
Kinase Inhibition in Multiple Myeloma: Current Scenario and Clinical Perspectives

 ,
,  , ,
, ,  and
and 
Abstract
:1. Introduction
2. Current Clinical MM Treatment Options
3. Tyrosine Kinase Inhibitors in MM
4. Ibrutinib: A BTK Inhibition Approach
5. Clinical Perspectives with Other TKIs
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tsang, M.; Le, M.; Ghazawi, F.M.; Cyr, J.; Alakel, A.; Rahme, E.; Lagacé, F.; Netchiporouk, E.; Moreau, L.; Zubarev, A.; et al. Multiple Myeloma Epidemiology and Patient Geographic Distribution in Canada: A Population Study. Cancer 2019, 125, 2435–2444. [Google Scholar] [CrossRef] [PubMed]
- Padala, S.A.; Barsouk, A.; Barsouk, A.; Rawla, P.; Vakiti, A.; Kolhe, R.; Kota, V.; Ajebo, G.H. Epidemiology, Staging, and Management of Multiple Myeloma. Med. Sci. 2021, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Bird, S.; Cairns, D.; Menzies, T.; Boyd, K.; Davies, F.; Cook, G.; Drayson, M.; Gregory, W.; Jenner, M.; Jones, J.; et al. Sex Differences in Multiple Myeloma Biology but Not Clinical Outcomes: Results from 3894 Patients in the Myeloma XI Trial. Clin. Lymphoma Myeloma Leuk. 2021, 21, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Marinac, C.R.; Ghobrial, I.M.; Birmann, B.M.; Soiffer, J.; Rebbeck, T.R. Dissecting Racial Disparities in Multiple Myeloma. Blood Cancer J. 2020, 10, 19. [Google Scholar] [CrossRef]
- Brigle, K.; Rogers, B. Pathobiology and Diagnosis of Multiple Myeloma. Semin. Oncol. Nurs. 2017, 33, 225–236. [Google Scholar] [CrossRef]
- Cowan, A.J.; Green, D.J.; Kwok, M.; Lee, S.; Coffey, D.G.; Holmberg, L.A.; Tuazon, S.; Gopal, A.K.; Libby, E.N. Diagnosis and Management of Multiple Myeloma: A Review. JAMA J. Am. Med. Assoc. 2022, 327, 464–477. [Google Scholar] [CrossRef]
- Firth, J. Haematology: Multiple Myeloma. Clin. Med. 2019, 19, 58–60. [Google Scholar] [CrossRef]
- Joshi, H.; Lin, S.; Fei, K.; Renteria, A.S.; Jacobs, H.; Mazumdar, M.; Jagannath, S.; Bickell, N.A. Multiple Myeloma, Race, Insurance and Treatment. Cancer Epidemiol. 2021, 73, 101974. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Multiple Myeloma: 2022 Update on Diagnosis, Risk Stratification, and Management. Am. J. Hematol. 2022, 97, 1086–1107. [Google Scholar] [CrossRef]
- Landgren, O.; Weiss, B.M. Patterns of Monoclonal Gammopathy of Undetermined Significance and Multiple Myeloma in Various Ethnic/Racial Groups: Support for Genetic Factors in Pathogenesis. Leukemia 2009, 23, 1691–1697. [Google Scholar] [CrossRef]
- Bunce, C.M.; Drayson, M.T. Dissecting Racial Disparities in Multiple Myeloma—Clues from Differential Immunoglobulin Levels. Blood Cancer J. 2020, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Joshua, D.E.; Bryant, C.; Dix, C.; Gibson, J.; Ho, J. Biology and Therapy of Multiple Myeloma. Med. J. Aust. 2019, 210, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Zhou, P.K. DNA Damage Repair: Historical Perspectives, Mechanistic Pathways and Clinical Translation for Targeted Cancer Therapy. Signal Transduct. Target. Ther. 2021, 6, 254. [Google Scholar] [CrossRef] [PubMed]
- Castaneda, O.; Baz, R. Multiple Myeloma Genomics—A Concise Review. Acta Med. Acad. 2019, 48, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Wallington-Beddoe, C.T.; Mynott, R.L. Prognostic and Predictive Biomarker Developments in Multiple Myeloma. J. Hematol. Oncol. 2021, 14, 151. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.-V.; Gay, F.; Anderson, K.C. Multiple Myeloma. Nat. Rev. Dis. Primers 2017, 3, 17046. [Google Scholar] [CrossRef]
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, I.M. Genomic Complexity of Multiple Myeloma and Its Clinical Implications. Nat. Rev. Clin. Oncol. 2017, 14, 100–113. [Google Scholar] [CrossRef]
- Zingone, A.; Kuehl, W.M. Pathogenesis of Monoclonal Gammopathy of Undetermined Significance and Progression to Multiple Myeloma. Semin. Hematol. 2011, 48, 4–12. [Google Scholar] [CrossRef]
- Furukawa, Y.; Kikuchi, J. Molecular Basis of Clonal Evolution in Multiple Myeloma. Int. J. Hematol. 2020, 111, 496–511. [Google Scholar] [CrossRef]
- Barwick, B.G.; Gupta, V.A.; Vertino, P.M.; Boise, L.H. Cell of Origin and Genetic Alterations in the Pathogenesis of Multiple Myeloma. Front. Immunol. 2019, 10, 1121. [Google Scholar] [CrossRef] [Green Version]
- Maura, F.; Rustad, E.H.; Boyle, E.M.; Morgan, G.J. Reconstructing the Evolutionary History of Multiple Myeloma. Best Pr. Res. Clin. Haematol. 2020, 33, 101145. [Google Scholar] [CrossRef] [PubMed]
- Dispenzieri, A.; Katzmann, J.A.; Kyle, R.A.; Larson, D.R.; Melton, L.J.; Colby, C.L.; Therneau, T.M.; Clark, R.; Kumar, S.K.; Bradwell, A.; et al. Prevalence and Risk of Progression of Light-Chain Monoclonal Gammopathy of Undetermined Significance: A Retrospective Population-Based Cohort Study. Lancet 2010, 375, 1721–1728. [Google Scholar] [CrossRef]
