
Modulating Immune Response in Viral Infection for Quantitative Forecasts of Drug Efficacy


Abstract
:1. Introduction
2. Therapeutic Options for Viral Infections
3. Mechanistic Models
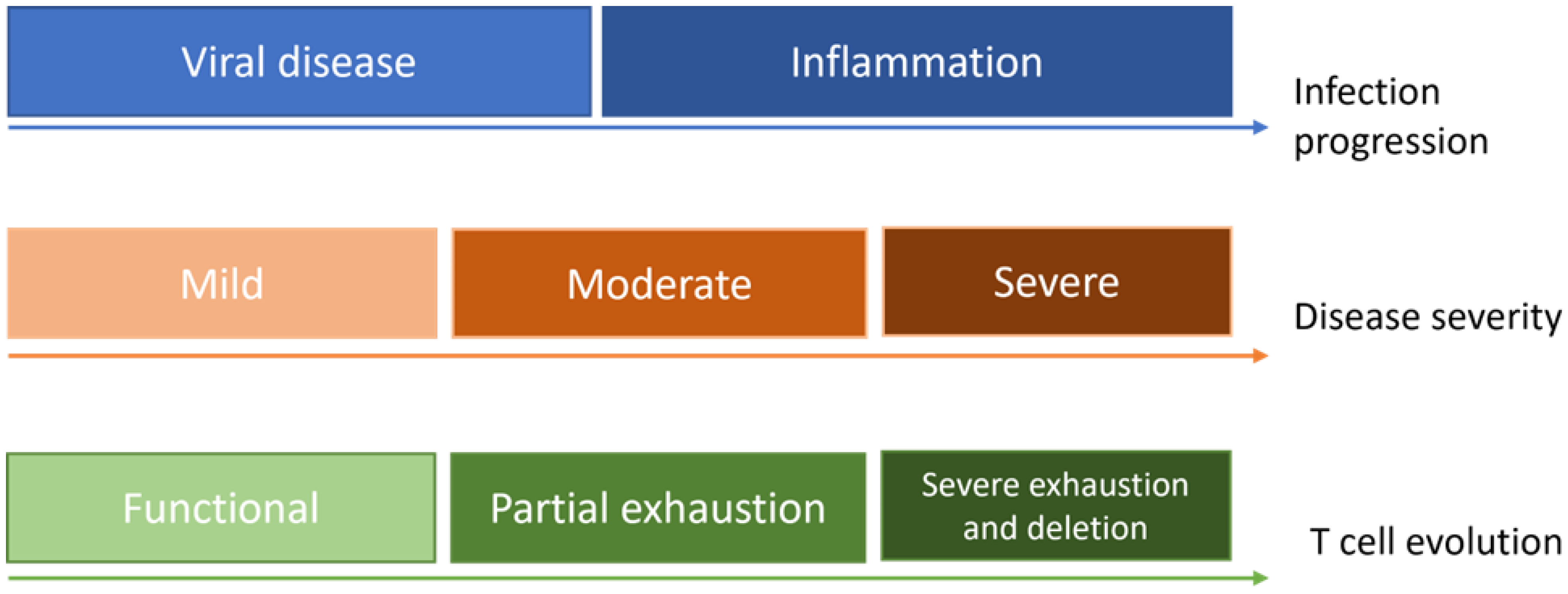
Interplay of Viral Infection Dynamics and the Immune System on Modelling
4. Strengthening the Immune System
4.1. Reverse T Cell Exhaustion
4.2. Measurement of Exhaustion and Prediction of the Drug Response by T Cell Subtype Profiling
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rai, K.R.; Shrestha, P.; Yang, B.; Chen, Y.; Liu, S.; Maarouf, M.; Chen, J.L. Acute Infection of Viral Pathogens and Their Innate Immune Escape. Front. Microbiol. 2021, 12, 672026. [Google Scholar] [CrossRef]
- Boldogh, I.; Albrecht, T.; Porter, D.D. Persistent Viral Infections. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996; Chapter 46. [Google Scholar]
- Lambert, P.F.; Sugden, B. Cancer issue: Viruses and Human Cancer. Yale J. Biol. Med. 2006, 79, 115. [Google Scholar]
- Smith, A.M. Validated models of immune response to virus infection. Curr. Opin. Syst. Biol. 2018, 12, 46–52. [Google Scholar] [CrossRef]
- Zitzmann, C.; Kaderali, L. Mathematical analysis of viral replication dynamics and antiviral treatment strategies: From basic models to age-based multi-scale modeling. Front. Microbiol. 2018, 9, 1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeitlinger, M.; Koch, B.C.P.; Bruggemann, R.; De Cock, P.; Felton, T.; Hites, M.; Le, J.; Luque, S.; MacGowan, A.P.; Marriott, D.J.E.; et al. Pharmacokinetics/Pharmacodynamics of Antiviral Agents Used to Treat SARS-CoV-2 and Their Potential Interaction with Drugs and Other Supportive Measures: A Comprehensive Review by the PK/PD of Anti-Infectives Study Group of the European Society of Antimicrobial Agents. Clin. Pharmacokinet. 2020, 59, 1195. [Google Scholar] [CrossRef] [PubMed]
- Kausar, S.; Khan, F.S.; Ur Rehman, M.I.M.; Akram, M.; Riaz, R.; Rasool, G.; Khan, A.H.; Saleem, I.; Shamim, S.; Malik, A. A review: Mechanism of action of antiviral drugs. Int. J. Immunopathol. Pharmacol. 2021, 35, 20587384211002621. [Google Scholar] [CrossRef]
- Meganck, R.M.; Baric, R.S. Developing therapeutic approaches for twenty-first-century emerging infectious viral diseases. Nat. Med. 2021, 27, 401. [Google Scholar] [CrossRef]
- Maginnis, M.S. Virus–Receptor Interactions: The Key to Cellular Invasion. J. Mol. Biol. 2018, 430, 2590. [Google Scholar] [CrossRef]
- Mahajan, S.; Choudhary, S.; Kumar, P.; Tomar, S. Antiviral strategies targeting host factors and mechanisms obliging +ssRNA viral pathogens. Bioorg. Med. Chem. 2021, 46, 116356. [Google Scholar] [CrossRef]
- Bauer, L.; Lyoo, H.; van der Schaar, H.M.; Strating, J.R.; van Kuppeveld, F.J. Direct-acting antivirals and host-targeting strategies to combat enterovirus infections. Curr. Opin. Virol. 2017, 24, 1–8. [Google Scholar] [CrossRef]
- Hope, J.L.; Bradley, L.M. Lessons in antiviral immunity. Science 2021, 371, 464–465. [Google Scholar] [CrossRef] [PubMed]
- Almocera, A.E.S.; Nguyen, V.K.; Hernandez-Vargas, E.A. Multiscale model within-host and between-host for viral infectious diseases. J. Math. Biol. 2018, 77, 1035–1057. [Google Scholar] [CrossRef] [PubMed]
- Zarnitsyna, V.I.; Gianlupi, J.F.; Hagar, A.; Sego, T.J.; Glazier, J.A. Advancing therapies for viral infections using mechanistic computational models of the dynamic interplay between the virus and host immune response. Curr. Opin. Virol. 2021, 50, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, N.; Bingham, R.; Chaplain, M.A.J.; Dosi, G.; Forni, G.; Knopoff, D.A.; Lowengrub, J.; Twarock, R.; Virgillito, M.E. A multiscale model of virus pandemic: Heterogeneous interactive entities in a globally connected world. Math. Model. Methods Appl. Sci. 2020, 30, 1591–1651. [Google Scholar] [CrossRef] [PubMed]
- Cicchese, J.M.; Evans, S.; Hult, C.; Joslyn, L.R.; Wessler, T.; Millar, J.A.; Marino, S.; Cilfone, N.A.; Mattila, J.T.; Linderman, J.J.; et al. Dynamic balance of pro- and anti-inflammatory signals controls disease and limits pathology. Immunol. Rev. 2018, 285, 147. [Google Scholar] [CrossRef]
- Waites, W.; Cavaliere, M.; Danos, V.; Datta, R.; Eggo, R.M.; Hallett, T.B.; Manheim, D.; Panovska-Griffiths, J.; Russell, T.W.; Zarnitsyna, V.I. Compositional modelling of immune response and virus transmission dynamics. Philos. Trans. R. Soc. A 2022, 380, 20210307. [Google Scholar] [CrossRef]
