
Immunogenic Cell Death Enhances Immunotherapy of Diffuse Intrinsic Pontine Glioma: From Preclinical to Clinical Studies

Abstract
:1. Introduction
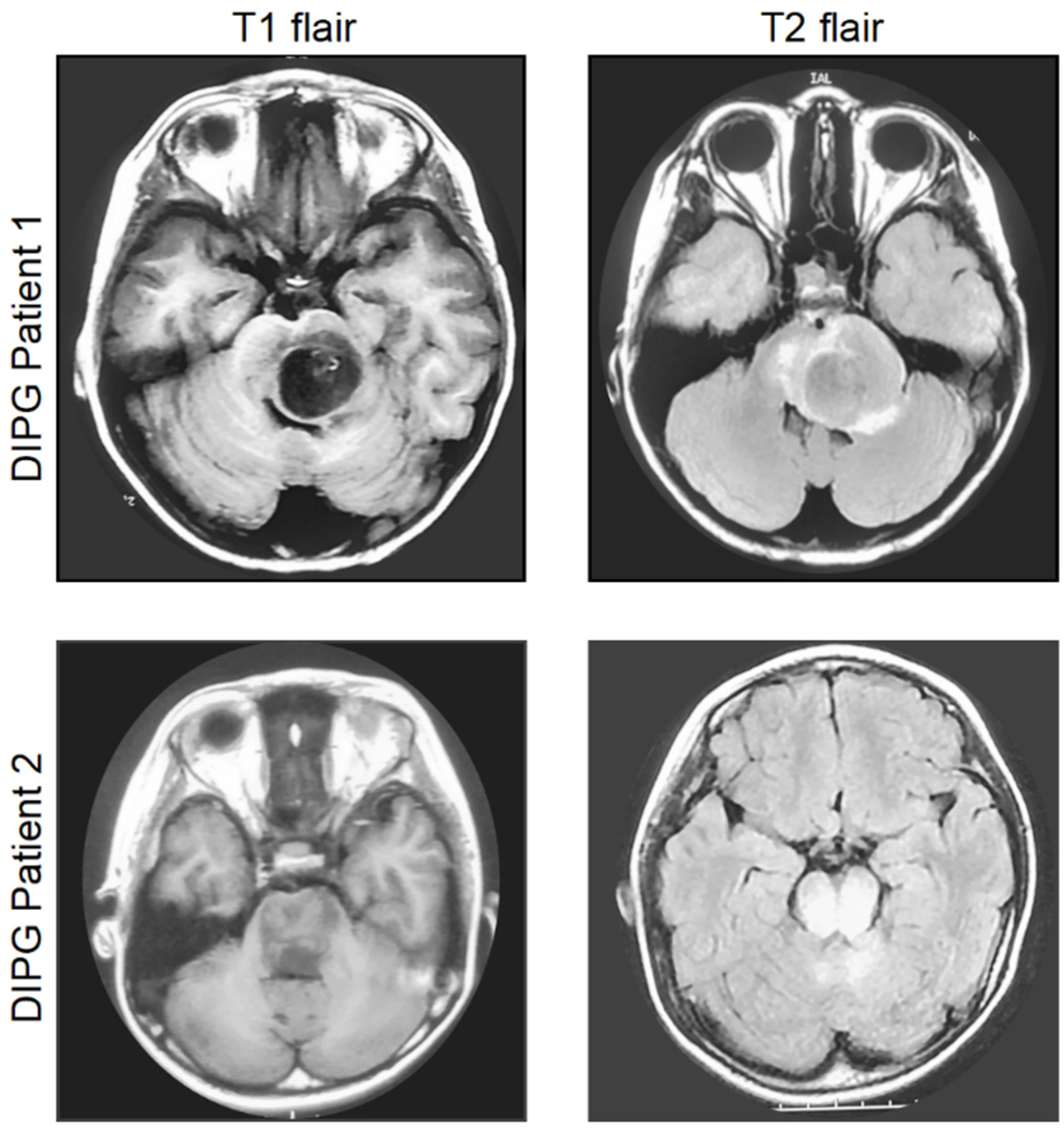
2. Diagnosis, Current Management, and Molecular Characteristics of DIPG
3. Immune Microenvironment of DIPG
3.1. Immunological Subgroups of DIPG
3.2. Tumor-Associated Macrophages (TAMs)
3.3. Tumor-Associated Lymphocytes
3.4. Natural Killer Cells
3.5. Tumor Immune-Related Molecules
4. Immunotherapy for DIPG
4.1. Checkpoint Inhibitors
4.2. CAR-T Cells
4.3. Vaccine Therapy
4.4. Oncolytic Viruses
5. Dilemma of Immunotherapy for DIPG
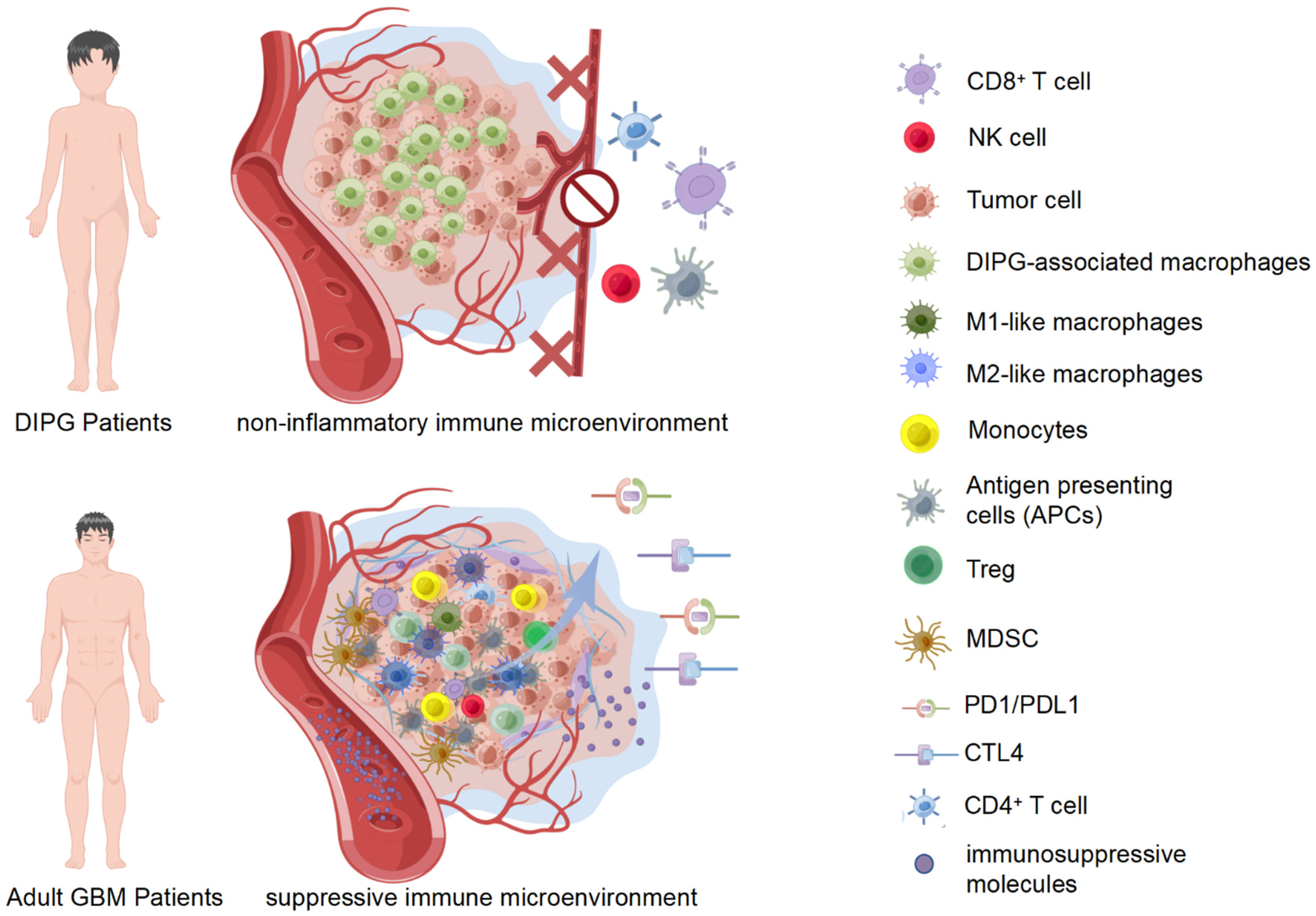
5.1. Non-Inflammatory Immune Microenvironment
5.2. Lower Mutational Load
5.3. Antigen Insufficiency, Attenuation, and Escape
5.4. Toxicity
5.5. Blood-Brain Barrier (BBB)
5.6. Cortisol Treatment
6. Immunogenic Cell Death (ICD)
6.1. Induction of ICD
6.2. Emission of DAMPs
6.2.1. HMGB1
6.2.2. CRT
6.2.3. ATP
7. The Diagnostic and Therapeutic Potential of Induced ICD in DIPG
7.1. Advances in DIPG Immunotherapy
7.2. Immunotherapeutic Modalities for Inducing ICD in DIPG
7.2.1. Induction of ICD in Adult GBM
7.2.2. Induction of ICD in DIPG
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grimm, S.A.; Chamberlain, M.C. Brainstem glioma: A review. Curr. Neurol. Neurosci. Rep. 2013, 13, 346. [Google Scholar] [CrossRef] [PubMed]
- Vitanza, N.A.; Monje, M. Diffuse Intrinsic Pontine Glioma: From Diagnosis to Next-Generation Clinical Trials. Curr. Treat Options Neurol. 2019, 21, 37. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Goumnerova, L.C.; Manley, P.; Chi, S.N.; Neuberg, D.; Puligandla, M.; Fangusaro, J.; Goldman, S.; Tomita, T.; Alden, T.; et al. Prospective feasibility and safety assessment of surgical biopsy for patients with newly diagnosed diffuse intrinsic pontine glioma. Neuro-Oncology 2018, 20, 1547–1555. [Google Scholar] [CrossRef]
- Cooney, T.; Lane, A.; Bartels, U.; Bouffet, E.; Goldman, S.; Leary, S.E.S.; Foreman, N.K.; Packer, R.J.; Broniscer, A.; Minturn, J.E.; et al. Contemporary survival endpoints: An International Diffuse Intrinsic Pontine Glioma Registry study. Neuro-Oncol. 2017, 19, 1279–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaye, E.C.; Baker, J.N.; Broniscer, A. Management of diffuse intrinsic pontine glioma in children: Current and future strategies for improving prognosis. CNS Oncol. 2014, 3, 421–431. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro-Oncology 2021, 23, iii1–iii105. [Google Scholar] [CrossRef]
- Yang, Y. Cancer immunotherapy: Harnessing the immune system to battle cancer. J. Clin. Investig. 2015, 125, 3335–3337. [Google Scholar] [CrossRef] [Green Version]
- Lieberman, N.A.P.; Vitanza, N.A.; Crane, C.A. Immunotherapy for brain tumors: Understanding early successes and limitations. Expert Rev. Neurother. 2018, 18, 251–259. [Google Scholar] [CrossRef]
- Lin, G.L.; Nagaraja, S.; Filbin, M.G.; Suva, M.L.; Vogel, H.; Monje, M. Non-inflammatory tumor microenvironment of diffuse intrinsic pontine glioma. Acta Neuropathol. Commun. 2018, 6, 51. [Google Scholar] [CrossRef]
- Lieberman, N.A.P.; DeGolier, K.; Kovar, H.M.; Davis, A.; Hoglund, V.; Stevens, J.; Winter, C.; Deutsch, G.; Furlan, S.N.; Vitanza, N.A.; et al. Characterization of the immune microenvironment of diffuse intrinsic pontine glioma: Implications for development of immunotherapy. Neuro-Oncology 2019, 21, 83–94. [Google Scholar] [CrossRef] [Green Version]
- Kline, C.; Felton, E.; Allen, I.E.; Tahir, P.; Mueller, S. Survival outcomes in pediatric recurrent high-grade glioma: Results of a 20-year systematic review and meta-analysis. J. Neurooncol. 2018, 137, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Cacciotti, C.; Choi, J.; Alexandrescu, S.; Zimmerman, M.A.; Cooney, T.M.; Chordas, C.; Clymer, J.; Chi, S.; Yeo, K.K. Immune checkpoint inhibition for pediatric patients with recurrent/refractory CNS tumors: A single institution experience. J. Neurooncol. 2020, 149, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Kline, C.; Liu, S.J.; Duriseti, S.; Banerjee, A.; Nicolaides, T.; Raber, S.; Gupta, N.; Haas-Kogan, D.; Braunstein, S.; Mueller, S. Reirradiation and PD-1 inhibition with nivolumab for the treatment of recurrent diffuse intrinsic pontine glioma: A single-institution experience. J. Neurooncol. 2018, 140, 629–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subashi, E.; Cordero, F.J.; Halvorson, K.G.; Qi, Y.; Nouls, J.C.; Becher, O.J.; Johnson, G.A. Tumor location, but not H3.3K27M, significantly influences the blood-brain-barrier permeability in a genetic mouse model of pediatric high-grade glioma. J. Neurooncol. 2016, 126, 243–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, L.B.; Salama, A.K.S. A review of cancer immunotherapy toxicity. CA Cancer J. Clin. 2020, 70, 86–104. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Lai, X.; Fu, S.; Ren, L.; Cai, H.; Zhang, H.; Gu, Z.; Ma, X.; Luo, K. Immunogenic Cell Death Activates the Tumor Immune Microenvironment to Boost the Immunotherapy Efficiency. Adv. Sci. 2022, 9, e2201734. [Google Scholar] [CrossRef]
- Mendez, F.; Kadiyala, P.; Nunez, F.J.; Carney, S.; Nunez, F.M.; Gauss, J.C.; Ravindran, R.; Pawar, S.; Edwards, M.; Garcia-Fabiani, M.B.; et al. Therapeutic Efficacy of Immune Stimulatory Thymidine Kinase and fms-like Tyrosine Kinase 3 Ligand (TK/Flt3L) Gene Therapy in a Mouse Model of High-Grade Brainstem Glioma. Clin. Cancer Res. 2020, 26, 4080–4092. [Google Scholar] [CrossRef] [Green Version]
- Fisher, P.G.; Breiter, S.N.; Carson, B.S.; Wharam, M.D.; Williams, J.A.; Weingart, J.D.; Foer, D.R.; Goldthwaite, P.T.; Tihan, T.; Burger, P.C. A clinicopathologic reappraisal of brain stem tumor classification. Identification of pilocystic astrocytoma and fibrillary astrocytoma as distinct entities. Cancer 2000, 89, 1569–1576. [Google Scholar] [CrossRef]
- Fischbein, N.J.; Prados, M.D.; Wara, W.; Russo, C.; Edwards, M.S.; Barkovich, A.J. Radiologic classification of brain stem tumors: Correlation of magnetic resonance imaging appearance with clinical outcome. Pediatr. Neurosurg. 1996, 24, 9–23. [Google Scholar] [CrossRef]
- Sharp, J.R.; Bouffet, E.; Stempak, D.; Gammon, J.; Stephens, D.; Johnston, D.L.; Eisenstat, D.; Hukin, J.; Samson, Y.; Bartels, U.; et al. A multi-centre Canadian pilot study of metronomic temozolomide combined with radiotherapy for newly diagnosed paediatric brainstem glioma. Eur. J. Cancer 2010, 46, 3271–3279. [Google Scholar] [CrossRef]
- Mandrell, B.N.; Baker, J.; Levine, D.; Gattuso, J.; West, N.; Sykes, A.; Gajjar, A.; Broniscer, A. Children with minimal chance for cure: Parent proxy of the child’s health-related quality of life and the effect on parental physical and mental health during treatment. J. Neurooncol. 2016, 129, 373–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hargrave, D.; Bartels, U.; Bouffet, E. Diffuse brainstem glioma in children: Critical review of clinical trials. Lancet Oncol. 2006, 7, 241–248. [Google Scholar] [CrossRef]
