
Bioadhesive 3D-Printed Skin Drug Delivery Polymeric Films: From the Drug Loading in Mesoporous Silica to the Manufacturing Process

 , , and
, , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Drug Loading
2.3. Drug Content
2.4. Scanning Electron Microscopy (SEM)
2.5. Thermogravimetric Analysis (TGA)
2.6. Nitrogen Adsorption-Desorption Isotherms
2.7. Differential Scanning Calorimetry (DSC)
2.8. X-ray Powder Diffraction (XRD)
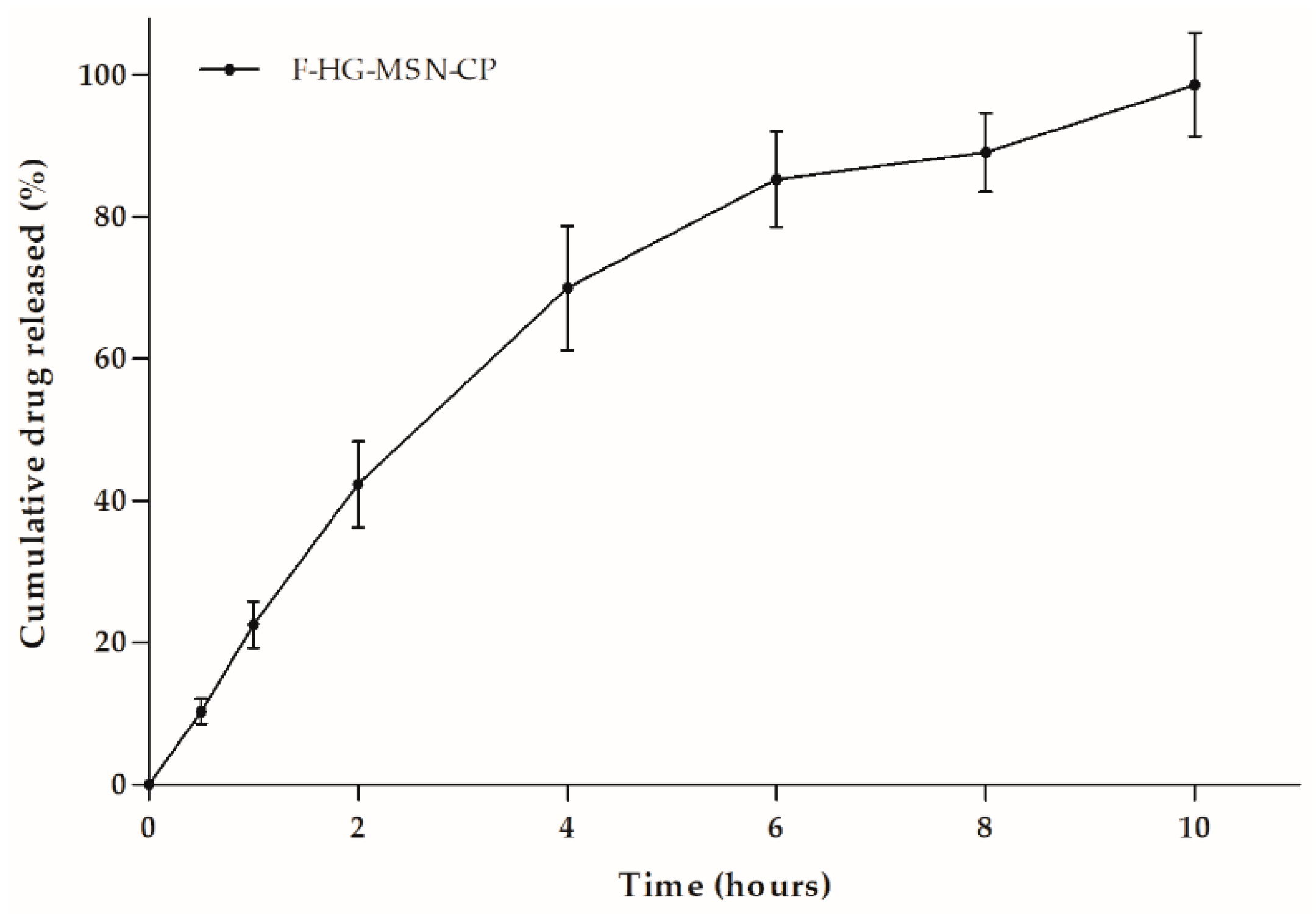
2.9. In Vitro Drug Dissolution Studies
2.10. Preparation of the 3D Printing Inks
2.11. Rheological Characterisation of the 3D Printing Inks
2.12. 3D Printing of the Skin Delivery Films
2.13. Physicochemical Characterisation of 3D-Printed Films
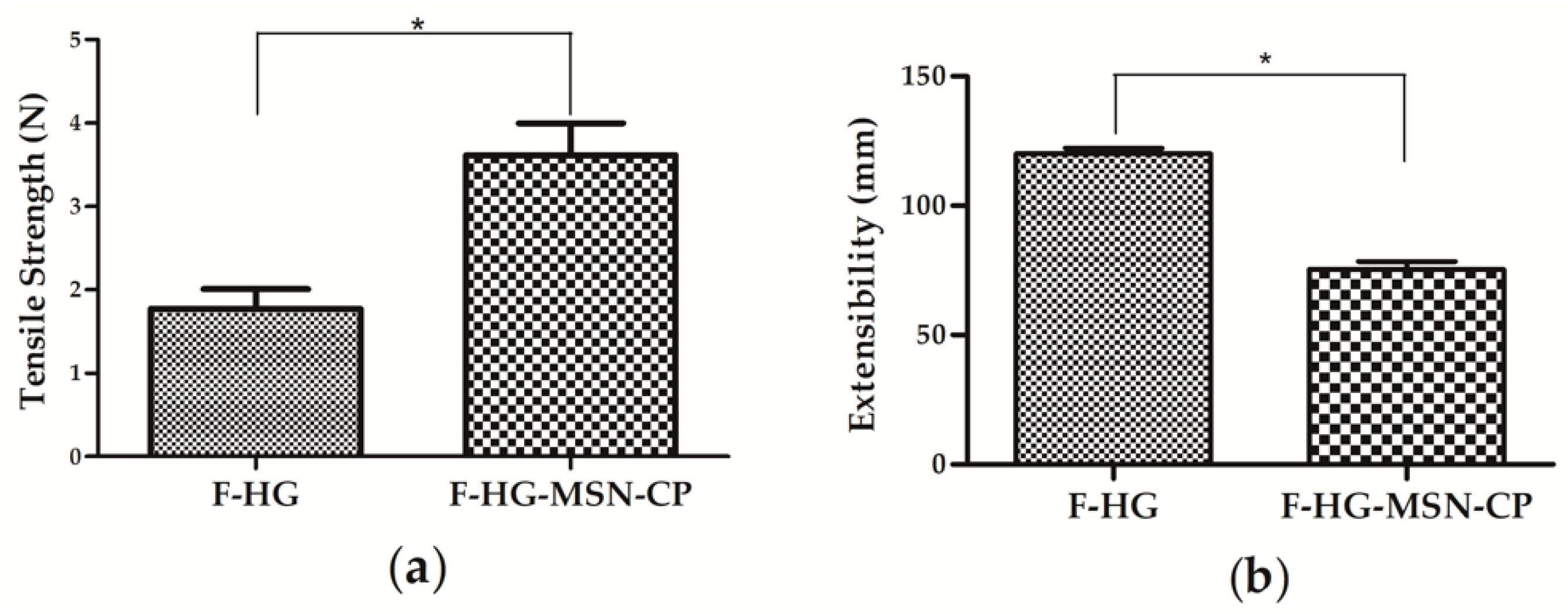
2.14. Mechanical Properties of 3D-Printed Films
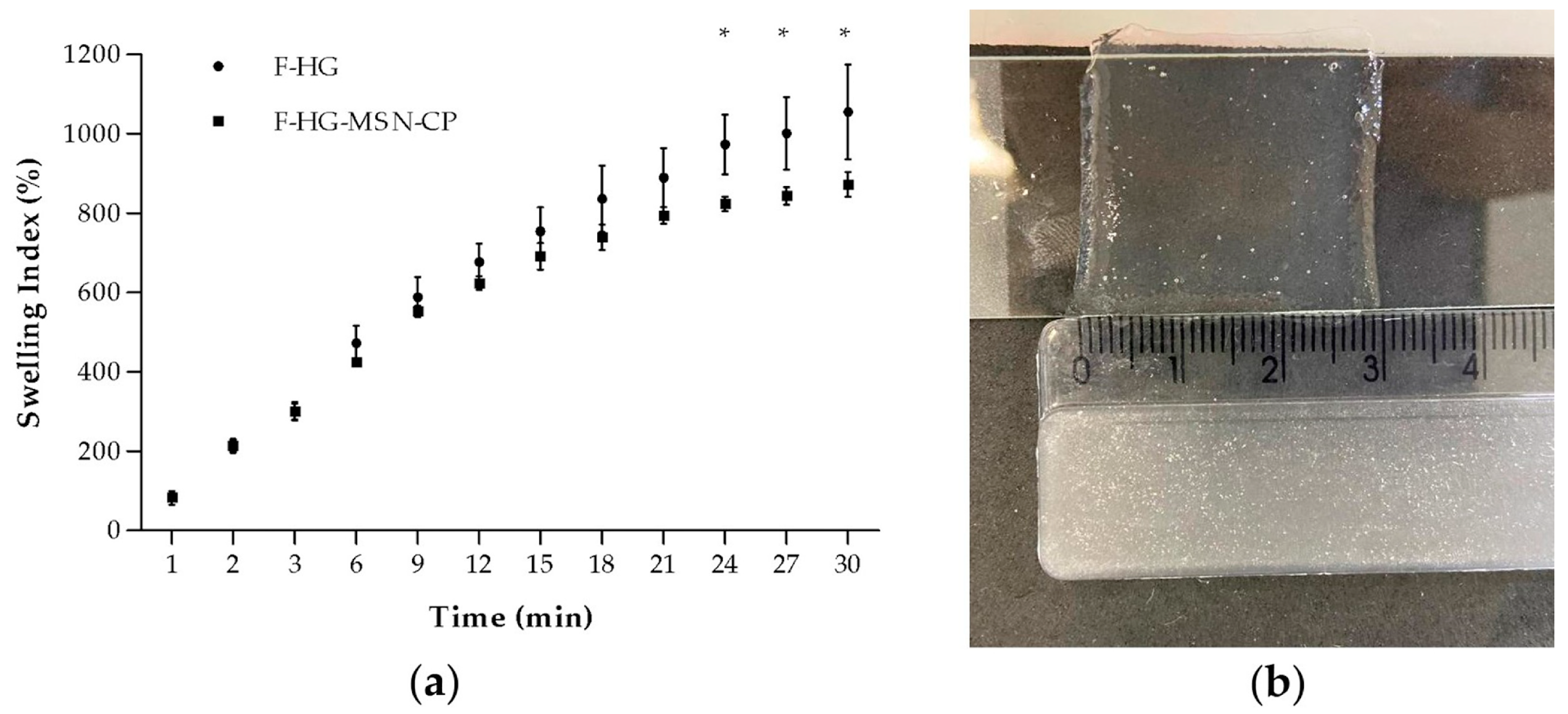
2.15. In Vitro Swelling Properties of 3D-Printed Films
2.16. In Vitro Drug Release from 3D-Printed Films
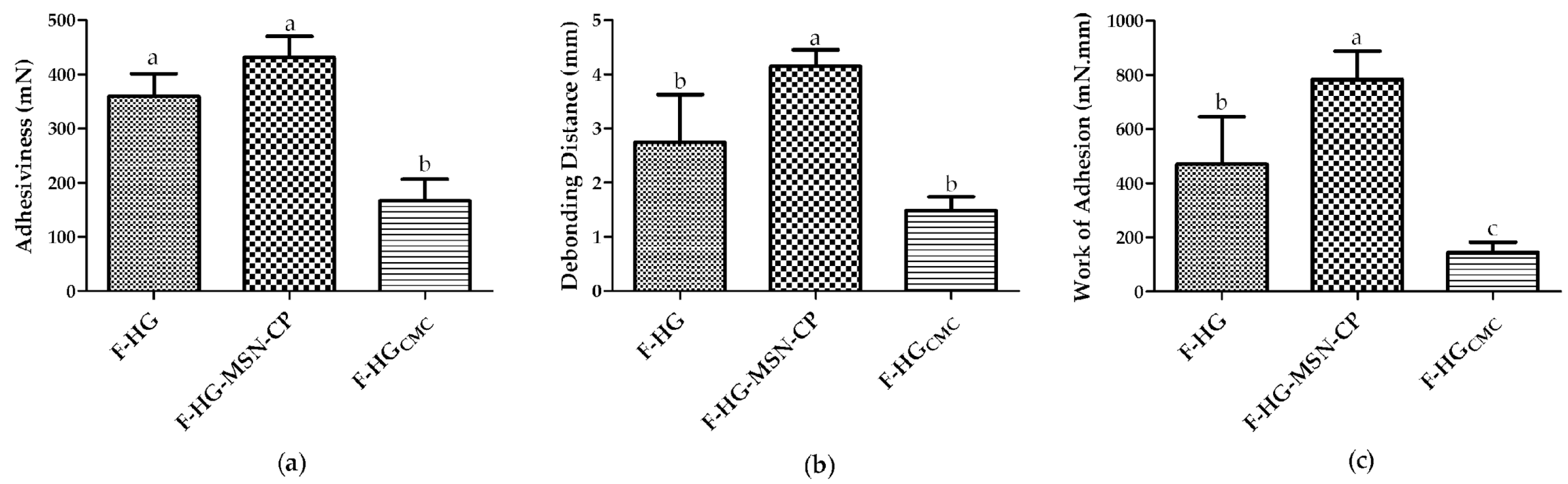
2.17. In Vitro Skin Adhesion Behaviour of the 3D-Printed Films
2.18. Statistical Analyses
3. Results and Discussion
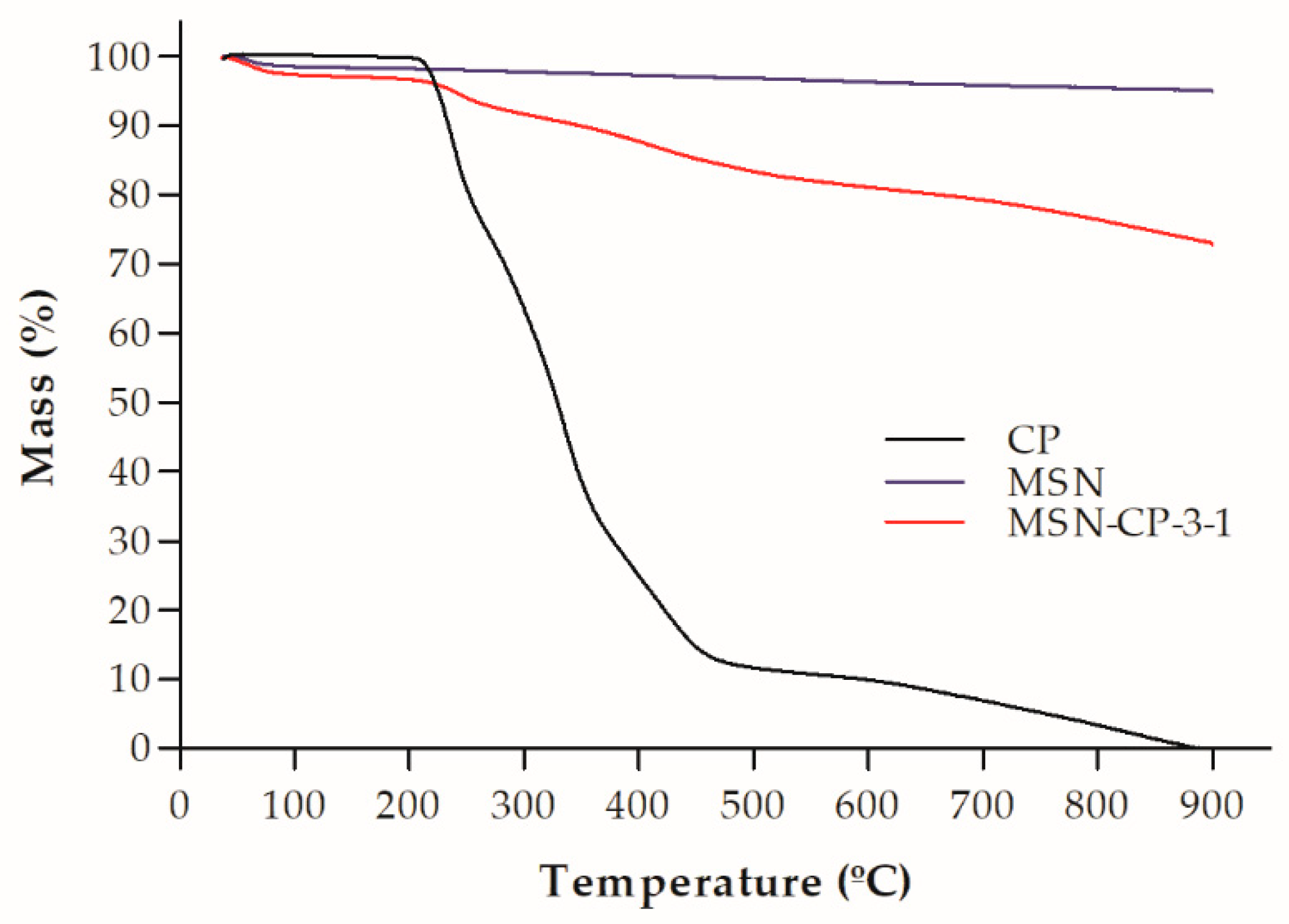
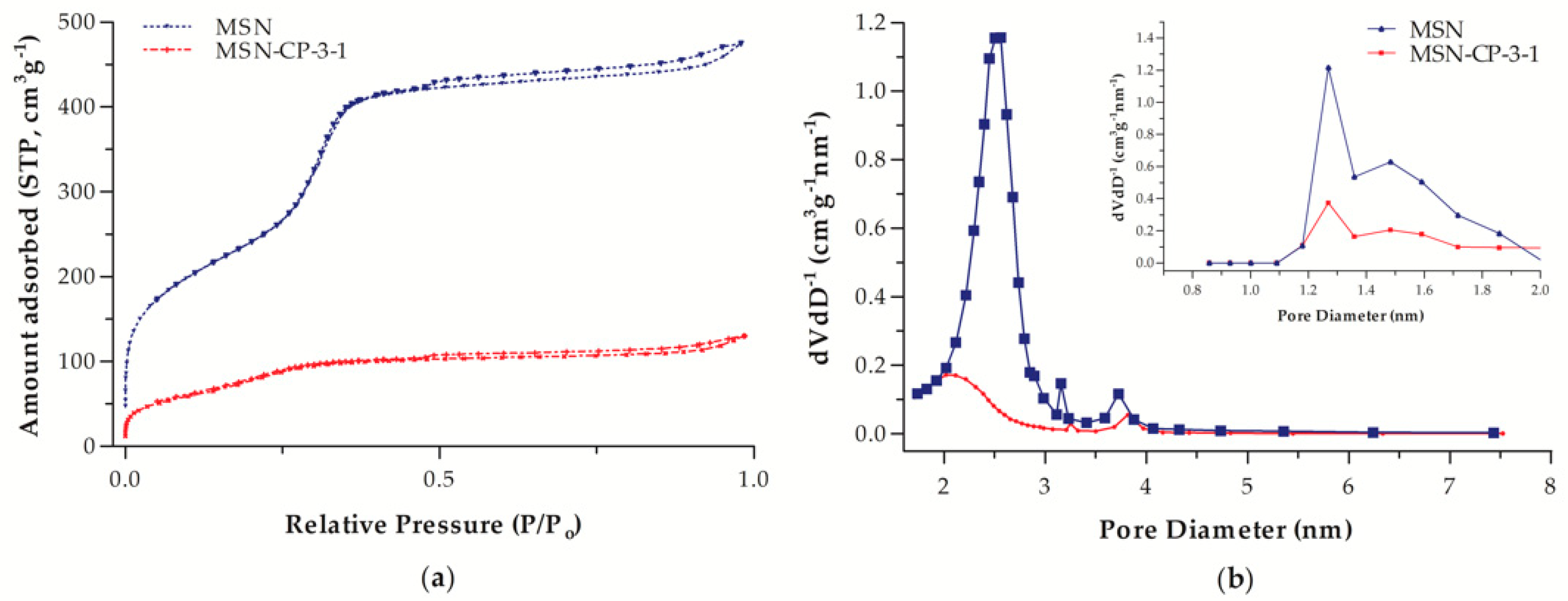
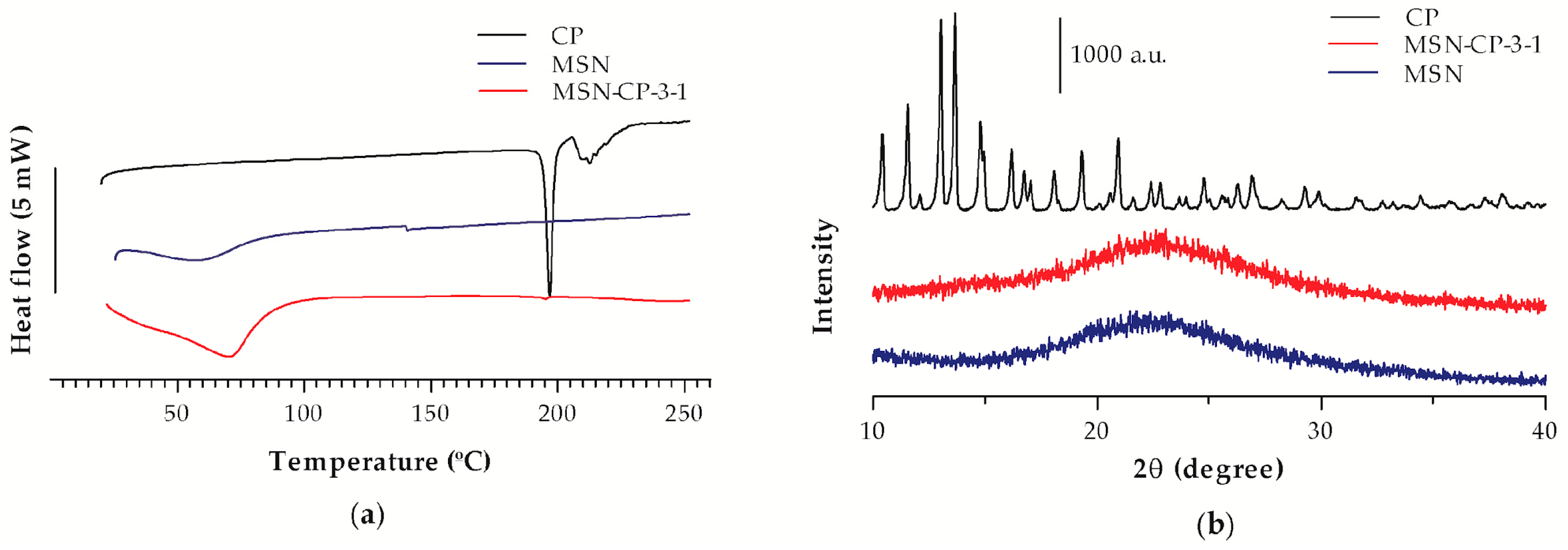
3.1. Physicochemical and Morphological Characterisation of Bare and CP-Loaded MSN
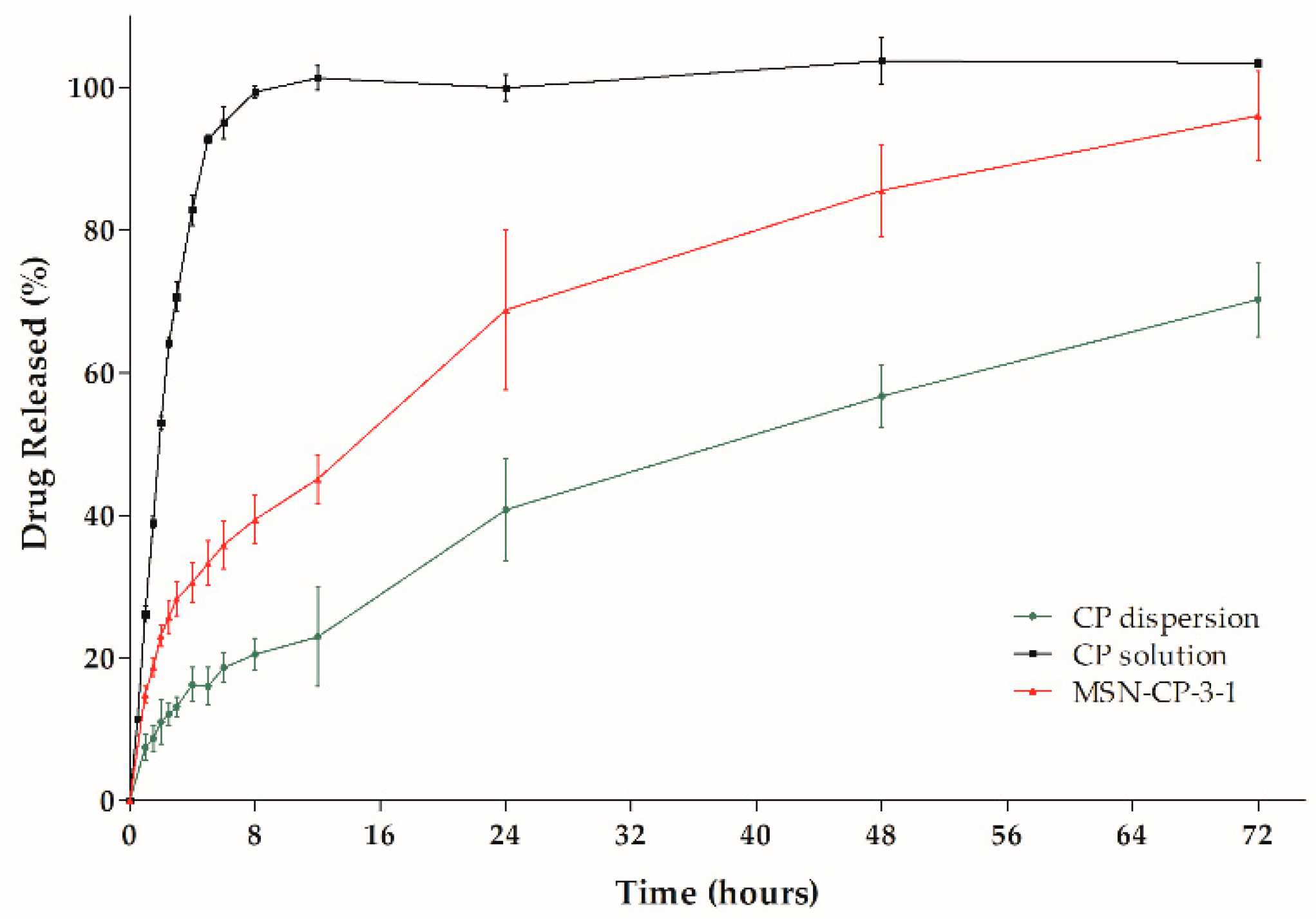
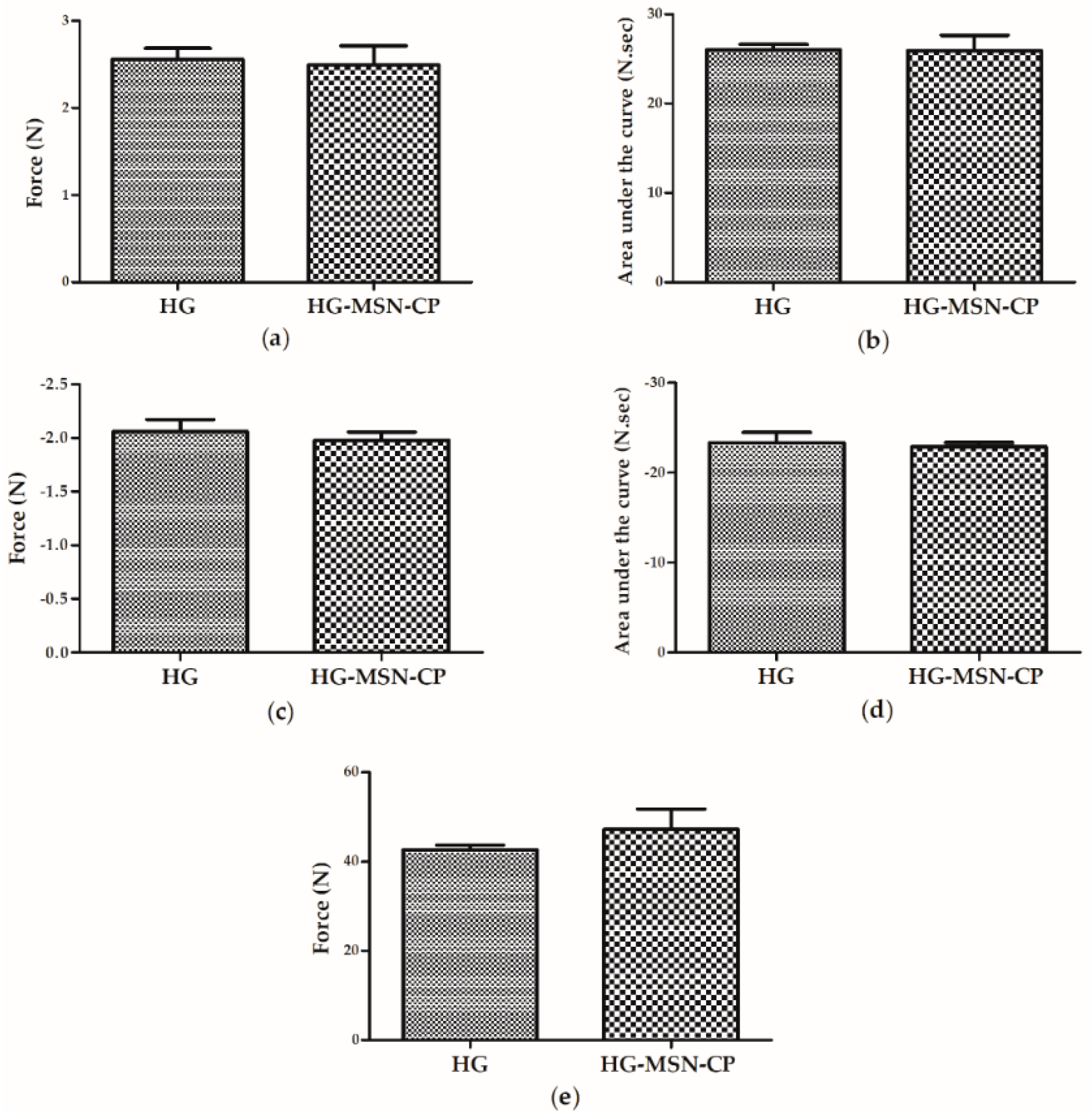
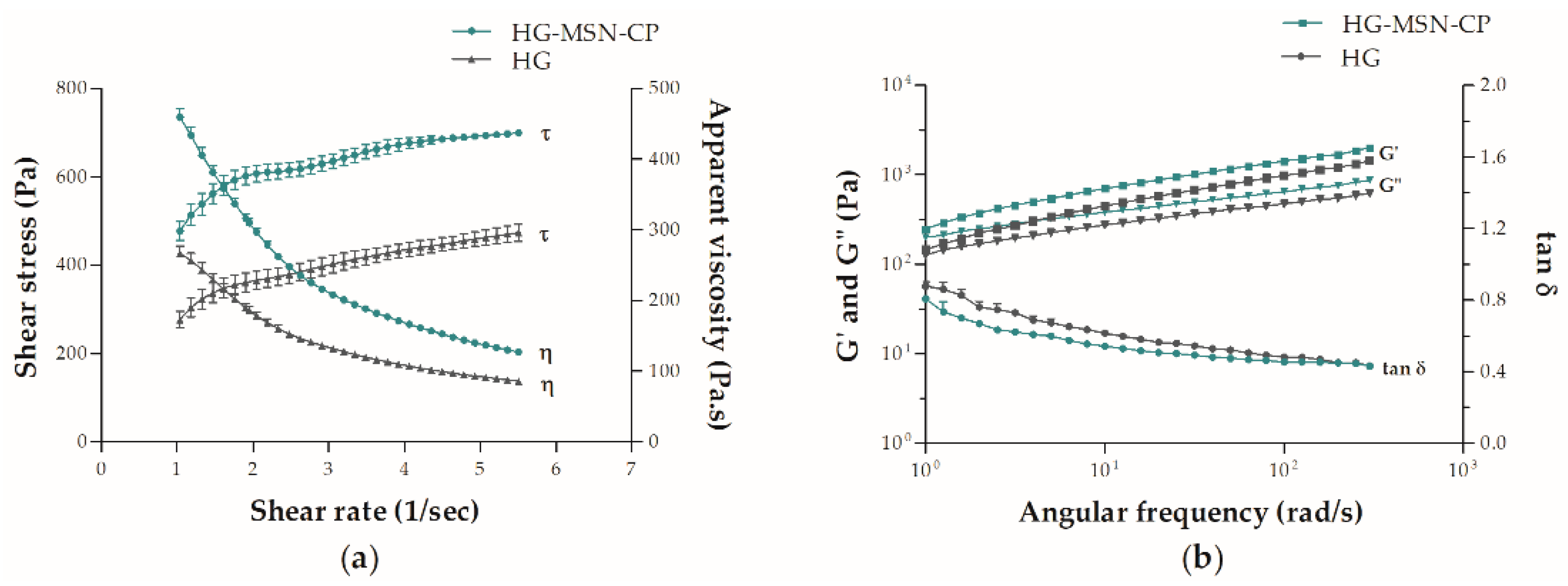
3.2. Hydrogel Printing Inks
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vallet-Regi, M.; Rámila, A.; Del Real, R.P.; Pérez-Pariente, J. A New Property of MCM-41: Drug Delivery System. Chem. Mater. 2000, 13, 308–311. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Y.; Feng, N. Mesoporous Silica Nanoparticles: Synthesis, Classification, Drug Loading, Pharmacokinetics, Biocompatibility, and Application in Drug Delivery. Expert Opin. Drug Deliv. 2019, 16, 219–237. [Google Scholar] [CrossRef] [PubMed]