- Kyle, R.A.; Therneau, T.M.; Rajkumar, S.V.; Larson, D.R.; Plevak, M.F.; Offord, J.R.; Dispenzieri, A.; Katzmann, J.A.; Melton, L.J. Prevalence of Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2006, 354, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Therneau, T.M.; Rajkumar, S.V.; Offord, J.R.; Larson, D.R.; Plevak, M.F.; Melton, L.J. A Long-Term Study of Prognosis in Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2002, 346, 564–569. [Google Scholar] [CrossRef]
- Greenberg, A.J.; Vachon, C.M.; Rajkumar, S. v Disparities in the Prevalence, Pathogenesis and Progression of Monoclonal Gammopathy of Undetermined Significance and Multiple Myeloma between Blacks and Whites. Leukemia 2012, 26, 609–614. [Google Scholar] [CrossRef]
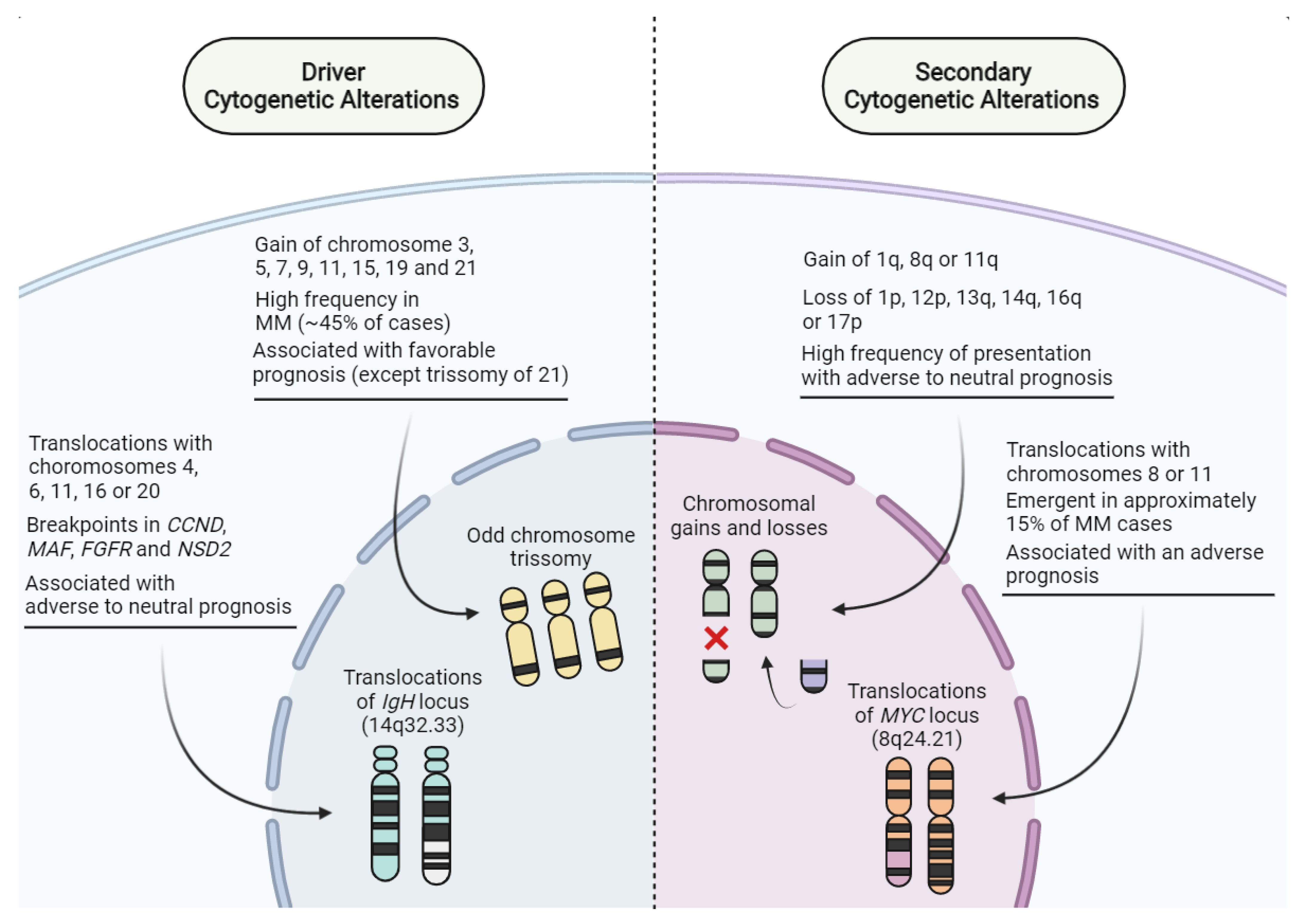
- Lakshman, A.; Paul, S.; Rajkumar, S.V.; Ketterling, R.P.; Greipp, P.T.; Dispenzieri, A.; Gertz, M.A.; Buadi, F.K.; Lacy, M.Q.; Dingli, D.; et al. Prognostic Significance of Interphase FISH in Monoclonal Gammopathy of Undetermined Significance. Leukemia 2018, 32, 1811–1815. [Google Scholar] [CrossRef]
- Neben, K.; Jauch, A.; Hielscher, T.; Hillengass, J.; Lehners, N.; Seckinger, A.; Granzow, M.; Raab, M.S.; Ho, A.D.; Goldschmidt, H.; et al. Progression in Smoldering Myeloma Is Independently Determined by the Chromosomal Abnormalities Del(17p), t(4;14), Gain 1q, Hyperdiploidy, and Tumor Load. J. Clin. Oncol. 2013, 31, 4325–4332. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Gupta, V.; Fonseca, R.; Dispenzieri, A.; Gonsalves, W.I.; Larson, D.; Ketterling, R.P.; Lust, J.A.; Kyle, R.A.; Kumar, S.K. Impact of Primary Molecular Cytogenetic Abnormalities and Risk of Progression in Smoldering Multiple Myeloma. Leukemia 2013, 27, 1738–1744. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; Attal, M.; Moreau, P.; Charbonnel, C.; Garban, F.; Hulin, C.; Leyvraz, S.; Michallet, M.; Yakoub-Agha, I.; Garderet, L.; et al. Genetic Abnormalities and Survival in Multiple Myeloma: The Experience of the Intergroupe Francophone Du Myélome. Blood 2007, 109, 3489–3495. [Google Scholar] [CrossRef]
- Chretien, M.-L.; Corre, J.; Lauwers-Cances, V.; Magrangeas, F.; Cleynen, A.; Yon, E.; Hulin, C.; Leleu, X.; Orsini-Piocelle, F.; Blade, J.-S.; et al. Understanding the Role of Hyperdiploidy in Myeloma Prognosis: Which Trisomies Really Matter? Blood 2015, 126, 2713–2719. [Google Scholar] [CrossRef] [Green Version]
- Willenbacher, W.; Seeber, A.; Steiner, N.; Willenbacher, E.; Gatalica, Z.; Swensen, J.; Kimbrough, J.; Vranic, S. Towards Molecular Profiling in Multiple Myeloma: A Literature Review and Early Indications of Its Efficacy for Informing Treatment Strategies. Int. J. Mol. Sci. 2018, 19, 2087. [Google Scholar] [CrossRef] [PubMed]
- Ziogas, D.C.; Dimopoulos, M.A.; Kastritis, E. Prognostic Factors for Multiple Myeloma in the Era of Novel Therapies. Expert Rev. Hematol. 2018, 11, 863–879. [Google Scholar] [CrossRef] [PubMed]
- Pawlyn, C.; Davies, F.E. Toward Personalized Treatment in Multiple Myeloma Based on Molecular Characteristics. Blood 2019, 133, 660–675. [Google Scholar] [CrossRef]
- Hanamura, I. [Advances in Multiple Myeloma Molecular Biology Research]. [Rinsho Ketsueki] Jpn. J. Clin. Hematol. 2019, 60, 1236–1242. [Google Scholar]
- Rajkumar, S.V. Multiple Myeloma: 2020 Update on Diagnosis, Risk-stratification and Management. Am. J. Hematol. 2020, 95, 548–567. [Google Scholar] [CrossRef] [PubMed]
- Usmani, S.Z.; Hoering, A.; Cavo, M.; Miguel, J.S.; Goldschimdt, H.; Hajek, R.; Turesson, I.; Lahuerta, J.J.; Attal, M.; Barlogie, B.; et al. Clinical Predictors of Long-Term Survival in Newly Diagnosed Transplant Eligible Multiple Myeloma—An IMWG Research Project. Blood Cancer J. 2018, 8, 123. [Google Scholar] [CrossRef] [PubMed]
- Child, J.A.; Morgan, G.J.; Davies, F.E.; Owen, R.G.; Bell, S.E.; Hawkins, K.; Brown, J.; Drayson, M.T.; Selby, P.J. High-Dose Chemotherapy with Hematopoietic Stem-Cell Rescue for Multiple Myeloma. N. Engl. J. Med. 2003, 348, 1875–1883. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Multiple Myeloma: Every Year a New Standard? Hematol. Oncol. 2019, 37, 62–65. [Google Scholar] [CrossRef]
- Gonsalves, W.I.; Buadi, F.K.; Ailawadhi, S.; Bergsagel, P.L.; Chanan Khan, A.A.; Dingli, D.; Dispenzieri, A.; Fonseca, R.; Hayman, S.R.; Kapoor, P.; et al. Utilization of Hematopoietic Stem Cell Transplantation for the Treatment of Multiple Myeloma: A Mayo Stratification of Myeloma and Risk-Adapted Therapy (MSMART) Consensus Statement. Bone Marrow Transplant. 2019, 54, 353–367. [Google Scholar] [CrossRef]