- Nguyen, L.K.N.; Megiddo, I.; Howick, S. Simulation models for transmission of health care–associated infection: A systematic review. Am J Infect Control. 2020, 48, 810–821. [Google Scholar] [CrossRef] [Green Version]
- Handel, A.; La Gruta, N.L. Thomas PGSimulation modelling for immunologists. Nat. Rev. Immunol. 2019, 20, 186–195. [Google Scholar] [CrossRef]
- Perelson, A.S. Ribeiro RMModeling the within-host dynamics of HIV infection. BMC Biol. 2013, 11, 96. [Google Scholar] [CrossRef] [Green Version]
- Perelson, A.S.; Ke, R. Mechanistic modelling of SARS-CoV-2 and other infectious diseases and the effects of therapeutics. Clin. Pharmacol. Ther. 2021, 109, 829. [Google Scholar] [CrossRef]
- Venugopal, V.; Padmanabhan, P.; Raja, R.; Dixit, N.M. Modelling how responsiveness to interferon improves interferon-free treatment of hepatitis C virus infection. PLoS Comput. Biol. 2018, 14, e1006335. [Google Scholar] [CrossRef] [Green Version]
- Bolouri, H.; Young, M.; Beilke, J.; Johnson, R.; Fox, B.; Huang, L.; Santini, C.C.; Hill, C.M.; Vries, A.R.V.; Shannon, P.T.; et al. Integrative network modeling reveals mechanisms underlying T cell exhaustion. Sci. Rep. 2020, 10, 1915. [Google Scholar] [CrossRef] [Green Version]
- Gorstein, E.; Martinello, M.; Churkin, A.; Dasgupta, S.; Walsh, K.; Applegate, T.L.; Yardeni, D.; Etzion, O.; Uprichard, S.L.; Barash, D.; et al. Modeling based response guided therapy in subjects with recent hepatitis C infection. Antivir. Res. 2020, 180, 104862. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Lurie, Y.; Meissner, E.G.; Major, M.; Sansone, N.; Uprichardag, S.L.; Cotler, S.J.; Dahar, H. Modeling HCV cure after an ultra-short duration of therapy with direct acting agents. Antivir. Res. 2017, 144, 281. [Google Scholar] [CrossRef] [PubMed]
- Baral, S.; Roy, R.; Dixit, N.M. Modeling how reversal of immune exhaustion elicits cure of chronic hepatitis C after the end of treatment with direct-acting antiviral agents. Immunol. Cell Biol. 2018, 96, 969. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.W.; Gilchrist, M.A.; Coombs, D.; Hyman, J.M.; Perelson, A.S. An age-structured model of hiv infection that allows for variations in the production rate of viral particles and the death rate of productively infected cells. Math. Biosci. Eng. 2004, 1, 267–288. [Google Scholar] [CrossRef]
- Rampersad, S.; Tennant, P. Replication and Expression Strategies of Viruses. In Viruses: Molecular Biology, Host Interactions and Applications to Biotechnology; Academic Press: Cambridge, MA, USA, 2018; pp. 55–82. [Google Scholar] [CrossRef]
- Kapitanov, G.I.; Chabot, J.R.; Narula, J.; Roy, M.; Neubert, H.; Palandra, J.; Farroki, V.; Johnson, J.S.; Webster, R.; Jones, H.M. A Mechanistic Site-Of-Action Model: A Tool for Informing Right Target, Right Compound, and Right Dose for Therapeutic Antagonistic Antibody Programs. Front. Bioinform. 2021, 1, 731340. [Google Scholar] [CrossRef]
- Weber, F. Antiviral Innate Immunity: Introduction. Encycl. Virol. 2021, 1, 577. [Google Scholar] [CrossRef]
- Stromberg, S.P.; Antia, R. Vaccination by Delayed Treatment of Infection. Vaccine 2011, 29, 9624. [Google Scholar] [CrossRef] [Green Version]
- Germain, R.N.; Meier-Schellersheim, M.; Nita-Lazar, A.; Fraser, I.D. Systems biology in immunology: A computational modeling perspective. Annu Rev Immunol. 2011, 29, 527–585. [Google Scholar] [CrossRef]
- Culos, A.; Tsai, A.S.; Stanley, N.; Becker, M.; Ghaemi, M.S.; McIlwain, D.R.; Fallahzadeh, R.; Tanada, A.; Nassar, H.; Espinosa, C.; et al. Integration of mechanistic immunological knowledge into a machine learning pipeline improves predictions. Nat. Mach. Intell. 2020, 2, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Basson, M.A. Signaling in cell differentiation and morphogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, M.A.; Bangham, C.R. Population dynamics of immune responses to persistent viruses. Science 1996, 272, 74–79. [Google Scholar] [CrossRef] [Green Version]
- Guedj, J.; Thiébaut, R.; Commenges, D. Maximum likelihood estimation in dynamical models of HIV. Biometrics 2007, 63, 1198–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, N.; Cummings, D.; Cauchemez, S.; Fraser, C.; Riley, S.; Meeyai, A.; Iamsirithaworn, S.; Burke, D.S. Strategies for containing an emerging influenza pandemic in Southeast Asia. Nature 2005, 437, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Thompson, W.W.; Shay, D.K.; Weintraub, E.; Brammer, L.; Cox, N.; Anderson, L.J.; Fukuda, K. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 2003, 289, 179–186. [Google Scholar] [CrossRef]
