1116. [Google Scholar] [CrossRef]
- Kurrikoff, K.; Vunk, B.; Langel, Ü. Status update in the use of cell-penetrating peptides for the delivery of macromolecular therapeutics. Expert Opin. Biol. Ther. 2021, 21, 361–370. [Google Scholar] [CrossRef]
- Falato, L.; Gestin, M.; Langel, Ü. Cell-penetrating peptides delivering siRNAs: An Overview. Methods Mol. Biol. 2021, 2282, 329–352. [Google Scholar]
- Zhang, C.; Yuan, W.; Wu, Y.; Wan, X.; Gong, Y. Co-delivery of EGFR and BRD4 siRNA by cell-penetrating peptides-modified redox-responsive complex in triple negative breast cancer cells. Life Sci. 2021, 266, 118886. [Google Scholar] [CrossRef]
- Feldman, K.S.; Pavlou, M.P.; Zahid, M. Cardiac targeting peptide: From identification to validation to mechanism of transduction. Methods Mol. Biol. 2021, 2211, 97–112. [Google Scholar] [PubMed]
- Ghorai, S.M.; Deep, A.; Magoo, D.; Gupta, C.; Gupta, N. Cell-penetrating and targeted peptides delivery systems as potential pharmaceutical carriers for enhanced delivery across the blood-brain barrier (BBB). Pharmaceutics 2023, 15, 1999. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Breton, A.L.; Préat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Wicki, A.; Witzigmann, D.; Balasubramanian, V.; Huwyler, J. Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications. J. Control. Release 2015, 200, 138–157. [Google Scholar] [CrossRef]
- Lin, W.J.; Kao, L.T. Cytotoxic enhancement of hexapeptide-conjugated micelles in EGFR high-expressed cancer cells. Expert Opin. Drug Deliv. 2014, 11, 1537–1550. [Google Scholar] [CrossRef]
- Huang, H.L.; Lin, W.J. Dual peptide-modified nanoparticles improve combination chemotherapy of etoposide and siPIK3CA against drug-resistant small cell lung carcinoma. Pharmaceutics 2020, 12, 254. [Google Scholar] [CrossRef]
- He, G.Z.; Lin, W.J. Peptide-functionalized nanoparticles-encapsulated cyclin-dependent kinases inhibitor seliciclib in transferrin receptor overexpressed cancer cells. Nanomaterial 2021, 11, 772. [Google Scholar] [CrossRef]
- Wu, R.; Tian, M.; Shu, C.; Zhou, C.; Guan, W. Determination of the critical micelle concentration of surfactants using fluorescence strategies. Soft Matter 2022, 18, 8920. [Google Scholar] [CrossRef]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef]
- Alai, M.; Lin, W.J. A novel once daily microparticulate dosage form comprising lansoprazole to prevent nocturnal acid breakthrough in the case of gastro-esophageal reflux disease: Preparation, pharmacokinetic and pharmacodynamic evaluation. J. Microencapsul. 2013, 30, 519–529. [Google Scholar] [CrossRef]
- Biemans, E.; Jakel, L.; de Waal, R.M.W.; Kuiperij, H.B.; Verbeek, M.M. Limitations of the hCMEC/D3 cell line as a model for Abeta clearance by the human blood-brain barrier. J. Neurosci. Res. 2017, 95, 1513–1522. [Google Scholar] [CrossRef]
- Goyal, R.; Macri, L.; Kohn, J. Formulation strategy for the delivery of cyclosporine a: Comparison of two polymeric nanospheres. Sci. Rep. 2015, 5, 13065. [Google Scholar] [CrossRef] [PubMed]
- Dorati, R.; DeTrizio, A.; Spalla, M.; Migliavacca, R.; Pagani, L.; Pisani, S.; Chiesa, E.; Conti, B.; Modena, T.; Genta, I. Gentamicin sulfate PEG-PLGA/PLGA-H nanoparticles: Screening design and antimicrobial effect evaluation toward clinic bacterial isolates. Nanomaterials 2018, 8, E37. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Monteiro-Riviere, N.A. Mechanisms of cell uptake, inflammatory potential and protein corona effects with gold nanoparticles. Nanomedicine 2016, 11, 3185–3203. [Google Scholar] [CrossRef]
- Dash, S.; Murthy, P.N.; Nath, L.; Chowdhury, P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol. Pharm. 2010, 67, 217–223. [Google Scholar] [PubMed]
- Du, J.; Lane, L.A.; Nie, S. Stimuli-responsive nanoparticles for targeting the tumor microenvironment. J. Control. Release 2015, 219, 205–214. [Google Scholar] [CrossRef]
- Creixell, M.; Peppas, N.A. Co-delivery of siRNA and therapeutic agents using nanocarriers to overcome cancer resistance. Nano Today 2012, 7, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Watanabe, R.; Choyke, P.L. Improving conventional enhanced permeability and retention (EPR) effects; what is the appropriate target? Theranostics 2014, 4, 81–89. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NPs | Yield (%) | Size (nm) | PdI | Zeta (mV) | EE (%) | DL (%) |
|---|---|---|---|---|---|---|
| PBC@PP5k/PP2k NPs | 65.1 ± 2.5 | 168.4 ± 4.3 | 0.15 ± 0.04 | −16.0 ± 1.4 | 60.6 ± 4.4 | 5.3 ± 0.4 |
| PBC@PPT5k/PP2k NPs | 65.7 ± 6.4 | 173.2 ± 7.8 | 0.13 ± 0.03 | −17.8 ± 1.4 | 62.0 ± 5.3 | 5.3 ± 0.1 |
| PBC@PPT5k/PPR2k NPs | 68.9 ± 2.5 | 185.8 ± 4.4 | 0.09 ± 0.07 | −14.3 ± 1.0 | 60.5 ± 3.6 | 5.0 ± 0.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lo, Y.-C.; Lin, W.-J. Improve BBB Penetration and Cytotoxicity of Palbociclib in U87-MG Glioblastoma Cells Delivered by Dual Peptide Functionalized Nanoparticles. Pharmaceutics 2023, 15, 2429. https://doi.org/10.3390/pharmaceutics15102429
Lo Y-C, Lin W-J. Improve BBB Penetration and Cytotoxicity of Palbociclib in U87-MG Glioblastoma Cells Delivered by Dual Peptide Functionalized Nanoparticles. Pharmaceutics. 2023; 15(10):2429. https://doi.org/10.3390/pharmaceutics15102429
Chicago/Turabian StyleLo, Yu-Chen, and Wen-Jen Lin. 2023. "Improve BBB Penetration and Cytotoxicity of Palbociclib in U87-MG Glioblastoma Cells Delivered by Dual Peptide Functionalized Nanoparticles" Pharmaceutics 15, no. 10: 2429. https://doi.org/10.3390/pharmaceutics15102429