- La Madrid, A.M.; Hashizume, R.; Kieran, M.W. Future Clinical Trials in DIPG: Bringing Epigenetics to the Clinic. Front. Oncol. 2015, 5, 148. [Google Scholar] [CrossRef] [Green Version]
- Hashizume, R.; Gupta, N. Patient-derived Tumor Models for Diffuse Intrinsic Pontine Gliomas. Curr. Neuropharmacol. 2017, 15, 98–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misuraca, K.L.; Cordero, F.J.; Becher, O.J. Pre-Clinical Models of Diffuse Intrinsic Pontine Glioma. Front. Oncol. 2015, 5, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caretti, V.; Zondervan, I.; Meijer, D.H.; Idema, S.; Vos, W.; Hamans, B.; Bugiani, M.; Hulleman, E.; Wesseling, P.; Vandertop, W.P.; et al. Monitoring of tumor growth and post-irradiation recurrence in a diffuse intrinsic pontine glioma mouse model. Brain Pathol. 2011, 21, 441–451. [Google Scholar] [CrossRef]
- Xi, G.; Rajaram, V.; Mania-Farnell, B.; Mayanil, C.S.; Soares, M.B.; Tomita, T.; Goldman, S. Efficacy of vincristine administered via convection-enhanced delivery in a rodent brainstem tumor model documented by bioluminescence imaging. Childs Nerv. Syst. 2012, 28, 565–574. [Google Scholar] [CrossRef]
- Kholosy, W.M.; Derieppe, M.; van den Ham, F.; Ober, K.; Su, Y.; Custers, L.; Schild, L.; van Zogchel, L.M.J.; Wellens, L.M.; Ariese, H.R.; et al. Neuroblastoma and DIPG Organoid Coculture System for Personalized Assessment of Novel Anticancer Immunotherapies. J. Pers. Med. 2021, 11, 869. [Google Scholar] [CrossRef]
- Roujeau, T.; Machado, G.; Garnett, M.R.; Miquel, C.; Puget, S.; Geoerger, B.; Grill, J.; Boddaert, N.; Di Rocco, F.; Zerah, M.; et al. Stereotactic biopsy of diffuse pontine lesions in children. J. Neurosurg. 2007, 107, 1–4. [Google Scholar] [CrossRef]
- Akay, A.; Islekel, S. MRI-guided frame-based stereotactic brainstem biopsy procedure: A single-center experience. Neurocirugia 2019, 30, 167–172. [Google Scholar] [CrossRef]
- Pincus, D.W.; Richter, E.O.; Yachnis, A.T.; Bennett, J.; Bhatti, M.T.; Smith, A. Brainstem stereotactic biopsy sampling in children. J. Neurosurg. 2006, 104, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, T.Y.; Hou, Y.; Bartom, E.; Lu, X.; Shilatifard, A.; Yue, F.; Saratsis, A. Epigenomic landscape and 3D genome structure in pediatric high-grade glioma. Sci. Adv. 2021, 7, eabg4126. [Google Scholar] [CrossRef] [PubMed]
- Surowiec, R.K.; Ferris, S.F.; Apfelbaum, A.; Espinoza, C.; Mehta, R.K.; Monchamp, K.; Sirihorachai, V.R.; Bedi, K.; Ljungman, M.; Galban, S. Transcriptomic Analysis of Diffuse Intrinsic Pontine Glioma (DIPG) Identifies a Targetable ALDH-Positive Subset of Highly Tumorigenic Cancer Stem-like Cells. Mol. Cancer Res. 2021, 19, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Broniscer, A.; Baker, J.N.; Baker, S.J.; Chi, S.N.; Geyer, J.R.; Morris, E.B.; Gajjar, A. Prospective collection of tissue samples at autopsy in children with diffuse intrinsic pontine glioma. Cancer 2010, 116, 4632–4637. [Google Scholar] [CrossRef] [Green Version]
- Aziz-Bose, R.; Monje, M. Diffuse intrinsic pontine glioma: Molecular landscape and emerging therapeutic targets. Curr. Opin. Oncol. 2019, 31, 522–530. [Google Scholar] [CrossRef]
- Wu, G.; Diaz, A.K.; Paugh, B.S.; Rankin, S.L.; Ju, B.; Li, Y.; Zhu, X.; Qu, C.; Chen, X.; Zhang, J.; et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 2014, 46, 444–450. [Google Scholar] [CrossRef]
- El-Hashash, A.H.K. Histone H3K27M Mutation in Brain Tumors. Adv. Exp. Med. Biol. 2021, 1283, 43–52. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tonjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef] [Green Version]
- Castel, D.; Philippe, C.; Calmon, R.; Le Dret, L.; Truffaux, N.; Boddaert, N.; Pages, M.; Taylor, K.R.; Saulnier, P.; Lacroix, L.; et al. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol. 2015, 130, 815–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wesseling, P.; Capper, D. WHO 2016 Classification of gliomas. Neuropathol. Appl. Neurobiol. 2018, 44, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.R.; Mackay, A.; Truffaux, N.; Butterfield, Y.; Morozova, O.; Philippe, C.; Castel, D.; Grasso, C.S.; Vinci, M.; Carvalho, D.; et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat. Genet. 2014, 46, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated Molecular Meta-Analysis of 1000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 32, 520–537.e5. [Google Scholar] [CrossRef] [Green Version]
- Paugh, B.S.; Zhu, X.; Qu, C.; Endersby, R.; Diaz, A.K.; Zhang, J.; Bax, D.A.; Carvalho, D.; Reis, R.M.; Onar-Thomas, A.; et al. Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res. 2013, 73, 6219–6229. [Google Scholar] [CrossRef] [Green Version]
- Paugh, B.S.; Broniscer, A.; Qu, C.; Miller, C.P.; Zhang, J.; Tatevossian, R.G.; Olson, J.M.; Geyer, J.R.; Chi, S.N.; da Silva, N.S.; et al. Genome-wide analyses identify recurrent amplifications of receptor tyrosine kinases and cell-cycle regulatory genes in diffuse intrinsic pontine glioma. J. Clin. Oncol. 2011, 29, 3999–4006. [Google Scholar] [CrossRef]
- Park, J.; Lee, W.; Yun, S.; Kim, S.P.; Kim, K.H.; Kim, J.I.; Kim, S.K.; Wang, K.C.; Lee, J.Y. STAT3 is a key molecule in the oncogenic behavior of diffuse intrinsic pontine glioma. Oncol. Lett. 2020, 20, 1989–1998. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, C.; Diplas, B.H.; Moure, C.J.; Chen, C.J.; Chen, L.H.; Du, C.; Zhu, H.; Greer, P.K.; Zhang, L.; et al. Targeting Mutant PPM1D Sensitizes Diffuse Intrinsic Pontine Glioma Cells to the PARP Inhibitor Olaparib. Mol. Cancer Res. 2020, 18, 968–980. [Google Scholar] [CrossRef] [Green Version]
- Damodharan, S.; Lara-Velazquez, M.; Williamsen, B.C.; Helgager, J.; Dey, M. Diffuse Intrinsic Pontine Glioma: Molecular Landscape, Evolving Treatment Strategies and Emerging Clinical Trials. J. Pers. Med. 2022, 12, 840. [Google Scholar] [CrossRef]
- Grasso, C.S.; Tang, Y.; Truffaux, N.; Berlow, N.E.; Liu, L.; Debily, M.A.; Quist, M.J.; Davis, L.E.; Huang, E.C.; Woo, P.J.; et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat. Med. 2015, 21, 555–559. [Google Scholar] [CrossRef]
- Hashizume, R.; Andor, N.; Ihara, Y.; Lerner, R.; Gan, H.; Chen, X.; Fang, D.; Huang, X.; Tom, M.W.; Ngo, V.; et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat. Med. 2014, 20, 1394–1396. [Google Scholar] [CrossRef]
- Kitange, G.J.; Mladek, A.C.; Carlson, B.L.; Schroeder, M.A.; Pokorny, J.L.; Cen, L.; Decker, P.A.; Wu, W.; Lomberk, G.A.; Gupta, S.K.; et al. Inhibition of histone deacetylation potentiates the evolution of acquired temozolomide resistance linked to MGMT upregulation in glioblastoma xenografts. Clin. Cancer Res. 2012, 18, 4070–4079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, C.P.; Figueroa, M.; Gangadharan, A.; Yang, Y.; Romero, M.M.; Kennis, B.A.; Yadavilli, S.; Henry, V.; Collier, T.; Monje, M.; et al. Pharmacologic inhibition of lysine-specific demethylase 1 as a therapeutic and immune-sensitization strategy in pediatric high-grade glioma. Neuro-Oncology 2020, 22, 1302–1314. [Google Scholar] [CrossRef] [PubMed]