- Saad, A.; Mahindra, A.; Zhang, M.-J.; Zhong, X.; Costa, L.J.; Dispenzieri, A.; Drobyski, W.R.; Freytes, C.O.; Gale, R.P.; Gasparetto, C.J.; et al. Hematopoietic Cell Transplant Comorbidity Index Is Predictive of Survival after Autologous Hematopoietic Cell Transplantation in Multiple Myeloma. Biol. Blood Marrow Transplant. 2014, 20, 402–408.e1. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Sonneveld, P. Front-Line Treatment in Younger Patients with Multiple Myeloma. Semin. Hematol. 2009, 46, 118–126. [Google Scholar] [CrossRef] [Green Version]
- Hus, I.; Mańko, J.; Jawniak, D.; Jurczyszyn, A.; Charliński, G.; Poniewierska-Jasak, K.; Usnarska-Zubkiewicz, L.; Sawicki, M.; Druzd-Sitek, A.; Świderska, A.; et al. High Efficacy and Safety of VTD as an Induction Protocol in Patients with Newly Diagnosed Multiple Myeloma Eligible for High Dose Therapy and Autologous Stem Cell Transplantation: A Report of the Polish Myeloma Study Group. Oncol. Lett. 2019, 18, 5811–5820. [Google Scholar] [CrossRef]
- Attal, M.; Lauwers-Cances, V.; Hulin, C.; Leleu, X.; Caillot, D.; Escoffre, M.; Arnulf, B.; Macro, M.; Belhadj, K.; Garderet, L.; et al. Lenalidomide, Bortezomib, and Dexamethasone with Transplantation for Myeloma. N. Engl. J. Med. 2017, 376, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- al Hamed, R.; Bazarbachi, A.H.; Malard, F.; Harousseau, J.-L.; Mohty, M. Current Status of Autologous Stem Cell Transplantation for Multiple Myeloma. Blood Cancer J. 2019, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Cavo, M.; Tacchetti, P.; Patriarca, F.; Petrucci, M.T.; Pantani, L.; Galli, M.; di Raimondo, F.; Crippa, C.; Zamagni, E.; Palumbo, A.; et al. Bortezomib with Thalidomide plus Dexamethasone Compared with Thalidomide plus Dexamethasone as Induction Therapy before, and Consolidation Therapy after, Double Autologous Stem-Cell Transplantation in Newly Diagnosed Multiple Myeloma: A Randomised Phase 3 Study. Lancet 2010, 376, 2075–2085. [Google Scholar] [CrossRef]
- Ghandili, S.; Weisel, K.C.; Bokemeyer, C.; Leypoldt, L.B. Current Treatment Approaches to Newly Diagnosed Multiple Myeloma. Oncol. Res. Treat. 2021, 44, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Attal, M.; Harousseau, J.-L.; Stoppa, A.-M.; Sotto, J.-J.; Fuzibet, J.-G.; Rossi, J.-F.; Casassus, P.; Maisonneuve, H.; Facon, T.; Ifrah, N.; et al. A Prospective, Randomized Trial of Autologous Bone Marrow Transplantation and Chemotherapy in Multiple Myeloma. N. Engl. J. Med. 1996, 335, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Ito, S. Proteasome Inhibitors for the Treatment of Multiple Myeloma. Cancers 2020, 12, 265. [Google Scholar] [CrossRef]
- Hideshima, T.; Mitsiades, C.; Akiyama, M.; Hayashi, T.; Chauhan, D.; Richardson, P.; Schlossman, R.; Podar, K.; Munshi, N.C.; Mitsiades, N.; et al. Molecular Mechanisms Mediating Antimyeloma Activity of Proteasome Inhibitor PS-341. Blood 2003, 101, 1530–1534. [Google Scholar] [CrossRef]
- Besse, A.; Besse, L.; Kraus, M.; Mendez-Lopez, M.; Bader, J.; Xin, B.T.; de Bruin, G.; Maurits, E.; Overkleeft, H.S.; Driessen, C. Proteasome Inhibition in Multiple Myeloma: Head-to-Head Comparison of Currently Available Proteasome Inhibitors. Cell Chem. Biol. 2019, 26, 340–351.e3. [Google Scholar] [CrossRef]
- Niewerth, D.; Jansen, G.; Assaraf, Y.G.; Zweegman, S.; Kaspers, G.J.L.; Cloos, J. Molecular Basis of Resistance to Proteasome Inhibitors in Hematological Malignancies. Drug Resist. Updates 2015, 18, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Adams, J. The Proteasome: A Suitable Antineoplastic Target. Nat. Rev. Cancer 2004, 4, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Nunes, A.T.; Annunziata, C.M. Proteasome Inhibitors: Structure and Function. Semin. Oncol. 2017, 44, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauhan, D.; Anderson, K.C.; Richardson, P.G. The Proteasome and Proteasome Inhibitors in Multiple Myeloma. Cancer Metastasis Rev. 2017, 36, 561–584. [Google Scholar] [CrossRef]
- Groen, K.; van de Donk, N.W.C.J.; Stege, C.A.M.; Zweegman, S.; Nijhof, I.S. Carfilzomib for Relapsed and Refractory Multiple Myeloma. Cancer Manag. Res. 2019, 11, 2663–2675. [Google Scholar] [CrossRef]
- Tan, C.R.C.; Abdul-Majeed, S.; Cael, B.; Barta, S.K. Clinical Pharmacokinetics and Pharmacodynamics of Bortezomib. Clin. Pharmacokinet. 2018, 58, 157–168. [Google Scholar] [CrossRef]
- Zinatizadeh, M.R.; Schock, B.; Chalbatani, G.M.; Zarandi, P.K.; Jalali, S.A.; Miri, S.R. The Nuclear Factor Kappa B (NF-KB) Signaling in Cancer Development and Immune Diseases. Genes Dis. 2021, 8, 287–297. [Google Scholar] [CrossRef]
- Chauhan, D.; Bianchi, G.; Anderson, K.C. Targeting the UPS as Therapy in Multiple Myeloma. BMC Biochem. 2008, 9, S1. [Google Scholar] [CrossRef]
- Richardson, P.G.; Sonneveld, P.; Schuster, M.W.; Irwin, D.; Stadtmauer, E.A.; Facon, T.; Harousseau, J.-L.; Ben-Yehuda, D.; Lonial, S.; Goldschmidt, H.; et al. Bortezomib or High-Dose Dexamethasone for Relapsed Multiple Myeloma. N. Engl. J. Med. 2005, 352, 2487–2498. [Google Scholar] [CrossRef]
- Richardson, P.G.; Zweegman, S.; O’Donnell, E.K.; Laubach, J.P.; Raje, N.; Voorhees, P.; Ferrari, R.H.; Skacel, T.; Kumar, S.K.; Lonial, S. Ixazomib for the Treatment of Multiple Myeloma. Expert Opin. Pharmacother. 2018, 19, 1949–1968. [Google Scholar] [CrossRef]
- Touzeau, C.; Moreau, P. Ixazomib in the Management of Relapsed Multiple Myeloma. Future Oncol. 2018, 14, 2013–2020. [Google Scholar] [CrossRef]
- Zanwar, S.; Abeykoon, J.P.; Kapoor, P. Ixazomib: A Novel Drug for Multiple Myeloma. Expert Rev. Hematol. 2018, 11, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Girona, A.; Heintel, D.; Zhang, L.-H.; Mendy, D.; Gaidarova, S.; Brady, H.; Bartlett, J.B.; Schafer, P.H.; Schreder, M.; Bolomsky, A.; et al. Lenalidomide Downregulates the Cell Survival Factor, Interferon Regulatory Factor-4, Providing a Potential Mechanistic Link for Predicting Response. Br. J. Haematol. 2011, 154, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Høyer, S.; Karsan, A. Mechanisms of Lenalidomide Sensitivity and Resistance. Exp. Hematol. 2020, 91, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Agnarelli, A.; Chevassut, T.; Mancini, E.J. IRF4 in Multiple Myeloma—Biology, Disease and Therapeutic Target. Leuk. Res. 2018, 72, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; O’Grady, S.; Tang, M.; Crown, J. MYC as a Target for Cancer Treatment. Cancer Treat. Rev. 2021, 94, 102154. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, E.P.; Sarno, E.N.; Galilly, R.; Cohn, Z.A.; Kaplan, G. Thalidomide Selectively Inhibits Tumor Necrosis Factor Alpha Production by Stimulated Human Monocytes. J. Exp. Med. 1991, 173, 699–703. [Google Scholar] [CrossRef]
- Davies, F.E.; Raje, N.; Hideshima, T.; Lentzsch, S.; Young, G.; Tai, Y.-T.; Lin, B.; Podar, K.; Gupta, D.; Chauhan, D.; et al. Thalidomide and Immunomodulatory Derivatives Augment Natural Killer Cell Cytotoxicity in Multiple Myeloma. Blood 2001, 98, 210–216. [Google Scholar] [CrossRef]
- Abe, Y.; Ishida, T. Immunomodulatory Drugs in the Treatment of Multiple Myeloma. Jpn. J. Clin. Oncol. 2019, 49, 695–702. [Google Scholar] [CrossRef]
- Goldsmith, S.R.; Foley, N.; Schroeder, M.A. Daratumumab for the Treatment of Multiple Myeloma. Drugs Today 2021, 57, 591. [Google Scholar] [CrossRef]
- Hosoya, H.; Sidana, S. Antibody-Based Treatment Approaches in Multiple Myeloma. Curr. Hematol. Malign. Rep. 2021, 16, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Nooka, A.K.; Kaufman, J.L.; Hofmeister, C.C.; Joseph, N.S.; Heffner, T.L.; Gupta, V.A.; Sullivan, H.C.; Neish, A.S.; Dhodapkar, M.V.; Lonial, S. Daratumumab in Multiple Myeloma. Cancer 2019, 125, 2364–2382. [Google Scholar] [CrossRef] [PubMed]
- Kastritis, E.; Palladini, G.; Minnema, M.C.; Wechalekar, A.D.; Jaccard, A.; Lee, H.C.; Sanchorawala, V.; Gibbs, S.; Mollee, P.; Venner, C.P.; et al. Daratumumab-Based Treatment for Immunoglobulin Light-Chain Amyloidosis. N. Engl. J. Med. 2021, 385, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Petrucci, M.T.; Vozella, F. The Anti-CD38 Antibody Therapy in Multiple Myeloma. Cells 2019, 8, 1629. [Google Scholar] [CrossRef]
- Vozella, F.; Fazio, F.; Lapietra, G.; Petrucci, M.T.; Martinelli, G.; Cerchione, C. Monoclonal Antibodies in Multiple Myeloma. Panminerva Med. 2021, 63, 21–27. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, M.; Boccadoro, M.; Smith, E.L. Novel Immunotherapies for Multiple Myeloma. Curr. Hematol. Malig. Rep. 2017, 12, 344–357. [Google Scholar] [CrossRef]
- Minnie, S.A.; Hill, G.R. Immunotherapy of Multiple Myeloma. J. Clin. Investig. 2020, 130, 1565–1575. [Google Scholar] [CrossRef]
- Tan, C.R.; Shah, U.A. Targeting BCMA in Multiple Myeloma. Curr. Hematol. Malig. Rep. 2021, 16, 367–383. [Google Scholar] [CrossRef]
- Mateos, M.-V.; Masszi, T.; Grzasko, N.; Hansson, M.; Sandhu, I.; Pour, L.; Viterbo, L.; Jackson, S.R.; Stoppa, A.-M.; Gimsing, P.; et al. Impact of Prior Therapy on the Efficacy and Safety of Oral Ixazomib-Lenalidomide-Dexamethasone vs. Placebo-Lenalidomide-Dexamethasone in Patients with Relapsed/Refractory Multiple Myeloma in TOURMALINE-MM1. Haematologica 2017, 102, 1767–1775. [Google Scholar] [CrossRef]
- Venezian Povoa, L.; Ribeiro, C.H.C.; Silva, I.T. da Machine Learning Predicts Treatment Sensitivity in Multiple Myeloma Based on Molecular and Clinical Information Coupled with Drug Response. PLoS ONE 2021, 16, e0254596. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteban-Villarrubia, J.; Soto-Castillo, J.J.; Pozas, J.; Román-Gil, M.S.; Orejana-Martín, I.; Torres-Jiménez, J.; Carrato, A.; Alonso-Gordoa, T.; Molina-Cerrillo, J. Tyrosine Kinase Receptors in Oncology. Int. J. Mol. Sci. 2020, 21, 8529. [Google Scholar] [CrossRef] [PubMed]
