
Targeting Lactate Dehydrogenase-B as a Strategy to Fight Cancer: Identification of Potential Inhibitors by In Silico Analysis and In Vitro Screening


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Virtual Screening
2.2.2. Molecular Dynamics
2.2.3. Molecular Docking
2.2.4. LDH-B Activity Assay
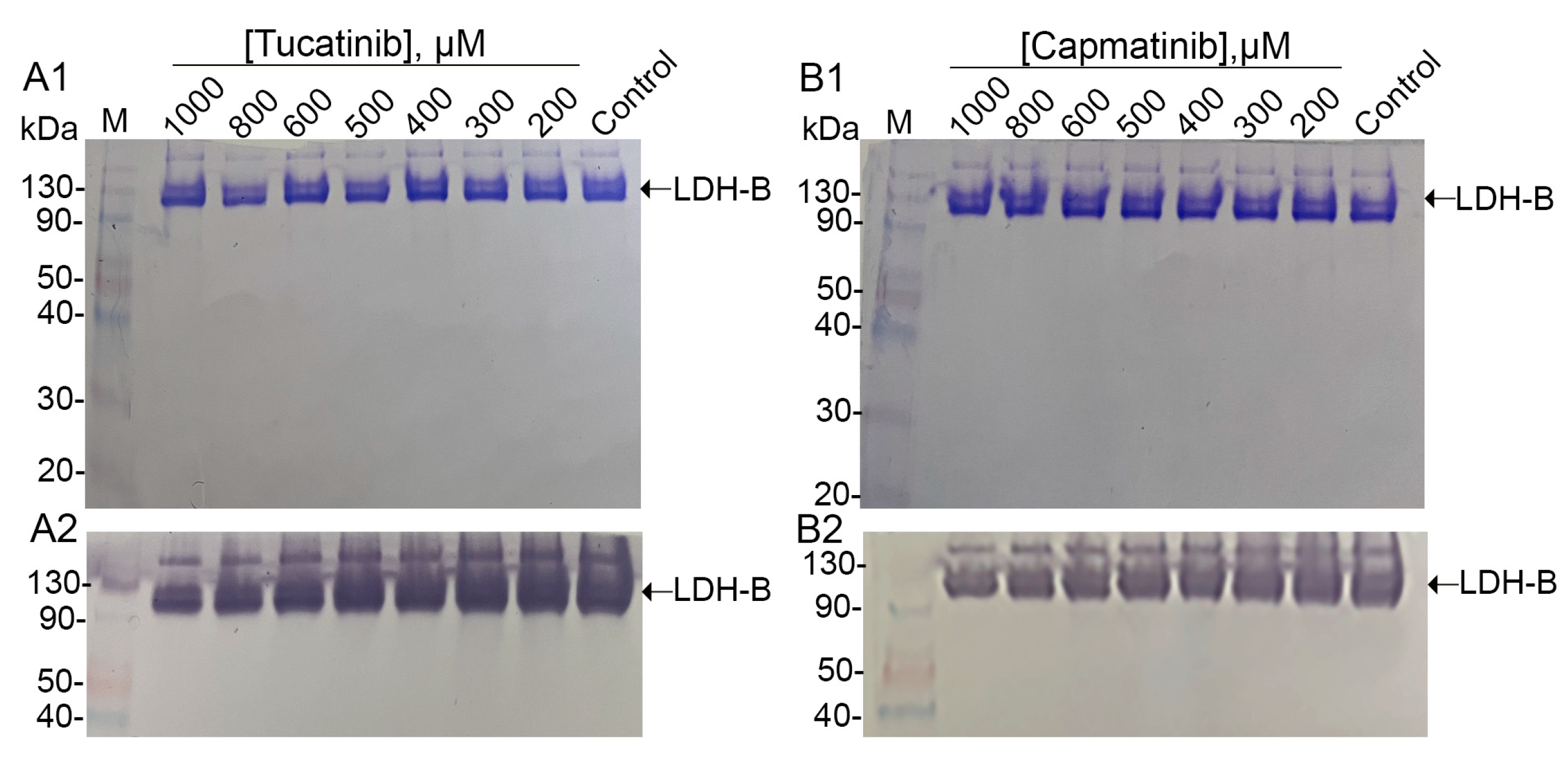
2.2.5. Zymogram Analysis
2.2.6. Kinetic Studies
2.2.7. Statistical Analysis
3. Results
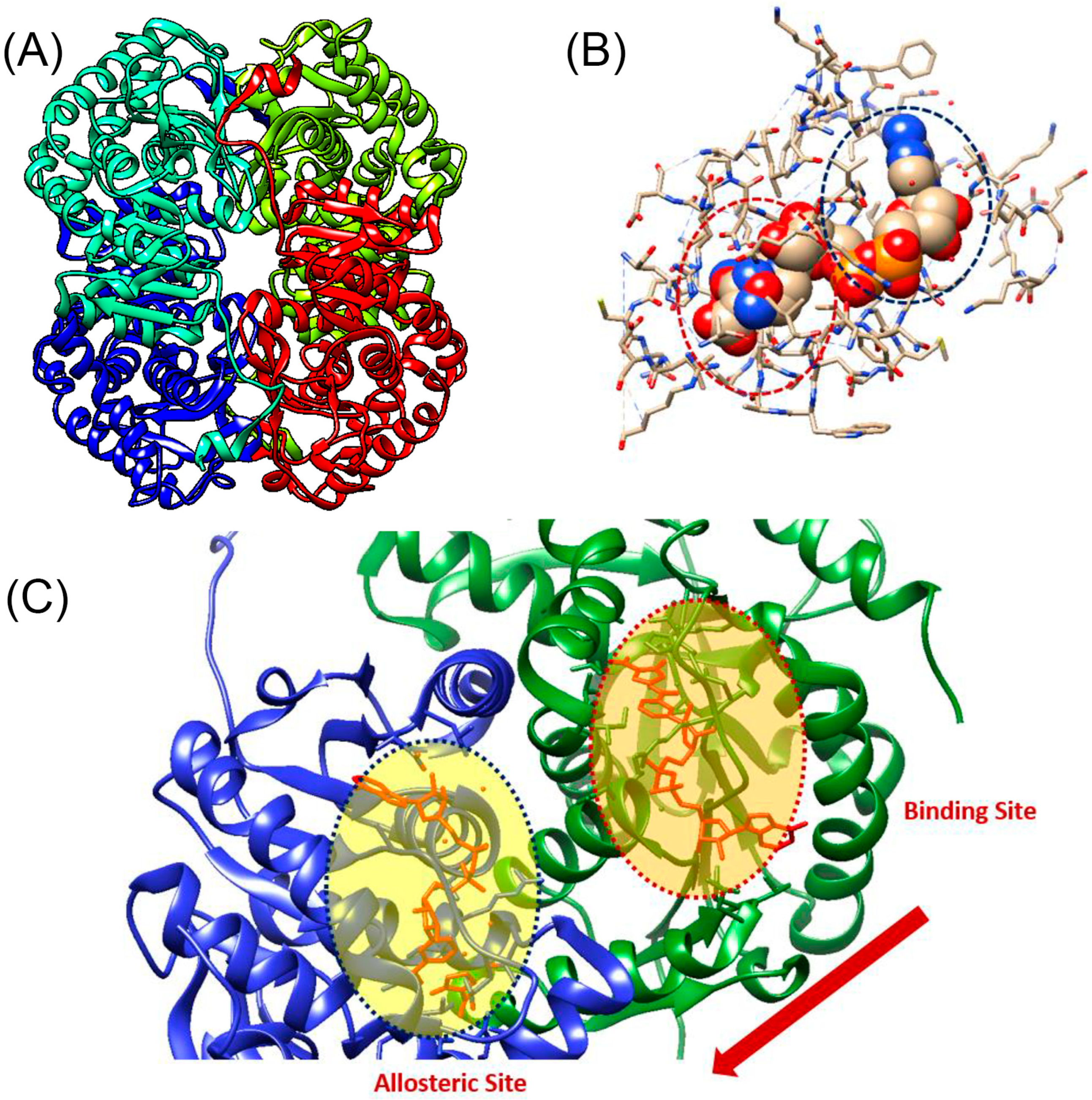
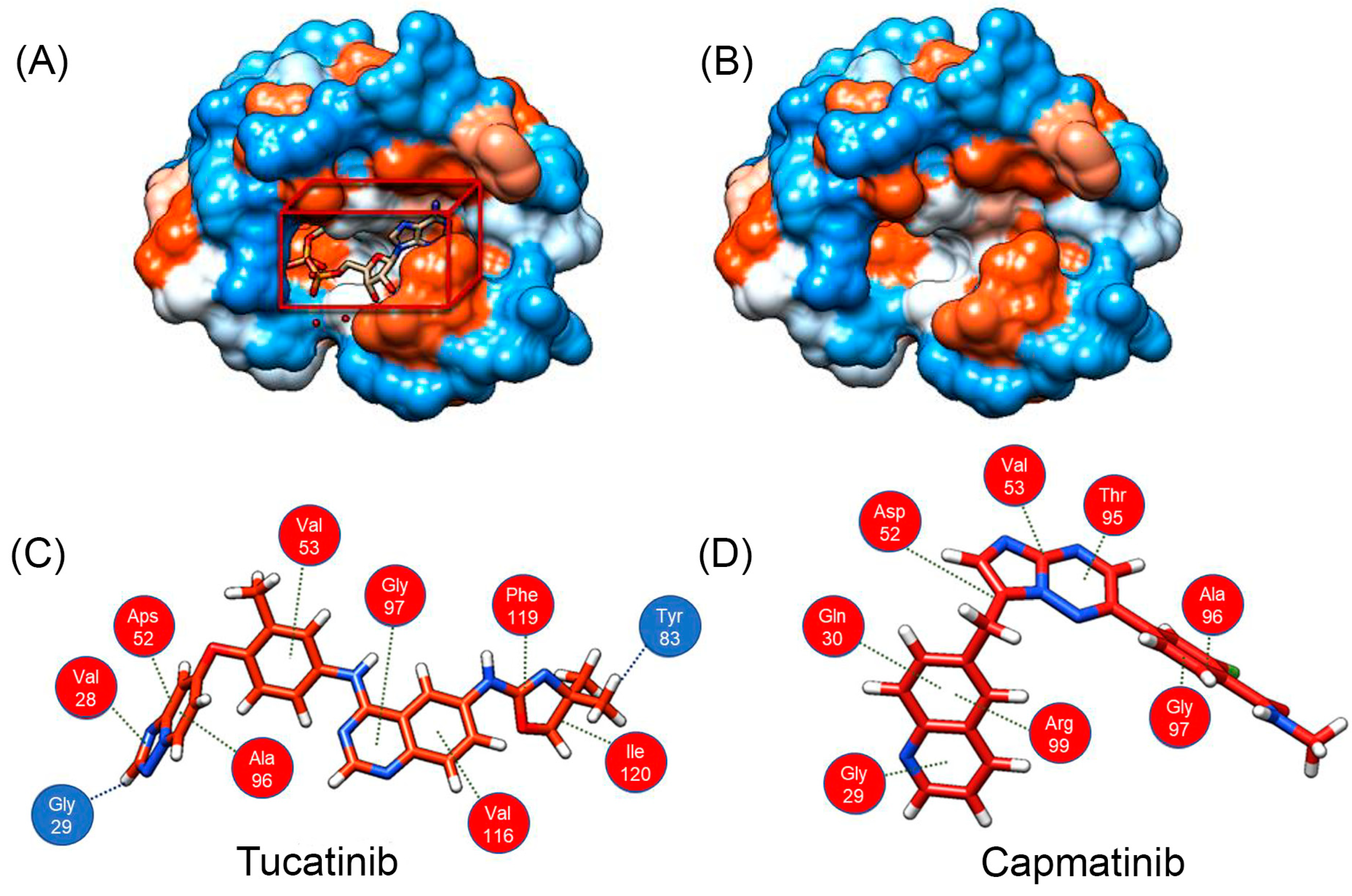
3.1. In Silico Screening of Potential LDH-B Inhibitors
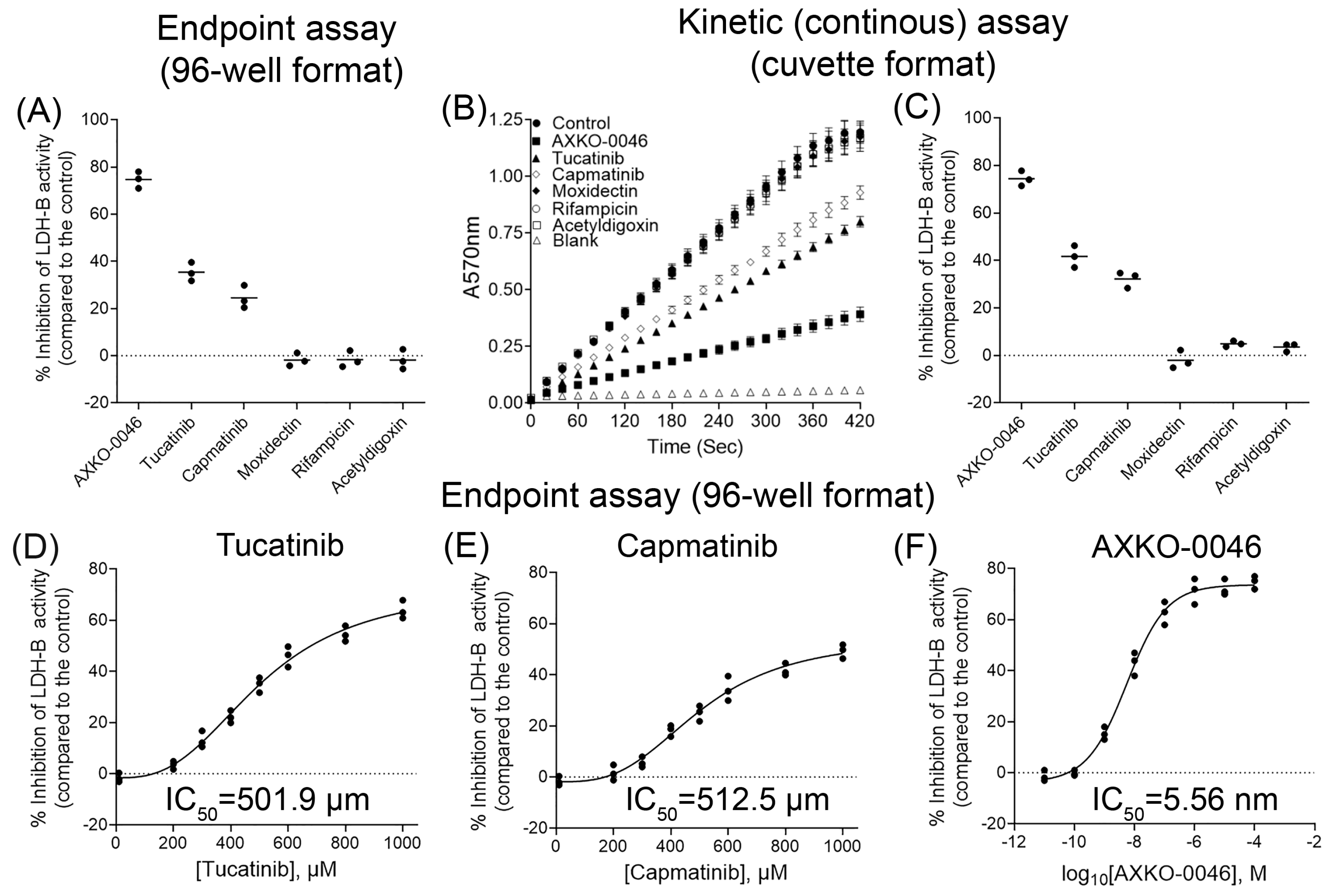
3.2. In Vitro Evaluation of Candidate LDH-B Inhibitors
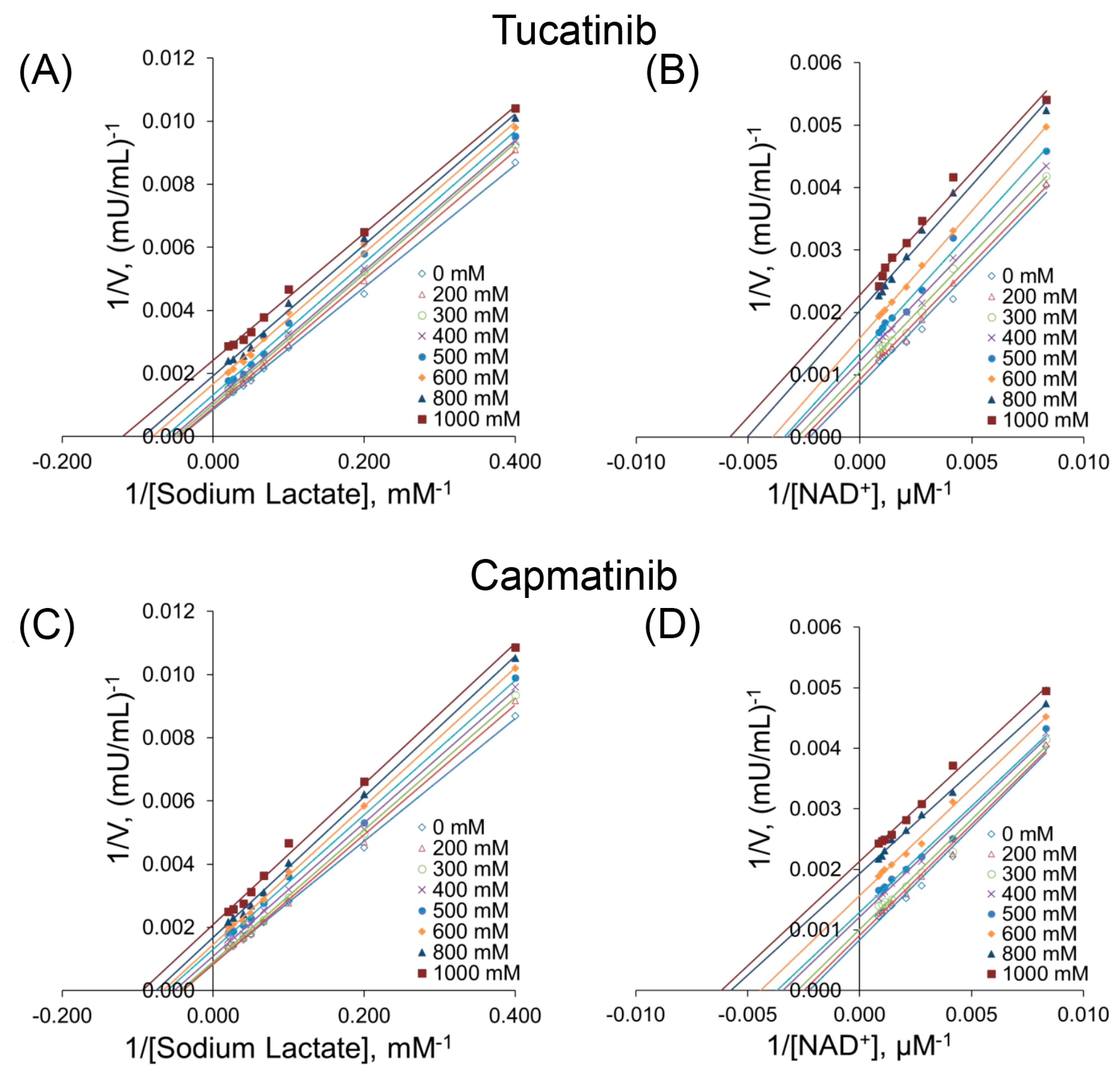
3.3. Kinetic Studies
3.4. Molecular Dynamics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mishra, D.; Banerjee, D. Lactate dehydrogenases as metabolic links between tumor and stroma in the tumor microenvironment. Cancers 2019, 11, 750. [Google Scholar] [CrossRef] [PubMed]
- Fadaka, A.; Ajiboye, B.; Ojo, O.; Adewale, O.; Olayide, I.; Emuowhochere, R. Biology of glucose metabolization in cancer cells. J. Oncol. Pract. 2017, 3, 45–51. [Google Scholar] [CrossRef]
- Lin, X.; Xiao, Z.; Chen, T.; Liang, S.H.; Guo, H. Glucose metabolism on tumor plasticity, diagnosis, and treatment. Front Oncol 2020, 10, 317. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Gao, W.-Q.; Liu, Y. Metabolic heterogeneity in cancer: An overview and therapeutic implications. Biochim. Biophys. Acta.-Reviews on Cancer 2020, 1874, 188421. [Google Scholar] [CrossRef] [PubMed]
- Brisson, L.; Bański, P.; Sboarina, M.; Dethier, C.; Danhier, P.; Fontenille, M.-J.; Van Hée, V.F.; Vazeille, T.; Tardy, M.; Falces, J.; et al. Lactate dehydrogenase B controls lysosome activity and autophagy in cancer. Cancer Cell 2016, 30, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Gallo, M.; Sapio, L.; Spina, A.; Naviglio, D.; Calogero, A.; Naviglio, S. Lactic dehydrogenase and cancer: An overview. FBL 2015, 20, 1234–1249. [Google Scholar] [CrossRef]
- Ding, J.; Karp, J.E.; Emadi, A. Elevated lactate dehydrogenase (LDH) can be a marker of immune suppression in cancer: Interplay between hematologic and solid neoplastic clones and their microenvironments. Cancer Biomark. 2017, 19, 353–363. [Google Scholar] [CrossRef]