- Hennika, T.; Hu, G.; Olaciregui, N.G.; Barton, K.L.; Ehteda, A.; Chitranjan, A.; Chang, C.; Gifford, A.J.; Tsoli, M.; Ziegler, D.S.; et al. Pre-Clinical Study of Panobinostat in Xenograft and Genetically Engineered Murine Diffuse Intrinsic Pontine Glioma Models. PLoS ONE 2017, 12, e0169485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiese, M.; Schill, F.; Sturm, D.; Pfister, S.; Hulleman, E.; Johnsen, S.A.; Kramm, C.M. No Significant Cytotoxic Effect of the EZH2 Inhibitor Tazemetostat (EPZ-6438) on Pediatric Glioma Cells with Wildtype Histone 3 or Mutated Histone 3.3. Klin. Padiatr. 2016, 228, 113–117. [Google Scholar] [CrossRef]
- Mohammad, F.; Weissmann, S.; Leblanc, B.; Pandey, D.P.; Hojfeldt, J.W.; Comet, I.; Zheng, C.; Johansen, J.V.; Rapin, N.; Porse, B.T.; et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 2017, 23, 483–492. [Google Scholar] [CrossRef]
- Keane, L.; Cheray, M.; Saidi, D.; Kirby, C.; Friess, L.; Gonzalez-Rodriguez, P.; Gerdes, M.E.; Grabert, K.; McColl, B.W.; Joseph, B. Inhibition of microglial EZH2 leads to anti-tumoral effects in pediatric diffuse midline gliomas. Neurooncol. Adv. 2021, 3, vdab096. [Google Scholar] [CrossRef]
- Piunti, A.; Hashizume, R.; Morgan, M.A.; Bartom, E.T.; Horbinski, C.M.; Marshall, S.A.; Rendleman, E.J.; Ma, Q.; Takahashi, Y.H.; Woodfin, A.R.; et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat. Med. 2017, 23, 493–500. [Google Scholar] [CrossRef]
- Wang, Z.J.; Ge, Y.; Altinok, D.; Poulik, J.; Sood, S.; Taub, J.W.; Edwards, H.; Kieran, M.W.; Steven, M. Concomitant Use of Panobinostat and Reirradiation in Progressive DIPG: Report of 2 Cases. J. Pediatr. Hematol./Oncol. 2017, 39, e332–e335. [Google Scholar] [CrossRef] [Green Version]
- Taylor, K.R.; Vinci, M.; Bullock, A.N.; Jones, C. ACVR1 mutations in DIPG: Lessons learned from FOP. Cancer Res. 2014, 74, 4565–4570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeman, C.M.; Cordero, F.J.; Hu, G.; Misuraca, K.; Romero, M.M.; Cardona, H.J.; Nazarian, J.; Hashizume, R.; McLendon, R.; Yu, P.; et al. ACVR1 R206H cooperates with H3.1K27M in promoting diffuse intrinsic pontine glioma pathogenesis. Nat. Commun. 2019, 10, 1023. [Google Scholar] [CrossRef]
- Carvalho, D.; Taylor, K.R.; Olaciregui, N.G.; Molinari, V.; Clarke, M.; Mackay, A.; Ruddle, R.; Henley, A.; Valenti, M.; Hayes, A.; et al. ALK2 inhibitors display beneficial effects in preclinical models of ACVR1 mutant diffuse intrinsic pontine glioma. Commun. Biol. 2019, 2, 156. [Google Scholar] [CrossRef]
- Werbrouck, C.; Evangelista, C.C.S.; Lobón-Iglesias, M.J.; Barret, E.; Le Teuff, G.; Merlevede, J.; Brusini, R.; Kergrohen, T.; Mondini, M.; Bolle, S.; et al. TP53 Pathway Alterations Drive Radioresistance in Diffuse Intrinsic Pontine Gliomas (DIPG). Clin. Cancer Res. 2019, 25, 6788–6800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, D.; Ruggiero, A.; Martini, M.; Rizzo, V.; Maurizi, P.; Riccardi, R. Molecular Biology in Pediatric High-Grade Glioma: Impact on Prognosis and Treatment. BioMed Res. Int. 2015, 2015, 215135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Khuong-Quang, D.A.; Buczkowicz, P.; Rakopoulos, P.; Liu, X.Y.; Fontebasso, A.M.; Bouffet, E.; Bartels, U.; Albrecht, S.; Schwartzentruber, J.; Letourneau, L.; et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012, 124, 439–447. [Google Scholar] [CrossRef] [Green Version]
- Saratsis, A.M.; Yadavilli, S.; Magge, S.; Rood, B.R.; Perez, J.; Hill, D.A.; Hwang, E.; Kilburn, L.; Packer, R.J.; Nazarian, J. Insights into pediatric diffuse intrinsic pontine glioma through proteomic analysis of cerebrospinal fluid. Neuro-Oncology 2012, 14, 547–560. [Google Scholar] [CrossRef]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.C.L.; Deshmukh, S.; Jessa, S.; Hadjadj, D.; Lisi, V.; Andrade, A.F.; Faury, D.; Jawhar, W.; Dali, R.; Suzuki, H.; et al. Histone H3.3G34-Mutant Interneuron Progenitors Co-opt PDGFRA for Gliomagenesis. Cell 2020, 183, 1617–1633.e22. [Google Scholar] [CrossRef] [PubMed]
- Asby, D.J.; Killick-Cole, C.L.; Boulter, L.J.; Singleton, W.G.; Asby, C.A.; Wyatt, M.J.; Barua, N.U.; Bienemann, A.S.; Gill, S.S. Combined use of CDK4/6 and mTOR inhibitors induce synergistic growth arrest of diffuse intrinsic pontine glioma cells via mutual downregulation of mTORC1 activity. Cancer Manag. Res. 2018, 10, 3483–3500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Peng, P.; Zhang, X.; Mania-Farnell, B.; Xi, G.; Wan, F. Advanced Pediatric Diffuse Pontine Glioma Murine Models Pave the Way towards Precision Medicine. Cancers 2021, 13, 1114. [Google Scholar] [CrossRef]
- Sturm, D.; Bender, S.; Jones, D.T.; Lichter, P.; Grill, J.; Becher, O.; Hawkins, C.; Majewski, J.; Jones, C.; Costello, J.F.; et al. Paediatric and adult glioblastoma: Multiform (epi)genomic culprits emerge. Nat. Rev. Cancer 2014, 14, 92–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinci, M.; Burford, A.; Molinari, V.; Kessler, K.; Popov, S.; Clarke, M.; Taylor, K.R.; Pemberton, H.N.; Lord, C.J.; Gutteridge, A.; et al. Functional diversity and cooperativity between subclonal populations of pediatric glioblastoma and diffuse intrinsic pontine glioma cells. Nat. Med. 2018, 24, 1204–1215. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e814. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Lazow, M.A.; Schafer, A.; Bartlett, A.; Senthil Kumar, S.; Mishra, D.K.; Dexheimer, P.; DeWire, M.; Fuller, C.; Leach, J.L.; et al. A pilot radiogenomic study of DIPG reveals distinct subgroups with unique clinical trajectories and therapeutic targets. Acta Neuropathol. Commun. 2021, 9, 14. [Google Scholar] [CrossRef]
- Glass, R.; Synowitz, M. CNS macrophages and peripheral myeloid cells in brain tumours. Acta Neuropathol. 2014, 128, 347–362. [Google Scholar] [CrossRef] [Green Version]
- Gutmann, D.H.; Kettenmann, H. Microglia/Brain Macrophages as Central Drivers of Brain Tumor Pathobiology. Neuron 2019, 104, 442–449. [Google Scholar] [CrossRef]
- Buonfiglioli, A.; Hambardzumyan, D. Macrophages and microglia: The cerberus of glioblastoma. Acta Neuropathol. Commun. 2021, 9, 54. [Google Scholar] [CrossRef]