- Wilta, F.; Chong, A.L.C.; Selvachandran, G.; Kotecha, K.; Ding, W. Generalized Susceptible-Exposed-Infectious-Recovered model and its contributing factors for analysing the death and recovery rates of the COVID-19 pandemic. Appl. Soft Comput. 2022, 123, 108973. [Google Scholar] [CrossRef]
- Cooper, I.; Mondal, A.; Antonopoulos, C.G. A SIR model assumption for the spread of COVID-19 in different communities. Chaos Solitons Fractals 2020, 139, 110057. [Google Scholar] [CrossRef]
- Schultze, J.L.; Aschenbrenner, A.C. COVID-19 and the human innate immune system. Cell 2021, 184, 1671–1692. [Google Scholar] [CrossRef]
- Pawelka, E.; Karolyi, M.; Mader, T.; Omid, S.; Kelani, H.; Baumgartner, S.; Ely, S.; Hoepler, W.; Jilma, B.; Koenig, F.; et al. COVID-19 is not “just another flu”: A real-life comparison of severe COVID-19 and influenza in hospitalized patients in Vienna, Austria. Infection 2021, 49, 907–916. [Google Scholar] [CrossRef]
- Liu, X.; Li, F.; Niu, H.; Ma, L.; Chen, J.; Zhang, Y.; Peng, L.; Gan, C.; Ma, X.; Zhu, B. IL-2 Restores T-Cell Dysfunction Induced by Persistent Mycobacterium tuberculosis Antigen Stimulation. Front. Immunol. 2019, 10, 2350, Erratum in Front. Immunol. 2020, 11, 1671. [Google Scholar] [CrossRef] [PubMed]
- Saeidi, A.; Zandi, K.; Cheok, Y.Y.; Saeidi, H.; Wong, W.F.; Lee, C.Y.Q.; Cheong, H.C.; Yong, Y.K.; Larsson, M.; Shankar, E.M. T-Cell Exhaustion in Chronic Infections: Reversing the State of Exhaustion and Reinvigorating Optimal Protective Immune Responses. Front. Immunol. 2018, 9, 2569. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S. Innate metabolic responses against viral infections. Nat. Metab. 2022, 4, 1245–1259. [Google Scholar] [CrossRef] [PubMed]
- Sumbria, D.; Berber, E.; Mathayan, M.; Rouse, B.T. Virus Infections and Host Metabolism—Can We Manage the Interactions? Front. Immunol. 2021, 11, 3727. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, X.; Wang, L.; Chen, S. Oncogenic virus-induced aerobic glycolysis and tumorigenesis. J. Cancer 2018, 9, 3699. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhao, A.; Li, M.; Shi, L.; Han, Q.; Hou, Z. Targeting T-cell metabolism to boost immune checkpoint inhibitor therapy. Front. Immunol. 2022, 13, 1046755. [Google Scholar] [CrossRef]
- Stirling, E.R.; Bronson, S.M.; Mackert, J.D.; Cook, K.L.; Triozzi, P.L.; Soto-Pantoja, D.R. Metabolic Implications of Immune Checkpoint Proteins in Cancer. Cells 2022, 11, 179. [Google Scholar] [CrossRef]
- Chaudhary, O.; Trotta, D.; Wang, K.; Wang, X.; Chu, X.; Bradley, C.; Okulicz, J.; Maves, R.C.; Kronmann, K.; Schofield, C.M.; et al. Patients with HIV-associated cancers have evidence of increased T cell dysfunction and exhaustion prior to cancer diagnosis. J. Immunother. Cancer 2022, 10, e004564. [Google Scholar] [CrossRef]
- Cai, H.; Liu, G.; Zhong, J.; Zheng, K.; Xiao, H.; Li, C.; Song, X.; Li, Y.; Xu, C.; Wu, H.; et al. Immune Checkpoints in Viral Infections. Viruses 2020, 12, 1051. [Google Scholar] [CrossRef]
- Bayati, F.; Mohammadi, M.; Valadi, M.; Jamshidi, S.; Foma, A.M.; Sharif-Paghaleh, E. The Therapeutic Potential of Regulatory T Cells: Challenges and Opportunities. Front. Immunol. 2021, 11, 3455. [Google Scholar] [CrossRef]
- Baron, M.; Soulié, C.; Lavolé, A.; Assoumou, L.; Abbar, B.; Fouquet, B.; Rousseau, A.; Veyri, M.; Samri, A.; Makinson, A.; et al. Impact of Anti PD-1 Immunotherapy on HIV Reservoir and Anti-Viral Immune Responses in People Living with HIV and Cancer. Cells 2022, 11, 1015. [Google Scholar] [CrossRef] [PubMed]
- Macatangay, B.J.C.; Gandhi, R.T.; Jones, R.B.; Mcmahon, D.K.; Lalama, C.M.; Bosch, R.J.; Cyktor, J.C.; Thomas, A.S.; Borowski, L.; Riddler, S.A.; et al. T cells with high PD-1 expression are associated with lower HIV-specific immune responses despite long-term antiretroviral therapy. AIDS 2020, 34, 15. [Google Scholar] [CrossRef] [PubMed]
- Velu, V.; Titanji, K.; Zhu, B.; Husain, S.; Pladevega, A.; Lai, L.; Vanderford, T.H.; Chennareddi, L.; Silvestri, G.; Freeman, G.J.; et al. Enhancing SIV-Specific Immunity In Vivo by PD-1 Blockade. Nature 2009, 458, 206. [Google Scholar] [CrossRef] [Green Version]