- Nigro, A.; Pellegrino, M.; Greco, M.; Comandè, A.; Sisci, D.; Pasqua, L.; Leggio, A.; Morelli, C. Dealing with Skin and Blood-Brain Barriers: The Unconventional Challenges of Mesoporous Silica Nanoparticles. Pharmaceutics 2018, 10, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kankala, R.K.; Han, Y.H.; Na, J.; Lee, C.H.; Sun, Z.; Wang, S.; Bin Kimura, T.; Ok, Y.S.; Yamauchi, Y.; Chen, A.Z.; et al. Nanoarchitectured Structure and Surface Biofunctionality of Mesoporous Silica Nanoparticles. Adv. Mater. 2020, 32, 1907035. [Google Scholar] [CrossRef] [PubMed]
- Farjadian, F.; Roointan, A.; Mohammadi-Samani, S.; Hosseini, M. Mesoporous Silica Nanoparticles: Synthesis, Pharmaceutical Applications, Biodistribution, and Biosafety Assessment. Chem. Eng. J. 2019, 359, 684–705. [Google Scholar] [CrossRef]
- Jeelani, P.G.; Mulay, P.; Venkat, R.; Ramalingam, C. Multifaceted Application of Silica Nanoparticles. A Review. Silicon 2020, 12, 1337–1354. [Google Scholar] [CrossRef]
- Chircov, C.; Spoială, A.; Păun, C.; Crăciun, L.; Ficai, D.; Ficai, A.; Andronescu, E.; Turculeƫ, S.C. Mesoporous Silica Platforms with Potential Applications in Release and Adsorption of Active Agents. Molecules 2020, 25, 3814. [Google Scholar] [CrossRef]
- Sapino, S.; Oliaro-Bosso, S.; Zonari, D.; Zattoni, A.; Ugazio, E. Mesoporous Silica Nanoparticles as a Promising Skin Delivery System for Methotrexate. Int. J. Pharm. 2017, 530, 239–248. [Google Scholar] [CrossRef]
- Maleki, A.; Kettiger, H.; Schoubben, A.; Rosenholm, J.M.; Ambrogi, V.; Hamidi, M. Mesoporous Silica Materials: From Physico-Chemical Properties to Enhanced Dissolution of Poorly Water-Soluble Drugs. J. Control. Release 2017, 262, 329–347. [Google Scholar] [CrossRef]