- Valvona, C.J.; Fillmore, H.L.; Nunn, P.B.; Pilkington, G.J. The regulation and function of lactate dehydrogenase A: Therapeutic potential in brain tumor. Brain Pathol. 2016, 26, 3–17. [Google Scholar] [CrossRef]
- Ždralević, M.; Brand, A.; Di Ianni, L.; Dettmer, K.; Reinders, J.; Singer, K.; Peter, K.; Schnell, A.; Bruss, C.; Decking, S.-M.; et al. Double genetic disruption of lactate dehydrogenases A and B is required to ablate the “Warburg effect” restricting tumor growth to oxidative metabolism. J. Biol. Chem. 2018, 293, 15947–15961. [Google Scholar] [CrossRef]
- Cheng, A.; Zhang, P.; Wang, B.; Yang, D.; Duan, X.; Jiang, Y.; Xu, T.; Jiang, Y.; Shi, J.; Ding, C.; et al. Aurora-A mediated phosphorylation of LDHB promotes glycolysis and tumor progression by relieving the substrate-inhibition effect. Nat. Commun. 2019, 10, 5566. [Google Scholar] [CrossRef]
- Khan, A.A.; Allemailem, K.S.; Alhumaydhi, F.A.; Gowder, S.J.T.; Rahmani, A.H. The biochemical and clinical perspectives of lactate dehydrogenase: An enzyme of active metabolism. Endocr Metab Immune Disord Drug Targets 2020, 20, 855–868. [Google Scholar] [CrossRef] [PubMed]
- Claps, G.; Faouzi, S.; Quidville, V.; Chehade, F.; Shen, S.; Vagner, S.; Robert, C. The multiple roles of LDH in cancer. Nat. Rev. Clin. Oncol. 2022, 19, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Talaiezadeh, A.; Shahriari, A.; Tabandeh, M.R.; Fathizadeh, P.; Mansouri, S. Kinetic characterization of lactate dehydrogenase in normal and malignant human breast tissues. Cancer Cell Int. 2015, 15, 19. [Google Scholar] [CrossRef]
- De Bari, L.; Chieppa, G.; Marra, E.; Passarella, S. L-lactate metabolism can occur in normal and cancer prostate cells via the novel mitochondrial L-lactate dehydrogenase. Int. J. Oncol. 2010, 37, 1607–1620. [Google Scholar] [CrossRef]
- Pizzuto, R.; Paventi, G.; Porcile, C.; Sarnataro, D.; Daniele, A.; Passarella, S. l-Lactate metabolism in HEP G2 cell mitochondria due to the l-lactate dehydrogenase determines the occurrence of the lactate/pyruvate shuttle and the appearance of oxaloacetate, malate and citrate outside mitochondria. Biochim. Biophys. Acta-Bioenergetics 2012, 1817, 1679–1690. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Fendt, S.-M. The metabolism of cancer cells during metastasis. Nat. Rev. Cancer 2021, 21, 162–180. [Google Scholar] [CrossRef]