- Tibes, R.; Trent, J.; Kurzrock, R. Tyrosine Kinase Inhibitors and the Dawn of Molecular Cancer Therapeutics. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 357–384. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.S.; van Etten, R.A. Tyrosine Kinases as Targets for Cancer Therapy. N. Engl. J. Med. 2005, 353, 172–187. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Cole, P.A. Catalytic Mechanisms and Regulation of Protein Kinases. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2014; Volume 548. [Google Scholar]
- Regad, T. Targeting RTK Signaling Pathways in Cancer. Cancers 2015, 7, 1758–1784. [Google Scholar] [CrossRef]
- Lind, J.; Czernilofsky, F.; Vallet, S.; Podar, K. Emerging Protein Kinase Inhibitors for the Treatment of Multiple Myeloma. Expert Opin. Emerg. Drugs 2019, 24, 133–152. [Google Scholar] [CrossRef]
- Knösel, T.; Kampmann, E.; Kirchner, T.; Altendorf-Hofmann, A. Tyrosinkinasen in Weichgewebstumoren. Pathologe 2014, 35, 198–201. [Google Scholar] [CrossRef]
- Drake, J.M.; Lee, J.K.; Witte, O.N. Clinical Targeting of Mutated and Wild-Type Protein Tyrosine Kinases in Cancer. Mol. Cell. Biol. 2014, 34, 1722–1732. [Google Scholar] [CrossRef]
- Jiao, Q.; Bi, L.; Ren, Y.; Song, S.; Wang, Q.; Wang, Y.S. Advances in Studies of Tyrosine Kinase Inhibitors and Their Acquired Resistance. Mol. Cancer 2018, 17, 1–12. [Google Scholar] [CrossRef]
- Chari, A.; Larson, S.; Holkova, B.; Cornell, R.F.; Gasparetto, C.; Karanes, C.; Matous, J.V.; Niesvizky, R.; Valent, J.; Lunning, M.; et al. Phase 1 Trial of Ibrutinib and Carfilzomib Combination Therapy for Relapsed or Relapsed and Refractory Multiple Myeloma. Leuk. Lymphoma 2018, 59, 2588–2594. [Google Scholar] [CrossRef]
- Chari, A.; Cornell, R.F.; Gasparetto, C.; Karanes, C.; Matous, J.V.; Niesvizky, R.; Lunning, M.; Usmani, S.Z.; Anderson, L.D.; Chhabra, S.; et al. Final Analysis of a Phase 1/2b Study of Ibrutinib Combined with Carfilzomib/Dexamethasone in Patients with Relapsed/Refractory Multiple Myeloma. Hematol. Oncol. 2020, 38, 353–362. [Google Scholar] [CrossRef]
- Hajek, R.; Pour, L.; Ozcan, M.; Martin Sánchez, J.; García Sanz, R.; Anagnostopoulos, A.; Oriol, A.; Cascavilla, N.; Terjung, A.; Lee, Y.; et al. A Phase 2 Study of Ibrutinib in Combination with Bortezomib and Dexamethasone in Patients with Relapsed/Refractory Multiple Myeloma. Eur. J. Haematol. 2020, 104, 435–442. [Google Scholar] [CrossRef]
- Richardson, P.G.; Bensinger, W.I.; Huff, C.A.; Costello, C.L.; Lendvai, N.; Berdeja, J.G.; Anderson, L.D.; Siegel, D.S.; Lebovic, D.; Jagannath, S.; et al. Ibrutinib Alone or with Dexamethasone for Relapsed or Relapsed and Refractory Multiple Myeloma: Phase 2 Trial Results. Br. J. Haematol. 2018, 180, 821–830. [Google Scholar] [CrossRef]
- Berenson, J.R.; To, J.; Spektor, T.M.; Martinez, D.; Turner, C.; Sanchez, A.; Ghermezi, M.; Eades, B.M.; Swift, R.A.; Schwartz, G.; et al. A Phase I Study of Ruxolitinib, Lenalidomide, and Steroids for Patients with Relapsed/Refractory Multiple Myeloma. Clin. Cancer Res. 2020, 26, 2346–2353. [Google Scholar] [CrossRef] [PubMed]
- Lendvai, N.; Yee, A.J.; Tsakos, I.; Alexander, A.; Devlin, S.M.; Hassoun, H.; Korde, N.; Lesokhin, A.M.; Landau, H.; Mailankody, S.; et al. Phase IB Study of Cabozantinib in Patients with Relapsed and/or Refractory Multiple Myeloma. Blood 2016, 127, 2355–2356. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, A.M.; Cook, R.J.; Bordoni, R.; Berenson, J.R.; Edenfield, W.J.; Mohan, S.; Zhou, G.; Asatiani, E.; Srinivas, N.; Savona, M.R. A Phase 1/2 Study of the Oral Janus Kinase 1 Inhibitors INCB052793 and Itacitinib Alone or in Combination with Standard Therapies for Advanced Hematologic Malignancies. Clin. Lymphoma Myeloma Leuk. 2022, 22, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Baljevic, M.; Zaman, S.; Baladandayuthapani, V.; Lin, Y.H.; de Partovi, C.M.; Berkova, Z.; Amini, B.; Thomas, S.K.; Shah, J.J.; Weber, D.M.; et al. Phase II Study of the C-MET Inhibitor Tivantinib (ARQ 197) in Patients with Relapsed or Relapsed/Refractory Multiple Myeloma. Ann. Hematol. 2017, 96, 977–985. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Patnaik, A.; Papadopoulos, K.P.; Rasco, D.W.; Becerra, C.R.; Allred, A.J.; Orford, K.; Aktan, G.; Ferron-Brady, G.; Ibrahim, N.; et al. Phase I Study of the MEK Inhibitor Trametinib in Combination with the AKT Inhibitor Afuresertib in Patients with Solid Tumors and Multiple Myeloma. Cancer Chemother. Pharmacol. 2015, 75, 183–189. [Google Scholar] [CrossRef]
- Yordanova, A.; Hose, D.; Neben, K.; Witzens-Harig, M.; Gütgemann, I.; Raab, M.S.; Moehler, T.; Goldschmidt, H.; Schmidt-Wolf, I.G. Sorafenib in Patients with Refractory or Recurrent Multiple Myeloma. Hematol. Oncol. 2013, 31, 197–200. [Google Scholar] [CrossRef]