- Herting, C.J.; Chen, Z.; Maximov, V.; Duffy, A.; Szulzewsky, F.; Shayakhmetov, D.M.; Hambardzumyan, D. Tumour-associated macrophage-derived interleukin-1 mediates glioblastoma-associated cerebral oedema. Brain 2019, 142, 3834–3851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Luo, Y.B.; Wu, W.; Zhang, L.; Wang, Z.; Dai, Z.; Feng, S.; Cao, H.; Cheng, Q.; Liu, Z. The molecular feature of macrophages in tumor immune microenvironment of glioma patients. Comput. Struct. Biotechnol. J. 2021, 19, 4603–4618. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S.; Locati, M.; Mantovani, A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: New molecules and patterns of gene expression. J. Immunol. 2006, 177, 7303–7311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Lee, J.; Choi, S.A.; Kim, S.K.; Wang, K.C.; Park, S.H.; Kim, S.H.; Lee, J.Y.; Phi, J.H. M1 macrophage recruitment correlates with worse outcome in SHH Medulloblastomas. BMC Cancer 2018, 18, 535. [Google Scholar] [CrossRef] [Green Version]
- Pages, F.; Kirilovsky, A.; Mlecnik, B.; Asslaber, M.; Tosolini, M.; Bindea, G.; Lagorce, C.; Wind, P.; Marliot, F.; Bruneval, P.; et al. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. J. Clin. Oncol. 2009, 27, 5944–5951. [Google Scholar] [CrossRef]
- Dieu-Nosjean, M.C.; Antoine, M.; Danel, C.; Heudes, D.; Wislez, M.; Poulot, V.; Rabbe, N.; Laurans, L.; Tartour, E.; de Chaisemartin, L.; et al. Long-term survival for patients with non-small-cell lung cancer with intratumoral lymphoid structures. J. Clin. Oncol. 2008, 26, 4410–4417. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Seo, J.Y.; Ahn, J.H.; Ahn, S.H.; Gong, G. Tumor-associated lymphocytes predict response to neoadjuvant chemotherapy in breast cancer patients. J. Breast Cancer 2013, 16, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef]
- Tan, A.H.; Goh, S.Y.; Wong, S.C.; Lam, K.P. T helper cell-specific regulation of inducible costimulator expression via distinct mechanisms mediated by T-bet and GATA-3. J. Biol. Chem. 2008, 283, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Yang, I.; Tihan, T.; Han, S.J.; Wrensch, M.R.; Wiencke, J.; Sughrue, M.E.; Parsa, A.T. CD8+ T-cell infiltrate in newly diagnosed glioblastoma is associated with long-term survival. J. Clin. Neurosci. 2010, 17, 1381–1385. [Google Scholar] [CrossRef] [Green Version]
- Plant, A.S.; Koyama, S.; Sinai, C.; Solomon, I.H.; Griffin, G.K.; Ligon, K.L.; Bandopadhayay, P.; Betensky, R.; Emerson, R.; Dranoff, G.; et al. Immunophenotyping of pediatric brain tumors: Correlating immune infiltrate with histology, mutational load, and survival and assessing clonal T cell response. J. Neurooncol. 2018, 137, 269–278. [Google Scholar] [CrossRef]
- Majzner, R.G.; Ramakrishna, S.; Yeom, K.W.; Patel, S.; Chinnasamy, H.; Schultz, L.M.; Richards, R.M.; Jiang, L.; Barsan, V.; Mancusi, R.; et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 2022, 603, 934–941. [Google Scholar] [CrossRef]
- Hashemi, E.; Malarkannan, S. Tissue-Resident NK Cells: Development, Maturation, and Clinical Relevance. Cancers 2020, 12, 1553. [Google Scholar] [CrossRef]
- Lakshmikanth, T.; Burke, S.; Ali, T.H.; Kimpfler, S.; Ursini, F.; Ruggeri, L.; Capanni, M.; Umansky, V.; Paschen, A.; Sucker, A.; et al. NCRs and DNAM-1 mediate NK cell recognition and lysis of human and mouse melanoma cell lines in vitro and in vivo. J. Clin. Investig. 2009, 119, 1251–1263. [Google Scholar] [CrossRef] [Green Version]
- Menard, C.; Blay, J.Y.; Borg, C.; Michiels, S.; Ghiringhelli, F.; Robert, C.; Nonn, C.; Chaput, N.; Taieb, J.; Delahaye, N.F.; et al. Natural killer cell IFN-gamma levels predict long-term survival with imatinib mesylate therapy in gastrointestinal stromal tumor-bearing patients. Cancer Res. 2009, 69, 3563–3569. [Google Scholar] [CrossRef] [Green Version]
- Bi, J.; Tian, Z. NK Cell Dysfunction and Checkpoint Immunotherapy. Front. Immunol. 2019, 10, 1999. [Google Scholar] [CrossRef]
- Haspels, H.N.; Rahman, M.A.; Joseph, J.V.; Gras Navarro, A.; Chekenya, M. Glioblastoma Stem-Like Cells Are More Susceptible Than Differentiated Cells to Natural Killer Cell Lysis Mediated Through Killer Immunoglobulin-Like Receptors-Human Leukocyte Antigen Ligand Mismatch and Activation Receptor-Ligand Interactions. Front. Immunol. 2018, 9, 1345. [Google Scholar] [CrossRef]
- Shaim, H.; Shanley, M.; Basar, R.; Daher, M.; Gumin, J.; Zamler, D.B.; Uprety, N.; Wang, F.; Huang, Y.; Gabrusiewicz, K.; et al. Targeting the alphav integrin/TGF-beta axis improves natural killer cell function against glioblastoma stem cells. J. Clin. Investig. 2021, 131, e142116. [Google Scholar] [CrossRef]
- Murad, S.; Michen, S.; Becker, A.; Fussel, M.; Schackert, G.; Tonn, T.; Momburg, F.; Temme, A. NKG2C+ NK Cells for Immunotherapy of Glioblastoma Multiforme. Int. J. Mol. Sci. 2022, 23, 5857. [Google Scholar] [CrossRef]
- Woan, K.V.; Kim, H.; Bjordahl, R.; Davis, Z.B.; Gaidarova, S.; Goulding, J.; Hancock, B.; Mahmood, S.; Abujarour, R.; Wang, H.; et al. Harnessing features of adaptive NK cells to generate iPSC-derived NK cells for enhanced immunotherapy. Cell Stem Cell 2021, 28, 2062–2075.e5. [Google Scholar] [CrossRef]
- Haberthur, K.; Brennan, K.; Hoglund, V.; Balcaitis, S.; Chinn, H.; Davis, A.; Kreuser, S.; Winter, C.; Leary, S.E.; Deutsch, G.H.; et al. NKG2D ligand expression in pediatric brain tumors. Cancer Biol. Ther. 2016, 17, 1253–1265. [Google Scholar] [CrossRef] [Green Version]
- Abbott, M.; Ustoyev, Y. Cancer and the Immune System: The History and Background of Immunotherapy. Semin. Oncol. Nurs. 2019, 35, 150923. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [Green Version]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [Green Version]
- Albiges, L.; Tannir, N.M.; Burotto, M.; McDermott, D.; Plimack, E.R.; Barthélémy, P.; Porta, C.; Powles, T.; Donskov, F.; George, S.; et al. Nivolumab plus ipilimumab versus sunitinib for first-line treatment of advanced renal cell carcinoma: Extended 4-year follow-up of the phase III CheckMate 214 trial. ESMO Open 2020, 5, e001079. [Google Scholar] [CrossRef]
- Regan, M.M.; Werner, L.; Rao, S.; Gupte-Singh, K.; Hodi, F.S.; Kirkwood, J.M.; Kluger, H.M.; Larkin, J.; Postow, M.A.; Ritchings, C.; et al. Treatment-Free Survival: A Novel Outcome Measure of the Effects of Immune Checkpoint Inhibition-A Pooled Analysis of Patients with Advanced Melanoma. J. Clin. Oncol. 2019, 37, 3350–3358. [Google Scholar] [CrossRef]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [Green Version]
- Sadelain, M.; Brentjens, R.; Riviere, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Cheng, J.; Mu, W.; Zhou, J.; Zhu, L. Advances in Universal CAR-T Cell Therapy. Front. Immunol. 2021, 12, 744823. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- Wang, D.; Starr, R.; Chang, W.C.; Aguilar, B.; Alizadeh, D.; Wright, S.L.; Yang, X.; Brito, A.; Sarkissian, A.; Ostberg, J.R.; et al. Chlorotoxin-directed CAR T cells for specific and effective targeting of glioblastoma. Sci. Transl. Med. 2020, 12, eaaw2672. [Google Scholar] [CrossRef]
- Berlow, N.E.; Svalina, M.N.; Quist, M.J.; Settelmeyer, T.P.; Zherebitskiy, V.; Kogiso, M.; Qi, L.; Du, Y.; Hawkins, C.E.; Hulleman, E.; et al. IL-13 receptors as possible therapeutic targets in diffuse intrinsic pontine glioma. PLoS ONE 2018, 13, e0193565. [Google Scholar] [CrossRef] [Green Version]
