
Preparation, Characterization and Pharmacokinetics of Tolfenamic Acid-Loaded Solid Lipid Nanoparticles
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. Preparation of the TA-SLN Suspension
2.4. Characterization of the TA-SLN Suspension
2.4.1. Determination of Particle Size, Polydispersity Index (PDI), and Zeta Potential
2.4.2. Loading Capacity (LC) and EE
2.4.3. Morphology Observation
2.4.4. Differential Scanning Calorimetry (DSC)
2.4.5. Fourier Transform Infrared (FT-IR) Spectroscopy
2.4.6. Settlement Rate, Redispersibility, and pH
2.4.7. Stability Studies
2.5. In Vitro Release
2.6. Pharmacokinetics
2.7. Determination of TA in Plasma Samples
2.8. Statistical Analysis
3. Results
3.1. Optimization of the TA-SLN Suspension
3.2. Properties of the TA-SLN Suspension
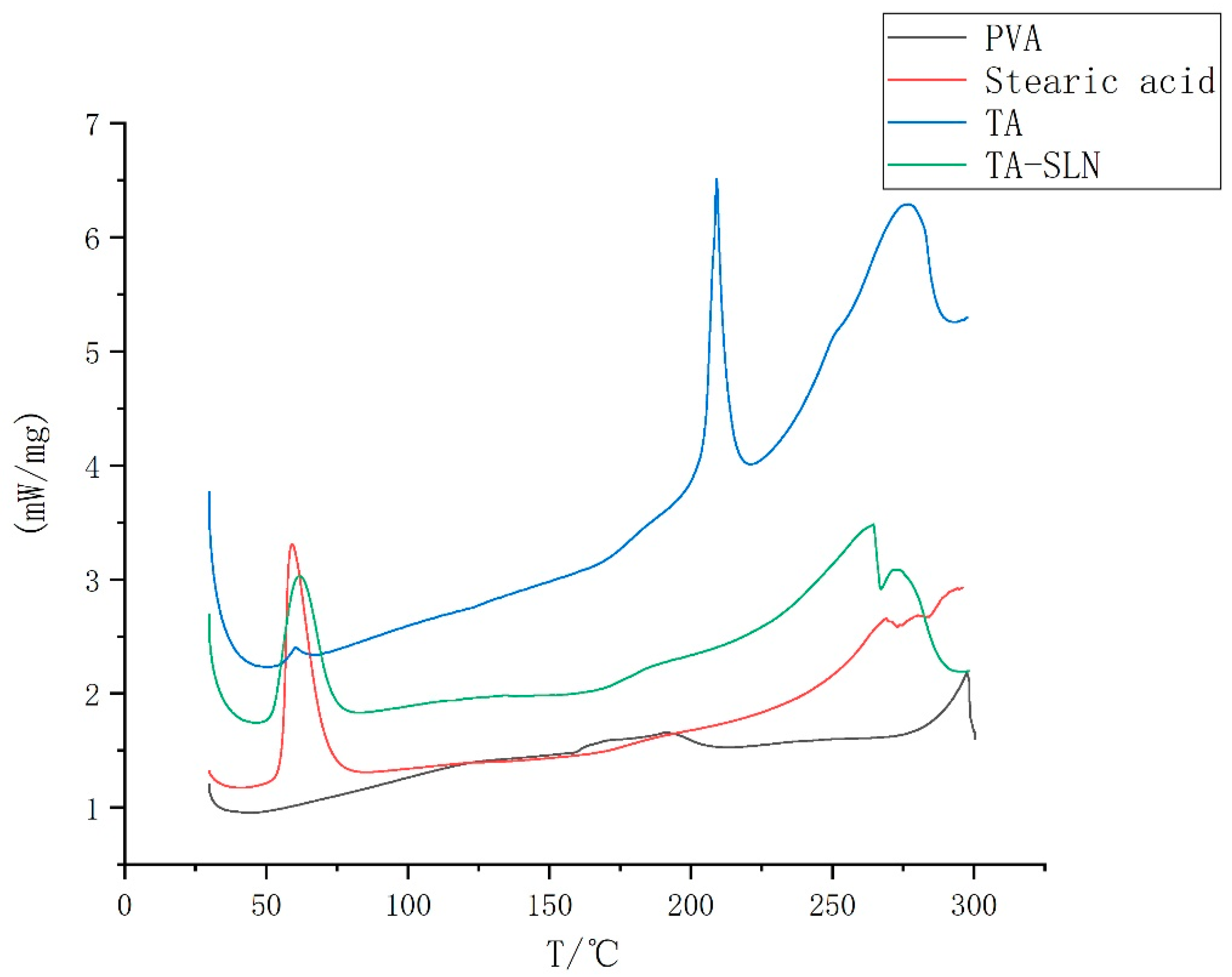
3.3. Thermal Characterization
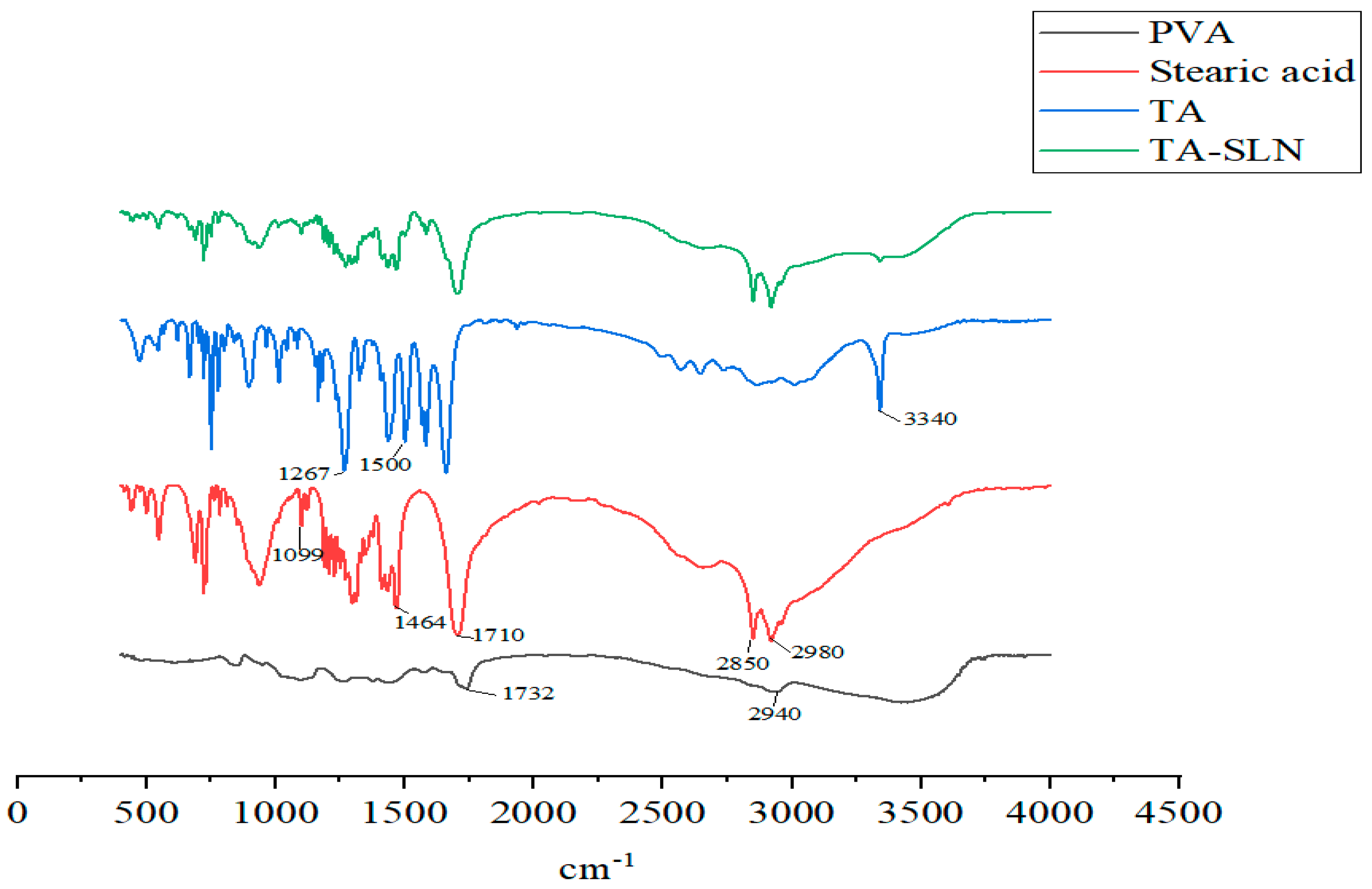
3.4. FT-IR Spectroscopy
3.5. Stability
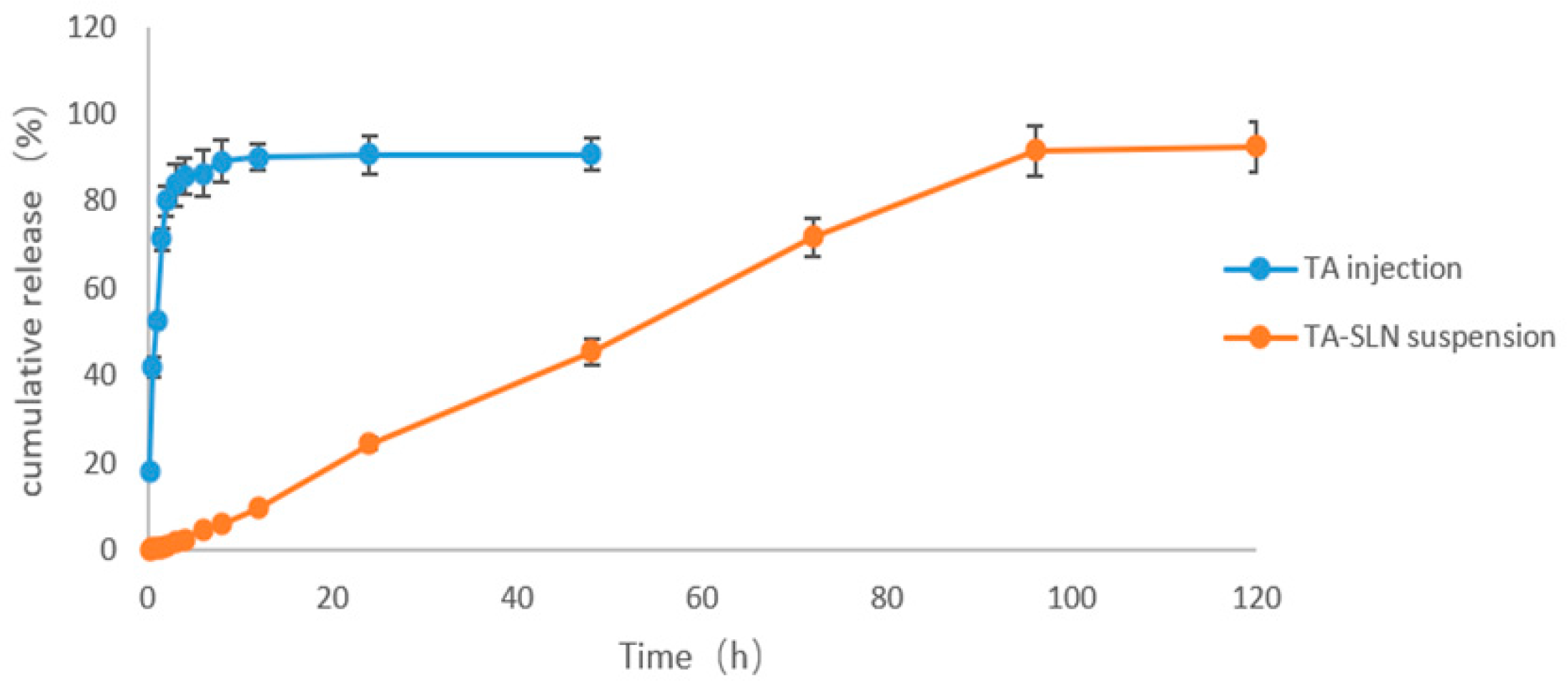
3.6. In Vitro Release
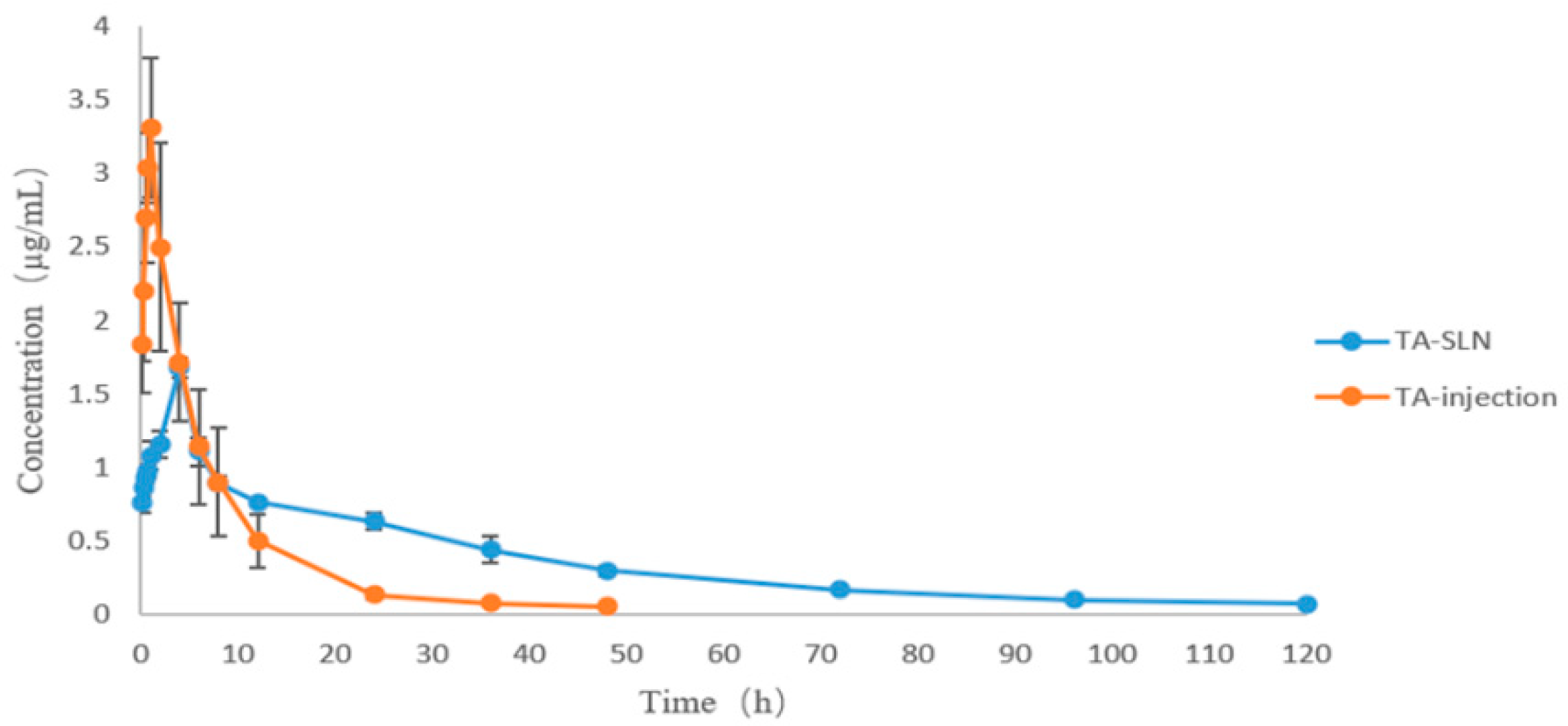
3.7. Pharmacokinetics
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Caporale, V.; Alessandrini, B.; Dalla Villa, P.; Del Papa, S. Global perspectives on animal welfare: Europe. Rev. Sci. Tech. 2005, 24, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Steagall, P.V.; Bustamante, H.; Johnson, C.B.; Turner, P.V. Pain Management in Farm Animals: Focus on Cattle, Sheep and Pigs. Animals 2021, 11, 1483. [Google Scholar] [CrossRef] [PubMed]
- Bacchi, S.; Palumbo, P.; Sponta, A.; Coppolino, M.F. Clinical pharmacology of non-steroidal anti-inflammatory drugs: A review. Antiinflamm. Antiallergy Agents Med. Chem. 2012, 11, 52–64. [Google Scholar] [CrossRef]
- Badri, W.; Miladi, K.; Nazari, Q.A.; Greige-Gerges, H.; Fessi, H.; Elaissari, A. Encapsulation of NSAIDs for inflammation management: Overview, progress, challenges and prospects. Int. J. Pharm. 2016, 515, 757–773. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Siril, P.F. Preparation and characterization of polyvinyl alcohol stabilized griseofulvin nanoparticles. Mater. Today Proc. 2016, 3, 2261–2267. [Google Scholar] [CrossRef]
- García-Rayado, G.; Navarro, M.; Lanas, A. NSAID induced gastrointestinal damage and designing GI-sparing NSAIDs. Expert Rev. Clin. Pharm. 2018, 11, 1031–1043. [Google Scholar] [CrossRef]
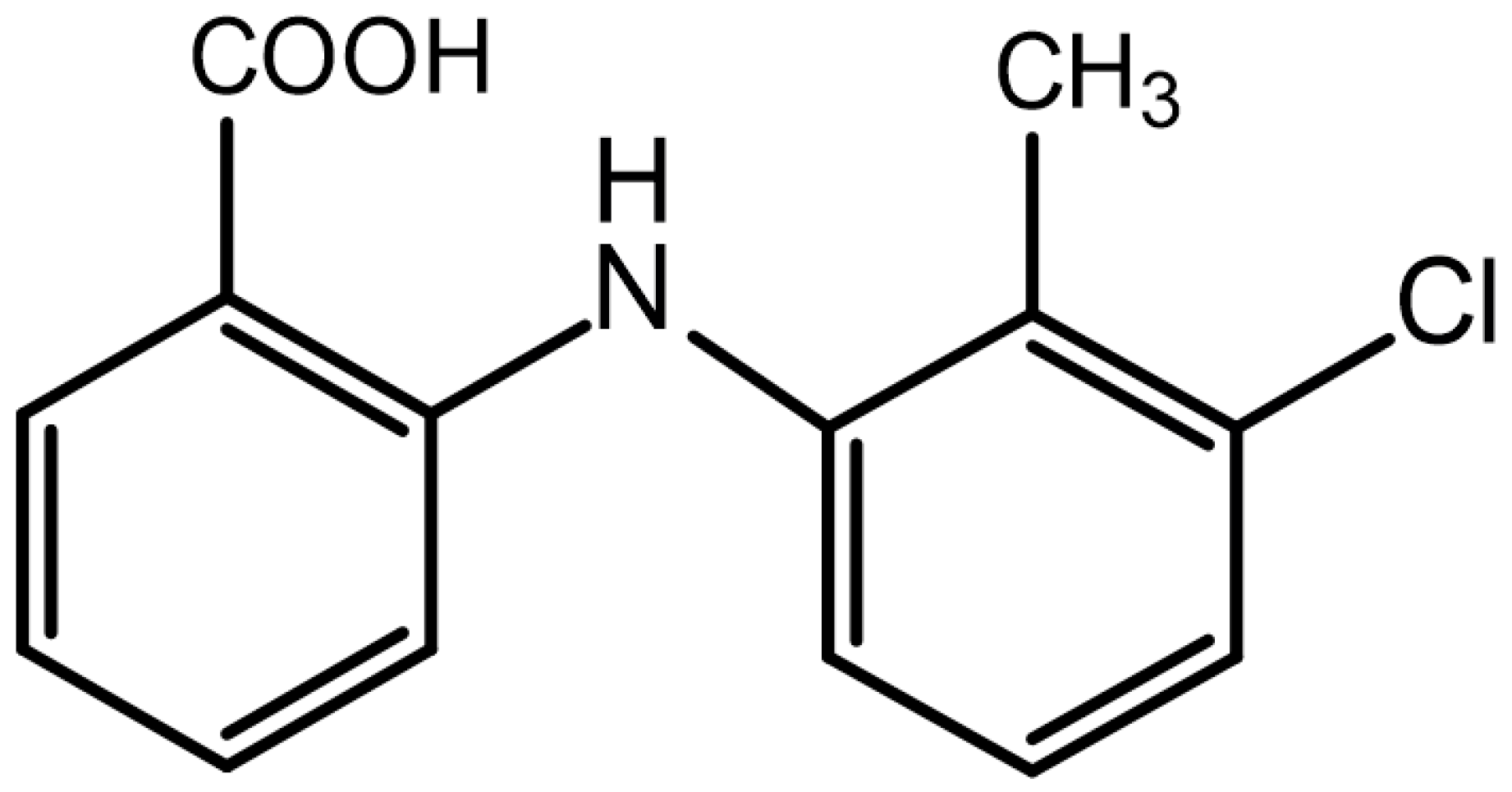
- Ahmed, S.; Sheraz, M.A.; Ahmad, I. Tolfenamic Acid. Profiles Drug. Subst. Excip. Relat. Methodol. 2018, 43, 255–319. [Google Scholar]
- Eskerod, O. Gastrointestinal Tolerance Studies on Tolfenamic Acid in Humans and Animals. Pharmacol. Toxicol. 1994, 75, 44–48. [Google Scholar] [CrossRef]
- EMEA/MRL/183/97–FINAL, Tolfenamic Acid Summary Report Committee Veterinary Medicinal Products. 1997. Available online: https://www.ema.europa.eu/en/documents/mrl-report/tolfenamic-acid-summary-report-committee-veterinary-medicinal-products_en.pdf (accessed on 5 August 2022).
