
Translational Considerations in the Development of Intranasal Treatments for Epilepsy
Abstract
:1. Introduction
2. Relationships between the Nose and Epilepsy
2.1. Historical and Epidemiological
2.2. Neurological
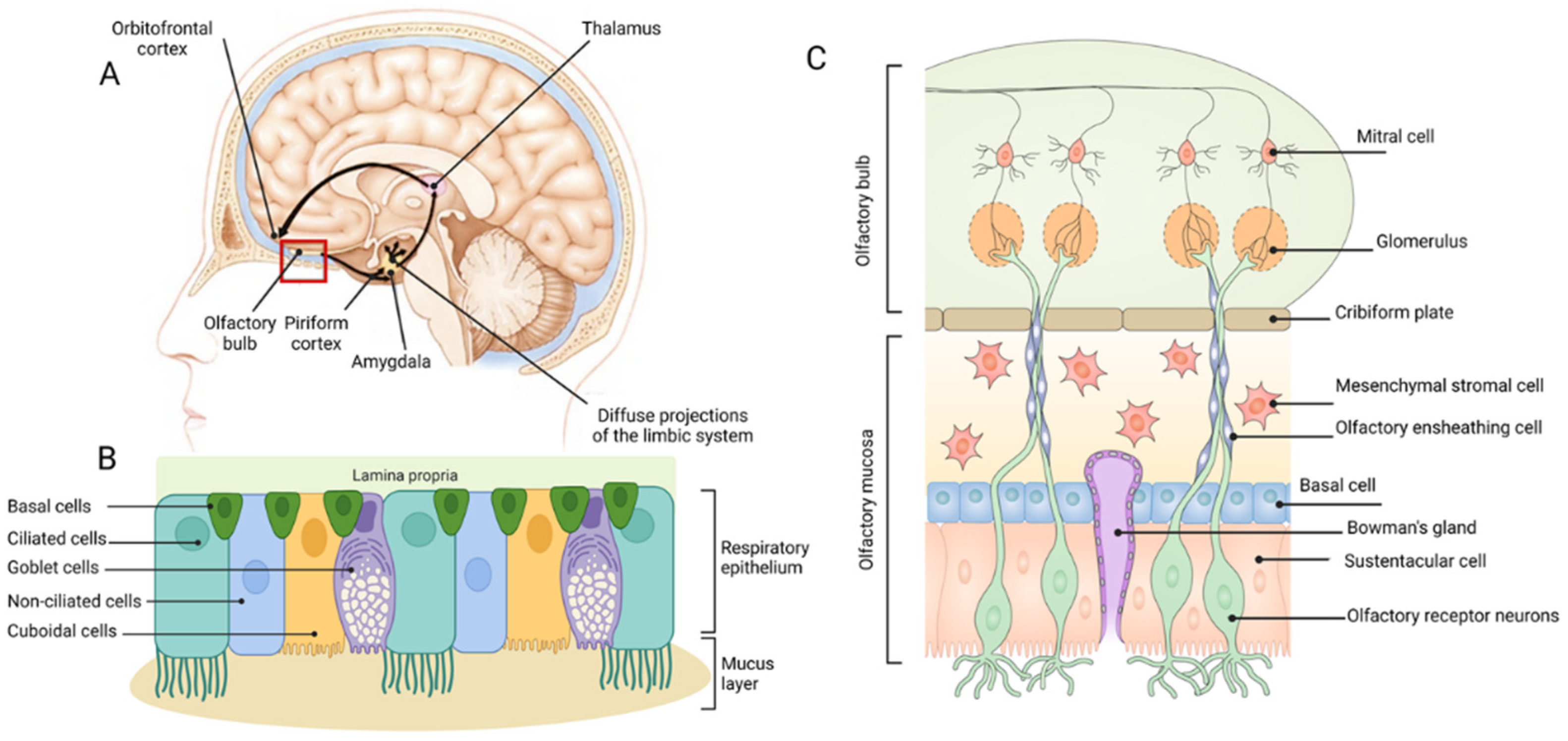
2.2.1. The Olfactory System
2.2.2. Epilepsy and the Olfactory System
2.2.3. Clinical and Social
3. The Anatomy and Physiology of Intranasal Administration to the Brain
3.1. The Nasal Passage and Epithelia
3.2. Respiratory Epithelium
3.3. Olfactory Epithelium
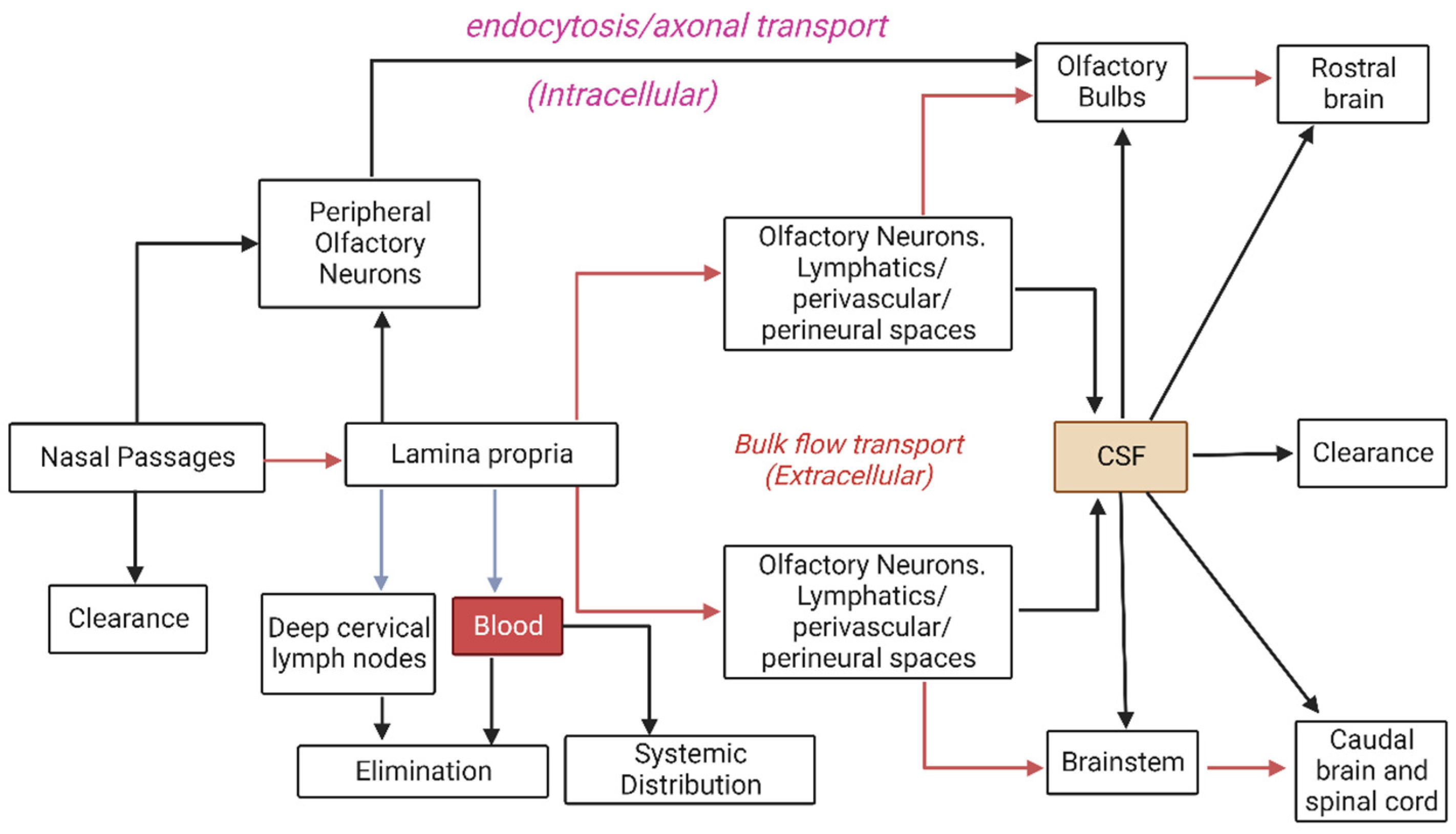
4. Nasal Routes of Absorption for Therapeutics
4.1. Systemic Transport
4.2. Intracellular Transport
4.3. Extracellular Transport
5. Animal Models for Intranasal Delivery
6. Animals as Seizure and Epilepsy Models for the Evaluation of Anti-Seizure Therapeutics
6.1. Overview of Key Models
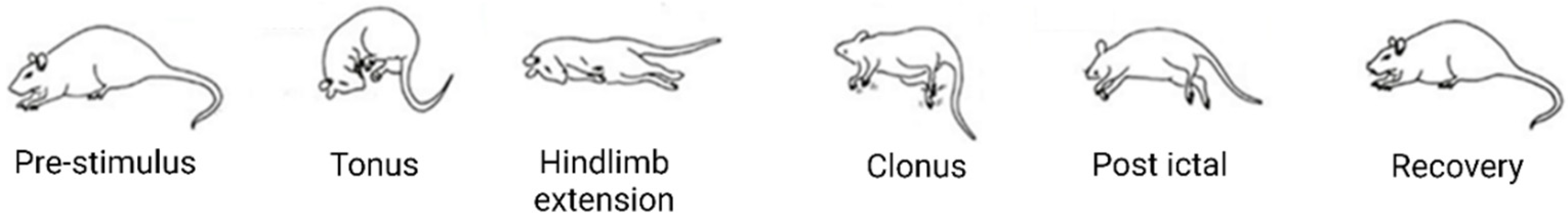
6.1.1. Maximal Electroshock Seizure Test
6.1.2. Maximal Electroshock Seizure Threshold Test
6.1.3. Pentylenetetrazole Test
6.1.4. 6-Hz “Psychomotor” Seizure Test
6.1.5. Kindling
6.2. Relevance to the Evaluation of Intranasal Delivery Pathways
7. Pharmaceutical Formulation of Anti-Seizure Therapeutics
7.1. Role of Pharmaceutical Formulation
7.2. Studies of Pharmaceutical Formulation for Anti-Seizure Therapeutic Delivery
7.2.1. Administration Technique

{kind=link}
{kind=link}
{kind=link}
| ASD | Delivery System | Materials | Tox. | PK | Efficacy | Ref. |
|---|---|---|---|---|---|---|
| CBZ | Gel | Carbopol 974P (mucoadhesive polymer, hypromellose, pH 7.4 | X | ✓ | X | [124] |
| Thermo-reversible gel | Carbopol 974P (mucoadhesive polymer), Pluronic F127 | X | ✓ | X | [125] | |
| Mucoadhesive o/w nanoemulgel | Oleic acid, Labrasol, xanthan gum (anionic mucoadhesive polymer) | X | X | ✓ | [125] | |
| Microemulsion | Oleic acid, Tween 80, Propylene glycol | ✓ | X | ✓ | [113] | |
| Oleoyl polyoylglycerides, Polyoxyl 40 hydrogenated castor oil, Diethylene glycol monoethyl ether, Polycarbopil (mucoadhesive) | ✓ | ✓ | X | [126] | ||
| Polymeric nanoparticles | Carboxymethyl chitosan | X | ✓ | X | [127] | |
| LMT | Thermo-reversible gel | Carbopol 974P (mucoadhesive polymer), Pluronic F127 | X | ✓ | X | [128] |
| Microemulsion | Glyceryl monostearate, Oleic acid, Tween 80, Pluronic P188 | X | ✓ | ✓ | [111] | |
| Microspheres (as suspension) | Chitosan, glutaraldehyde | ✓ | X | ✓ | [129] | |
| Nanoliposomes | Phospholipon 90G, cholesterol | ✓ (in vitro) | X | X | [130] | |
| Polymeric nanoparticles | PLGA and Poloxamer 407 | ✓ | ✓ | ✓ | [131] | |
| PHT | Microemulsion | Capmul MCM (glyceryl monocaprylate), Labrasol, PEG-8 caprylic/capric glycerides and Transcutol (diethylene glycol monoethyl ether) | ✓ | ✓ | ✓ | [112] |
| Nanoparticles | Lecithin-chitosan | X | ✓ | ✓ | [132] | |
| PBT | Gel | Carbopol 974P (mucoadhesive polymer, hypromellose, pH 9.5 | X | ✓ | ✓ | [123] |
| LZM | Lipid nanoparticles in a gel | Glycerol monostearate, oleic acid and Tween 80, chitosan, Pluronic F127, β-glycerol phosphate disodium salt pentahydrate | X | ✓ | X | [133] |
| DZP | Polymeric nanoparticles | PLGA (Poly(D,L-lactide-co-glycolide), Pluronic F127 | X | ✓ | X | [134] |
| TRH | Polymeric nanoparticles | PLA (Polylactide) | X | X | ✓ | [17,18] |
| VA | Lipid nanoparticles | Cetyl palmitate, soy lecithin, octyldodecanol | X | ✓ | ✓ | [135] |
| LVT | Thermo-reversible gel | Pluronic F127; Carbopol 974P and Noveon® Polycarbophil. | ✓ | ✓ | X | [136] |
| ZNA | Thermo-reversible gel | Pluronic F127; Carbopol 974P and Noveon® Polycarbophil | X | ✓ | X | [137] |
| Drug | Animal Model | Dose (µg) | Volume | Anaesthesia | Method | Ref. |
|---|---|---|---|---|---|---|
| CBZ | Mouse | 12 to 16 | 12 to 16 µL in one nostril | Ketamine and xylazine (i.p.) | Tubing | [128] |
| 625 | 100 µL in one nostril | Diethyl ether | Cannula strengthened by jacketed non-protruding needle | [125] | ||
| 40 to 60 | 25 µL in each nostril | Not reported | Tubing | [127] | ||
| Rat | 35 to 40 | 10 µL in each nostril | Ketamine (i.m.) | Tubing | [126] | |
| 50 (administered) 40 (accepted) | 50 mg gel into one nostril. Estimated that 80% was accepted | None | Tubing | [124] | ||
| 1600 to 2000 | 55 µL in each nostril | Not reported | Tubing | [113] | ||
| LMT | Mouse | 110 to 125 | Not reported Both nostrils. | Ketamine and xylazine (route not stated) | Tubing | [129] |
| 120 to 160 | 12 to 16 µL in one nostril | Ketamine and xylazine (i.p.) | Tubing | [138] | ||