- Lian, X.; Kats, D.; Rasmussen, S.; Martin, L.R.; Karki, A.; Keller, C.; Berlow, N.E. Design considerations of an IL13Ralpha2 antibody-drug conjugate for diffuse intrinsic pontine glioma. Acta Neuropathol. Commun. 2021, 9, 88. [Google Scholar] [CrossRef]
- Mount, C.W.; Majzner, R.G.; Sundaresh, S.; Arnold, E.P.; Kadapakkam, M.; Haile, S.; Labanieh, L.; Hulleman, E.; Woo, P.J.; Rietberg, S.P.; et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M(+) diffuse midline gliomas. Nat. Med. 2018, 24, 572–579. [Google Scholar] [CrossRef]
- Choi, B.D.; Suryadevara, C.M.; Gedeon, P.C.; Herndon, J.E., 2nd; Sanchez-Perez, L.; Bigner, D.D.; Sampson, J.H. Intracerebral delivery of a third generation EGFRvIII-specific chimeric antigen receptor is efficacious against human glioma. J. Clin. Neurosci. 2014, 21, 189–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhavan, D.; Alizadeh, D.; Wang, D.; Weist, M.R.; Shepphird, J.K.; Brown, C.E. CAR T cells for brain tumors: Lessons learned and road ahead. Immunol. Rev. 2019, 290, 60–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Luther, N.; Ibrahim, G.M.; Hawkins, C.; Vibhakar, R.; Handler, M.H.; Souweidane, M.M. B7-H3, a potential therapeutic target, is expressed in diffuse intrinsic pontine glioma. J. Neurooncol. 2013, 111, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef] [PubMed]
- Morandi, F.; Sabatini, F.; Podesta, M.; Airoldi, I. Immunotherapeutic Strategies for Neuroblastoma: Present, Past and Future. Vaccines 2021, 9, 43. [Google Scholar] [CrossRef]
- Chulanetra, M.; Morchang, A.; Sayour, E.; Eldjerou, L.; Milner, R.; Lagmay, J.; Cascio, M.; Stover, B.; Slayton, W.; Chaicumpa, W.; et al. GD2 chimeric antigen receptor modified T cells in synergy with sub-toxic level of doxorubicin targeting osteosarcomas. Am. J. Cancer Res. 2020, 10, 674–687. [Google Scholar]
- Navid, F.; Santana, V.M.; Barfield, R.C. Anti-GD2 antibody therapy for GD2-expressing tumors. Curr. Cancer Drug Targets 2010, 10, 200–209. [Google Scholar] [CrossRef]
- Razavi, A.; Keshavarz-Fathi, M.; Pawelek, J.; Rezaei, N. Chimeric antigen receptor T-cell therapy for melanoma. Expert Rev. Clin. Immunol. 2021, 17, 209–223. [Google Scholar] [CrossRef]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic cancer vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Manjili, M.H.; Subjeck, J.R.; Sarkar, D.; Fisher, P.B.; Wang, X.Y. Therapeutic cancer vaccines: Past, present, and future. Adv. Cancer Res. 2013, 119, 421–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollack, I.F.; Jakacki, R.I.; Butterfield, L.H.; Hamilton, R.L.; Panigrahy, A.; Potter, D.M.; Connelly, A.K.; Dibridge, S.A.; Whiteside, T.L.; Okada, H. Antigen-specific immune responses and clinical outcome after vaccination with glioma-associated antigen peptides and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in children with newly diagnosed malignant brainstem and nonbrainstem gliomas. J. Clin. Oncol. 2014, 32, 2050–2058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollack, I.F.; Jakacki, R.I.; Butterfield, L.H.; Hamilton, R.L.; Panigrahy, A.; Normolle, D.P.; Connelly, A.K.; Dibridge, S.; Mason, G.; Whiteside, T.L.; et al. Antigen-specific immunoreactivity and clinical outcome following vaccination with glioma-associated antigen peptides in children with recurrent high-grade gliomas: Results of a pilot study. J. Neurooncol. 2016, 130, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Ochs, K.; Ott, M.; Bunse, T.; Sahm, F.; Bunse, L.; Deumelandt, K.; Sonner, J.K.; Keil, M.; von Deimling, A.; Wick, W.; et al. K27M-mutant histone-3 as a novel target for glioma immunotherapy. Oncoimmunology 2017, 6, e1328340. [Google Scholar] [CrossRef] [Green Version]
- Mueller, S.; Taitt, J.M.; Villanueva-Meyer, J.E.; Bonner, E.R.; Nejo, T.; Lulla, R.R.; Goldman, S.; Banerjee, A.; Chi, S.N.; Whipple, N.S.; et al. Mass cytometry detects H3.3K27M-specific vaccine responses in diffuse midline glioma. J. Clin. Investig. 2020, 130, 6325–6337. [Google Scholar] [CrossRef]
- Benitez-Ribas, D.; Cabezon, R.; Florez-Grau, G.; Molero, M.C.; Puerta, P.; Guillen, A.; Paco, S.; Carcaboso, A.M.; Santa-Maria Lopez, V.; Cruz, O.; et al. Immune Response Generated with the Administration of Autologous Dendritic Cells Pulsed with an Allogenic Tumoral Cell-Lines Lysate in Patients with Newly Diagnosed Diffuse Intrinsic Pontine Glioma. Front. Oncol. 2018, 8, 127. [Google Scholar] [CrossRef] [Green Version]
- Ylosmaki, E.; Cerullo, V. Design and application of oncolytic viruses for cancer immunotherapy. Curr. Opin. Biotechnol. 2020, 65, 25–36. [Google Scholar] [CrossRef]
- Lawler, S.E.; Speranza, M.C.; Cho, C.F.; Chiocca, E.A. Oncolytic Viruses in Cancer Treatment: A Review. JAMA Oncol. 2017, 3, 841–849. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Kang, X.; Chen, K.S.; Jehng, T.; Jones, L.; Chen, J.; Huang, X.F.; Chen, S.Y. An engineered oncolytic virus expressing PD-L1 inhibitors activates tumor neoantigen-specific T cell responses. Nat. Commun. 2020, 11, 1395. [Google Scholar] [CrossRef]
- Melcher, A.; Harrington, K.; Vile, R. Oncolytic virotherapy as immunotherapy. Science 2021, 374, 1325–1326. [Google Scholar] [CrossRef]
- Martinez-Velez, N.; Marigil, M.; Garcia-Moure, M.; Gonzalez-Huarriz, M.; Aristu, J.J.; Ramos-Garcia, L.I.; Tejada, S.; Diez-Valle, R.; Patino-Garcia, A.; Becher, O.J.; et al. Delta-24-RGD combined with radiotherapy exerts a potent antitumor effect in diffuse intrinsic pontine glioma and pediatric high grade glioma models. Acta Neuropathol. Commun. 2019, 7, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Velez, N.; Garcia-Moure, M.; Marigil, M.; Gonzalez-Huarriz, M.; Puigdelloses, M.; Gallego Perez-Larraya, J.; Zalacain, M.; Marrodan, L.; Varela-Guruceaga, M.; Laspidea, V.; et al. The oncolytic virus Delta-24-RGD elicits an antitumor effect in pediatric glioma and DIPG mouse models. Nat. Commun. 2019, 10, 2235. [Google Scholar] [CrossRef] [PubMed]
- Tejada, S.; Diez-Valle, R.; Dominguez, P.D.; Patino-Garcia, A.; Gonzalez-Huarriz, M.; Fueyo, J.; Gomez-Manzano, C.; Idoate, M.A.; Peterkin, J.; Alonso, M.M. DNX-2401, an Oncolytic Virus, for the Treatment of Newly Diagnosed Diffuse Intrinsic Pontine Gliomas: A Case Report. Front. Oncol. 2018, 8, 61. [Google Scholar] [CrossRef] [Green Version]
- Cockle, J.V.; Bruning-Richardson, A.; Scott, K.J.; Thompson, J.; Kottke, T.; Morrison, E.; Ismail, A.; Carcaboso, A.M.; Rose, A.; Selby, P.; et al. Oncolytic Herpes Simplex Virus Inhibits Pediatric Brain Tumor Migration and Invasion. Mol. Ther. Oncolytics 2017, 5, 75–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritterhouse, L.L. Tumor mutational burden. Cancer Cytopathol. 2019, 127, 735–736. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Jardim, D.L.; Goodman, A.; de Melo Gagliato, D.; Kurzrock, R. The Challenges of Tumor Mutational Burden as an Immunotherapy Biomarker. Cancer Cell 2021, 39, 154–173. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Findlay, I.J.; De Iuliis, G.N.; Duchatel, R.J.; Jackson, E.R.; Vitanza, N.A.; Cain, J.E.; Waszak, S.M.; Dun, M.D. Pharmaco-proteogenomic profiling of pediatric diffuse midline glioma to inform future treatment strategies. Oncogene 2022, 41, 461–475. [Google Scholar] [CrossRef]