- Binder, B. Thimme RCD4+ T cell responses in human viral infection: Lessons from hepatitis C. J. Clin. Investig. 2020, 130, 595–597. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, N.; Kaplan, D.E.; Coleclough, J.; Li, Y.; Valiga, M.E.; Kaminski, M.; Shaked, A.; Olthoff, K.; Gostick, E.; Price, D.A.; et al. Functional restoration of HCV-specific CD8 T cells by PD-1 blockade is defined by PD-1 expression and compartmentalization. Gastroenterology 2008, 134, 1927–1937.e2. [Google Scholar] [CrossRef] [Green Version]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic insufficiencies due to metabolic alterations regulated by PD-1 are an early driver of CD8+ T cell exhaustion. Immunity 2016, 45, 358. [Google Scholar] [CrossRef] [Green Version]
- Puigserver, P.; Spiegelman, B.M. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): Transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schank, M.; Zhao, J.; Moorman, J.P.; Yao, Z.Q. The Impact of HIV- and ART-Induced Mitochondrial Dysfunction in Cellular Senescence and Aging. Cells 2021, 10, 174. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Schurich, A.; Pallett, L.J.; Jajbhay, D.; Wijngaarden, J.; Otano, I.; Gill, U.S.; Hansi, N.; Kennedy, P.T.; Nastouli, E.; Gilson, R.; et al. Distinct Metabolic Requirements of Exhausted and Functional Virus-Specific CD8 T Cells in the Same Host. Cell Rep. 2016, 16, 1243–1252. [Google Scholar] [CrossRef]
- Barili, V.; Boni, C.; Rossi, M.; Vecchi, A.; Zecca, A.; Penna, A.; Missale, G.; Ferrari, C.; Fisicaro, P. Metabolic regulation of the HBV-specific T cell function. Antivir. Res. 2021, 185, 104989. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Wei, F.; Ren, X. Exhausted T cells and epigenetic status. Cancer Biol. Med. 2020, 17, 923–936. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Yao, D.; Tan, J.; He, Z.; Yu, Z.; Chen, J.; Luo, G.; Wang, C.; Zhou, F.; Zha, X.; et al. Memory T cells skew toward terminal differentiation in the CD8+ T cell population in patients with acute myeloid leukemia. J. Hematol. Oncol. 2018, 11, 93. [Google Scholar] [CrossRef] [PubMed]
- Pennock, N.D.; White, J.T.; Cross, E.W.; Cheney, E.E.; Tamburini, B.A.; Kedl, R.M. T cell responses: Naïve to memory and everything in between. Adv. Physiol. Educ. 2013, 37, 273. [Google Scholar] [CrossRef] [Green Version]
- Schillebeeckx, I.; Earls, J.; Flanagan, K.C.; Hiken, J.; Bode, A.; Armstrong, J.R.; Messina, D.N.; Adkins, D.; Ley, J.; Alborelli, I.; et al. T cell subtype profiling measures exhaustion and predicts anti-PD-1 response. Sci Rep. 2022, 12, 1342. [Google Scholar] [CrossRef]
- Li, H.; van der Merwe, P.A.; Sivakumar, S. Biomarkers of response to PD-1 pathway blockade. Br. J. Cancer 2022, 126, 1663–1675. [Google Scholar] [CrossRef]
- Davis, J.E.; Sharpe, C.; Mason, K.; Tam, C.S.; Koldej, R.M.; Ritchie, D.S. Ibrutinib protects T cells in patients with CLL from proliferation-induced senescence. J. Transl. Med. 2021, 19, 473. [Google Scholar] [CrossRef]
- Porichis, F.; Hart, M.G.; Zupkosky, J.; Barblu, L.; Kwon, D.S.; McMullen, A.; Brennan, T.; Ahmed, R.; Freeman, G.J.; Kavanagh, D.G.; et al. Differential impact of PD-1 and/or interleukin-10 blockade on HIV-1-specific CD4 T cell and antigen-presenting cell functions. J. Virol. 2014, 88, 2508–2518. [Google Scholar] [CrossRef] [Green Version]
- Ejrnaes, M.; Filippi, C.M.; Martinic, M.M.; Ling, E.M.; Togher, L.M.; Crotty, S.; von Herrath, M.G. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J. Exp. Med. 2006, 203, 2461–2472. [Google Scholar] [CrossRef]
- Braun, M.Y. The Natural History of T Cell Metabolism. Int. J. Mol. Sci. 2021, 22, 6779. [Google Scholar] [CrossRef]
- Corrado, M.; Pearce, E.L. Targeting memory T cell metabolism to improve immunity. J Clin Investig. 2022, 132, e148546. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Type | Antiviral Drug | Therapeutic Indication | Mechanism of Action |
|---|---|---|---|
| Direct acting antiviral | Convalescent plasma | Commonly used to treat viral infections such as influenza and COVID-19 | tHe exact mechanisms of action of convalescent plasma will depend on the specific virus and the specific immune responses that are being targeted. It works by:
|
| Nucleoside analogs | Acyclovir: Used to treat herpes simplex virus infections, including genital herpes and cold sores. | It is converted into its active form, acyclovir triphosphate, inside infected cells. Once inside the cells, acyclovir triphosphate inhibits the activity of a viral enzyme called herpesvirus-specific DNA polymerase, which is responsible for replicating the herpesvirus’s DNA. By inhibiting this enzyme, acyclovir helps to prevent the virus from replicating and spreading to other cells. | |