| Rat | 720 to 970 | 100 µL in each nostril | Ketamine (i.m.) | Not reported | [111] | |
| 166 to 291 | Not reported | Not reported | Not reported | [131] | ||
| PHT | Rat | 3520 | 88 µL in each nostril | Not reported | Tubing | [112] |
| Mouse | 280 to 420 | 60 µL (number of nostrils not reported) | None | Dropper | [132] | |
| PBT | Rat | 1100 to 1200 2000 to 2200 6000 to 6600 | 7 to 40 µL in each nostril | Propofol (i.v.) | Deposited at opening of nares or using tubing | [123] |
| LZM | Rat | 200 | 50 µL in each nostril | Not reported | Tubing | [133] |
| VA | Rat | 720 to 840 | 100 µL in each nostril over a few minutes | Light ether | Tubing | [135] |
| DZP | Rat | 40 to 50 | 10 µL each nostril | Ketamine (i.p.) | Tubing | [134] |
| TRH | Rat | 20 | 25 µL in each nostril (chronic administration) | Isoflurane | Surgically inserted cannulae | [17,18] |
| LVT | Mouse | 625 | 25 µL (left nostril only) | Ketamine and xylazine (i.p.) | MicroSprayer® Aerosolizer coupled to a high-pressure syringe | [136] |
| ZNA | Mouse | 418 to 501 | 50 µL (left nostril only) | Ketamine and xylazine (i.p.) | MicroSprayer® Aerosolizer coupled to a high-pressure syringe | [137] |
7.2.2. Adverse Effects and Toxicity
7.2.3. Quantification of Drug in Tissues
7.2.4. Qualitative Distribution in Tissue
7.2.5. Efficacy
7.2.6. In the Pipeline
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fisher, R.S.; van Emde Boas, W.; Blume, W.; Elger, C.; Genton, P.; Lee, P.; Engel, J., Jr. Epileptic Seizures and Epilepsy: Definitions Proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 2005, 46, 470–472. [Google Scholar] [CrossRef]
- Huff, J.S.; Fountain, N.B. Pathophysiology and definitions of seizures and status epilepticus. Emerg. Med. Clin. N. Am. 2011, 29, 1–13. [Google Scholar] [CrossRef]
- Fisher, R.S.; Cross, J.H.; French, J.A.; Higurashi, N.; Hirsch, E.; Jansen, F.E.; Lagae, L.; Moshé, S.L.; Peltola, J.; Roulet Perez, E. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 522–530. [Google Scholar]
- Janmohamed, M.; Brodie, M.J.; Kwan, P. Pharmacoresistance Epidemiology, mechanisms, and impact on epilepsy treatment. Neuropharmacology 2020, 168, 107790. [Google Scholar] [PubMed]
- Spiciarich, M.C.; von Gaudecker, J.R.; Jurasek, L.; Clarke, D.F.; Burneo, J.; Vidaurre, J. Global Health and Epilepsy: Update and Future Directions. Curr. Neurol. Neurosci. Rep. 2019, 19, 30. [Google Scholar] [PubMed]
- Beghi, E. The Epidemiology of Epilepsy. Neuroepidemiology 2020, 54, 185–191. [Google Scholar] [CrossRef]
- Jacoby, A.; Snape, D.; Baker, G.A. Epilepsy and social identity: The stigma of a chronic neurological disorder. Lancet Neurol. 2005, 4, 171–178. [Google Scholar] [CrossRef]
- Sperling, M.R. The consequences of uncontrolled epilepsy. CNS Spectr. 2004, 9, 98–101, 106–109. [Google Scholar] [CrossRef]
- Wilcox, K.S.; Dixon-Salazar, T.; Sills, G.J.; Ben-Menachem, E.; White, H.S.; Porter, R.J.; Dichter, M.A.; Moshé, S.L.; Noebels, J.L.; Privitera, M.D.; et al. Issues related to development of new antiseizure treatments. Epilepsia 2013, 54 (Suppl. S4), 24–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fattorusso, A.; Matricardi, S.; Mencaroni, E.; Dell’Isola, G.B.; Di Cara, G.; Striano, P.; Verrotti, A. The Pharmacoresistant Epilepsy: An overview on existant and new emerging therapies. Front. Neurol. 2021, 12, 1030. [Google Scholar] [CrossRef]
- Engel, J., Jr. Etiology as a risk factor for medically refractory epilepsy: A case for early surgical intervention. Neurology 1998, 51, 1243–1244. [Google Scholar] [CrossRef]
- Kwan, P.; Arzimanoglou, A.; Berg, A.T.; Brodie, M.J.; Allen Hauser, W.; Mathern, G.; Moshé, S.L.; Perucca, E.; Wiebe, S.; French, J. Definition of drug resistant epilepsy: Consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 2010, 51, 1069–1077. [Google Scholar] [CrossRef]
- Löscher, W.; Potschka, H.; Sisodiya, S.M.; Vezzani, A. Drug Resistance in Epilepsy: Clinical Impact, Potential Mechanisms, and New Innovative Treatment Options. Pharmacol. Rev. 2020, 72, 606–638. [Google Scholar] [PubMed]
- Kwan, P.; Schachter, S.C.; Brodie, M.J. Drug-Resistant Epilepsy. N. Engl. J. Med. 2011, 365, 919–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löscher, W. Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs. Seizure 2011, 20, 359–368. [Google Scholar] [CrossRef] [Green Version]