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; de Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting from Germline Biallelic Mismatch Repair Deficiency. J. Clin. Oncol. 2016, 34, 2206–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [Green Version]
- Paugh, B.S.; Qu, C.; Jones, C.; Liu, Z.; Adamowicz-Brice, M.; Zhang, J.; Bax, D.A.; Coyle, B.; Barrow, J.; Hargrave, D.; et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J. Clin. Oncol. 2010, 28, 3061–3068. [Google Scholar] [CrossRef] [PubMed]
- Pollack, I.F.; Hamilton, R.L.; James, C.D.; Finkelstein, S.D.; Burnham, J.; Yates, A.J.; Holmes, E.J.; Zhou, T.; Finlay, J.L. Rarity of PTEN deletions and EGFR amplification in malignant gliomas of childhood: Results from the Children’s Cancer Group 945 cohort. J. Neurosurg. 2006, 105, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Bax, D.A.; Gaspar, N.; Little, S.E.; Marshall, L.; Perryman, L.; Regairaz, M.; Viana-Pereira, M.; Vuononvirta, R.; Sharp, S.Y.; Reis-Filho, J.S.; et al. EGFRvIII deletion mutations in pediatric high-grade glioma and response to targeted therapy in pediatric glioma cell lines. Clin. Cancer Res. 2009, 15, 5753–5761. [Google Scholar] [CrossRef] [Green Version]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- Thompson, J.A. New NCCN Guidelines: Recognition and Management of Immunotherapy-Related Toxicity. J. Natl. Compr. Cancer Netw. 2018, 16, 594–596. [Google Scholar] [CrossRef]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef]
- Villadolid, J.; Amin, A. Immune checkpoint inhibitors in clinical practice: Update on management of immune-related toxicities. Transl. Lung Cancer Res. 2015, 4, 560–575. [Google Scholar] [CrossRef]
- Gupta, A.; De Felice, K.M.; Loftus, E.V., Jr.; Khanna, S. Systematic review: Colitis associated with anti-CTLA-4 therapy. Aliment. Pharmacol. Ther. 2015, 42, 406–417. [Google Scholar] [CrossRef]
- Suzman, D.L.; Pelosof, L.; Rosenberg, A.; Avigan, M.I. Hepatotoxicity of immune checkpoint inhibitors: An evolving picture of risk associated with a vital class of immunotherapy agents. Liver Int. 2018, 38, 976–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barroso-Sousa, R.; Barry, W.T.; Garrido-Castro, A.C.; Hodi, F.S.; Min, L.; Krop, I.E.; Tolaney, S.M. Incidence of Endocrine Dysfunction Following the Use of Different Immune Checkpoint Inhibitor Regimens: A Systematic Review and Meta-analysis. JAMA Oncol. 2018, 4, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Ryder, M.; Callahan, M.; Postow, M.A.; Wolchok, J.; Fagin, J.A. Endocrine-related adverse events following ipilimumab in patients with advanced melanoma: A comprehensive retrospective review from a single institution. Endocr. Relat. Cancer 2014, 21, 371–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puzanov, I.; Diab, A.; Abdallah, K.; Bingham, C.O., 3rd; Brogdon, C.; Dadu, R.; Hamad, L.; Kim, S.; Lacouture, M.E.; LeBoeuf, N.R.; et al. Managing toxicities associated with immune checkpoint inhibitors: Consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J. Immunother. Cancer 2017, 5, 95. [Google Scholar] [CrossRef] [Green Version]
- Cappelli, L.C.; Brahmer, J.R.; Forde, P.M.; Le, D.T.; Lipson, E.J.; Naidoo, J.; Zheng, L.; Bingham, C.O., 3rd; Shah, A.A. Clinical presentation of immune checkpoint inhibitor-induced inflammatory arthritis differs by immunotherapy regimen. Semin. Arthritis Rheum. 2018, 48, 553–557. [Google Scholar] [CrossRef]
- Frey, N.; Porter, D. Cytokine Release Syndrome with Chimeric Antigen Receptor T Cell Therapy. Biol. Blood Marrow Transplant. 2019, 25, e123–e127. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef] [Green Version]
- Brudno, J.N.; Kochenderfer, J.N. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 2019, 34, 45–55. [Google Scholar] [CrossRef]
- Shimabukuro-Vornhagen, A.; Godel, P.; Subklewe, M.; Stemmler, H.J.; Schlosser, H.A.; Schlaak, M.; Kochanek, M.; Boll, B.; von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. Immunother. Cancer 2018, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Brown, B.D.; Tambaro, F.P.; Kohorst, M.; Chi, L.; Mahadeo, K.M.; Tewari, P.; Petropoulos, D.; Slopis, J.M.; Sadighi, Z.; Khazal, S. Immune Effector Cell Associated Neurotoxicity (ICANS) in Pediatric and Young Adult Patients Following Chimeric Antigen Receptor (CAR) T-Cell Therapy: Can We Optimize Early Diagnosis? Front. Oncol. 2021, 11, 634445. [Google Scholar] [CrossRef] [PubMed]
- Ygberg, S.; Nilsson, A. The developing immune system—From foetus to toddler. Acta Paediatr. 2012, 101, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Schuelke, M.R.; Wongthida, P.; Thompson, J.; Kottke, T.; Driscoll, C.B.; Huff, A.L.; Shim, K.G.; Coffey, M.; Pulido, J.; Evgin, L.; et al. Diverse immunotherapies can effectively treat syngeneic brainstem tumors in the absence of overt toxicity. J. Immunother. Cancer 2019, 7, 188. [Google Scholar] [CrossRef] [PubMed]
- Aspelund, A.; Antila, S.; Proulx, S.T.; Karlsen, T.V.; Karaman, S.; Detmar, M.; Wiig, H.; Alitalo, K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015, 212, 991–999. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Wilson, E.H.; Weninger, W.; Hunter, C.A. Trafficking of immune cells in the central nervous system. J. Clin. Investig. 2010, 120, 1368–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, V.H. A revised view of the central nervous system microenvironment and major histocompatibility complex class II antigen presentation. J. Neuroimmunol. 1998, 90, 113–121. [Google Scholar] [CrossRef]
- Matyszak, M.K.; Perry, V.H. The potential role of dendritic cells in immune-mediated inflammatory diseases in the central nervous system. Neuroscience 1996, 74, 599–608. [Google Scholar] [CrossRef]
- McMahon, D.; Oakden, W.; Hynynen, K. Investigating the effects of dexamethasone on blood-brain barrier permeability and inflammatory response following focused ultrasound and microbubble exposure. Theranostics 2020, 10, 1604–1618. [Google Scholar] [CrossRef]
- Shapiro, W.R.; Hiesiger, E.M.; Cooney, G.A.; Basler, G.A.; Lipschutz, L.E.; Posner, J.B. Temporal effects of dexamethasone on blood-to-brain and blood-to-tumor transport of 14C-alpha-aminoisobutyric acid in rat C6 glioma. J. Neurooncol. 1990, 8, 197–204. [Google Scholar] [CrossRef]