| Lamivudine: Used to treat HIV and hepatitis B virus infections. | It is converted into its active form, lamivudine triphosphate, inside infected cells. Once inside the cells, lamivudine triphosphate inhibits the activity of a viral enzyme called reverse transcriptase, which is responsible for replicating the HIV virus’s RNA into DNA. By inhibiting this enzyme, lamivudine helps to prevent the virus from replicating and spreading to other cells. | ||
| Tenofovir: Used to treat HIV and hepatitis B virus infections. | It is converted into its active form, tenofovir diphosphate, inside infected cells. Once inside the cells, tenofovir diphosphate inhibits the activity of a viral enzyme called reverse transcriptase, which is responsible for replicating the HIV virus’s RNA into DNA. By inhibiting this enzyme, tenofovir helps to prevent the virus from replicating and spreading to other cells. | ||
| Valacyclovir: Used to treat herpes simplex virus infections, including genital herpes and cold sores. | It is converted into its active form, tenofovir diphosphate, inside infected cells. Once inside the cells, tenofovir diphosphate inhibits the activity of a viral enzyme called reverse transcriptase, which is responsible for replicating the HIV virus’s RNA into DNA. By inhibiting this enzyme, tenofovir helps to prevent the virus from replicating and spreading to other cells. | ||
| Zidovudine: Used to treat HIV infections | It is converted into its active form, zidovudine triphosphate, inside infected cells. Once inside the cells, zidovudine triphosphate inhibits the activity of a viral enzyme called reverse transcriptase, which is responsible for replicating the HIV virus’s RNA into DNA. By inhibiting this enzyme, zidovudine helps to prevent the virus from replicating and spreading to other cells. | ||
| Monoclonal antibodies | Palivizumab: Used to prevent respiratory syncytial virus (RSV) infections in high-risk individuals, such as premature infants and individuals with compromised immune systems. | It is directed against a specific protein called the F protein, which is found on the surface of the respiratory syncytial virus (RSV). Palivizumab binds to the F protein and prevents the virus from attaching to and infecting cells in the respiratory tract. This helps to prevent the development of RSV infection and its associated complications. | |
| Remdesivir: Used to treat COVID-19. | It is converted into its active form, remdesivir triphosphate, inside infected cells. Once inside the cells, remdesivir triphosphate inhibits the activity of a viral enzyme called RNA-dependent RNA polymerase, which is responsible for replicating the viral RNA. By inhibiting this enzyme, remdesivir helps to prevent the virus from replicating and spreading to other cells. | ||
| Casirivimab/imdevimab: Used to treat COVID-19. | Are monoclonal antibodies that are directed against a specific protein called the spike protein, which is found on the surface of SARS-CoV-2, the virus that causes COVID-19. Casirivimab and imdevimab bind to the spike protein and prevent the virus from attaching to and infecting cells in the respiratory tract. This helps to prevent the development of COVID-19 and its associated complications. | ||
| Peginterferon lambda: Used to treat hepatitis C virus infection. | Interferons are proteins that are produced by the body’s immune system in response to viral infections. They help to stimulate the immune system to fight the virus and also inhibit the replication of the virus. Peginterferon lambda is a long-acting form of interferon that is given by injection once a week. It is thought to work by stimulating the production of proteins that inhibit the replication of HCV and by activating immune cells to attack the virus. | ||
| Fusion inhibitors | Enfuvirtide: Used to treat HIV infections | It works by binding to a specific protein called gp41, which is found on the surface of the HIV virus and is involved in the process of viral fusion. By binding to gp41, enfuvirtide helps to prevent the virus from fusing with and infecting host cells. | |