- Löscher, W.; Schmidt, D. Which animal models should be used in the search for new antiepileptic drugs? A proposal based on experimental and clinical considerations. Epilepsy Res. 1988, 2, 145–181. [Google Scholar]
- Kubek, M.J.; Domb, A.J.; Veronesi, M.C. Attenuation of kindled seizures by intranasal delivery of neuropeptide-loaded nanoparticles. Neurotherapeutics 2009, 6, 359–371. [Google Scholar] [CrossRef] [Green Version]
- Veronesi, M.C.; Aldouby, Y.; Domb, A.J.; Kubek, M.J. Thyrotropin-releasing hormone d,l polylactide nanoparticles (TRH-NPs) protect against glutamate toxicity in vitro and kindling development in vivo. Brain Res. 2009, 1303, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.S.; Ho, J. Potential new methods for antiepileptic drug delivery. CNS Drugs 2002, 16, 579–593. [Google Scholar] [CrossRef]
- Bennewitz, M.F.; Saltzman, W.M. Nanotechnology for delivery of drugs to the brain for epilepsy. Neurotherapeutics 2009, 6, 323–336. [Google Scholar] [CrossRef]
- Ruiz, M.; Castro, G. Nanoformulations of Antiepileptic Drugs: In Vitro and In Vivo Studies. In Antiepileptic Drug Discovery; Humana Press: New York, NY, USA, 2016. [Google Scholar]
- Kapoor, M.; Cloyd, J.C.; Siegel, R.A. A review of intranasal formulations for the treatment of seizure emergencies. J. Control Release 2016, 237, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Costantino, H.R.; Illum, L.; Brandt, G.; Johnson, P.H.; Quay, S.C. Intranasal delivery: Physicochemical and therapeutic aspects. Int. J. Pharm. 2007, 337, 1–24. [Google Scholar] [PubMed]
- Jaseja, H. Scientific basis behind traditional practice of application of “shoe-smell” in controlling epileptic seizures in the eastern countries. Clin. Neurol. Neurosurg. 2008, 110, 535–538. [Google Scholar] [CrossRef]
- Betts, T.; Betts, H. John hall and his epileptic patients—epilepsy management in early 17th century england. Seizure 1998, 7, 411–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gowers, W.R. Epilepsy and Other Chronic Convulsive Diseases: Their Causes, Symptoms, and Treatment; Old Hickory Bookshop: Washington, CT, USA, 1901. [Google Scholar]
- Efron, R. The Effect Of Olfactory Stimuli In Arresting Uncinate Fits. Brain 1956, 79, 267–281. [Google Scholar] [CrossRef]
- Efron, R. The conditioned inhibition of uncinate fits. Brain 1957, 80, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Betts, T. Use of aromatherapy (with or without hypnosis) in the treatment of intractable epilepsy—A two-year follow-up study. Seizure 2003, 12, 534–538. [Google Scholar] [CrossRef] [Green Version]
- Khurshid, K.; Crow, A.J.D.; Rupert, P.E.; Minniti, N.L.; Carswell, M.A.; Mechanic-Hamilton, D.J.; Kamath, V.; Doty, R.L.; Moberg, P.J.; Roalf, D.R. A Quantitative Meta-analysis of Olfactory Dysfunction in Epilepsy. Neuropsychol. Rev. 2019, 29, 328–337. [Google Scholar] [CrossRef]
- Acharya, V.; Acharya, J.; Lüders, H. Olfactory epileptic auras. Neurology 1998, 51, 56–61. [Google Scholar] [CrossRef]
- Chen, C.; Shih, Y.-H.; Yen, D.-J.; Lirng, J.-F.; Guo, Y.-C.; Yu, H.-Y.; Yiu, C.-H. Olfactory Auras in Patients with Temporal Lobe Epilepsy. Epilepsia 2003, 44, 257–260. [Google Scholar] [CrossRef]
- Vaughan, D.N.; Jackson, G.D. The piriform cortex and human focal epilepsy. Front. Neurol. 2014, 5, 259. [Google Scholar] [CrossRef] [Green Version]
- Blair, R.D. Temporal lobe epilepsy semiology. Epilepsy Res. Treat. 2012, 2012, 751510. [Google Scholar] [CrossRef] [PubMed]
- Ache, B.W.; Young, J.M. Olfaction: Diverse species, conserved principles. Neuron 2005, 48, 417–430. [Google Scholar] [CrossRef] [Green Version]
- Lochhead, J.J.; Thorne, R.G. Intranasal delivery of biologics to the central nervous system. Adv. Drug. Deliv. Rev. 2012, 64, 614–628. [Google Scholar] [CrossRef] [PubMed]
- Bekkers, J.M.; Suzuki, N. Neurons and circuits for odor processing in the piriform cortex. Trends Neurosci. 2013, 36, 429–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courtiol, E.; Wilson, D.A. The olfactory thalamus: Unanswered questions about the role of the mediodorsal thalamic nucleus in olfaction. Front. Neural. Circuits 2015, 9, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.; Minko, T. Nanotherapeutics for Nose-to-Brain Drug Delivery: An Approach to Bypass the Blood Brain Barrier. Pharmaceutics 2021, 13, 2049. [Google Scholar] [CrossRef] [PubMed]
- Hummel, T.; Henkel, S.; Negoias, S.; Galván, J.R.; Bogdanov, V.; Hopp, P.; Hallmeyer-Elgner, S.; Gerber, J.; Reuner, U.; Haehner, A. Olfactory bulb volume in patients with temporal lobe epilepsy. J. Neurol. 2013, 260, 1004–1008. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves Pereira, P.M.; Insausti, R.; Artacho-Pérula, E.; Salmenperä, T.; Kälviäinen, R.; Pitkänen, A. MR volumetric analysis of the piriform cortex and cortical amygdala in drug-refractory temporal lobe epilepsy. AJNR Am. J. Neuroradiol. 2005, 26, 319–332. [Google Scholar]
- Bensafi, M.; Sobel, N.; Khan, R.M. Hedonic-specific activity in piriform cortex during odor imagery mimics that during odor perception. J. Neurophysiol. 2007, 98, 3254–3262. [Google Scholar] [CrossRef] [Green Version]
- Ebert, U.; Löscher, W. Strong olfactory stimulation reduces seizure susceptibility in amygdala-kindled rats. Neurosci. Lett. 2000, 287, 199–202. [Google Scholar] [CrossRef]
- Vismer, M.S.; Forcelli, P.A.; Skopin, M.D.; Gale, K.; Koubeissi, M.Z. The piriform, perirhinal, and entorhinal cortex in seizure generation. Front. Neural. Circuits 2015, 9, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwabe, K.; Ebert, U.; Löscher, W. Bilateral lesions of the central but not anterior or posterior parts of the piriform cortex retard amygdala kindling in rats. Neuroscience 2000, 101, 513–521. [Google Scholar] [CrossRef]
- Löscher, W.; Ebert, U. The role of the piriform cortex in kindling. Prog. Neurobiol. 1996, 50, 427–481. [Google Scholar] [CrossRef] [PubMed]
- Wahnschaffe, U.; Ebert, U.; Löscher, W. The effects of lesions of the posterior piriform cortex on amygdala kindling in the rat. Brain Res. 1993, 615, 295–303. [Google Scholar] [CrossRef]
- Lehmann, H.; Ebert, U.; Löscher, W. Amygdala-kindling induces a lasting reduction of GABA-immunoreactive neurons in a discrete area of the ipsilateral piriform cortex. Synapse 1998, 29, 299–309. [Google Scholar] [CrossRef]
- West, S.E.; Doty, R.L. Influence of epilepsy and temporal lobe resection on olfactory function. Epilepsia 1995, 36, 531–542. [Google Scholar] [CrossRef]
- Laufs, H.; Richardson, M.P.; Salek-Haddadi, A.; Vollmar, C.; Duncan, J.S.; Gale, K.; Lemieux, L.; Löscher, W.; Koepp, M.J. Converging PET and fMRI evidence for a common area involved in human focal epilepsies. Neurology 2011, 77, 904–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, A.; Fortuna, A.; Alves, G.; Falcão, A. Intranasal drug delivery: How, why and what for? J. Pharm. Pharm. Sci. 2009, 12, 288–311. [Google Scholar] [CrossRef] [Green Version]
- Rech, M.A.; Barbas, B.; Chaney, W.; Greenhalgh, E.; Turck, C. When to Pick the Nose: Out-of-Hospital and Emergency Department Intranasal Administration of Medications. Ann. Emerg. Med. 2017, 70, 203–211. [Google Scholar] [CrossRef]
- Sutter, R.; Kaplan, P.W.; Rüegg, S. Outcome predictors for status epilepticus—What really counts. Nat. Rev. Neurol. 2013, 9, 525–534. [Google Scholar] [CrossRef] [PubMed]
- O’Regan, M.E.; Brown, J.K.; Clarke, M. Nasal rather than rectal benzodiazepines in the management of acute childhood seizures? Dev. Med. Child. Neurol. 1996, 38, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Jeannet, P.Y.; Roulet, E.; Maeder-Ingvar, M.; Gehri, M.; Jutzi, A.; Deonna, T. Home and hospital treatment of acute seizures in children with nasal midazolam. Eur. J. Paediatr. Neurol. 1999, 3, 73–77. [Google Scholar] [CrossRef]
- Kutlu, N.O.; Yakinci, C.; Dogrul, M.; Durmaz, Y. Intranasal midazolam for prolonged convulsive seizures. Brain Dev. 2000, 22, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Fişgin, T.; Gürer, Y.; Senbil, N.; Teziç, T.; Zorlu, P.; Okuyaz, C.; Akgün, D. Nasal midazolam effects on childhood acute seizures. J. Child Neurol. 2000, 15, 833–835. [Google Scholar] [CrossRef]
- Harbord, M.G.; Kyrkou, N.E.; Kyrkou, M.R.; Kay, D.; Coulthard, K.P. Use of intranasal midazolam to treat acute seizures in paediatric community settings. J. Paediatr. Child Health 2004, 40, 556–558. [Google Scholar] [CrossRef]
- Wilson, M.T.; Macleod, S.; O’Regan, M.E. Nasal/buccal midazolam use in the community. Arch. Dis. Child. 2004, 89, 50–51. [Google Scholar] [CrossRef] [Green Version]
- Kälviäinen, R. Intranasal therapies for acute seizures. Epilepsy Behav. 2015, 49, 303–306. [Google Scholar] [CrossRef]
- Elliott, W.; Chan, J. Midazolam Nasal Spray (Nayzilam) CIV. Intern. Med. Alert 2019, 41. Available online: https://www.proquest.com/openview/2f707c9e87f4057f9e8c26ea15878cb3/1?pq-origsite=gscholar&cbl=136155 (accessed on 1 June 2022).