- Hue, C.D.; Cho, F.S.; Cao, S.; Dale Bass, C.R.; Meaney, D.F.; Morrison, B., 3rd. Dexamethasone potentiates in vitro blood-brain barrier recovery after primary blast injury by glucocorticoid receptor-mediated upregulation of ZO-1 tight junction protein. J. Cereb. Blood Flow Metab. 2015, 35, 1191–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fauquette, W.; Amourette, C.; Dehouck, M.P.; Diserbo, M. Radiation-induced blood-brain barrier damages: An in vitro study. Brain Res. 2012, 1433, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Giles, A.J.; Hutchinson, M.N.D.; Sonnemann, H.M.; Jung, J.; Fecci, P.E.; Ratnam, N.M.; Zhang, W.; Song, H.; Bailey, R.; Davis, D.; et al. Dexamethasone-induced immunosuppression: Mechanisms and implications for immunotherapy. J. Immunother. Cancer 2018, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kummel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef]
- Schmid, P.; Salgado, R.; Park, Y.H.; Munoz-Couselo, E.; Kim, S.B.; Sohn, J.; Im, S.A.; Foukakis, T.; Kuemmel, S.; Dent, R.; et al. Pembrolizumab plus chemotherapy as neoadjuvant treatment of high-risk, early-stage triple-negative breast cancer: Results from the phase 1b open-label, multicohort KEYNOTE-173 study. Ann. Oncol. 2020, 31, 569–581. [Google Scholar] [CrossRef]
- D’Amico, L.; Menzel, U.; Prummer, M.; Muller, P.; Buchi, M.; Kashyap, A.; Haessler, U.; Yermanos, A.; Gebleux, R.; Briendl, M.; et al. A novel anti-HER2 anthracycline-based antibody-drug conjugate induces adaptive anti-tumor immunity and potentiates PD-1 blockade in breast cancer. J. Immunother. Cancer 2019, 7, 16. [Google Scholar] [CrossRef]
- Zappasodi, R.; Pupa, S.M.; Ghedini, G.C.; Bongarzone, I.; Magni, M.; Cabras, A.D.; Colombo, M.P.; Carlo-Stella, C.; Gianni, A.M.; Di Nicola, M. Improved clinical outcome in indolent B-cell lymphoma patients vaccinated with autologous tumor cells experiencing immunogenic death. Cancer Res. 2010, 70, 9062–9072. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Shim, M.K.; Kim, W.J.; Choi, J.; Nam, G.H.; Kim, J.; Kim, J.; Moon, Y.; Kim, H.Y.; Park, J.; et al. Cancer-activated doxorubicin prodrug nanoparticles induce preferential immune response with minimal doxorubicin-related toxicity. Biomaterials 2021, 272, 120791. [Google Scholar] [CrossRef]
- Menger, L.; Vacchelli, E.; Adjemian, S.; Martins, I.; Ma, Y.; Shen, S.; Yamazaki, T.; Sukkurwala, A.Q.; Michaud, M.; Mignot, G.; et al. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci. Transl. Med. 2012, 4, 143ra199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fucikova, J.; Kralikova, P.; Fialova, A.; Brtnicky, T.; Rob, L.; Bartunkova, J.; Spisek, R. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res. 2011, 71, 4821–4833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Yin, G.; Le, V.; Zhang, A.; Chen, S.; Liang, X.; Liu, J. Photodynamic-therapy Activates Immune Response by disrupting Immunity Homeostasis of Tumor Cells, which Generates Vaccine for Cancer Therapy. Int J Biol Sci. 2016, 12, 120–132. [Google Scholar] [CrossRef] [Green Version]
- Fucikova, J.; Kepp, O.; Kasikova, L.; Petroni, G.; Yamazaki, T.; Liu, P.; Zhao, L.; Spisek, R.; Kroemer, G.; Galluzzi, L. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020, 11, 1013. [Google Scholar] [CrossRef]
- Chen, J.; Xie, J.; Jiang, Z.; Wang, B.; Wang, Y.; Hu, X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene 2011, 30, 4297–4306. [Google Scholar] [CrossRef] [Green Version]
- Schiavoni, G.; Sistigu, A.; Valentini, M.; Mattei, F.; Sestili, P.; Spadaro, F.; Sanchez, M.; Lorenzi, S.; D’Urso, M.T.; Belardelli, F.; et al. Cyclophosphamide synergizes with type I interferons through systemic dendritic cell reactivation and induction of immunogenic tumor apoptosis. Cancer Res. 2011, 71, 768–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, A.D.; Dudek, A.M.; Ferreira, G.B.; Verfaillie, T.; Vandenabeele, P.; Krysko, D.V.; Mathieu, C.; Agostinis, P. ROS-induced autophagy in cancer cells assists in evasion from determinants of immunogenic cell death. Autophagy 2013, 9, 1292–1307. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Enlightening the impact of immunogenic cell death in photodynamic cancer therapy. EMBO J. 2012, 31, 1055–1057. [Google Scholar] [CrossRef] [Green Version]
- Krysko, O.; Love Aaes, T.; Bachert, C.; Vandenabeele, P.; Krysko, D.V. Many faces of DAMPs in cancer therapy. Cell Death Dis. 2013, 4, e631. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Krysko, D.V.; Verfaillie, T.; Kaczmarek, A.; Ferreira, G.B.; Marysael, T.; Rubio, N.; Firczuk, M.; Mathieu, C.; Roebroek, A.J.; et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012, 31, 1062–1079. [Google Scholar] [CrossRef] [Green Version]
- Thorburn, J.; Horita, H.; Redzic, J.; Hansen, K.; Frankel, A.E.; Thorburn, A. Autophagy regulates selective HMGB1 release in tumor cells that are destined to die. Cell Death Differ. 2009, 16, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Franz, S.; Herrmann, K.; Furnrohr, B.G.; Sheriff, A.; Frey, B.; Gaipl, U.S.; Voll, R.E.; Kalden, J.R.; Jack, H.M.; Herrmann, M. After shrinkage apoptotic cells expose internal membrane-derived epitopes on their plasma membranes. Cell Death Differ. 2007, 14, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.-C.; Chen, H.-M.; Chen, S.-S.; Huang, L.-T.; Chang, W.-T.; Wei, W.-C.; Chou, L.-C.; Arulselvan, P.; Wu, J.-B.; Kuo, S.-C.; et al. Specific microtubule-depolymerizing agents augment efficacy of dendritic cell-based cancer vaccines. J. Biomed. Sci. 2011, 18, 44. [Google Scholar] [CrossRef] [Green Version]
- Trabanelli, S.; Ocadlikova, D.; Gulinelli, S.; Curti, A.; Salvestrini, V.; Vieira, R.P.; Idzko, M.; Di Virgilio, F.; Ferrari, D.; Lemoli, R.M. Extracellular ATP exerts opposite effects on activated and regulatory CD4+ T cells via purinergic P2 receptor activation. J. Immunol. 2012, 189, 1303–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diederich, M. Natural compound inducers of immunogenic cell death. Arch. Pharm. Res. 2019, 42, 629–645. [Google Scholar] [CrossRef]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef]
- Park, J.S.; Gamboni-Robertson, F.; He, Q.; Svetkauskaite, D.; Kim, J.Y.; Strassheim, D.; Sohn, J.W.; Yamada, S.; Maruyama, I.; Banerjee, A.; et al. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am. J. Physiol. Cell Physiol. 2006, 290, C917–C924. [Google Scholar] [CrossRef]