- Müller, R.H.; Hespeler, D.; Jin, N.; Pyo, S.M. SmartPearls—Novel Physically Stable Amorphous Delivery System for Poorly Soluble Dermal Actives. Int. J. Pharm. 2019, 555, 314–321. [Google Scholar] [CrossRef]
- Fontana, M.C.; Laureano, J.V.; Forgearini, B.; dos Santos, J.; Pohlmann, A.R.; Guterres, S.S.; de Araujo, B.V.; Beck, R.C.R. Spray-Dried Raloxifene Submicron Particles for Pulmonary Delivery: Development and in Vivo Pharmacokinetic Evaluation in Rats. Int. J. Pharm. 2020, 585, 119429. [Google Scholar] [CrossRef] [PubMed]
- Hespeler, D.; Pyo, S.M.; Müller, R.H. Dermal SmartPearls—Optimized Silica Particles for Commercial Products & Mechanistic Considerations. Int. J. Pharm. 2020, 574, 118757. [Google Scholar] [CrossRef] [PubMed]
- Parmar, P.K.; Wadhawan, J.; Bansal, A.K. Pharmaceutical Nanocrystals: A Promising Approach for Improved Topical Drug Delivery. Drug Discov. Today 2021, 26, 2329–2349. [Google Scholar] [CrossRef] [PubMed]
- Lio, D.C.S.; Liu, C.; Oo, M.M.S.; Wiraja, C.; Teo, M.H.Y.; Zheng, M.; Chew, S.W.T.; Wang, X.; Xu, C. Transdermal Delivery of Small Interfering RNAs with Topically Applied Mesoporous Silica Nanoparticles for Facile Skin Cancer Treatment. Nanoscale 2019, 11, 17041–17051. [Google Scholar] [CrossRef] [PubMed]
- Nafisi, S.; Samadi, N.; Houshiar, M.; Maibach, H.I. Mesoporous Silica Nanoparticles for Enhanced Lidocaine Skin Delivery. Int. J. Pharm. 2018, 550, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Valetti, S.; Thomsen, H.; Wankar, J.; Falkman, P.; Manet, I.; Feiler, A.; Ericson, M.B.; Engblom, J. Can Mesoporous Nanoparticles Promote Bioavailability of Topical Pharmaceutics? Int. J. Pharm. 2021, 602, 120609. [Google Scholar] [CrossRef]