- Markert, C.L. Lactate dehydrogenase isozymes: Dissociation and recombination of subunits. Science 1963, 140, 1329–1330. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Giatromanolaki, A.; Simopoulos, C.; Polychronidis, A.; Sivridis, E. Lactate dehydrogenase 5 (LDH5) relates to up-regulated hypoxia inducible factor pathway and metastasis in colorectal cancer. Clin. Exp. Metastasis 2005, 22, 25–30. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Kontomanolis, E.; Giatromanolaki, A.; Sivridis, E.; Liberis, V. Serum and tissue LDH levels in patients with breast/gynaecological cancer and benign diseases. Gynecol. Obstet. Invest. 2009, 67, 162–168. [Google Scholar] [CrossRef]
- Balinsky, D.; Platz, C.E.; Lewis, J.W. Isozyme patterns of normal, benign, and malignant human breast tissues. Cancer Res. 1983, 43, 5895–5901. [Google Scholar]
- Rosso, M.; Lapyckyj, L.; Besso, M.J.; Monge, M.; Reventós, J.; Canals, F.; Quevedo Cuenca, J.O.; Matos, M.L.; Vazquez-Levin, M.H. Characterization of the molecular changes associated with the overexpression of a novel epithelial cadherin splice variant mRNA in a breast cancer model using proteomics and bioinformatics approaches: Identification of changes in cell metabolism and an increased expression of lactate dehydrogenase B. Cancer Metab. 2019, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- McCleland, M.L.; Adler, A.S.; Shang, Y.; Hunsaker, T.; Truong, T.; Peterson, D.; Torres, E.; Li, L.; Haley, B.; Stephan, J.P.; et al. An integrated genomic screen identifies LDHB as an essential gene for triple-negative breast cancer. Cancer Res 2012, 72, 5812–5823. [Google Scholar] [CrossRef]
- Shi, L.; Yan, H.; An, S.; Shen, M.; Jia, W.; Zhang, R.; Zhao, L.; Huang, G.; Liu, J. SIRT5-mediated deacetylation of LDHB promotes autophagy and tumorigenesis in colorectal cancer. Mol. Oncol. 2019, 13, 358–375. [Google Scholar] [CrossRef] [PubMed]
- Hirschhaeuser, F.; Sattler, U.G.; Mueller-Klieser, W. Lactate: A metabolic key player in cancer. Cancer Res. 2011, 71, 6921–6925. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Gao, Y.; Trappetti, V.; Hertig, D.; Karatkevich, D.; Losmanova, T.; Urzi, C.; Ge, H.; Geest, G.A.; Bruggmann, R.; et al. Targeting lactate dehydrogenase B-dependent mitochondrial metabolism affects tumor initiating cells and inhibits tumorigenesis of non-small cell lung cancer by inducing mtDNA damage. Cell. Mol. Life Sci. 2022, 79, 445. [Google Scholar] [CrossRef]