- Andersson, U.; Wang, H.; Palmblad, K.; Aveberger, A.C.; Bloom, O.; Erlandsson-Harris, H.; Janson, A.; Kokkola, R.; Zhang, M.; Yang, H.; et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J. Exp. Med. 2000, 192, 565–570. [Google Scholar] [CrossRef]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Criollo, A.; Ortiz, C.; Lidereau, R.; Mariette, C.; Chaput, N.; Mira, J.P.; Delaloge, S.; et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol. Rev. 2007, 220, 47–59. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Hong, C.; Qiu, X.; Li, Y.; Huang, Q.; Zhong, Z.; Zhang, Y.; Liu, X.; Sun, L.; Lv, P.; Gao, X.M. Functional analysis of recombinant calreticulin fragment 39-272: Implications for immunobiological activities of calreticulin in health and disease. J. Immunol. 2010, 185, 4561–4569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- la Sala, A.; Ferrari, D.; Corinti, S.; Cavani, A.; Di Virgilio, F.; Girolomoni, G. Extracellular ATP induces a distorted maturation of dendritic cells and inhibits their capacity to initiate Th1 responses. J. Immunol. 2001, 166, 1611–1617. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, D.; La Sala, A.; Chiozzi, P.; Morelli, A.; Falzoni, S.; Girolomoni, G.; Idzko, M.; Dichmann, S.; Norgauer, J.; Di Virgilio, F. The P2 purinergic receptors of human dendritic cells: Identification and coupling to cytokine release. FASEB J. 2000, 14, 2466–2476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beavis, P.A.; Stagg, J.; Darcy, P.K.; Smyth, M.J. CD73: A potent suppressor of antitumor immune responses. Trends Immunol. 2012, 33, 231–237. [Google Scholar] [CrossRef]
- Lee, S.; Kambhampati, M.; Yadavilli, S.; Gordish-Dressman, H.; Santi, M.; Cruz, C.R.; Packer, R.J.; Almira-Suarez, M.I.; Hwang, E.I.; Nazarian, J. Differential Expression of Wilms’ Tumor Protein in Diffuse Intrinsic Pontine Glioma. J. Neuropathol. Exp. Neurol. 2019, 78, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Wongthida, P.; Schuelke, M.R.; Driscoll, C.B.; Kottke, T.; Thompson, J.M.; Tonne, J.; Stone, C.; Huff, A.L.; Wetmore, C.; Davies, J.A.; et al. Ad-CD40L mobilizes CD4 T cells for the treatment of brainstem tumors. Neuro-Oncology 2020, 22, 1757–1770. [Google Scholar] [CrossRef] [PubMed]
- Khatua, S.; Cooper, L.J.N.; Sandberg, D.I.; Ketonen, L.; Johnson, J.M.; Rytting, M.E.; Liu, D.D.; Meador, H.; Trikha, P.; Nakkula, R.J.; et al. Phase I study of intraventricular infusions of autologous ex vivo expanded NK cells in children with recurrent medulloblastoma and ependymoma. Neuro-Oncology 2020, 22, 1214–1225. [Google Scholar] [CrossRef]
- Chastkofsky, M.I.; Pituch, K.C.; Katagi, H.; Zannikou, M.; Ilut, L.; Xiao, T.; Han, Y.; Sonabend, A.M.; Curiel, D.T.; Bonner, E.R.; et al. Mesenchymal Stem Cells Successfully Deliver Oncolytic Virotherapy to Diffuse Intrinsic Pontine Glioma. Clin. Cancer Res. 2021, 27, 1766–1777. [Google Scholar] [CrossRef]
- Alli, S.; Figueiredo, C.A.; Golbourn, B.; Sabha, N.; Wu, M.Y.; Bondoc, A.; Luck, A.; Coluccia, D.; Maslink, C.; Smith, C.; et al. Brainstem blood brain barrier disruption using focused ultrasound: A demonstration of feasibility and enhanced doxorubicin delivery. J. Control. Release 2018, 281, 29–41. [Google Scholar] [CrossRef]
- Bredlau, A.L.; Dixit, S.; Chen, C.; Broome, A.M. Nanotechnology Applications for Diffuse Intrinsic Pontine Glioma. Curr. Neuropharmacol. 2017, 15, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Himes, B.T.; Zhang, L.; Daniels, D.J. Treatment Strategies in Diffuse Midline Gliomas with the H3K27M Mutation: The Role of Convection-Enhanced Delivery in Overcoming Anatomic Challenges. Front. Oncol. 2019, 9, 31. [Google Scholar] [CrossRef] [Green Version]
- Bander, E.D.; Ramos, A.D.; Wembacher-Schroeder, E.; Ivasyk, I.; Thomson, R.; Morgenstern, P.F.; Souweidane, M.M. Repeat convection-enhanced delivery for diffuse intrinsic pontine glioma. J. Neurosurg. Pediatr. 2020, 26, 661–666. [Google Scholar] [CrossRef]
- Riss, T.; Niles, A.; Moravec, R.; Karassina, N.; Vidugiriene, J. Cytotoxicity Assays: In Vitro Methods to Measure Dead Cells. In Assay Guidance Manual; Markossian, S., Grossman, A., Brimacombe, K., Arkin, M., Auld, D., Austin, C., Baell, J., Chung, T.D.Y., Coussens, N.P., Dahlin, J.L., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Wesierska-Gadek, J.; Gueorguieva, M.; Ranftler, C.; Zerza-Schnitzhofer, G. A new multiplex assay allowing simultaneous detection of the inhibition of cell proliferation and induction of cell death. J. Cell Biochem. 2005, 96, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Stallard, S.; Savelieff, M.G.; Wierzbicki, K.; Mullan, B.; Miklja, Z.; Bruzek, A.; Garcia, T.; Siada, R.; Anderson, B.; Singer, B.H.; et al. CSF H3F3A K27M circulating tumor DNA copy number quantifies tumor growth and in vitro treatment response. Acta Neuropathol. Commun. 2018, 6, 80. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; See, A.P.; Phallen, J.; Jackson, C.M.; Belcaid, Z.; Ruzevick, J.; Durham, N.; Meyer, C.; Harris, T.J.; Albesiano, E.; et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riva, M.; Wouters, R.; Sterpin, E.; Giovannoni, R.; Boon, L.; Himmelreich, U.; Gsell, W.; Van Ranst, M.; Coosemans, A. Radiotherapy, Temozolomide, and Antiprogrammed Cell Death Protein 1 Treatments Modulate the Immune Microenvironment in Experimental High-Grade Glioma. Neurosurgery 2021, 88, E205–E215. [Google Scholar] [CrossRef]
- Koks, C.A.; Garg, A.D.; Ehrhardt, M.; Riva, M.; Vandenberk, L.; Boon, L.; De Vleeschouwer, S.; Agostinis, P.; Graf, N.; Van Gool, S.W. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int. J. Cancer 2015, 136, E313–E325. [Google Scholar] [CrossRef]
- Suryadevara, C.M.; Riccione, K.A.; Sampson, J.H. Immunotherapy Gone Viral: Bortezomib and oHSV Enhance Antitumor NK-Cell Activity. Clin. Cancer Res. 2016, 22, 5164–5166. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, R.; Zullo, S.A.; Tanaka, R.; Ramsey, J.; Blaese, M.; Xanthopoulos, K.G. Induction of a therapeutic antitumor immunological response by intratumoral injection of genetically engineered Semliki Forest virus to produce interleukin-12. Neurosurg. Focus 2000, 9, e7. [Google Scholar] [CrossRef]
- Efimova, I.; Catanzaro, E.; Van der Meeren, L.; Turubanova, V.D.; Hammad, H.; Mishchenko, T.A.; Vedunova, M.V.; Fimognari, C.; Bachert, C.; Coppieters, F.; et al. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. J Immunother. Cancer 2020, 8, e001369. [Google Scholar] [CrossRef]
- Kim, J.E.; Patel, M.A.; Mangraviti, A.; Kim, E.S.; Theodros, D.; Velarde, E.; Liu, A.; Sankey, E.W.; Tam, A.; Xu, H.; et al. Combination Therapy with Anti-PD-1, Anti-TIM-3, and Focal Radiation Results in Regression of Murine Gliomas. Clin. Cancer Res. 2017, 23, 124–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, S.; Mazzoccoli, L.; Jash, A.; Govero, J.; Bais, S.S.; Hu, T.; Fontes-Garfias, C.R.; Shan, C.; Okada, H.; Shresta, S.; et al. Zika virus oncolytic activity requires CD8+ T cells and is boosted by immune checkpoint blockade. JCI Insight 2021, 6, e144619. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zheng, M.; Zhang, Z.; Tang, X.; Chen, Y.; Peng, A.; Peng, X.; Tong, A.; Zhou, L. Interleukin-7-loaded oncolytic adenovirus improves CAR-T cell therapy for glioblastoma. Cancer Immunol. Immunother. 2021, 70, 2453–2465. [Google Scholar] [CrossRef] [PubMed]
- Maczynska, J.; Raes, F.; Da Pieve, C.; Turnock, S.; Boult, J.K.R.; Hoebart, J.; Niedbala, M.; Robinson, S.P.; Harrington, K.J.; