
Fragment-Based and Structural Investigation for Discovery of JNK3 Inhibitors

Abstract
:1. Introduction
2. Materials and Methods
2.1. Fragment Library
2.2. Preparation of JNK3
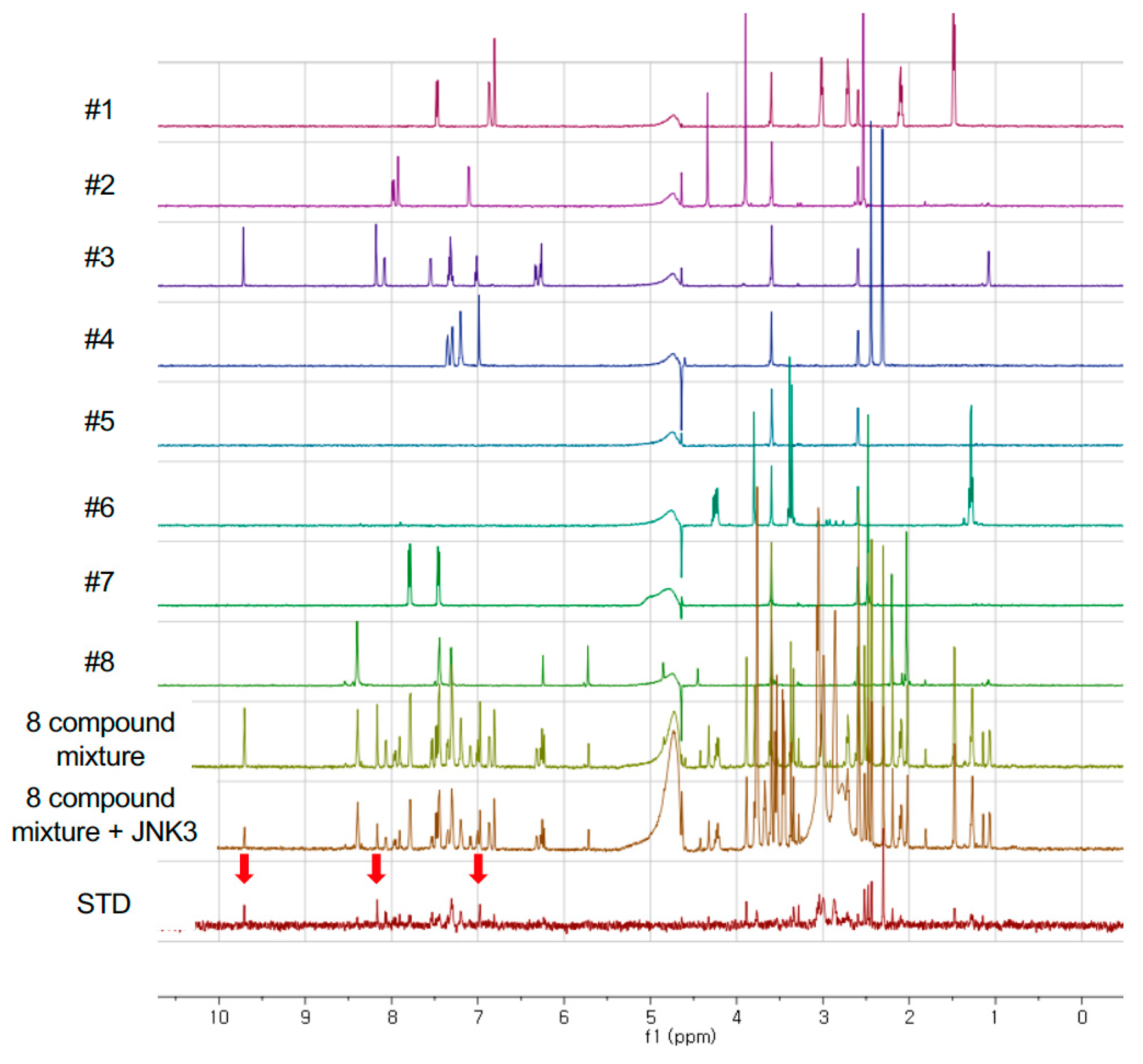
2.3. NMR Experiments
2.4. Crystallization and Data Collection
2.5. Structure Determination and Refinement

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystals | 3A8 | Inhibitor |
|---|---|---|
| PDB ID | 7YL1 | 4KKH |
| Data collection | ||
| Space group | P212121 | P212121 |
| Cell dimensions | ||
| a, b, c (Å) | 50.15, 71.98, 107.16 | 52.32, 71.50, 107.08 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Wavelength (Å) | 0.97951 | 0.97951 |
| Resolution (Å) * | 59.75–2.48 (2.55–2.49) | 50–2.0 (2.03–2.00) |
| Redundancy * | 13.3 (12.5) | 13.5 (12.8) |
| Completeness (%) * | 99.1 (99.4) | 100 (100) |
| I/σI * | 33.6 (3.7) | 58.9 (9.4) |
| Rmerge (%) a,* | 9.0 (35.3) | 9.0 (44.2) |
| Refinement | ||
| Resolution (Å) | 36.61–2.49 | 37.45–2.0 |
| No. of reflections | 13,406 | 26,471 |
| Rwork/Rfree (%) b | 20.4/29.4 | 22.9/27.7 |
| No. of atoms | 2925 | 2942 |
| Average B-factors | Protein/water/3A8 43.139/42.040/48.722 | Protein/water/inhibitor 43.774/47.582/35.827 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.007 | 0.007 |
| Bond angle (°) | 1.157 | 1.109 |
| Ramachandran plot | ||
| Residues in favored/allowed/ disallowed regions (%) c | 93.9/5.5/0.6 | 97.4/2.6/0/0 |
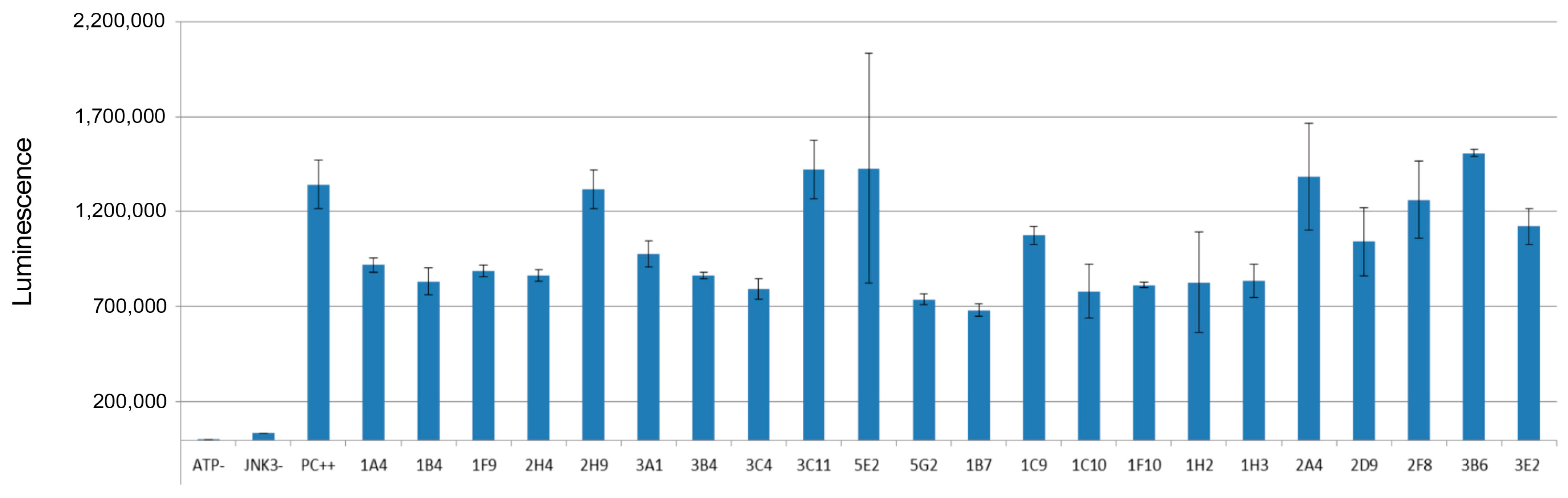
2.6. In Vitro Kinase Assay
3. Results
3.1. Virtual Screening
3.2. Fragment Screening
3.3. In Vitro Kinase Assay
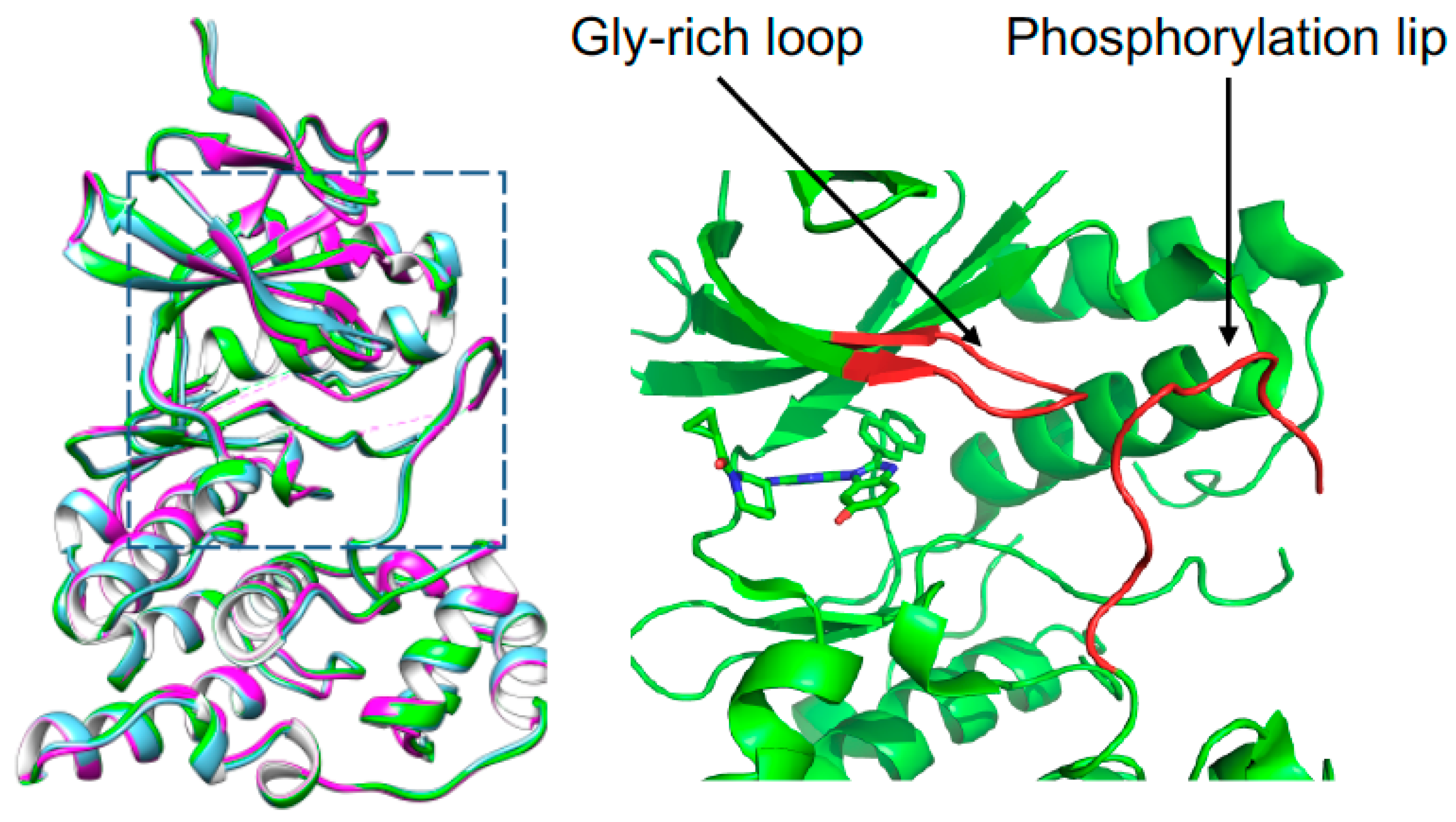
3.4. Overall Structure of JNK3 in Complex with Fragment and Inhibitor
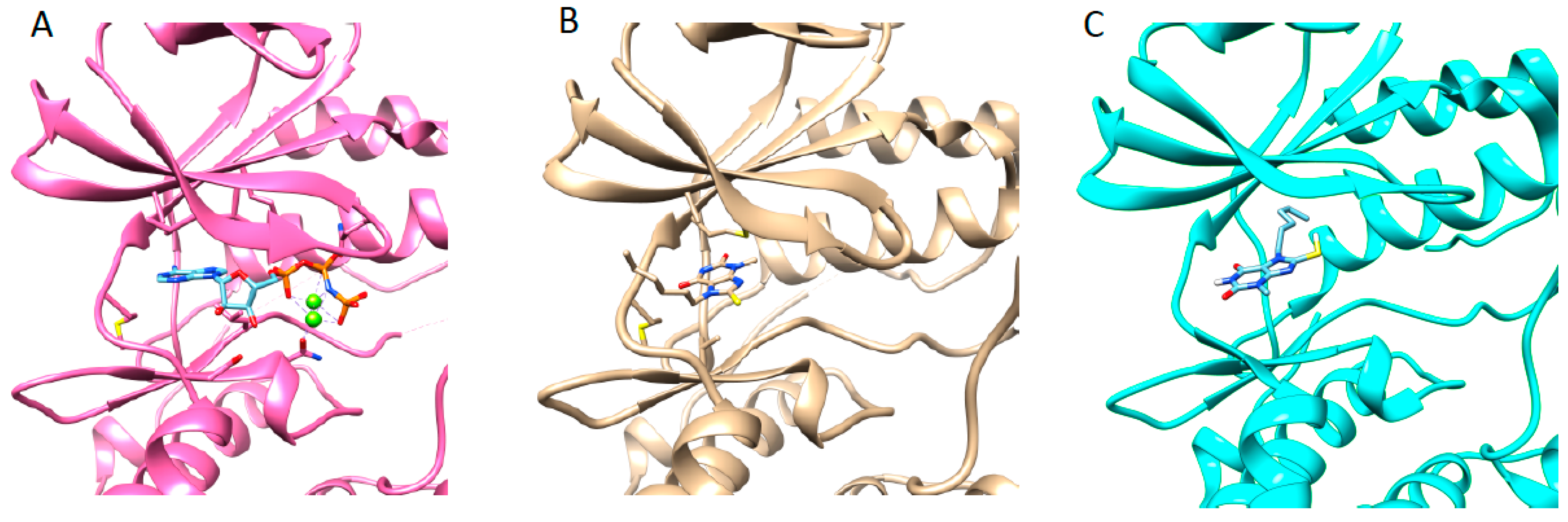
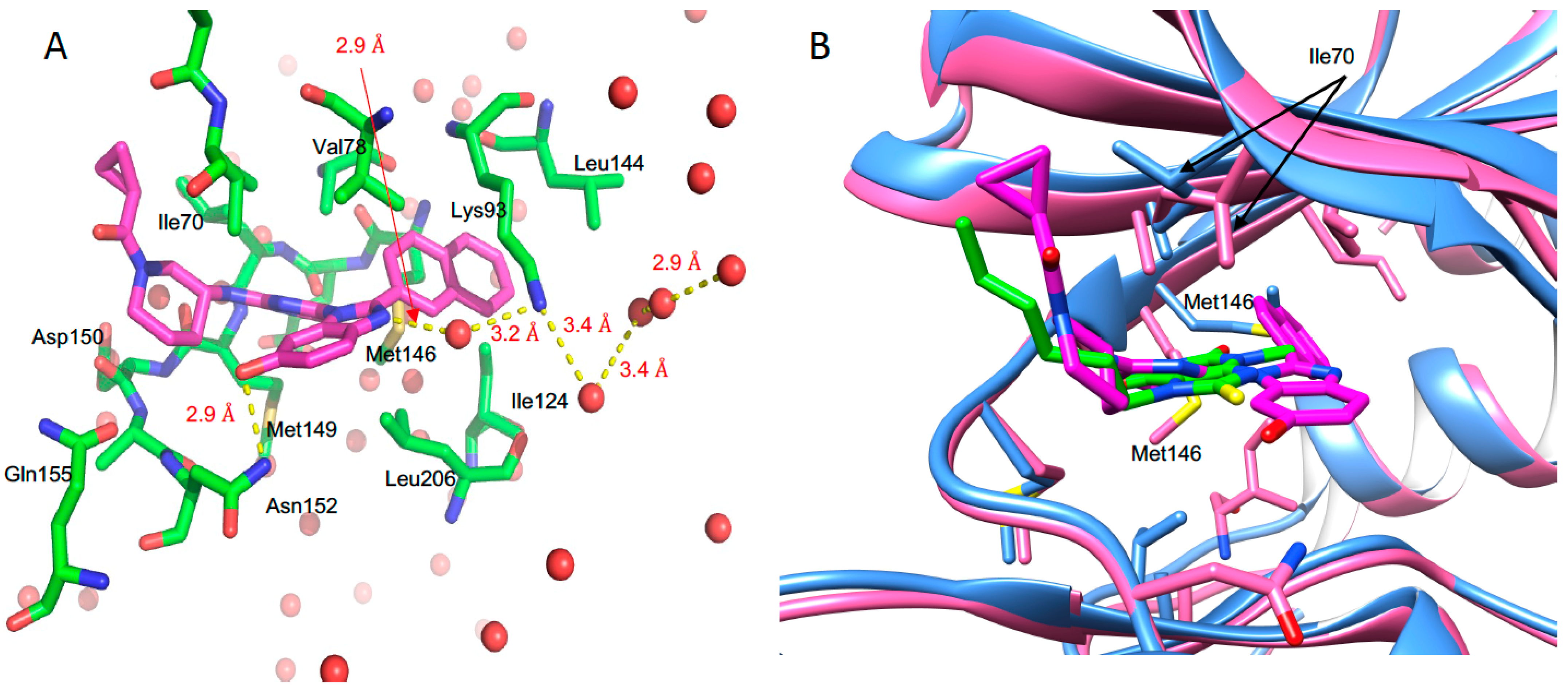
3.5. Structure of JNK3 in Complex with Fragment 3A8
3.6. Structure JNK3 in Complex with a Potent Inhibitor
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef]
- Ip, Y.T.; Davis, R.J. Signal transduction by the c-Jun N-terminal kinase (JNK)—From inflammation to development. Curr. Opin. Cell Biol. 1998, 10, 205–219. [Google Scholar] [CrossRef]
- Mohit, A.A.; Martin, J.H.; Miller, C.A. p493F12 kinase: A novel MAP kinase expressed in a subset of neurons in the human nervous system. Neuron 1995, 14, 67–78. [Google Scholar] [CrossRef]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Zeke, A.; Misheva, M.; Remenyi, A.; Bogoyevitch, M.A. JNK Signaling: Regulation and Functions Based on Complex Protein-Protein Partnerships. Microbiol. Mol. Biol. Rev. 2016, 80, 793–835. [Google Scholar] [CrossRef] [PubMed]
- Tournier, C.; Dong, C.; Turner, T.K.; Jones, S.N.; Flavell, R.A.; Davis, R.J. MKK7 is an essential component of the JNK signal transduction pathway activated by proinflammatory cytokines. Genes Dev. 2001, 15, 1419–1426. [Google Scholar] [CrossRef]
- Graczyk, P.P. JNK inhibitors as anti-inflammatory and neuroprotective agents. Future Med. Chem. 2013, 5, 539–551. [Google Scholar] [CrossRef]
- Assi, K.; Pillai, R.; Gomez-Munoz, A.; Owen, D.; Salh, B. The specific JNK inhibitor SP600125 targets tumour necrosis factor-alpha production and epithelial cell apoptosis in acute murine colitis. Immunology 2006, 118, 112–121. [Google Scholar] [CrossRef]
- Hefetz-Sela, S.; Stein, I.; Klieger, Y.; Porat, R.; Sade-Feldman, M.; Zreik, F.; Nagler, A.; Pappo, O.; Quagliata, L.; Dazert, E.; et al. Acquisition of an immunosuppressive protumorigenic macrophage phenotype depending on c-Jun phosphorylation. Proc. Natl. Acad. Sci. USA 2014, 111, 17582–17587. [Google Scholar] [CrossRef]
- Das, M.; Garlick, D.S.; Greiner, D.L.; Davis, R.J. The role of JNK in the development of hepatocellular carcinoma. Genes Dev. 2011, 25, 634–645. [Google Scholar] [CrossRef] [Green Version]
- Shibata, W.; Maeda, S.; Hikiba, Y.; Yanai, A.; Sakamoto, K.; Nakagawa, H.; Ogura, K.; Karin, M.; Omata, M. c-Jun NH2-terminal kinase 1 is a critical regulator for the development of gastric cancer in mice. Cancer Res. 2008, 68, 5031–5039. [Google Scholar] [CrossRef] [PubMed]
- Han, M.S.; Barrett, T.; Brehm, M.A.; Davis, R.J. Inflammation Mediated by JNK in Myeloid Cells Promotes the Development of Hepatitis and Hepatocellular Carcinoma. Cell Rep. 2016, 15, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Raina, A.K.; Rottkamp, C.A.; Aliev, G.; Perry, G.; Boux, H.; Smith, M.A. Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer’s disease. J. Neurochem. 2001, 76, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Gourmaud, S.; Paquet, C.; Dumurgier, J.; Pace, C.; Bouras, C.; Gray, F.; Laplanche, J.-L.; Meurs, E.F.; Mouton-Liger, F.; Hugon, J. Increased levels of cerebrospinal fluid JNK3 associated with amyloid pathology: Links to cognitive decline. J. Psychiatry Neurosci. 2015, 40, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Li, H.; Zhang, B.; Xiong, R.; Zhang, Y.; Kang, W.-Y.; Chen, W.; Zhao, Z.-B.; Chen, S.D. Small peptide inhibitor of JNK3 protects dopaminergic neurons from MPTP induced injury via inhibiting the ASK1-JNK3 signaling pathway. PLoS ONE 2015, 10, e0119204. [Google Scholar] [CrossRef]
- Badshah, H.; Ali, T.; Rehman, S.-U.; Amin, F.-U.; Ullah, F.; Kim, T.H.; Kim, M.O. Protective Effect of Lupeol Against Lipopolysaccharide-Induced Neuroinflammation via the p38/c-Jun N-Terminal Kinase Pathway in the Adult Mouse Brain. J. Neuroimmune Pharmacol. 2016, 11, 48–60. [Google Scholar] [CrossRef]
- Zhang, S.; Gui, X.-H.; Huang, L.-P.; Deng, M.-Z.; Fang, R.-M.; Ke, X.-H.; He, Y.-P.; Li, L.; Fang, Y.-Q. Neuroprotective Effects of beta-Asarone Against 6-Hydroxy Dopamine-Induced Parkinsonism via JNK/Bcl-2/Beclin-1 Pathway. Mol. Neurobiol. 2016, 53, 83–94. [Google Scholar] [CrossRef]
- Wang, W.; Ma, C.; Mao, Z.; Li, M. JNK inhibition as a potential strategy in treating Parkinson’s disease. Drug News Perspect. 2004, 17, 646–654. [Google Scholar] [CrossRef]
- Duong, M.T.H.; Lee, J.-H.; Ahn, H.-C. C-Jun N-terminal kinase inhibitors: Structural insight into kinase-inhibitor complexes. Comput. Struct. Biotechnol. J. 2020, 18, 1440–1457. [Google Scholar] [CrossRef]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schioth, H.B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discov. 2021, 20, 839–861. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2021 update. Pharmacol. Res. 2021, 165, 105463. [Google Scholar] [CrossRef] [PubMed]
- Hajduk, P.J.; Greer, J. A decade of fragment-based drug design: Strategic advances and lessons learned. Nat. Rev. Drug Discov. 2007, 6, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Erlanson, D.A.; Fesik, S.W.; Hubbard, R.E.; Jahnke, W.; Jhoti, H. Twenty years on: The impact of fragments on drug discovery. Nat. Rev. Drug Discov. 2016, 15, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.-F.; Qiu, H.-Y.; Wang, Z.-F.; Zhang, Y.-J.; Wang, Z.-C.; Li, D.-D.; Zhu, H.-L. Identification of novel B-Raf(V600E) inhibitors employing FBDD strategy. Biochem. Pharmacol. 2017, 132, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Lamoree, B.; Hubbard, R.E. Current perspectives in fragment-based lead discovery (FBLD). Essays Biochem. 2017, 61, 453–464. [Google Scholar] [CrossRef]
- Irie, T.; Sawa, M. 7-Azaindole: A Versatile Scaffold for Developing Kinase Inhibitors. Chem. Pharm. Bull. 2018, 66, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Mortenson, P.N.; Berdini, V.; O’Reilly, M. Fragment-based approaches to the discovery of kinase inhibitors. Methods Enzym. 2014, 548, 69–92. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef]
- Meyer, B.; Peters, T. NMR spectroscopy techniques for screening and identifying ligand binding to protein receptors. Angew. Chem. Int. Ed. Engl. 2003, 42, 864–890. [Google Scholar] [CrossRef]
- Mayer, M.; Meyer, B. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J. Am. Chem. Soc. 2001, 123, 6108–6117. [Google Scholar] [CrossRef]
- Streiff, J.H.; Juranic, N.O.; Macura, S.I.; Warner, D.O.; Jones, K.A.; Perkins, W.J. Saturation transfer difference nuclear magnetic resonance spectroscopy as a method for screening proteins for anesthetic binding. Mol. Pharmacol. 2004, 66, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Frommlet, A.; Perez, M.; Cobas, C.; Blechschmidt, A.; Dominguez, S.; Lingel, A. Fast and Efficient Fragment-Based Lead Generation by Fully Automated Processing and Analysis of Ligand-Observed NMR Binding Data. J. Med. Chem. 2016, 59, 3303–3310. [Google Scholar] [CrossRef]
- Johnson, J.A.; Olson, N.M.; Tooker, M.J.; Bur, S.K.; Pomerantz, W.C.K. Combined Protein- and Ligand-Observed NMR Workflow to Screen Fragment Cocktails against Multiple Proteins: A Case Study Using Bromodomains. Molecules 2020, 25, 3949. [Google Scholar] [CrossRef] [PubMed]
- Dalvit, C.; Fogliatto, G.; Stewart, A.; Veronesi, M.; Stockman, B. WaterLOGSY as a method for primary NMR screening: Practical aspects and range of applicability. J. Biomol. NMR 2001, 21, 349–359. [Google Scholar] [CrossRef]