- Kankaanranta, H.; Moilanen, E.; Vapaatalo, H. Tolfenamic acid inhibits leukotriene B4-induced chemotaxis of polymorphonuclear leukocytes in vitro. Inflammation 1991, 15, 137–143. [Google Scholar] [CrossRef]
- Proudman, K.E.; McMillan, R.M. Are tolfenamic acid and tenidap dual inhibitors of 5-lipoxygenase and cyclo-oxygenase? Agents Actions 1991, 34, 121–124. [Google Scholar] [CrossRef]
- Pandey, S.; Patel, P.; Gupta, A. Novel Solid Lipid Nanocarrier of Glibenclamide: A Factorial Design Approach with Response Surface Methodology. Curr. Pharm. Des. 2018, 24, 1811–1820. [Google Scholar] [CrossRef] [PubMed]
- Turk, E.; Tekeli, I.O.; Durna Corum, D.; Corum, O.; Altinok Yipel, F.; Ilhan, A.; Emiroglu, S.B.; Uguz, H.; Uney, K. Pharmacokinetics of tolfenamic acid in goats after different administration routes. J. Vet. Pharmacol. Ther. 2021, 44, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.H.; Mäder, K.; Gohla, S. Solid lipid nanoparticles (SLN) for controlled drug delivery—A review of the state of the art. Eur. J. Pharm. Biopharm. 2000, 50, 161–177. [Google Scholar] [CrossRef]
- Ali, H.; Singh, S.K. Biological voyage of solid lipid nanoparticles: A proficient carrier in nanomedicine. Ther. Deliv. 2016, 7, 691–709. [Google Scholar] [CrossRef] [PubMed]
- Gordillo-Galeano, A.; Mora-Huertas, C.E. Solid lipid nanoparticles and nanostructured lipid carriers: A review emphasizing on particle structure and drug release. Eur. J. Pharm. Biopharm. 2018, 133, 285–308. [Google Scholar] [CrossRef]
- Cavalli, R.; Marengo, E.; Rodriguez, L.; Gasco, M.R. Effects of some experimental factors on the production process of solid lipid nanoparticles. Eur. J. Pharm. Biopharm. 1996, 42, 110–115. [Google Scholar]
- Tian, H.; Lu, Z.; Li, D.; Hu, J. Preparation and characterization of citral-loaded solid lipid nanoparticles. Food Chem. 2018, 248, 78–85. [Google Scholar] [CrossRef]
- Kheradmandnia, S.; Vasheghani-Farahani, E.; Nosrati, M.; Atyabi, F. Preparation and characterization of ketoprofen-loaded solid lipid nanoparticles made from beeswax and carnauba wax. Nanomedicine 2010, 6, 753–759. [Google Scholar] [CrossRef]
- Mohammadi, P.; Mahjub, R.; Mohammadi, M.; Derakhshandeh, K.; Ghaleiha, A.; Mahboobian, M.M. Pharmacokinetics and brain distribution studies of perphenazine-loaded solid lipid nanoparticles. Drug Dev. Ind. Pharm. 2021, 47, 146–152. [Google Scholar] [CrossRef]
- Trotta, M.; Debernardi, F.; Caputo, O. Preparation of solid lipid nanoparticles by a solvent emulsification-diffusion technique. Int. J. Pharm. 2003, 257, 153–160. [Google Scholar] [CrossRef]
- Xie, S.; Pan, B.; Wang, M.; Zhu, L.; Wang, F.; Dong, Z.; Wang, X.; Zhou, W. Formulation, characterization and pharmacokinetics of praziquantel-loaded hydrogenated castor oil solid lipid nanoparticles. Nanomedicine 2010, 5, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, H.; Yao, H.; Mu, Q.; Zhao, G.; Li, Y.; Hu, H.; Niu, X. Pharmacokinetic and anti-inflammatory effects of sanguinarine solid lipid nanoparticles. Inflammation 2014, 37, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Parveen, R.; Ahmad, F.J.; Iqbal, Z.; Samim, M.; Ahmad, S. Solid lipid nanoparticles of anticancer drug andrographolide: Formulation, in vitro and in vivo studies. Drug Dev. Ind. Pharm. 2014, 40, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Attama, A.A.; Umeyor, C.E. The use of solid lipid nanoparticles for sustained drug release. Ther. Deliv. 2015, 6, 669–684. [Google Scholar] [CrossRef]
- Rostami, E.; Kashanian, S.; Azandaryani, A.H.; Faramarzi, H.; Dolatabadi, J.E.; Omidfar, K. Drug targeting using solid lipid nanoparticles. Chem. Phys. Lipids. 2014, 181, 56–61. [Google Scholar] [CrossRef]
- Banerjee, S.; Pillai, J. Solid lipid matrix mediated nanoarchitectonics for improved oral bioavailability of drugs. Expert. Opin. Drug Metab. Toxicol. 2019, 15, 499–515. [Google Scholar] [CrossRef]
- Kaur, R.; Sharma, N.; Tikoo, K.; Sinha, V.R. Development of mirtazapine loaded solid lipid nanoparticles for topical delivery: Optimization, characterization and cytotoxicity evaluation. Int. J. Pharm. 2020, 586, 119439. [Google Scholar] [CrossRef]
- Xu, H.; Zeiger, B.W.; Suslick, K.S. Sonochemical synthesis of nanomaterials. Chem. Soc. Rev. 2013, 42, 2555–2567. [Google Scholar] [CrossRef]
- Gedanken, A. Using sonochemistry for the fabrication of nanomaterials. Ultrason. Sonochemistry 2004, 11, 47–55. [Google Scholar] [CrossRef]
- Brotchie, A.; Grieser, F.; Ashokkumar, M. Effect of power and frequency on bubble-size distributions in acoustic cavitation. Phys. Rev. Lett. 2009, 102, 084302. [Google Scholar] [CrossRef]
- Jangid, A.K.; Patel, K.; Jain, P.; Patel, S.; Gupta, N.; Pooja, D.; Kulhari, H. Inulin-pluronic-stearic acid based double folded nanomicelles for pH-responsive delivery of resveratrol. Carbohydr. Polym. 2020, 247, 116730. [Google Scholar] [CrossRef]
- Al-Sahaf, Z.; Raimi-Abraham, B.; Licciardi, M.; de Mohac, L.M. Influence of Polyvinyl Alcohol (PVA) on PVA-Poly-N-hydroxyethyl-aspartamide (PVA-PHEA) Microcrystalline Solid Dispersion Films. AAPS PharmSciTech 2020, 21, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Gilpin, R.K.; Zhou, W. Infrared studies of the polymorphic states of the fenamates. J. Pharm. Biomed. Anal. 2005, 37, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Jabeen, S.; Dines, T.J.; Leharne, S.A.; Chowdhry, B.Z. Raman and IR spectroscopic studies of fenamates--conformational differences in polymorphs of flufenamic acid, mefenamic acid and tolfenamic acid. Spectrochim. Acta Mol. Biomol. Spectrosc. 2012, 96, 972–985. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Sheraz, M.A.; Rehman, I.U. Studies on tolfenamic acid-chitosan intermolecular interactions: Effect of pH, polymer concentration and molecular weight. AAPS PharmSciTech 2013, 14, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Kuskov, A.N.; Luss, A.L.; Gritskova, I.A.; Shtilman, M.I.; Motyakin, M.V.; Levina, I.I.; Nechaeva, A.M.; Sizova, O.Y.; Tsatsakis, A.M.; Mezhuev, Y.O. Kinetics and Mechanism of Synthesis of Carboxyl-Containing N-Vinyl-2-Pyrrolidone Telehelics for Pharmacological Use. Polymers 2021, 13, 2569. [Google Scholar] [CrossRef] [PubMed]
- Bagde, A.; Patel, K.; Kutlehria, S.; Chowdhury, N.; Singh, M. Formulation of topical ibuprofen solid lipid nanoparticle (SLN) gel using hot melt extrusion technique (HME) and determining its anti-inflammatory strength. Drug Deliv. Transl. Res. 2019, 9, 816–827. [Google Scholar] [CrossRef]
- Shah, M.; Agrawal, Y. Ciprofloxacin hydrochloride-loaded glyceryl monostearate nanoparticle: Factorial design of Lutrol F68 and Phospholipon 90G. J. Microencapsul. 2012, 29, 331–343. [Google Scholar] [CrossRef]
- Golfomitsou, I.; Mitsou, E.; Xenakis, A.; Papadimitriou, V. Development of food grade O/W nanoemulsions as carriers of vitamin D for the fortification of emulsion based food matrices: A structural and activity study. J. Mol. Liq. 2018, 268, 734–742. [Google Scholar] [CrossRef]
- Kaneko, J.J.; Harvey, W.J.; Bruss, M.L. Clinical Biochemistry of Domestic Animals; Academic Press: London, UK, 2008. [Google Scholar]
- Cortés, H.; Hernández-Parra, H.; Bernal-Chávez, S.A.; Prado-Audelo, M.L.D.; Caballero-Florán, I.H.; Borbolla-Jiménez, F.V.; González-Torres, M.; Magaña, J.J.; Leyva-Gómez, G. Non-Ionic Surfactants for Stabilization of Polymeric Nanoparticles for Biomedical Uses. Materials 2021, 14, 3197. [Google Scholar] [CrossRef]
- Kumar, V.V.; Chandrasekar, D.; Ramakrishna, S.; Kishan, V.; Rao, Y.M.; Diwan, P.V. Development and evaluation of nitrendipine loaded solid lipid nanoparticles: Influence of wax and glyceride lipids on plasma pharmacokinetics. Int. J. Pharm. 2007, 335, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Jenning, V.; Gohla, S. Comparison of wax and glyceride solid lipid nanoparticles (SLN). Int. J. Pharm. 2000, 196, 219–222. [Google Scholar] [CrossRef]
- Schöler, N.; Olbrich, C.; Tabatt, K.; Müller, R.H.; Hahn, H.; Liesenfeld, O. Surfactant, but not the size of solid lipid nanoparticles (SLN) influences viability and cytokine production of macrophages. Int. J. Pharm. 2001, 221, 57–67. [Google Scholar] [CrossRef]
- Schwarz, C.; Mehnert, W. Solid lipid nanoparticles (SLN) for controlled drug delivery. II. Drug incorporation and physicochemical characterization. J. Microencapsul. 1999, 16, 205–213. [Google Scholar] [CrossRef]
- Sharifi, F.; Jahangiri, M.; Nazir, I.; Asim, M.H.; Ebrahimnejad, P.; Hupfauf, A.; Gust, R.; Bernkop-Schnürch, A. Zeta potential changing nanoemulsions based on a simple zwitterion. J. Colloid Interface Sci. 2021, 585, 126–137. [Google Scholar] [CrossRef]
- Zhang, Y.; Jing, Q.; Hu, H.; He, Z.; Wu, T.; Guo, T.; Feng, N. Sodium dodecyl sulfate improved stability and transdermal delivery of salidroside-encapsulated niosomes via effects on zeta potential. Int. J. Pharm. 2020, 580, 119183. [Google Scholar] [CrossRef]
- He, C.; Hu, Y.; Yin, L.; Tang, C.; Yin, C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 2010, 31, 3657–3666. [Google Scholar] [CrossRef]
- Khan, M.K.; Nigavekar, S.S.; Minc, L.D.; Kariapper, M.S.; Nair, B.M.; Lesniak, W.G.; Balogh, L.P. In vivo biodistribution of dendrimers and dendrimer nanocomposites—Implications for cancer imaging and therapy. Technol. Cancer Res. Treat. 2005, 4, 603–613. [Google Scholar] [CrossRef]
- Mehnert, W.; Mäder, K. Solid lipid nanoparticles: Production, characterization and applications. Adv. Drug Deliv. Rev. 2001, 47, 165–196. [Google Scholar] [CrossRef]
- Pattadar, D.K.; Zamborini, F.P. Effect of Size, Coverage, and Dispersity on the Potential-Controlled Ostwald Ripening of Metal Nanoparticles. Langmuir 2019, 35, 16416–16426. [Google Scholar] [CrossRef]
- Schultheiss, N.; Newman, A. Pharmaceutical Cocrystals and Their Physicochemical Properties. Cryst. Growth Des. 2009, 9, 2950–2967. [Google Scholar] [CrossRef] [PubMed]
- Manin, A.N.; Voronin, A.P.; Drozd, K.V.; Manin, N.G.; Bauer-Brandl, A.; Perlovich, G.L. Cocrystal screening of hydroxybenzamides with benzoic acid derivatives: A comparative study of thermal and solution-based methods. Eur. J. Pharm. Sci. 2014, 65, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Hirakura, Y.; Yuda, M.; Terada, K. Coformer screening using thermal analysis based on binary phase diagrams. Pharm. Res. 2014, 31, 1946–1957. [Google Scholar] [CrossRef] [PubMed]
- Berthomieu, C.; Hienerwadel, R. Fourier transform infrared (FTIR) spectroscopy. Photosynth. Res. 2009, 101, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Reddy, L.H.; Murthy, R.S. Etoposide-loaded nanoparticles made from glyceride lipids: Formulation, characterization, in vitro drug release, and stability evaluation. AAPS PharmSciTech 2005, 6, E158–E166. [Google Scholar] [CrossRef]
- McCartan, A.J.S.; Curran, D.W.; Mrsny, R.J. Evaluating parameters affecting drug fate at the intramuscular injection site. J. Control. Release 2021, 336, 322–335. [Google Scholar] [CrossRef]
- Luo, Y.; Chen, D.; Ren, L.; Zhao, X.; Qin, J. Solid lipid nanoparticles for enhancing vinpocetine’s oral bioavailability. J. Control. Release 2006, 114, 53–59. [Google Scholar] [CrossRef]
- Hu, L.; Tang, X.; Cui, F. Solid lipid nanoparticles (SLNs) to improve oral bioavailability of poorly soluble drugs. J. Pharm. Pharmacol. 2004, 56, 1527–1535. [Google Scholar] [CrossRef]
- Azhar Shekoufeh Bahari, L.; Hamishehkar, H. The Impact of Variables on Particle Size of Solid Lipid Nanoparticles and Nanostructured Lipid Carriers; A Comparative Literature Review. Adv. Pharm. Bull. 2016, 6, 143–151. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Level | ||
|---|---|---|---|
| 1 | 2 | 3 | |
| Type | Poloxamer 188 | PVA | PVPK30 |
| Concentration | 2% | 3% | 4% |
| Volume (mL) | 20 | 25 | 30 |
| No | Type (A) | Concentration (B) | Volume (C) | LC (%) | Size (nm) | Zeta Potential (mV) | PDI |
|---|---|---|---|---|---|---|---|
| 1 | 3 | 3 | 1 | 6.14 ± 0.12 | 417.63 ± 7.44 | −24.17 | 0.27 ± 0.03 |
| 2 | 1 | 2 | 3 | 23.48 ± 0.58 | 458.83 ± 1.62 | −22.77 | 0.35 ± 0.02 |
| 3 | 3 | 1 | 3 | 4.64 ± 0.09 | 577.51 ± 9.97 | −25.43 | 0.40 ± 0.01 |
| 4 | 1 | 3 | 2 | 6.47 ± 0.14 | 508.53 ± 8.11 | −22.60 | 0.27 ± 0.01 |
| 5 | 2 | 3 | 3 | 26.39 ± 0.37 | 668.30 ± 1.25 | −11.17 | 0.34 ± 0.04 |
| 6 | 3 | 2 | 2 | 5.17 ± 0.14 | 1225.21 ± 5.29 | −22.00 | 0.38 ± 0.04 |
| 7 | 2 | 2 | 1 | 16.59 ± 0.12 | 541.57 ± 4.72 | −14.77 | 0.39 ± 0.02 |
| 8 | 2 | 1 | 2 | 26.61 ± 0.20 | 634.72 ± 4.81 | −13.67 | 0.37 ± 0.01 |
| 9 | 1 | 1 | 1 | 20.81 ± 0.24 | 483.03 ± 9.17 | −23.20 | 0.34 ± 0.01 |
| K1 | 16.92 | 17.35 | 14.51 | ||||
| K2 | 23.19 | 15.08 | 12.75 | ||||
| K3 | 5.31 | 13.00 | 18.17 | ||||
| R | 17.88 | 4.35 | 5.42 | ||||
| Optimum | A2 | B1 | C3 |
| Influence Factors | Time (d) | |||
|---|---|---|---|---|
| 0 | 5 | 10 | ||
| High temperature | Appearance | Milky white | Milky white | Milky white |
| Sedimentation rate | 1 | 1 | 1 | |
| Redispersibility | Good | Good | Good | |
| Content labelling amount (%) | 100.19 | 99.36 | 99.22 | |
| Relative substance | ND | ND | ND | |
| Size | 460 | 482 | 479 | |
| High humidity | Appearance | Milky white | Milky white | Milky white |
| Sedimentation rate | 1 | 1 | 1 | |
| Redispersibility | Good | Good | Good | |
| Content labelling amount (%) | 100.19 | 99.22 | 99.83 | |
| Relative substance | ND | ND | ND | |
| Size | 460 | 466 | 471 | |
| High light | Appearance | Milky white | Milky white | Milky white |
| Sedimentation rate | 1 | 1 | 1 | |
| Redispersibility | Good | Good | Good | |
| Content labelling amount (%) | 100.19 | 99.04 | 99.35 | |
| Relative substance | ND | ND | ND | |
| Size | 460 | 485 | 491 | |
| Fit Equation | TA Injection | TA-SLN Suspension |
|---|---|---|
| Zero-order equation | Mt = 66.15t + 0.78 (R2 = 0.1422) | Mt = 0.87t + 0.43 (R2 = 0.9792) |
| Formula One | Mt = 89.02 (1 − e−1.043t) (R2 = 0.9835) | - |
| Higuchi | Mt = 8.05X1/2 + 53.84 (R2 = 0.3648) | Mt = 9.66X1/2 − 14.01 (R2 = 0.9592) |
| Parameter | Unit | TA Injection | TA-SLN Suspension |
|---|---|---|---|
| Tmax | h | 0.91 ± 0.21 | 4.00 b |
| Cmax | µg/mL | 3.37 ± 0.36 | 1.68 ± 0.07 b |
| AUC0–∞ | µg h/mL | 23.26 ± 4.92 | 43.11 ± 1.16 b |
| Vd | mL/kg | 3220.21 ± 1323.89 | 4295.76 ± 508.01 |
| CL | mL/h/kg | 168.13 ± 27.11 | 85.60 ± 2.57 b |
| MRT0–∞ | h | 8.49 ± 1.36 | 32.13 ± 0.75 b |
| T1/2 | h | 5.88 ± 0.94 | 22.26 ± 0.52 b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, W.; Deng, Z.; Xiang, Y.; Zhu, D.; Yi, D.; Mo, Y.; Liu, Y.; Qin, L.; Huang, L.; Wan, B.; et al. Preparation, Characterization and Pharmacokinetics of Tolfenamic Acid-Loaded Solid Lipid Nanoparticles. Pharmaceutics 2022, 14, 1929. https://doi.org/10.3390/pharmaceutics14091929
Xu W, Deng Z, Xiang Y, Zhu D, Yi D, Mo Y, Liu Y, Qin L, Huang L, Wan B, et al. Preparation, Characterization and Pharmacokinetics of Tolfenamic Acid-Loaded Solid Lipid Nanoparticles. Pharmaceutics. 2022; 14(9):1929. https://doi.org/10.3390/pharmaceutics14091929
Chicago/Turabian StyleXu, Wei, Zhaoyou Deng, Yifei Xiang, Dujuan Zhu, Dandan Yi, Yihao Mo, Yu Liu, Lanqian Qin, Ling Huang, Bingjie Wan, and et al. 2022. "Preparation, Characterization and Pharmacokinetics of Tolfenamic Acid-Loaded Solid Lipid Nanoparticles" Pharmaceutics 14, no. 9: 1929. https://doi.org/10.3390/pharmaceutics14091929