| Maraviroc: Used to treat HIV infections | It works by binding to a specific protein called CCR5, which is found on the surface of host cells and is used by the HIV virus to enter cells. By binding to CCR5, maraviroc helps to prevent the virus from fusing with and infecting host cells. | ||
| Polymerase inhibitors | Ribavirin: Used to treat hepatitis C virus infections and certain types of respiratory virus infections. | It works by inhibiting the activity of viral polymerases, which are enzymes that the virus uses to replicate its genetic material. Ribavirin is thought to work by inhibiting the synthesis of the virus’s genetic material, which helps to prevent the virus from replicating and spreading to other cells. | |
| Oseltamivir: Used to treat influenza virus infections. | Is a prodrug, meaning that it is converted into its active form, oseltamivir carboxylate, inside infected cells. Once inside the cells, oseltamivir carboxylate inhibits the activity of a viral enzyme called neuraminidase, which is responsible for releasing newly formed viruses from infected cells. By inhibiting this enzyme, oseltamivir helps to prevent the virus from spreading to other cells. | ||
| Zanamivir: Used to treat influenza virus infections. | It works by inhibiting the activity of a viral enzyme called neuraminidase, which is responsible for releasing newly formed viruses from infected cells. By inhibiting this enzyme, zanamivir helps to prevent the virus from spreading to other cells. | ||
| Sofosbuvir: Used to treat hepatitis C virus infections. | It works by inhibiting the activity of a viral enzyme called NS5B polymerase, which is responsible for replicating the HCV virus’s RNA. By inhibiting this enzyme, sofosbuvir helps to prevent the virus from replicating and spreading to other cells. | ||
| Receptor decoys | Maraviroc: Used to treat HIV infections | The mechanisms of this drugs are described above. Receptor decoys and fusion inhibitors are similar in that they both work by blocking the interaction between a virus and its target cell. Receptor decoys are a class of direct-acting antiviral drugs that work by blocking the interaction between a virus and its target cell. They do this by providing a “decoy” receptor that the virus can bind to, which prevents the virus from attaching to and infecting the target cell. Fusion inhibitors are a class of direct-acting antiviral drugs that work by inhibiting the process of viral fusion | |
| Enfuvirtide: Used to treat HIV infections | |||
| Host-factor antiviral | Endocytosis inhibitors | Amantadine: Used to treat influenza virus infections | It works by inhibiting the activity of a viral enzyme called M2, which is responsible for releasing newly formed viruses from infected cells. By inhibiting this enzyme, amantadine helps to prevent the virus from spreading to other cells. |
| Rimantadine: Used to treat influenza virus infections | |||
| Oseltamivir: Used to treat influenza virus infections | Oseltamivir is a prodrug, meaning that it is converted into its active form, oseltamivir carboxylate, inside infected cells. Once inside the cells, oseltamivir carboxylate inhibits the activity of a viral enzyme called neuraminidase, which is responsible for releasing newly formed viruses from infected cells. By inhibiting this enzyme, oseltamivir helps to prevent the virus from spreading to other cells. | ||
| Interferons | Alpha interferon (IFN-α) is produced by leukocytes (white blood cells) and is effective against a wide range of viruses, including herpesvirus, hepatitis B and C viruses, and human papillomavirus (HPV). | Interferons (IFN) work by activating interferon receptors on the surface of infected cells, which triggers a signaling pathway that leads to the production of proteins called antiviral effectors. These effectors inhibit the replication of viruses by blocking the production of viral proteins, disrupting viral assembly and release, and activating the immune system to clear the infection. | |
| Beta interferon (IFN-β) is produced by fibroblasts (a type of cell found in connective tissue) and is effective against certain viruses, including herpes simplex virus and HIV. | |||
| Gamma interfero (IFN-γ) is produced by immune cells called T cells and natural killer cells, and is effective against a variety of viruses, including HIV and hepatitis C virus. | |||
| Kinase inhibitors | Maraviroc is an HIV-1 entry inhibitor | Targets the CCR5 co-receptor on immune cells. It blocks the interaction between HIV-1 and CCR5, preventing the virus from entering the cell. | |