- Sperling, M.R.; Haas, K.F.; Krauss, G.; Seif Eddeine, H.; Henney, H.R.; Rabinowicz, A.L., 3rd; Bream, G.; Squillacote, D.; Carrazana, E.J. Dosing feasibility and tolerability of intranasal diazepam in adults with epilepsy. Epilepsia 2014, 55, 1544–1550. [Google Scholar] [CrossRef]
- Ivaturi, V.; Kriel, R.; Brundage, R.; Loewen, G.; Mansbach, H.; Cloyd, J. Bioavailability of intranasal vs. rectal diazepam. Epilepsy Res. 2013, 103, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Hogan, R.E.; Tarquinio, D.; Sperling, M.R.; Klein, P.; Miller, I.; Segal, E.B.; Rabinowicz, A.L.; Carrazana, E. Pharmacokinetics and safety of VALTOCO (NRL-1; diazepam nasal spray) in patients with epilepsy during seizure (ictal/peri-ictal) and nonseizure (interictal) conditions: A phase 1, open-label study. Epilepsia 2020, 61, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Wermeling, D.P.; Miller, J.L.; Archer, S.M.; Manaligod, J.M.; Rudy, A.C. Bioavailability and pharmacokinetics of lorazepam after intranasal, intravenous, and intramuscular administration. J. Clin. Pharmacol. 2001, 41, 1225–1231. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.; Tambe, P.; Sammons, H.; Mulla, H.; Cole, R.; Choonara, I. Pharmacokinetics of buccal and intranasal lorazepam in healthy adult volunteers. Eur. J. Clin. Pharmacol. 2012, 68, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Arya, R.; Gulati, S.; Kabra, M.; Sahu, J.K.; Kalra, V. Intranasal versus intravenous lorazepam for control of acute seizures in children: A randomized open-label study. Epilepsia 2011, 52, 788–793. [Google Scholar] [CrossRef] [PubMed]
- Rudra, N.; Ghosh, T.; Roy, U.K. A comparative study on intranasal versus intravenous lorazepam in the management of acute seizure in children. Folia Med. 2021, 63, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D. Rescue therapies for seizure emergencies: Current and future landscape. Neurol. Sci. 2021, 42, 4017–4027. [Google Scholar] [CrossRef] [PubMed]
- Harkema, J.R.; Carey, S.A.; Wagner, J.G. The nose revisited: A brief review of the comparative structure, function, and toxicologic pathology of the nasal epithelium. Toxicol. Pathol. 2006, 34, 252–269. [Google Scholar] [CrossRef]
- Mistry, A.; Stolnik, S.; Illum, L. Nanoparticles for direct nose-to-brain delivery of drugs. Int. J. Pharm. 2009, 379, 146–157. [Google Scholar] [CrossRef]
- Illum, L. Transport of drugs from the nasal cavity to the central nervous system. Eur. J. Pharm. Sci. 2000, 11, 1–18. [Google Scholar] [CrossRef]
- Dhuria, S.V.; Hanson, L.R.; Frey, W.H., 2nd. Intranasal delivery to the central nervous system: Mechanisms and experimental considerations. J. Pharm. Sci. 2010, 99, 1654–1673. [Google Scholar] [CrossRef]
- Uraih, L.C.; Maronpot, R.R. Normal histology of the nasal cavity and application of special techniques. Environ. Health Perspect. 1990, 85, 187–208. [Google Scholar]
- Illum, L. Is nose-to-brain transport of drugs in man a reality? J. Pharm. Pharmacol. 2004, 56, 3–17. [Google Scholar] [CrossRef]
- Crowe, T.P.; Greenlee, M.H.W.; Kanthasamy, A.G.; Hsu, W.H. Mechanism of intranasal drug delivery directly to the brain. Life Sci. 2018, 195, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Gänger, S.; Schindowski, K. Tailoring Formulations for Intranasal Nose-to-Brain Delivery: A Review on Architecture, Physico-Chemical Characteristics and Mucociliary Clearance of the Nasal Olfactory Mucosa. Pharmaceutics 2018, 10, 116. [Google Scholar] [CrossRef]
- van Riel, D.; Verdijk, R.; Kuiken, T. The olfactory nerve: A shortcut for influenza and other viral diseases into the central nervous system. J. Pathol. 2015, 235, 277–287. [Google Scholar] [CrossRef]
- Yang, Z.; Huang, Y.; Gan, G.; Sawchuk, R.J. Microdialysis evaluation of the brain distribution of stavudine following intranasal and intravenous administration to rats. J. Pharm. Sci. 2005, 94, 1577–1588. [Google Scholar] [CrossRef] [PubMed]
- Luzzati, V.; Benoit, E.; Charpentier, G.; Vachette, P. X-ray Scattering Study of Pike Olfactory Nerve: Elastic, Thermodynamic and Physiological Properties of the Axonal Membrane. J. Mol. Biol. 2004, 343, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, J.J.; Wolak, D.J.; Pizzo, M.E.; Thorne, R.G. Rapid transport within cerebral perivascular spaces underlies widespread tracer distribution in the brain after intranasal administration. J. Cereb. Blood Flow Metab. 2015, 35, 371–381. [Google Scholar] [CrossRef]
- Hadaczek, P.; Yamashita, Y.; Mirek, H.; Tamas, L.; Bohn, M.C.; Noble, C.; Park, J.W.; Bankiewicz, K. The “perivascular pump” driven by arterial pulsation is a powerful mechanism for the distribution of therapeutic molecules within the brain. Mol. Ther. 2006, 14, 69–78. [Google Scholar] [CrossRef]
- Illum, L. Nasal delivery. The use of animal models to predict performance in man. J. Drug Target. 1996, 3, 427–442. [Google Scholar] [CrossRef]
- Kozlovskaya, L.; Abou-Kaoud, M.; Stepensky, D. Quantitative analysis of drug delivery to the brain via nasal route. J. Control. Release 2014, 189, 133–140. [Google Scholar] [CrossRef] [PubMed]
- McGann, J.P. Poor human olfaction is a 19th-century myth. Science 2017, 356, eaam7263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djupesland, P.G.; Mahmoud, R.A.; Messina, J.C. Accessing the brain: The nose may know the way. J. Cereb. Blood Flow Metab. 2013, 33, 793–794. [Google Scholar] [CrossRef] [Green Version]
- Löscher, W. Fit for purpose application of currently existing animal models in the discovery of novel epilepsy therapies. Epilepsy Res. 2016, 126, 157–184. [Google Scholar] [CrossRef]
- Putnam, T.J.; Merritt, H.H. Experimental Determination of The Anticonvulsant Properties Of Some Phenyl Derivatives. Science 1937, 85, 525–526. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.L.; Albertson, T.E. Neuropharmacology Methods in Epilepsy Research; CRC Press: Boca Raton, FL, USA, 2019. [Google Scholar]
- Browning, R.A.; Nelson, D.K. Variation in threshold and pattern of electroshock-induced seizures in rats depending on site of stimulation. Life Sci. 1985, 37, 2205–2211. [Google Scholar] [CrossRef]
- Löscher, W.; Fassbender, C.P.; Nolting, B. The role of technical, biological and pharmacological factors in the laboratory evaluation of anticonvulsant drugs. II. Maximal electroshock seizure models. Epilepsy Res. 1991, 8, 79–94. [Google Scholar] [CrossRef]
- Löscher, W.; Hönack, D.; Fassbender, C.P.; Nolting, B. The role of technical, biological and pharmacological factors in the laboratory evaluation of anticonvulsant drugs. III. Pentylenetetrazole seizure models. Epilepsy Res. 1991, 8, 171–189. [Google Scholar] [CrossRef] [PubMed]