- McCleland, M.L.; Adler, A.S.; Deming, L.; Cosino, E.; Lee, L.; Blackwood, E.M.; Solon, M.; Tao, J.; Li, L.; Shames, D.; et al. Lactate dehydrogenase B is required for the growth of KRAS-dependent lung adenocarcinomas. Clin. Cancer Res. 2013, 19, 773–784. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, H.; Xu, A.; Li, N.; Liu, J.; Liu, C.; Lv, D.; Wu, S.; Huang, L.; Yang, S.; et al. Elevation of serum l-lactate dehydrogenase B correlated with the clinical stage of lung cancer. Lung Cancer 2006, 54, 95–102. [Google Scholar] [CrossRef]
- Wang, R.; Li, J.; Zhang, C.; Guan, X.; Qin, B.; Jin, R.; Qin, L.; Xu, S.; Zhang, X.; Liu, R.; et al. Lactate dehydrogenase B is required for pancreatic cancer cell immortalization through activation of telomerase activity. Front. Oncol. 2022, 12, 821620. [Google Scholar] [CrossRef]
- Dennison, J.B.; Molina, J.R.; Mitra, S.; González-Angulo, A.M.; Balko, J.M.; Kuba, M.G.; Sanders, M.E.; Pinto, J.A.; Gómez, H.L.; Arteaga, C.L.; et al. Lactate dehydrogenase B: A metabolic marker of response to neoadjuvant chemotherapy in breast cancer. Clin. Cancer Res. 2013, 19, 3703–3713. [Google Scholar] [CrossRef]
- Manerba, M.; Di Ianni, L.; Fiume, L.; Roberti, M.; Recanatini, M.; Di Stefano, G. Lactate dehydrogenase inhibitors sensitize lymphoma cells to cisplatin without enhancing the drug effects on immortalized normal lymphocytes. Eur. J. Pharm. Sci. 2015, 74, 95–102. [Google Scholar] [CrossRef]
- Valvona, C.J.; Fillmore, H.L. Oxamate, but not selective targeting of LDH-A, inhibits medulloblastoma cell glycolysis, growth and motility. Brain Sci. 2018, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zhao, Y.; Ding, Y.; Liu, H.; Liu, Z.; Fodstad, O.; Riker, A.I.; Kamarajugadda, S.; Lu, J.; Owen, L.B.; et al. Warburg effect in chemosensitivity: Targeting lactate dehydrogenase-A re-sensitizes taxol-resistant cancer cells to taxol. Mol. Cancer. 2010, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liu, H.; Liu, Z.; Ding, Y.; Ledoux, S.P.; Wilson, G.L.; Voellmy, R.; Lin, Y.; Lin, W.; Nahta, R.; et al. Overcoming trastuzumab resistance in breast cancer by targeting dysregulated glucose metabolism. Cancer Res. 2011, 71, 4585–4597. [Google Scholar] [CrossRef] [PubMed]
- Qiao, T.; Xiong, Y.; Feng, Y.; Guo, W.; Zhou, Y.; Zhao, J.; Jiang, T.; Shi, C.; Han, Y. Inhibition of LDH-A by oxamate enhances the efficacy of anti-PD-1 treatment in an NSCLC humanized mouse model. Front. Oncol. 2021, 11, 632364. [Google Scholar] [CrossRef]
- Xiang, J.; Zhou, L.; He, Y.; Wu, S. LDH-A inhibitors as remedies to enhance the anticancer effects of PARP inhibitors in ovarian cancer cells. Aging 2021, 13, 25920–25930. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S.; Sogabe, S.; Miwa, M.; Fujimoto, T.; Takakura, N.; Naotsuka, A.; Kitamura, S.; Kawamoto, T.; Soga, T. Identification of the first highly selective inhibitor of human lactate dehydrogenase B. Sci. Rep. 2021, 11, 21353. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New docking methods, expanded force field, and python bindings. J. Chem. Inf. Model 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Cameron, A.; Read, J.; Tranter, R.; Winter, V.J.; Sessions, R.B.; Brady, R.L.; Vivas, L.; Easton, A.; Kendrick, H.; Croft, S.L.; et al. Identification and activity of a series of azole-based compounds with lactate dehydrogenase-directed anti-malarial activity. J. Biol. Chem. 2004, 279, 31429–31439. [Google Scholar] [CrossRef]
- Vlasiou, M.C.; Petrou, C.C.; Sarigiannis, Y.; Pafiti, K.S. Density functional theory studies and molecular docking on xanthohumol, 8-prenylnaringenin and their symmetric substitute diethanolamine derivatives as inhibitors for colon cancer-related proteins. Symmetry 2021, 13, 948. [Google Scholar] [CrossRef]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA-a self-parameterizing force field. Proteins 2002, 47, 393–402. [Google Scholar] [CrossRef]
- Yang, J.-M.; Chen, C.-C. GEMDOCK: A generic evolutionary method for molecular docking. Proteins 2004, 55, 288–304. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.-C.; Chen, Y.-F.; Lin, S.-R.; Yang, J.-M. iGEMDOCK: A graphical environment of enhancing GEMDOCK using pharmacological interactions and post-screening analysis. BMC Bioinform. 2011, 12, S33. [Google Scholar] [CrossRef] [PubMed]
- Bender, B.J.; Gahbauer, S.; Luttens, A.; Lyu, J.; Webb, C.M.; Stein, R.M.; Fink, E.A.; Balius, T.E.; Carlsson, J.; Irwin, J.J.; et al. A practical guide to large-scale docking. Nat. Protoc. 2021, 16, 4799–4832. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Papaneophytou, C.; Zervou, M.E.; Theofanous, A. Optimization of a colorimetric assay to determine lactate dehydrogenase B activity using design of experiments. SLAS Discov. 2021, 26, 383–399. [Google Scholar] [CrossRef]
- Mayer, K.M. Colorimetric dehydrogenase screen based on NAD(P)H generation. Methods Mol. Biol. 2003, 230, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, S.I.; Jin, L.; Gasparovich, S.R.; Tao, L. Multiple alcohol dehydrogenases but no functional acetaldehyde dehydrogenase causing excessive acetaldehyde production from ethanol by oral streptococci. Microbiology 2013, 159, 1437–1446. [Google Scholar] [CrossRef]
- Copeland, R.A. Reversible inhibitors. In Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis, 2nd ed.; Wiley-VCH: Hoboken, NJ, USA, 2000; pp. 266–304. [Google Scholar]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Sonveaux, P.; Végran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Invest. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [PubMed]
- Okumura, N.; Terasawa, F.; Ueno, I.; Oki, K.; Yamauchi, K.; Hidaka, H.; Tozuka, M.; Okura, M.; Katsuyama, T. Genetic analyses in homozygous and heterozygous variants of lactate dehydrogenase-B (H) subunit-LD-B Matsumoto I and II (LD-B W323R). Clin. Chim. Acta 1999, 287, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Sudo, K.; Maekawa, M.; Houki, N.; Okuda, T.; Akizuki, S.; Magara, T.; Kawano, K. A novel in-frame deletion mutation in a case of lactate dehydrogenase (LD) H subunit deficiency showing an atypical LD isoenzyme pattern in serum and erythrocytes. Clin. Biochem. 1999, 32, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.; Schafer, X.L.; Ambeskovic, A.; Spencer, C.M.; Land, H.; Munger, J. Addiction to coupling of the Warburg effect with glutamine catabolism in cancer cells. Cell Rep. 2016, 17, 821–836. [Google Scholar] [CrossRef]
- Schlam, I.; Swain, S.M. HER2-positive breast cancer and tyrosine kinase inhibitors: The time is now. npj Breast Cancer 2021, 7, 56. [Google Scholar] [CrossRef]