- Kim, M.-H.; Lee, J.; Jung, K.; Kim, M.; Park, Y.-J.; Ahn, H.; Kwon, Y.H.; Hah, J.-M. Syntheses and biological evaluation of 1-heteroaryl-2-aryl-1H-benzimidazole derivatives as c-Jun N-terminal kinase inhibitors with neuroprotective effects. Bioorganic Med. Chem. 2013, 21, 2271–2285. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Gu, Y.; Fox, T.; Coll, J.T.; Fleming, M.A.; Markland, W.; Caron, P.R.; Wilson, K.P.; Su, M.S. Crystal structure of JNK3: A kinase implicated in neuronal apoptosis. Structure 1998, 6, 983–991. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Scapin, G.; Patel, S.B.; Lisnock, J.; Becker, J.W.; LoGrasso, P.V. The structure of JNK3 in complex with small molecule inhibitors: Structural basis for potency and selectivity. Chem. Biol. 2003, 10, 705–712. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Probst, G.D.; Bowers, S.; Sealy, J.M.; Truong, A.P.; Hom, R.K.; Galemmo, R.A., Jr.; Konradi, A.W.; Sham, H.L.; Quincy, D.A.; Pan, H.; et al. Highly selective c-Jun N-terminal kinase (JNK) 2 and 3 inhibitors with in vitro CNS-like pharmacokinetic properties prevent neurodegeneration. Bioorg. Med. Chem. Lett. 2011, 21, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Cryst. D 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Cryst. D 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.W.; Leaver-Fay, A.; Chen, V.B.; Block, J.N.; Kapral, G.J.; Wang, X.; Murray, L.W.; Arendall, W.B., 3rd; Snoeyink, J.; Richardson, J.S.; et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35, W375–W383. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, L.; DeLano, W. PyMOL. Available online: http://www.pymol.org/pymol (accessed on 28 July 2022).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Nguyen, P.L.; Bui, B.P.; Duong, M.T.H.; Lee, K.; Ahn, H.-C.; Cho, J. Suppression of LPS-Induced Inflammation and Cell Migration by Azelastine through Inhibition of JNK/NF-kappaB Pathway in BV2 Microglial Cells. Int. J. Mol. Sci. 2021, 22, 9061. [Google Scholar] [CrossRef]





 | |||
| Code | R1 | R2 | R3 |
|---|---|---|---|
| 1A4 | -CH3 | -SCH2COOH | -H |
| 2H4 | -CH2-CH=CH2 | -SCH2COOH | -H |
| 1F9 | -CH3 |  | -CH3 |
| 2D5 | -CH2-CH(CH3)2 | -NH-NH2 | -H |
| 2D8 | -CH2-CH=CH-CH3 | -SH | -H |
| 3A1 | -CH3 | -SH | -H |
| 3A8 | -CH2-CH2- CH2-CH2-CH2-CH3 | -SH | -H |
| 3B11 | -CH2-CO-CH3 | -H | -CH3 |
| 3B7 |  | -H | -H |
| 1C9 | -CH(CH3)2 | -S-CH2-CO-NH2 | -H |
| 2A4 | -CH2-CH2-CH3 | -S-CH2-CO-NH2 | -H |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duong, M.T.H.; Ahn, H.-C. Fragment-Based and Structural Investigation for Discovery of JNK3 Inhibitors. Pharmaceutics 2022, 14, 1900. https://doi.org/10.3390/pharmaceutics14091900
Duong MTH, Ahn H-C. Fragment-Based and Structural Investigation for Discovery of JNK3 Inhibitors. Pharmaceutics. 2022; 14(9):1900. https://doi.org/10.3390/pharmaceutics14091900
Chicago/Turabian StyleDuong, Men Thi Hoai, and Hee-Chul Ahn. 2022. "Fragment-Based and Structural Investigation for Discovery of JNK3 Inhibitors" Pharmaceutics 14, no. 9: 1900. https://doi.org/10.3390/pharmaceutics14091900