- Kimball, A.W.; Burnett, W.T., Jr.; Doherty, D.G. Chemical protection against ionizing radiation. I. Sampling methods for screening compounds in radiation protection studies with mice. Radiat. Res. 1957, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bankstahl, M.; Bankstahl, J.P.; Bloms-Funke, P.; Löscher, W. Striking differences in proconvulsant-induced alterations of seizure threshold in two rat models. Neurotoxicology 2012, 33, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Browning, R.A.; Nelson, D.K. Modification of electroshock and pentylenetetrazol seizure patterns in rats after precollicular transections. Exp. Neurol. 1986, 93, 546–556. [Google Scholar] [CrossRef]
- Piredda, S.G.; Woodhead, J.H.; Swinyard, E.A. Effect of stimulus intensity on the profile of anticonvulsant activity of phenytoin, ethosuximide and valproate. J. Pharmacol. Exp. Ther. 1985, 232, 741–745. [Google Scholar] [PubMed]
- Brown, W.C.; Schiffman, D.O.; Swinyard, E.A.; Goodman, L.S. Comparative assay of an antiepileptic drugs by psychomotor seizure test and minimal electroshock threshold test. J. Pharmacol. Exp. Ther. 1953, 107, 273–283. [Google Scholar] [PubMed]
- Barton, M.E.; Klein, B.D.; Wolf, H.H.; White, H.S. Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res. 2001, 47, 217–227. [Google Scholar] [CrossRef]
- Barton, M.E.; Peters, S.C.; Shannon, H.E. Comparison of the effect of glutamate receptor modulators in the 6 Hz and maximal electroshock seizure models. Epilepsy Res. 2003, 56, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, C.S.; West, P.J.; Thomson, K.E.; Edwards, S.F.; Smith, M.D.; White, H.S.; Wilcox, K.S. Development and pharmacologic characterization of the rat 6 Hz model of partial seizures. Epilepsia 2017, 58, 1073–1084. [Google Scholar] [CrossRef] [Green Version]
- Purpura, D.P. Experimental Models of Epilepsy: A Manual for the Laboratory Worker; Dominick, P., Penry, J.K., Eds.; Raven Press: New York, NY, USA, 1972. [Google Scholar]
- Löscher, W.; Klitgaard, H.; Twyman, R.E.; Schmidt, D. New avenues for anti-epileptic drug discovery and development. Nat. Rev. Drug Discov. 2013, 12, 757–776. [Google Scholar] [CrossRef]
- Racine, R.J. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 1972, 32, 281–294. [Google Scholar] [CrossRef]
- Sato, M.; Racine, R.J.; McIntyre, D.C. Kindling: Basic mechanisms and clinical validity. Electroencephalogr. Clin. Neurophysiol. 1990, 76, 459–472. [Google Scholar] [CrossRef]
- Töllner, K.; Wolf, S.; Löscher, W.; Gernert, M. The anticonvulsant response to valproate in kindled rats is correlated with its effect on neuronal firing in the substantia nigra pars reticulata: A new mechanism of pharmacoresistance. J. Neurosci. 2011, 31, 16423–16434. [Google Scholar] [CrossRef] [Green Version]
- Ebert, U.; Löscher, W. Characterization of phenytoin-resistant kindled rats, a new model of drug-resistant partial epilepsy: Influence of genetic factors. Epilepsy Res. 1999, 33, 217–226. [Google Scholar] [CrossRef]
- Srivastava, A.K.; White, H.S. Carbamazepine, but not valproate, displays pharmacoresistance in lamotrigine-resistant amygdala kindled rats. Epilepsy Res. 2013, 104, 26–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potschka, H.; Volk, H.A.; Löscher, W. Pharmacoresistance and expression of multidrug transporter P-glycoprotein in kindled rats. Neuroreport 2004, 15, 1657–1661. [Google Scholar] [CrossRef] [PubMed]
- Dichter, M. Mechanisms of Epileptogenesis: The Transition to Seizure; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Araki, Y.; Ueki, S. Changes in Sensitivity to Convulsion in Mice with Olfactory Bulb Ablation. Jpn. J. Pharmacol. 1972, 22, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Alam, T.; Pandit, J.; Vohora, D.; Aqil, M.; Ali, A.; Sultana, Y. Optimization of nanostructured lipid carriers of lamotrigine for brain delivery: In vitro characterization and in vivo efficacy in epilepsy. Expert. Opin. Drug Deliv. 2015, 12, 181–194. [Google Scholar] [CrossRef]
- Acharya, S.P.; Pundarikakshudu, K.; Upadhyay, P.; Shelat, P.K.; Lalwani, A. Development of phenytoin intranasal microemulsion for treatment of epilepsy. J. Pharm. Investig. 2015, 45, 375–384. [Google Scholar] [CrossRef]
- Acharya, S.P.; Pundarikakshudu, K.; Panchal, A.; Lalwani, A. Preparation and evaluation of transnasal microemulsion of carbamazepine. Asian J. Pharm. Sci. 2013, 8, 64–70. [Google Scholar] [CrossRef] [Green Version]
- Löscher, W. Valproate: A reappraisal of its pharmacodynamic properties and mechanisms of action. Prog. Neurobiol. 1999, 58, 31–59. [Google Scholar] [CrossRef]
- Houghton, G.W.; Richens, A.; Toseland, P.A.; Davidson, S.; Falconer. Brain concentrations of phenytoin, phenobarbitone and primidone in epileptic patients. Eur. J. Clin. Pharmacol. 1975, 9, 73–78. [Google Scholar] [CrossRef]
- Kwan, P.; Brodie, M.J. Potential role of drug transporters in the pathogenesis of medically intractable epilepsy. Epilepsia 2005, 46, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Potschka, H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat. Rev. Neurosci. 2005, 6, 591–602. [Google Scholar] [CrossRef]
- Löscher, W.; Nolting, B.; Fassbender, C.P. The role of technical, biological and pharmacological factors in the laboratory evaluation of anticonvulsant drugs. I. The influence of administration vehicles. Epilepsy Res. 1990, 7, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Ong, W.Y.; Shalini, S.M.; Costantino, L. Nose-to-brain drug delivery by nanoparticles in the treatment of neurological disorders. Curr. Med. Chem. 2014, 21, 4247–4256. [Google Scholar] [CrossRef] [PubMed]
- Ugwoke, M.I.; Agu, R.U.; Verbeke, N.; Kinget, R. Nasal mucoadhesive drug delivery: Background, applications, trends and future perspectives. Adv. Drug Deliv. Rev. 2005, 57, 1640–1665. [Google Scholar] [CrossRef] [PubMed]
- Mayor, S.H.; Illum, L. Investigation of the effect of anaesthesia on nasal absorption of insulin in rats. Int. J. Pharm. 1997, 149, 123–129. [Google Scholar] [CrossRef]
- Thorne, R.G.; Pronk, G.J.; Padmanabhan, V.; Frey, W.H., 2nd. Delivery of insulin-like growth factor-I to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience 2004, 127, 481–496. [Google Scholar] [CrossRef]
- Czapp, M.; Bankstahl, J.P.; Zibell, G.; Potschka, H. Brain penetration and anticonvulsant efficacy of intranasal phenobarbital in rats. Epilepsia 2008, 49, 1142–1150. [Google Scholar] [CrossRef]
- Barakat, N.S.; Omar, S.A.; Ahmed, A.A. Carbamazepine uptake into rat brain following intra-olfactory transport. J. Pharm. Pharmacol. 2006, 58, 63–72. [Google Scholar] [CrossRef]
- Samia, O.; Hanan, R.; Kamal, E.T. Carbamazepine mucoadhesive nanoemulgel (MNEG) as brain targeting delivery system via the olfactory mucosa. Drug Deliv. 2012, 19, 58–67. [Google Scholar] [CrossRef]
- Patel, R.B.; Patel, M.R.; Bhatt, K.K.; Patel, B.G.; Gaikwad, R.V. Microemulsion-based drug delivery system for transnasal delivery of Carbamazepine: Preliminary brain-targeting study. Drug Deliv. 2016, 23, 207–213. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Yang, S.; Ho, P.C. Intranasal administration of carbamazepine-loaded carboxymethyl chitosan nanoparticles for drug delivery to the brain. Asian J. Pharm. Sci. 2018, 13, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Serralheiro, A.; Alves, G.; Fortuna, A.; Falcão, A. Intranasal administration of carbamazepine to mice: A direct delivery pathway for brain targeting. Eur. J. Pharm. Sci. 2014, 60, 32–39. [Google Scholar] [CrossRef]