- Zhang, Y.; Xia, M.; Jin, K.; Wang, S.; Wei, H.; Fan, C.; Wu, Y.; Li, X.; Li, X.; Li, G.; et al. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol. Cancer 2018, 17, 45. [Google Scholar] [CrossRef]
- Thabault, L.; Brisson, L.; Brustenga, C.; Martinez Gache, S.A.; Prévost, J.R.C.; Kozlova, A.; Spillier, Q.; Liberelle, M.; Benyahia, Z.; Messens, J.; et al. Interrogating the lactate dehydrogenase tetramerization site using (Stapled) peptides. J. Med. Chem. 2020, 63, 4628–4643. [Google Scholar] [CrossRef]
- Eventoff, W.; Rossmann, M.G.; Taylor, S.S.; Torff, H.J.; Meyer, H.; Keil, W.; Kiltz, H.H. Structural adaptations of lactate dehydrogenase isozymes. Proc. Natl. Acad. Sci. USA 1977, 74, 2677–2681. [Google Scholar] [CrossRef]
- Zhai, X.; Yang, Y.; Wan, J.; Zhu, R.; Wu, Y. Inhibition of LDH-A by oxamate induces G2/M arrest, apoptosis and increases radiosensitivity in nasopharyngeal carcinoma cells. Oncol. Rep. 2013, 30, 2983–2991. [Google Scholar] [CrossRef]
- Kanno, T.; Sudo, K.; Maekawa, M.; Nishimura, Y.; Ukita, M.; Fukutake, K. Lactate dehydrogenase M-subunit deficiency: A new type of hereditary exertional myopathy. Clin. Chim. Acta 1988, 173, 89–98. [Google Scholar] [CrossRef]
- Granchi, C.; Roy, S.; Giacomelli, C.; Macchia, M.; Tuccinardi, T.; Martinelli, A.; Lanza, M.; Betti, L.; Giannaccini, G.; Lucacchini, A.; et al. Discovery of N-hydroxyindole-based inhibitors of human lactate dehydrogenase isoform A (LDH-A) as starvation agents against cancer cells. J. Med. Chem. 2011, 54, 1599–1612. [Google Scholar] [CrossRef] [PubMed]
- Vanderporten, E.; Frick, L.; Turincio, R.; Thana, P.; Lamarr, W.; Liu, Y. Label-free high-throughput assays to screen and characterize novel lactate dehydrogenase inhibitors. Anal. Biochem. 2013, 441, 115–122. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
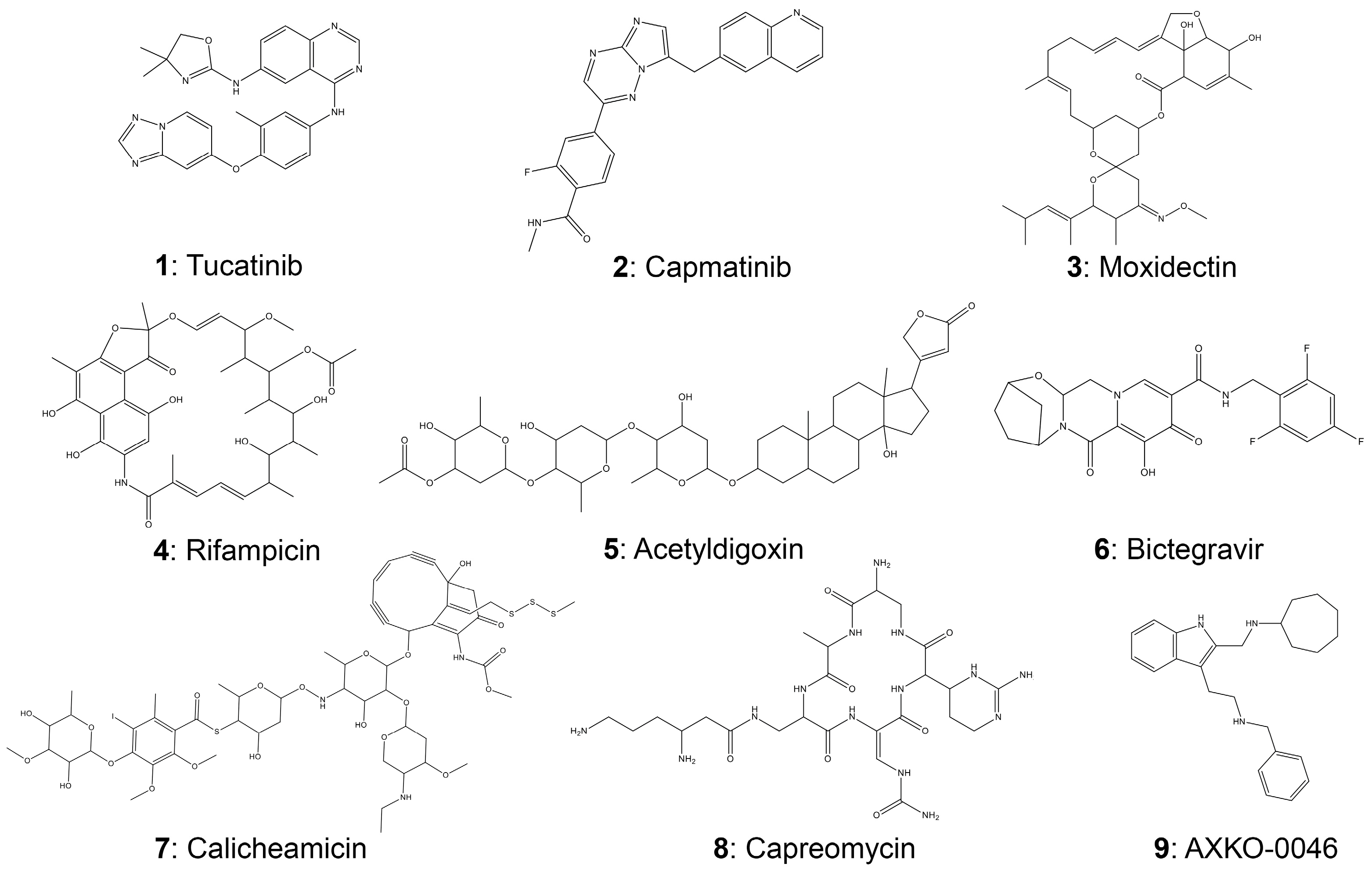
| # | Compound | CLogP 1 | Molecular Mass (g/mole) | Binding Affinity 2 |
|---|---|---|---|---|
| 1 | Tucatinib | 5.32 | 480.52 | −9.2 |
| 2 | Capmatinib | 1.88 | 412.42 | −9.3 |
| 3 | Moxidectin | 6.65 | 639.80 | −9.5 |
| 4 | Rifampicin | 3.86 | 697.77 | −9.5 |
| 5 | Acetyldigoxin | 3.70 | 806.98 | −10.0 |
| 6 | Bictegravir | −0.01 | 449.38 | −9.0 |
| 7 | Calicheamicin | 2.00 | 1368.65 | −9.3 |
| 8 | Capreomycin | N.A. 3 | 1321.41 | −9.1 |
| 9 | AΧKO-0046 | 4.74 | 448.47 | −6.6 |
| Tucatinib, (μΜ) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Control | 200 | 300 | 400 | 500 | 600 | 800 | 1000 | |
| Lactate | ||||||||
| Vmax (mU/mL) | 1052 ± 36 | 971 ± 26 | 916 ± 22 | 858 ± 17 | 772 ± 26 | 644 ± 19 | 527 ± 16 | 421 ± 23 |
| Km (mΜ) | 18.5 ± 1.6 | 17.6 ± 0.9 | 17.2 ± 1.2 | 16.5 ± 1.1 | 15.9 ± 1.3 | 14.3 ± 0.9 | 10.9 ± 0.7 | 8.7 ± 0.8 |
| NAD+ | ||||||||
| Vmax (mU/mL) | 1035 ± 42 | 983 ± 41 | 893 ± 31 | 784 ± 41 | 723 ± 32 | 624 ± 26 | 521 ± 21 | 474 ± 25 |
| Km (μΜ) | 311 ± 21 | 305 ± 12 | 271 ± 21 | 254 ± 12 | 220 ± 17 | 209 ± 14 | 178 ± 16 | 168 ± 18 |
| Capmatinib, (μM) | ||||||||
| Control | 200 | 300 | 400 | 500 | 600 | 800 | 1000 | |
| Lactate | ||||||||
| Vmax (mU/mL) | 1052 ± 36 | 987 ± 42 | 927 ± 33 | 839 ± 28 | 725 ± 26 | 653 ± 21 | 587 ± 16 | 513 ± 19 |
| Km (mΜ) | 18.5 ± 1.6 | 17.1 ± 1.4 | 16.5 ± 1.2 | 15.8 ± 1.1 | 14.5 ± 0.9 | 13.3 ± 0.8 | 12.6 ± 0.9 | 12.4 ± 1.2 |
| NAD+ | ||||||||
| Vmax (mU/mL) | 1035 ± 42 | 973 ± 23 | 892 ± 26 | 799 ± 22 | 733 ± 31 | 629 ± 26 | 526 ± 27 | 469 ± 32 |
| Km (μΜ) | 311 ± 21 | 308 ± 23 | 298 ± 18 | 286 ± 21 | 279 ± 15 | 256 ± 16 | 245 ± 14 | 211 ± 18 |
| Parameter | Tucatinib | Capmatinib | ||
|---|---|---|---|---|
| NAD+ | Lactate | NAD+ | Lactate | |
| Κi (mM) | 4.16 | 2.96 | 1.53 | 2.69 |
| α a | 0.20 | 0.28 | 0.58 | 0.36 |
| r2 b | 0.9648 | 0.9743 | 0.9630 | 0.9815 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vlasiou, M.; Nicolaidou, V.; Papaneophytou, C. Targeting Lactate Dehydrogenase-B as a Strategy to Fight Cancer: Identification of Potential Inhibitors by In Silico Analysis and In Vitro Screening. Pharmaceutics 2023, 15, 2411. https://doi.org/10.3390/pharmaceutics15102411
Vlasiou M, Nicolaidou V, Papaneophytou C. Targeting Lactate Dehydrogenase-B as a Strategy to Fight Cancer: Identification of Potential Inhibitors by In Silico Analysis and In Vitro Screening. Pharmaceutics. 2023; 15(10):2411. https://doi.org/10.3390/pharmaceutics15102411
Chicago/Turabian StyleVlasiou, Manos, Vicky Nicolaidou, and Christos Papaneophytou. 2023. "Targeting Lactate Dehydrogenase-B as a Strategy to Fight Cancer: Identification of Potential Inhibitors by In Silico Analysis and In Vitro Screening" Pharmaceutics 15, no. 10: 2411. https://doi.org/10.3390/pharmaceutics15102411