- De Oliveira, R.S.; Fantaus, S.S.; Jos, A.; Melero, A.; Carlos, R.; Beck, R. 3D-Printed Products for Topical Skin Applications: From Personalized Dressings to Drug Delivery. Pharmaceutics 2021, 13, 1946. [Google Scholar] [CrossRef]
- dos Santos, J.; de Oliveira, R.S.; de Oliveira, T.V.; Velho, M.C.; Konrad, M.V.; da Silva, G.S.; Deon, M.; Beck, R.C.R. 3D Printing and Nanotechnology: A Multiscale Alliance in Personalized Medicine. Adv. Funct. Mater. 2021, 31, 2009691. [Google Scholar] [CrossRef]
- Beck, R.C.R.; Chaves, P.S.; Goyanes, A.; Vukosavljevic, B.; Buanz, A.; Windbergs, M.; Basit, A.W.; Gaisford, S. 3D Printed Tablets Loaded with Polymeric Nanocapsules: An Innovative Approach to Produce Customized Drug Delivery Systems. Int. J. Pharm. 2017, 528, 268–279. [Google Scholar] [CrossRef]
- de Oliveira, T.V.; de Oliveira, R.S.; Funk, N.L.; Petzhold, C.L.; Beck, R.C.R. Redispersible 3D Printed Nanomedicines: An Original Application of the Semisolid Extrusion Technique. Int. J. Pharm. 2022, 624, 122029. [Google Scholar] [CrossRef]
- Schmidt, L.M.; de Oliveira, T.V.; dos Santos, J.; Funk, N.L.; Petzhold, C.L.; Benvenutti, E.V.; Deon, M.; Beck, R.C.R. Drug-Loaded Mesoporous Silica on Carboxymethyl Cellulose Hydrogel: Development of Innovative 3D Printed Hydrophilic Films. Int. J. Pharm. 2022, 620, 121750. [Google Scholar] [CrossRef] [PubMed]
- Fontana, M.C.; Rezer, J.F.P.; Coradini, K.; Leal, D.B.R.; Beck, R.C.R. Improved Efficacy in the Treatment of Contact Dermatitis in Rats by a Dermatological Nanomedicine Containing Clobetasol Propionate. Eur. J. Pharm. Biopharm. 2011, 79, 241–249. [Google Scholar] [CrossRef] [PubMed]
- De Andrade, D.F.; Fontana, M.C.; Beck, R.C.R. Evaluation of Three Synthetic Membranes as Limiting Barrier for in Vitro Drug Release Studies from Hydrogels Containing Polymeric Nanocapsules. Curr. Nanosci. 2014, 10, 367–373. [Google Scholar] [CrossRef]
- De Andrade, D.F.; Fontana, M.C.; Pohlmann, A.R.; Guterres, S.S.; Beck, R.C.R. Nanoencapsulation of Clobetasol Propionate Decreases Its Penetration to Skin Layers without Changing Its Relative Skin Distribution. J. Nanosci. Nanotechnol. 2015, 15, 875–879. [Google Scholar] [CrossRef]
- Fülöpová, N.; Pavloková, S.; DeBono, I.; Vetchý, D.; Franc, A. Development and Comparison of Various Coated Hard Capsules Suitable for Enteric Administration to Small Patient Cohorts. Pharmaceutics 2022, 14, 1577. [Google Scholar] [CrossRef] [PubMed]
- Chaves, P.D.S.; Frank, L.A.; Frank, A.G.; Pohlmann, A.R.; Guterres, S.S.; Beck, R.C.R. Mucoadhesive Properties of Eudragit®RS100, Eudragit®S100, and Poly(ε-Caprolactone) Nanocapsules: Influence of the Vehicle and the Mucosal Surface. AAPS PharmSciTech 2018, 19, 1637–1646. [Google Scholar] [CrossRef] [PubMed]
- Arriagada, F.; Nonell, S.; Morales, J. Silica-Based Nanosystems for Therapeutic Applications in the Skin. Nanomedicine 2019, 14, 2243–2267. [Google Scholar] [CrossRef]
- Hassan, A.F.; Helmy, S.A.; Donia, A. MCM-41 for Meloxicam Dissolution Improvement: In Vitro Release and in Vivo Bioavailability Studies. J. Braz. Chem. Soc. 2015, 26, 1367–1378. [Google Scholar] [CrossRef]
- Shariatinia, Z.; Zahraee, Z. Controlled Release of Metformin from Chitosan–Based Nanocomposite Films Containing Mesoporous MCM-41 Nanoparticles as Novel Drug Delivery Systems. J. Colloid Interface Sci. 2017, 501, 60–76. [Google Scholar] [CrossRef]
- Kruk, M.; Jaroniec, M.; Kim, J.M.; Ryoo, R. Characterization of Highly Ordered MCM-41 Silicas Using X-ray Diffraction and Nitrogen Adsorption. Langmuir 1999, 15, 5279–5284. [Google Scholar] [CrossRef]
- de Andrade, D.F.; Vukosavljevic, B.; Hoppe, J.B.; Pohlmann, A.R.; Guterres, S.S.; Windbergs, M.; Külkamp-Guerreiro, I.; Salbego, C.G.; Beck, R.C.R. Redispersible Spray-Dried Powder Containing Nanoencapsulated Curcumin: The Drying Process Does Not Affect Neuroprotection In Vitro. AAPS PharmSciTech 2019, 20, 1–10. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, E.G.; Cardoso, A.M.; Paese, K.; Coradini, K.; de Oliveira, C.V.; Pohlmann, A.R.; Oliveira, M.S.; Guterres, S.S.; Beck, R.C.R. Reconstituted Spray-Dried Phenytoin-Loaded Nanocapsules Improve the in Vivo Phenytoin Anticonvulsant Effect and the Survival Time in Mice. Int. J. Pharm. 2018, 551, 121–132. [Google Scholar] [CrossRef]
- Ambrogi, V.; Perioli, L.; Pagano, C.; Marmottini, F.; Ricci, M.; Sagnella, A.; Rossi, C. Use of SBA-15 for Furosemide Oral Delivery Enhancement. Eur. J. Pharm. Sci. 2012, 46, 43–48. [Google Scholar] [CrossRef]
- Price, R.R.; Gaber, B.P.; Lvov, Y. In-Vitro Release Characteristics of Tetracycline HCl, Khellin and Nicotinamide Adenine Dineculeotide from Halloysite; a Cylindrical Mineral. J. Microencapsul. 2008, 18, 713–722. [Google Scholar] [CrossRef]
- Jaroniec, C.P.; Gilpin, R.K.; Jaroniec, M. Adsorption and Thermogravimetric Studies of Silica-Based Amide Bonded Phases. J. Phys. Chem. B 1997, 101, 6861–6866. [Google Scholar] [CrossRef]
- La-Salvia, N.; Lovón-Quintana, J.J.; Lovón, A.S.P.; Valença, G.P. Influence of Aluminum Addition in the Framework of MCM-41 Mesoporous Molecular Sieve Synthesized by Non-Hydrothermal Method in an Alkali-Free System. Mater. Res. 2017, 20, 1461–1469. [Google Scholar] [CrossRef] [Green Version]
- Hate, S.S.; Reutzel-Edens, S.M.; Taylor, L.S. Interplay of Adsorption, Supersaturation and the Presence of an Absorptive Sink on Drug Release from Mesoporous Silica-Based Formulations. Pharm. Res. 2020, 37, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.A.; Schmitt, P.D.; Toth, S.J.; Deng, F.; Zhang, S.; Simpson, G.J. Parts per Million Powder X-ray Diffraction. Anal. Chem. 2015, 87, 10950–10955. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.; Keck, C.M.; Müller, R.H. Oral Hesperidin—Amorphization and Improved Dissolution Properties by Controlled Loading onto Porous Silica. Int. J. Pharm. 2017, 518, 253–263. [Google Scholar] [CrossRef]
- Rowe, R.C.; Sheskey, P.J.; Quinn, M.E. Handbook of Pharmaceutical Excipients, 6th ed.; Pharmaceutical Press: London, UK; American Pharmacists Association: Washington, DC, USA, 2009. [Google Scholar]
- Markov, P.A.; Krachkovsky, N.S.; Durnev, E.A.; Martinson, E.A.; Litvinets, S.G.; Popov, S.V. Mechanical Properties, Structure, Bioadhesion, and Biocompatibility of Pectin Hydrogels. J. Biomed. Mater. Res. Part A 2017, 105, 2572–2581. [Google Scholar] [CrossRef]
- Saidin, N.M.; Anuar, N.K.; Meor Mohd Affandi, M.M.R. Roles of Polysaccharides in Transdermal Drug Delivery System and Future Prospects. J. Appl. Pharm. Sci. 2018, 8, 141–157. [Google Scholar] [CrossRef] [Green Version]
- Seoane-Viaño, I.; Januskaite, P.; Alvarez-Lorenzo, C.; Basit, A.W.; Goyanes, A. Semi-Solid Extrusion 3D Printing in Drug Delivery and Biomedicine: Personalised Solutions for Healthcare Challenges. J. Control. Release 2021, 332, 367–389. [Google Scholar] [CrossRef] [PubMed]
- Sjöholm, E.; Sandler, N. Additive Manufacturing of Personalized Orodispersible Warfarin Films. Int. J. Pharm. 2019, 564, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Khan, B.A.; Karim, F.; Khan, M.K.; Haider, F.; Khan, S. Synthesis and Characterization of Polymeric Responsive CMC/Pectin Hydrogel Films Loaded with Tamarix Aphylla Extract as Potential Wound Dressings. Biocell 2021, 45, 1273–1285. [Google Scholar] [CrossRef]