- Taksande, J.B. Formulation and Pharmacodynamic Investigations of Lamotrigine Microspheres in Pentylenetetrazole-Induced Seizures in Mice. Asian J. Pharm. (AJP) 2017, 11, S215–S224. [Google Scholar]
- Praveen, A.; Aqil, M.; Imam, S.S.; Ahad, A.; Moolakkadath, T.; Ahmad, F.J. Lamotrigine encapsulated intra-nasal nanoliposome formulation for epilepsy treatment: Formulation design, characterization and nasal toxicity study. Colloids Surf. B Biointerfaces 2019, 174, 553–562. [Google Scholar] [CrossRef]
- Shah, P.; Dubey, P.; Vyas, B.; Kaul, A.; Mishra, A.K.; Chopra, D.; Patel, P. Lamotrigine loaded PLGA nanoparticles intended for direct nose to brain delivery in epilepsy: Pharmacokinetic, pharmacodynamic and scintigraphy study. Artif. Cells Nanomed. Biotechnol. 2021, 49, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Yousfan, A.; Rubio, N.; Al-Ali, M.; Nattouf, A.H.; Kafa, H. Intranasal delivery of phenytoin-loaded nanoparticles to the brain suppresses pentylenetetrazol-induced generalized tonic clonic seizures in an epilepsy mouse model. Biomater Sci. 2021, 9, 7547–7564. [Google Scholar] [CrossRef]
- Taymouri, S.; Minaiyan, M.; Ebrahimi, F.; Tavakoli, N. In-vitro and in-vivo evaluation of chitosan-based thermosensitive gel containing lorazepam NLCs for the treatment of status epilepticus. IET Nanobiotechnol. 2020, 14, 148–154. [Google Scholar] [CrossRef]
- Sharma, D.; Sharma, R.K.; Sharma, N.; Gabrani, R.; Sharma, S.K.; Ali, J.; Dang, S. Nose-To-Brain Delivery of PLGA-Diazepam Nanoparticles. AAPS PharmSciTech 2015, 16, 1108–1121. [Google Scholar] [CrossRef] [Green Version]
- Eskandari, S.; Varshosaz, J.; Minaiyan, M.; Tabbakhian, M. Brain delivery of valproic acid via intranasal administration of nanostructured lipid carriers: In vivo pharmacodynamic studies using rat electroshock model. Int. J. Nanomed. 2011, 6, 363–371. [Google Scholar]
- Gonçalves, J.; Bicker, J.; Gouveia, F.; Liberal, J.; Oliveira, R.C.; Alves, G.; Falcão, A.; Fortuna, A. Nose-to-brain delivery of levetiracetam after intranasal administration to mice. Int. J. Pharm. 2019, 564, 329–339. [Google Scholar] [CrossRef]
- Gonçalves, J.; Alves, G.; Carona, A.; Bicker, J.; Vitorino, C.; Falcão, A.; Fortuna, A. Pre-Clinical Assessment of the Nose-to-Brain Delivery of Zonisamide After Intranasal Administration. Pharm. Res. 2020, 37, 74. [Google Scholar] [CrossRef] [PubMed]
- Serralheiro, A.; Alves, G.; Fortuna, A.; Falcão, A. Direct nose-to-brain delivery of lamotrigine following intranasal administration to mice. Int. J. Pharm. 2015, 490, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Illum, L. Nanoparticulate systems for nasal delivery of drugs: A real improvement over simple systems? J. Pharm. Sci. 2007, 96, 473–483. [Google Scholar] [CrossRef]
- Renne, R.; Brix, A.; Harkema, J.; Herbert, R.; Kittel, B.; Lewis, D.; March, T.; Nagano, K.; Pino, M.; Rittinghausen, S.; et al. Proliferative and nonproliferative lesions of the rat and mouse respiratory tract. Toxicol. Pathol. 2009, 37, 5s–73s. [Google Scholar] [CrossRef] [Green Version]
- Young, J.T. Histopathologic examination of the rat nasal cavity. Fundam. Appl. Toxicol. 1981, 1, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.B.; Patel, M.R.; Bhatt, K.K.; Patel, B.G. Formulation consideration and characterization of microemulsion drug delivery system for transnasal administration of carbamazepine. Bull. Fac. Pharm. Cairo Univ. 2013, 51, 243–253. [Google Scholar] [CrossRef]
- Löscher, W. The pharmacokinetics of antiepileptic drugs in rats: Consequences for maintaining effective drug levels during prolonged drug administration in rat models of epilepsy. Epilepsia 2007, 48, 1245–1258. [Google Scholar] [CrossRef]
- Markowitz, G.J.; Kadam, S.D.; Boothe, D.M.; Irving, N.D.; Comi, A.M. The pharmacokinetics of commonly used antiepileptic drugs in immature CD1 mice. Neuroreport 2010, 21, 452. [Google Scholar] [CrossRef]
- Löscher, W.; Hönack, D. Intravenous carbamazepine: Comparison of different parenteral formulations in a mouse model of convulsive status epilepticus. Epilepsia 1997, 38, 106–113. [Google Scholar] [CrossRef]
- Miller, A.A.; Wheatley, P.; Sawyer, D.A.; Baxter, M.G.; Roth, B. Pharmacological Studies on Lamotrigine, A Novel Potential Antiepileptic Drug. Epilepsia 1986, 27, 483–489. [Google Scholar] [CrossRef]
- Kudriakova, T.B.; Sirota, L.A.; Rozova, G.I.; Gorkov, V.A. Autoinduction and steady-state pharmacokinetics of carbamazepine and its major metabolites. Br. J. Clin. Pharmacol. 1992, 33, 611–615. [Google Scholar] [CrossRef] [Green Version]
- Greenspan, P.; Fowler, S.D. Spectrofluorometric studies of the lipid probe, nile red. J. Lipid Res. 1985, 26, 781–789. [Google Scholar] [CrossRef]
- Manocha, A.; Sharma, K.K.; Mediratta, P.K. Possible mechanism of anticonvulsant effect of ketamine in mice. Indian J. Exp. Biol. 2001, 39, 1002–1008. [Google Scholar] [PubMed]
- Synowiec, A.S.; Singh, D.S.; Yenugadhati, V.; Valeriano, J.P.; Schramke, C.J.; Kelly, K.M. Ketamine use in the treatment of refractory status epilepticus. Epilepsy Res. 2013, 105, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Borgeat, A. Propofol: Pro- or anticonvulsant? Eur. J. Anaesthesiol. Suppl. 1997, 15, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Borowicz, K.K.; Łuszczki, J.; Czuczwar, S.J. Interactions between non-barbiturate injectable anesthetics and conventional antiepileptic drugs in the maximal electroshock test in mice—An isobolographic analysis. Eur. Neuropsychopharmacol. 2004, 14, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Business Wire. Virpax to Develop Intranasal Cannabidiol Product for the Management of Epilepsy in Adults and Children; Business Wire: San Francisco, CA, USA, 2021. [Google Scholar]
- de Carvalho Reis, R.; Almeida, K.J.; da Silva Lopes, L.; de Melo Mendes, C.M.; Bor-Seng-Shu, E. Efficacy and adverse event profile of cannabidiol and medicinal cannabis for treatment-resistant epilepsy: Systematic review and meta-analysis. Epilepsy Behav. 2020, 102, 106635. [Google Scholar] [CrossRef]
- Palrasu, M.; Wright, L.; Patel, M.; Leech, L.; Branch, S.; Harrelson, S.; Khan, S. Perspectives on Challenges in Cannabis Drug Delivery Systems: Where Are We? Med. Cannabis Cannabinoids 2022, 5, 102–119. [Google Scholar] [CrossRef]
- Esposito, E.; Drechsler, M.; Cortesi, R.; Nastruzzi, C. Encapsulation of cannabinoid drugs in nanostructured lipid carriers. Eur. J. Pharm. Biopharm. 2016, 102, 87–91. [Google Scholar] [CrossRef]



| Parameter | Human | Rat |
|---|---|---|
| Nasal cavity volume | 25 cm3 | 0.26 to 0.4 cm3 |
| Nasal cavity surface area | 150 to160 cm2 | 13.4 to 14 cm2 |
| Surface area per unit volume | 6.4 | 51.5 |
| Olfactory epithelium area (area, %) | 12.5 cm2, 8% | 6.75 cm2, 50% |
| CSF volume | 160 mL | 150 µL |