- Scheid, C.; Reece, D.; Beksac, M.; Spencer, A.; Callander, N.; Sonneveld, P.; Kalimi, G.; Cai, C.; Shi, M.; Scott, J.W.; et al. Phase 2 Study of Dovitinib in Patients with Relapsed or Refractory Multiple Myeloma with or without t(4;14) Translocation. Eur. J. Haematol. 2015, 95, 316–324. [Google Scholar] [CrossRef]
- Estupiñán, H.Y.; Bouderlique, T.; He, C.; Berglöf, A.; Gupta, D.; Saher, O.; Daza Cruz, M.Á.; Peña-Perez, L.; Yu, L.; Zain, R.; et al. Novel Mouse Model Resistant to Irreversible BTK Inhibitors: A Tool Identifying New Therapeutic Targets and Side Effects. Blood Adv. 2020, 4, 2439–2450. [Google Scholar] [CrossRef] [PubMed]
- Ahn, I.E.; Brown, J.R. Targeting Bruton’s Tyrosine Kinase in CLL. Front. Immunol. 2021, 12, 687458. [Google Scholar] [CrossRef] [PubMed]
- Munir, T.; Brown, J.R.; O’Brien, S.; Barrientos, J.C.; Barr, P.M.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; et al. Final Analysis from RESONATE: Up to Six Years of Follow-up on Ibrutinib in Patients with Previously Treated Chronic Lymphocytic Leukemia or Small Lymphocytic Lymphoma. Am. J. Hematol. 2019, 94, 1353–1363. [Google Scholar] [CrossRef]
- Lipsky, A.; Lamanna, N. Managing Toxicities of Bruton Tyrosine Kinase Inhibitors. Hematology 2020, 2020, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Scheerens, H.; Li, S.-J.; Schultz, B.E.; Sprengeler, P.A.; Burrill, L.C.; Mendonca, R.V.; Sweeney, M.D.; Scott, K.C.K.; Grothaus, P.G.; et al. Discovery of Selective Irreversible Inhibitors for Bruton’s Tyrosine Kinase. ChemMedChem 2007, 2, 58–61. [Google Scholar] [CrossRef]
- Bender, A.T.; Gardberg, A.; Pereira, A.; Johnson, T.; Wu, Y.; Grenningloh, R.; Head, J.; Morandi, F.; Haselmayer, P.; Liu-Bujalski, L. Ability of Bruton’s Tyrosine Kinase Inhibitors to Sequester Y551 and Prevent Phosphorylation Determines Potency for Inhibition of Fc Receptor but Not B-Cell Receptor Signaling. Mol. Pharmacol. 2017, 91, 208–219. [Google Scholar] [CrossRef]
- Pal Singh, S.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s Tyrosine Kinase in B Cells and Malignancies. Mol. Cancer 2018, 17, 57, Erratum in Mol. Cancer 2019, 18, 79. [Google Scholar] [CrossRef]
- Burger, J.A. Bruton Tyrosine Kinase Inhibitors. Cancer J. 2019, 25, 386–393. [Google Scholar] [CrossRef]
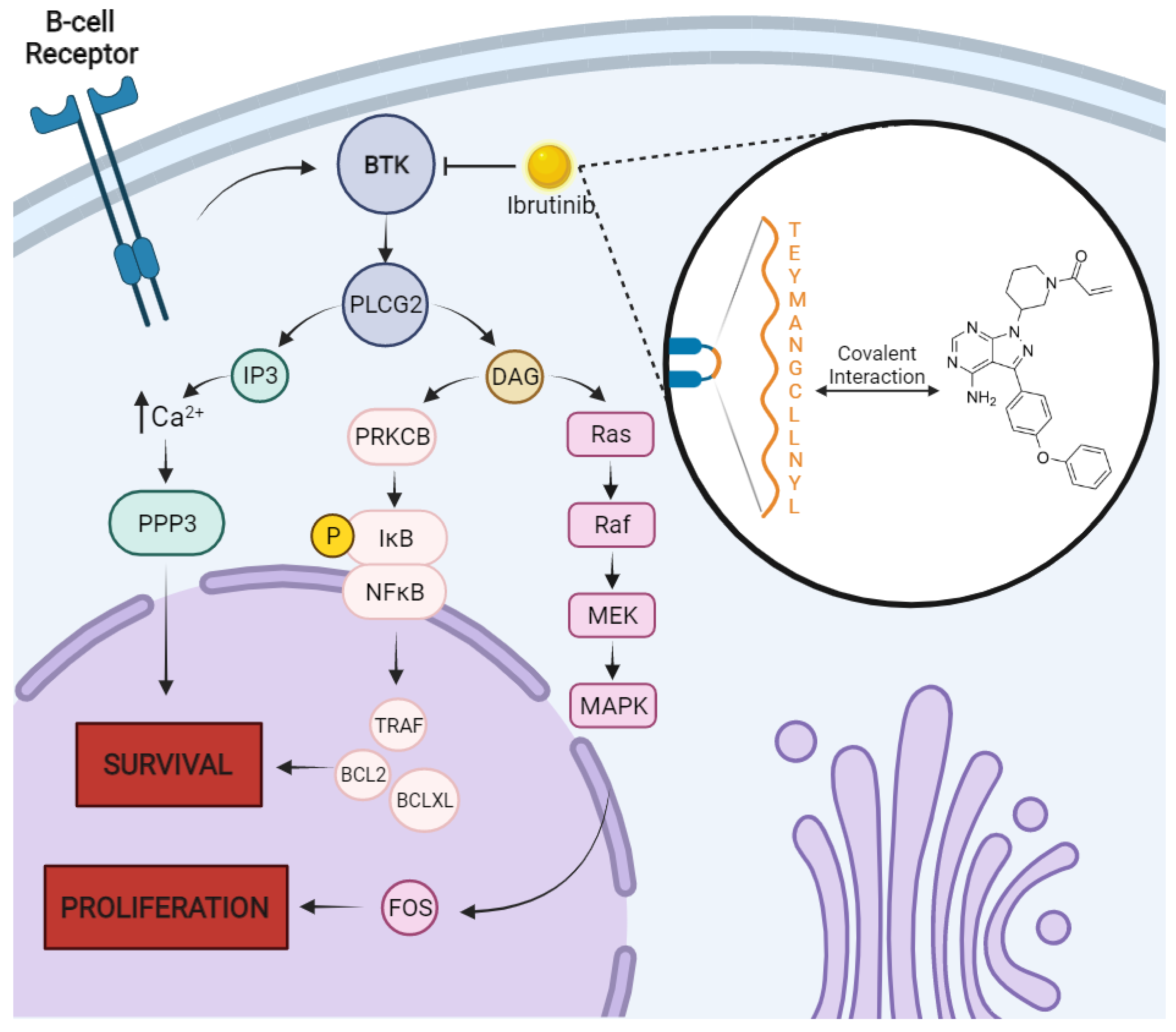
- von Suskil, M.; Sultana, K.N.; Elbezanti, W.O.; Al-Odat, O.S.; Chitren, R.; Tiwari, A.K.; Challagundla, K.B.; Srivastava, S.K.; Jonnalagadda, S.C.; Budak-Alpdogan, T.; et al. Bruton’s Tyrosine Kinase Targeting in Multiple Myeloma. Int. J. Mol. Sci. 2021, 22, 5707. [Google Scholar] [CrossRef]
- Lampson, B.L.; Yu, L.; Glynn, R.J.; Barrientos, J.C.; Jacobsen, E.D.; Banerji, V.; Jones, J.A.; Walewska, R.; Savage, K.J.; Michaud, G.F.; et al. Ventricular Arrhythmias and Sudden Death in Patients Taking Ibrutinib. Blood 2017, 129, 2581–2584. [Google Scholar] [CrossRef]
- Edwards, C.M. BTK Inhibition in Myeloma: Targeting the Seed and the Soil. Blood 2012, 120, 1757–1759. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.T.; Chang, B.Y.; Kong, S.Y.; Fulciniti, M.; Yang, G.; Calle, Y.; Hu, Y.; Lin, J.; Zhao, J.J.; Cagnetta, A.; et al. Bruton Tyrosine Kinase Inhibition Is a Novel Therapeutic Strategy Targeting Tumor in the Bone Marrow Microenvironment in Multiple Myeloma. Blood 2012, 120, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.T.; Anderson, K.C. Bruton’s Tyrosine Kinase: Oncotarget in Myeloma. Oncotarget 2012, 3, 913–914. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Dong, Y.; Jiang, Q.L.; Zhang, B.; Hu, A.M. Bruton’s Tyrosine Kinase: Potential Target in Human Multiple Myeloma. Leuk. Lymphoma 2014, 55, 177–181. [Google Scholar] [CrossRef]
- Campbell, R.; Chong, G.; Hawkes, E.A. Novel Indications for Bruton’s Tyrosine Kinase Inhibitors, beyond Hematological Malignancies. J. Clin. Med. 2018, 7, 62. [Google Scholar] [CrossRef]
- Mohamed, A.J.; Yu, L.; Bäckesjö, C.M.; Vargas, L.; Faryal, R.; Aints, A.; Christensson, B.; Berglöf, A.; Vihinen, M.; Nore, B.F.; et al. Bruton’s Tyrosine Kinase (Btk): Function, Regulation, and Transformation with Special Emphasis on the PH Domain. Immunol. Rev. 2009, 228, 58–73. [Google Scholar] [CrossRef]
- de Weerdt, I.; Koopmans, S.M.; Kater, A.P.; van Gelder, M. Incidence and Management of Toxicity Associated with Ibrutinib and Idelalisib: A Practical Approach. Haematologica 2017, 102, 1629–1639. [Google Scholar] [CrossRef]
- Tang, C.P.S.; McMullen, J.; Tam, C. Cardiac Side Effects of Bruton Tyrosine Kinase (BTK) Inhibitors. Leukemia and Lymphoma 2018, 59. [Google Scholar] [CrossRef]
- Ahn, I.E. Cardiovascular Adverse Events of Ibrutinib. Blood 2019, 134, 1881–1882. [Google Scholar] [CrossRef]
- Dickerson, T.; Wiczer, T.; Waller, A.; Philippon, J.; Porter, K.; Haddad, D.; Guha, A.; Rogers, K.A.; Bhat, S.; Byrd, J.C.; et al. Hypertension and Incident Cardiovascular Events Following Ibrutinib Initiation. Blood 2019, 134, 1919–1928. [Google Scholar] [CrossRef]