- Gazzi, R.P.; Frank, L.A.; Onzi, G.; Pohlmann, A.R.; Guterres, S.S. New Pectin-Based Hydrogel Containing Imiquimod-Loaded Polymeric Nanocapsules for Melanoma Treatment. Drug Deliv. Transl. Res. 2020, 10, 1829–1840. [Google Scholar] [CrossRef]
- Thirawong, N.; Nunthanid, J.; Puttipipatkhachorn, S.; Sriamornsak, P. Mucoadhesive Properties of Various Pectins on Gastrointestinal Mucosa: An in Vitro Evaluation Using Texture Analyzer. Eur. J. Pharm. Biopharm. 2007, 67, 132–140. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | BET Surface Area (m2 g−1) | Pore Volume (cm3 g−1) |
|---|---|---|
| MSN | 899 | 0.731 |
| MSN-CP-3-1 | 291 | 0.198 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Oliveira, R.S.; Funk, N.L.; dos Santos, J.; de Oliveira, T.V.; de Oliveira, E.G.; Petzhold, C.L.; Costa, T.M.H.; Benvenutti, E.V.; Deon, M.; Beck, R.C.R. Bioadhesive 3D-Printed Skin Drug Delivery Polymeric Films: From the Drug Loading in Mesoporous Silica to the Manufacturing Process. Pharmaceutics 2023, 15, 20. https://doi.org/10.3390/pharmaceutics15010020
de Oliveira RS, Funk NL, dos Santos J, de Oliveira TV, de Oliveira EG, Petzhold CL, Costa TMH, Benvenutti EV, Deon M, Beck RCR. Bioadhesive 3D-Printed Skin Drug Delivery Polymeric Films: From the Drug Loading in Mesoporous Silica to the Manufacturing Process. Pharmaceutics. 2023; 15(1):20. https://doi.org/10.3390/pharmaceutics15010020
Chicago/Turabian Stylede Oliveira, Rafaela Santos, Nadine Lysyk Funk, Juliana dos Santos, Thayse Viana de Oliveira, Edilene Gadelha de Oliveira, Cesar Liberato Petzhold, Tania Maria Haas Costa, Edilson Valmir Benvenutti, Monique Deon, and Ruy Carlos Ruver Beck. 2023. "Bioadhesive 3D-Printed Skin Drug Delivery Polymeric Films: From the Drug Loading in Mesoporous Silica to the Manufacturing Process" Pharmaceutics 15, no. 1: 20. https://doi.org/10.3390/pharmaceutics15010020