| CSF volume replacement frequency | 5 h | Hourly |
| Shape of upper airways | L-shaped | Linear |
| Type of breathing at rest | Oronasal | Obligate nose |
| Connection between nasal cavity and oral cavity | No (incisive canal is not patent) | Yes (nasopalatine canal is patent) |
| Vascular swell bodies in septum | No | Yes |
| Turbinates (number and shape) | 3; comma shaped | 3; t-shaped with elaborate scrolls |
| Presence of ethmoid sinuses (air cells) and spheroid sinuses | Yes | No |
| Maxillary sinuses | Large; open | Small; closed |
| Nasal secretion movement | Mostly posteriorly (to nasopharynx) | Mostly anteriorly (towards nostril) |
| Inspiratory airflow route | Close to floor of nasal passage | Upward and laterally |
| ASD | Tissues Analysed | PK Parameters Reported | Time Points after Administration | Routes/Formulations Compared | Test | Endpoint | Time of Test | Anaesthesia | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| CBZ | Brain—olfactory bulbs, frontal cortex, remainder; plasma; liver | DTE; brain, plasma and liver concentration; B:P; Tmax; Cmax; AUC; kel; t1/2, MRT; F | 5, 10, 15, 30, 60 min | i.n. (form.); i.v. (form.) | - | - | - | Ketamine and xylazine (i.p.) | [128] |
| Brain; plasma | Brain and plasma concentration; AUC; Tmax; Cmax; kel; t1/2; %DTE; %DTP | 30, 60, 120, 240, 480 min | i.n. (form. x 2); i.n. (sol.); i.v. (form.) | - | - | Ketamine (i.m.) | [126] | ||
| Brain; plasma | Brain and plasma concentration; B:P ratio; AUC; Cmax; Tmax; MRT, AUC (B:P) | 5, 10, 15, 20, 30, 45, 60, 90, 120 min | No treatment; i.n. (sol.); i.n. (form.); p.o. (sol.) | - | - | - | None | [124] | |
| - | - | - | i.n. (sol.); i.n. (form.); p.o. (form.); i.n. (sol.); No treatment | MES (auricular) | Duration of HLE | 60 min | None reported | [113] | |
| - | - | - | i.n. (form.); i.n. (sol.); No treatment | MES variant (auricular) PTZ (i.p.) | MES variant: number of trials until death PTZ: onset to convulsion, time until death | 5 min (MES variant) 15 min (PTZ) | Diethyl ether | [125] | |
| Brain; plasma | Brain and plasma concentration; AUC; Tmax; Cmax; MRT | 0.25, 0.5, 1, 2, 3, 4 h | i.n. (form); i.n. (sol.) | - | - | - | None | [127] | |
| DZP | Brain, plasma | %DTE; brain and plasma concentration; Cmax; Tmax; AUC | 30, 60, 120, 240, 480 min | i.n. (sol.); i.n. (form.); i.v. (sol.) | - | - | - | Ketamine (i.p.) | [134] |
| VA | Brain; plasma | Brain and plasma concentration; B:P | 60 min | i.n. (form. no drug); i.n. (form.); i.n. (sol.); i.p. (form. no drug); i.p. (form.); i.p. (sol.) | MES variation (auricular) | E:F ratio of hindlimbs | 15, 30, 60, 90, 120 min | Light ether | [135] |
| PHT | Brain | Brain concentration | 15 and 30 min | No treatment; i.n. (form.); p.o. (form.); i.p. (sol.) | MES (auricular) | Duration of HLE | 60 min | None reported | [112] |
| Brain, serum, liver, spleen and kidneys | DTE% and DTP%; Brain, plasma and liver concentration; Cmax; Tmax; AUCbrain/AUCplasma ratio; t1/2 | 5, 15, 60, 240, 1440, 2880, 4320, 5760, 7200 min | i.n. (form); i.p. (sol) | PTZ (s.c.) | Duration; frequency; total number of EEG signal | 1, 4, 48 h | Ketamine and xylazine (i.p.) | [132] | |
| PBT | Whole brain. OB, frontal cortex, piriform cortex, amygdala, hippocampus, parahippocampal cortex, caudal cortex, cerebellum, pons. Frontal cortex dialysate; plasma. | D:P; (microdialysis in frontal cortex); brain and plasma concentration (homogenate); B:P (homogenate) | 10 min (microdissected regions) 2, 5, 10, 20, 30, 60, 200, 240 min (whole brain and plasma) 15, 30, 60, 90, 120, 180, 240 min (dialysate) | i.n. (form. no drug); i.n. (form.); i.v. (form. no drug); i.v. (form.) | Amygdala kindling | ADT; seizure severity and duration; ADD; GST | 60 min | Propofol (i.v.) | [123] |
| LMT | - | - | - | Saline (route not reported); i.n. (form.); i.p. (form.) | PTZ (s.c.) | Onset to clonic convulsion Protection against mortality | 30 min | Ketamine and xylazine (route not reported) | [129] |
| Brain—olfactory bulbs, frontal cortex, remainder; plasma; liver | DTE; Brain, plasma and liver concentration; B:P; Cmax; Tmax; AUC; kel; k; t1/2; MRT; Absolute i.n. F; AUC ratio (L:P) | 5, 10, 15, 30, 60, 120, 240 min | i.n. (form.); i.v. (form.) | - | - | - | Ketamine and xylazine (i.p.) | [138] | |
| Brain; plasma | Brain and plasma concentration | 24 h | i.n. (sol.); i.n. (form.); p.o. | MES (auricular) | HLE incidence; Latency to HLE; Duration of HLE | 60 min 24 h | Ketamine (i.m.) | [111] | |
| Brain; plasma | Brain, plasma concentration; B:P; Tmax; Cmax; AUC; i.n. F; t1/2, MRT; F | 15, 30, 60, 120, 240, 480 min | i.n. (form.); i.n. (sol.); i.v. (sol) | PTZ (route of administration not reported) | Onset to seizure | 15, 30 and 60 min | Not reported | [131] | |
| THR | - | - | - | i.n. (form.); i.n. (form. no drug) | Amygdala kindling | ADD Number of seizures until first Stage 5; Number of seizures until fully kindled | Daily stimulations until fully kindled; Doses administered at both 60 and 30 min before stimulation | Isoflurane | [17,18] |
| LZM | - | - | - | i.n. (form.); i.n. (form. no drug); i.p. (sol.) | PTZ (s.c.) | Lag time of incidence; severity of symptoms in trunk (0–3); severity of symptoms in hands and feet (0–3); duration of symptoms | - | Not reported | [13] |
| LVT | Brain, plasm, lung and kidney | tmax; Cmax; AUCt; AUCinf; AUCextrap; kel; t1/2el; MRT; F; AUCt; AUCbrain/AUCplasma; AUClung/AUCplasma; AUCkidney/AUCplasma | 5, 15, 30, 60, 90, 120 and 240 min | i.n. (form.); i.v. (sol) | Ketamine and xylazine (i.p.) | [136] | |||
| ZNA | Brain, plasm, lung and kidney | tmax; Cmax; AUCt; AUCinf; AUCextrap; kel; t1/2el; MRT; F; AUCt; AUCbrain/AUCplasma; AUClung/AUCplasma; AUCkidney/AUCplasma | 5, 15, 30, 60, 90, 120, 240, 360, 480 and 720 min | i.n. (form.); i.n. (form. no drug); i.v. (sol); p.o. (sol.) | - | - | Ketamine and xylazine (i.p.) | [137] |
| ASM | Plasma Half-Life (h) [138,143,144,145] | Time (min) to Peak Effect after Single Parenteral Dose [92,146] | |||
|---|---|---|---|---|---|
| Rats | Mice | Human | Rats | Mice | |
| Carbamazepine | 1.2–3.5 | 30–60 | 25–50 | 30 | 15 |
| Phenobarbital | 9–20 | 7.5 | 70–100 | 60 | 30 |
| Lamotrigine | 12–30 | 8 * | 21–50 | 60 | 120 |
| Phenytoin | 1–8 | 16 | 15–20 | 30 | 120 |
| Valproic acid | 1–5 | 0.8 | 8–15 | 15 | 5 |
| Diazepam | 1.4 | 7.7 | 24–72 | 15 | 15 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prentice, R.N.; Rizwan, S.B. Translational Considerations in the Development of Intranasal Treatments for Epilepsy. Pharmaceutics 2023, 15, 233. https://doi.org/10.3390/pharmaceutics15010233
Prentice RN, Rizwan SB. Translational Considerations in the Development of Intranasal Treatments for Epilepsy. Pharmaceutics. 2023; 15(1):233. https://doi.org/10.3390/pharmaceutics15010233
Chicago/Turabian StylePrentice, Richard N., and Shakila B. Rizwan. 2023. "Translational Considerations in the Development of Intranasal Treatments for Epilepsy" Pharmaceutics 15, no. 1: 233. https://doi.org/10.3390/pharmaceutics15010233