- Castillo, J.J.; Meid, K.; Gustine, J.N.; Leventoff, C.; White, T.; Flynn, C.A.; Sarosiek, S.; Demos, M.G.; Guerrera, M.L.; Kofides, A.; et al. Long-Term Follow-up of Ibrutinib Monotherapy in Treatment-Naive Patients with Waldenstrom Macroglobulinemia. Leukemia 2022, 36, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Srkalovic, G.; Hussein, M.A.; Hoering, A.; Zonder, J.A.; Popplewell, L.L.; Trivedi, H.; Mazzoni, S.; Sexton, R.; Orlowski, R.Z.; Barlogie, B. A Phase II Trial of BAY 43-9006 (Sorafenib) (NSC-724772) in Patients with Relapsing and Resistant Multiple Myeloma: SWOG S0434. Cancer Med. 2014, 3, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, R.Z.; Zaman, S.; Thomas, S.K.; Alexanian, R.; Shah, J.J.; Weber, D.M.; Wang, M.; Holloway, A.N.; Baladandayuthapani, V.; Lin, H.Y.; et al. Phase II Study of The C-MET Inhibitor ARQ 197 (Tivantinib) In Patients With Relapsed Or Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2013, 122, 1953. [Google Scholar] [CrossRef]
- Spencer, A.; Yoon, S.S.; Harrison, S.J.; Morris, S.R.; Smith, D.A.; Brigandi, R.A.; Gauvin, J.; Kumar, R.; Opalinska, J.B.; Chen, C. The Novel AKT Inhibitor Afuresertib Shows Favorable Safety, Pharmacokinetics, and Clinical Activity in Multiple Myeloma. Blood 2014, 124, 2190–2195. [Google Scholar] [CrossRef] [PubMed]
- Trudel, S.; Bahlis, N.J.; Venner, C.P.; Hay, A.E.; Kis, O.; Chow, S.; Li, Z.H.; Wei, E.N.; Wang, L.; Tran, C.; et al. Biomarker Driven Phase II Clinical Trial of Trametinib in Relapsed/Refractory Multiple Myeloma with Sequential Addition of the AKT Inhibitor, GSK2141795 at Time of Disease Progression to Overcome Treatment Failure: A Trial of the Princess Margaret Phase II Consortium. Blood 2016, 128, 4526. [Google Scholar] [CrossRef]
- Hoffner, B.; Benchich, K. Trametinib: A Targeted Therapy in Metastatic Melanoma. J. Adv. Pract. Oncol. 2018, 9, 741. [Google Scholar] [CrossRef]
- Blagden, S.P.; Hamilton, A.L.; Mileshkin, L.; Wong, S.; Michael, A.; Hall, M.; Goh, J.C.; Lisyanskaya, A.S.; DeSilvio, M.; Frangou, E.; et al. Phase IB Dose Escalation and Expansion Study of Akt Inhibitor Afuresertib with Carboplatin and Paclitaxel in Recurrent Platinum-Resistant Ovarian Cancer. Clin. Cancer Res. 2019, 25, 1472–1478. [Google Scholar] [CrossRef]
- Moreau, P.; Attal, M.; Garban, F.; Hulin, C.; Facon, T.; Marit, G.; Michallet, M.; Doyen, C.; Leyvraz, S.; Mohty, M.; et al. Heterogeneity of t(4;14) in Multiple Myeloma. Long-Term Follow-up of 100 Cases Treated with Tandem Transplantation in IFM99 Trials. Leukemia 2007, 21, 2020–2024. [Google Scholar] [CrossRef]
- Hart, K.C.; Robertson, S.C.; Donoghue, D.J. Identification of Tyrosine Residues in Constitutively Activated Fibroblast Growth Factor Receptor 3 Involved in Mitogenesis, Stat Activation, and Phosphatidylinositol 3-Kinase Activation. Mol. Biol. Cell 2001, 12, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Kanai, M.; Göke, M.; Tsunekawa, S.; Podolsky, D.K. Signal Transduction Pathway of Human Fibroblast Growth Factor Receptor 3. J. Biol. Chem. 1997, 272, 6621–6628. [Google Scholar] [CrossRef] [PubMed]
- Xin, X.; Abrams, T.J.; Hollenbach, P.W.; Rendahl, K.G.; Tang, Y.; Oei, Y.A.; Embry, M.G.; Swinarski, D.E.; Garrett, E.N.; Pryer, N.K.; et al. CHIR-258 Is Efficacious in a Newly Developed Fibroblast Growth Factor Receptor 3-Expressing Orthotopic Multiple Myeloma Model in Mice. Clin. Cancer Res. 2006, 12, 4908–4915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chell, V.; Balmanno, K.; Little, A.S.; Wilson, M.; Andrews, S.; Blockley, L.; Hampson, M.; Gavine, P.R.; Cook, S.J. Tumour Cell Responses to New Fibroblast Growth Factor Receptor Tyrosine Kinase Inhibitors and Identification of a Gatekeeper Mutation in FGFR3 as a Mechanism of Acquired Resistance. Oncogene 2013, 32, 3059–3070. [Google Scholar] [CrossRef] [PubMed]
- Chesi, M.; Brents, L.A.; Ely, S.A.; Bais, C.; Robbiani, D.F.; Mesri, E.A.; Kuehl, W.M.; Bergsagel, P.L. Activated Fibroblast Growth Factor Receptor 3 Is an Oncogene That Contributes to Tumor Progression in Multiple Myeloma. Blood 2001, 97, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Chesi, M.; Bergsagel, P.L. Advances in the Pathogenesis and Diagnosis of Multiple Myeloma. Int. J. Lab. Hematol. 2015, 37, 108–114. [Google Scholar] [CrossRef]
- Cardona-Benavides, I.J.; de Ramón, C.; Gutiérrez, N.C. Genetic Abnormalities in Multiple Myeloma: Prognostic and Therapeutic Implications. Cells 2021, 10, 336. [Google Scholar] [CrossRef]
- Richter, J.; Ramasamy, K.; Rasche, L.; Bladé, J.; Zweegman, S.; Davies, F.; Dimopoulos, M. Management of Patients with Difficult-to-Treat Multiple Myeloma. Futur. Oncol. 2021, 17, 2089–2105. [Google Scholar] [CrossRef]