| Selzentry (maraviroc) is an HIV-1 entry inhibitor | Targets the CCR5 co-receptor on immune cells. It blocks the interaction between HIV-1 and CCR5, preventing the virus from entering the cell. | ||
| Acyclovir is an antiviral drug that is effective against herpesvirus infections. | It works by inhibiting the viral enzyme thymidine kinase, which is required for the synthesis of viral DNA | ||
| Ganciclovir is an antiviral drug that is effective against cytomegalovirus (CMV) infections. | It works by inhibiting the viral enzyme DNA polymerase, which is required for the synthesis of viral DNA | ||
| Valganciclovir is a prodrug of ganciclovir that is used to treat CMV infections. | It is converted to ganciclovir by the enzyme valacyclovir hydrolyase, which is expressed in infected cells | ||
| Raltegravir is an HIV-1 integrase inhibitor | Blocks the integration of HIV-1 DNA into the host genome. It works by inhibiting the activity of the HIV-1 integrase enzyme | ||
| Lipidomic drugs | Amantadine is an antiviral drug that is effective against influenza A virus. | It works by blocking the ion channel M2, which is required for the release of viral particles from infected cells. | |
| Oseltamivir is an antiviral drug that is effective against influenza A and B viruses. | It works by inhibiting the viral enzyme neuraminidase, which is required for the release of viral particles from infected cells. | ||
| Zanamivir is an antiviral drug that is effective against influenza A and B viruses. | It works by inhibiting the viral enzyme neuraminidase, which is required for the release of viral particles from infected cells. | ||
| Remdesivir is an antiviral drug that is effective against a wide range of RNA viruses, including SARS-CoV-2 (the virus that causes COVID-19). | It works by inhibiting the viral RNA polymerase enzyme, which is required for the synthesis of viral RNA. | ||
| Favipiravir is an antiviral drug that is effective against RNA viruses including influenza A and B viruses and Ebola virus. | It works by inhibiting the viral RNA polymerase enzyme, which is required for the synthesis of viral RNA. | ||
| Sofosbuvir is an antiviral drug that is effective against hepatitis C virus (HCV). | It works by inhibiting the viral NS5B RNA polymerase enzyme, which is required for the synthesis of viral RNA. | ||
| Direct-acting and host-factor antiviral | Protease inhibitors | Saquinavir is an HIV protease inhibitor | Blocks the protease enzyme, which is required for the processing of viral proteins. |
| Ritonavir is an HIV protease inhibitor | |||
| Nelfinavir is an HIV protease inhibitor | |||
| Lopinavir is an HIV protease inhibitor | |||
| Boceprevir is an HCV protease inhibitor | |||
| Telaprevir is an HCV protease inhibitor | |||
| Camostat mesilate is an antiviral drug that is effective against a variety of viruses, including influenza A and B viruses, respiratory syncytial virus, and norovirus. | It works by inhibiting the protease enzyme trypsin, which is required for the release of viral particles from infected cells. | ||
| Fosamprenavir is an HIV protease inhibitor that is converted to its active form, amprenavir, by the enzyme CYP3A4. | It blocks the protease enzyme, which is required for the processing of viral proteins | ||
| Indinavir is an HIV protease inhibitor | That blocks the protease enzyme, which is required for the processing of viral proteins | ||
| Nelfinavir is an HIV protease inhibitor | |||
| Ritonavir is an HIV protease inhibitor | |||
| Saquinavir is an HIV protease inhibitor | |||
| Translation inhibitors | Efavirenz is an HIV reverse transcriptase inhibitor that blocks the synthesis of viral DNA from viral RNA. | Blocks the synthesis of viral DNA from viral RNA. It works by inhibiting the activity of the HIV reverse transcriptase enzyme, which is required for the synthesis of viral DNA from viral RNA. | |
| Emtricitabine is an HIV reverse transcriptase inhibitor | Blocks the synthesis of viral DNA from viral RNA. It works by inhibiting the activity of the HIV reverse transcriptase enzyme, which is required for the synthesis of viral DNA from viral RNA. | ||
| Tenofovir is an HIV reverse transcriptase inhibitor that blocks the synthesis of viral DNA from viral RNA. | It works by inhibiting the activity of the HIV reverse transcriptase enzyme, which is required for the synthesis of viral DNA from viral RNA. | ||