- Palma, M.; Mulder, T.A.; Österborg, A. BTK Inhibitors in Chronic Lymphocytic Leukemia: Biological Activity and Immune Effects. Front. Immunol. 2021, 12, 686768. [Google Scholar] [CrossRef]
- Brullo, C.; Villa, C.; Tasso, B.; Russo, E.; Spallarossa, A. Btk Inhibitors: A Medicinal Chemistry and Drug Delivery Perspective. Int. J. Mol. Sci. 2021, 22, 7641. [Google Scholar] [CrossRef]
- Tasso, B.; Spallarossa, A.; Russo, E.; Brullo, C. The Development of Btk Inhibitors: A Five-Year Update. Molecules 2021, 26, 7411. [Google Scholar] [CrossRef]
- Lewis, K.L.; Cheah, C.Y. Non-Covalent Btk Inhibitors—The New Btkids on the Block for b-Cell Malignancies. J. Pers. Med. 2021, 11, 764. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Clinical Study Phase | Targeted Kinase | Kinase Inhibitor | Associated Treatment | Clinical Outcome | Adverse Events | References |
|---|---|---|---|---|---|---|
| I | BTK | Ibrutinib | Carfilzomib/Dex-methasone | ORR of 67% and a PFS of 7.2 months. | Hypertension, anemia, pneumonia, fatigue, diarrhea, and thrombocytopenia. | [91] |
| I/IIb | BTK | Ibrutinib | Carfilzomib/Dexamethasone | Acceptable safety profiles with PFS and ORR of 7.7 months and 71%, respectively. The average one-year OS rate was 77%. | Thrombocytopenia, anemia, diarrhea, fatigue, nausea, and hypertension. | [92] |
| II | BTK | Ibrutinib | Bortezomib/Dexamethasone | The drug combination initially increased the levels of infections and risk minimization measures were necessary. Clinical response was observed in 57% of patients with a duration of 9.5 months. | Thrombocytopenia, diarrhea, anemia, asthenia, and pneumonia. | [93] |
| II | BTK | Ibrutinib | Dexamethasone | The highest CBR was achieved in the combination of ibrutinib 840 mg with dexamethasone 40 mg. With CBR of 28%, ORR of 5%, sustained SD of 23%, and median PFS of 4.6 months. | Diarrhea, fatigue, nausea, anemia, and thrombocytopenia. | [94] |
| I | JAK | Ruxolitinib | Lenalidomide and metilprednisolone | The drug showed the ability to abrogate resistance to lenalidomide. Featuring CBR of 46% and ORR of 38%. | Anemia, thrombocytopenia and lymphopenia, sepsis, and pneumonia. | [95] |
| Ib | HGF and MET | Cabozantinib | NR | The drug alone has no significant activity in patients with refractory MM. The study was interrupted, and the rates were not calculated. | Grade 2 congestive heart failure and grade 3 APN. The remaining AEs were related to intestinal events. | [96] |
| Ib | JAK1 | INCB052793 | NR | No significant responses were observed. ORR of 24% and OS of 6.7 months. | Thrombocytopenia, anemia, fatigue, nausea, and vomiting. | [97] |
| II | c-MET | Tivantinib | NR | In isolation, the drug did not present a satisfactory response in refractory patients. SD of 36% and PD of 63% were obtained. | Neutropenia, hypertension, syncope, infection, and pain. | [98] |
| II | MEK and AKT | Trametinib and Afuresertib | NR | MTDs were found, being concentrations that are below the monotherapy concentration of each drug. However, these doses were considered subtherapeutic. | Diarrhea, acneiform dermatitis, maculopapular rash, fatigue, dry skin, nausea, dyspnea, and vomiting. | [99] |
| II | VEGF | Sorafenib | NR | Only one patient completed the 13 cycles of treatment and achieved PR, another 7 patients remained in PD. | Fatigue, nausea, hypertension, dermal toxicity, hematologic toxicity, and heart attack. | [100] |
| II | FGFR3 | Dovitinib | NR | The SD rate in t(4;14)-positive patients was higher, being 61.5%, compared with 34.6% rates for those translocation-negative | Diarrhea, nausea, vomiting, and fatigue. | [101] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barreto, I.V.; Machado, C.B.; Almeida, D.B.; Pessoa, F.M.C.d.P.; Gadelha, R.B.; Pantoja, L.d.C.; Oliveira, D.d.S.; Ribeiro, R.M.; Lopes, G.S.; de Moraes Filho, M.O.; et al. Kinase Inhibition in Multiple Myeloma: Current Scenario and Clinical Perspectives. Pharmaceutics 2022, 14, 1784. https://doi.org/10.3390/pharmaceutics14091784
Barreto IV, Machado CB, Almeida DB, Pessoa FMCdP, Gadelha RB, Pantoja LdC, Oliveira DdS, Ribeiro RM, Lopes GS, de Moraes Filho MO, et al. Kinase Inhibition in Multiple Myeloma: Current Scenario and Clinical Perspectives. Pharmaceutics. 2022; 14(9):1784. https://doi.org/10.3390/pharmaceutics14091784
Chicago/Turabian StyleBarreto, Igor Valentim, Caio Bezerra Machado, Davi Benevides Almeida, Flávia Melo Cunha de Pinho Pessoa, Renan Brito Gadelha, Laudreísa da Costa Pantoja, Deivide de Sousa Oliveira, Rodrigo Monteiro Ribeiro, Germison Silva Lopes, Manoel Odorico de Moraes Filho, and et al. 2022. "Kinase Inhibition in Multiple Myeloma: Current Scenario and Clinical Perspectives" Pharmaceutics 14, no. 9: 1784. https://doi.org/10.3390/pharmaceutics14091784