| Zidovudine is an HIV reverse transcriptase inhibitor | It works by inhibiting the activity of the HIV reverse transcriptase enzyme, which is required for the synthesis of viral DNA from viral RNA. | ||
| Baricitinib is an antiviral drug that is effective against a variety of RNA viruses, including influenza A and B viruses, respiratory syncytial virus, and norovirus. | It works by inhibiting the translation of viral mrna into viral proteins. It does this by inhibiting the activity of the ribosomes, which are the cellular machinery that reads the genetic code in mrna and synthesizes proteins based on that code. | ||
| Ruxolitinib is an antiviral drug that is effective against a variety of RNA viruses, including influenza A and B viruses, respiratory syncytial virus, and norovirus. | It works by inhibiting the translation of viral mrna into viral proteins. It does this by inhibiting the activity of the ribosomes, which are the cellular machinery that reads the genetic code in mrna |
| Disease | How It Can Be Modulated? |
|---|---|
| Acquired immunodeficiency syndrome (AIDS), which is caused HIV | Example 1: HIV-1 immune dynamics model, which was developed by Nowak and May in 1996 [35]. This model describes the interactions between HIV, CD4+ T cells, and antiviral drugs. The model includes equations that describe the rate of HIV replication, the rate of CD4+ T cell production and loss, and the rate at which antiviral drugs inhibit HIV replication. Example 2: HIV-1 treatment intensification model, which was developed by Guedj et al. in 2007 [36]. This model is based on the HIV-1 immune dynamics model and incorporates additional equations to describe the effects of different ART regimens on the virus. |
| Influenza caused by the influenza virus | Example 1: influenza transmission model, which was developed by Ferguson et al. in 2005 [37]. This model describes the transmission of influenza from person to person, as well as the effect of antiviral drugs on the virus. The model includes equations that describe the rate of influenza transmission, the rate of antiviral drug-induced reduction in the number of infectious individuals, and the rate at which individuals become immune to the virus. Example 2: influenza vaccine effectiveness model, which was developed by Thompson et al. in 2006 [38]. This model is based on the influenza transmission model and incorporates additional equations to describe the effects of vaccination on the virus and the immune system. The model is based on the influenza transmission model, which was developed by Ferguson et al. in 2005, and incorporates additional equations to describe the effects of vaccination on the virus and the immune system. It’s important to note that the model is based on a specific set of assumptions and may not be applicable to all situations |
| Severe acute respiratory syndrome (SARS), which is caused by the SARS-CoV-2 virus | Example 1: SEIR (susceptible-exposed-infectious-recovered) model, which is a type of compartmental model that describes the transmission of infectious diseases. The SEIR model includes equations that describe the rate of transmission of SARS-CoV-2, the rate of recovery, and the rate at which individuals become immune to the virus [39]. Example 2: SIR (susceptible-infectious-recovered) model, which is a variant of the SEIR model that does not include an exposed compartment. The SIR model has been used to study the effectiveness of various interventions, such as antiviral drugs and vaccines, on the transmission of SARS-CoV-2 [40]. |
| Treatment Approach | Target |
|---|---|
| Antibody blockade | PD-1/PD-L1 |
| Altering metabolic pathways | PCK1 GLS1 Pyruvate kinase |
| Decrease PD-1 expression | FoxOT TOX |
| Improve TCR signaling | AKT IL-10 or IL10R IL.2R |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, B.; Vale, N. Modulating Immune Response in Viral Infection for Quantitative Forecasts of Drug Efficacy. Pharmaceutics 2023, 15, 167. https://doi.org/10.3390/pharmaceutics15010167
Costa B, Vale N. Modulating Immune Response in Viral Infection for Quantitative Forecasts of Drug Efficacy. Pharmaceutics. 2023; 15(1):167. https://doi.org/10.3390/pharmaceutics15010167
Chicago/Turabian StyleCosta, Bárbara, and Nuno Vale. 2023. "Modulating Immune Response in Viral Infection for Quantitative Forecasts of Drug Efficacy" Pharmaceutics 15, no. 1: 167. https://doi.org/10.3390/pharmaceutics15010167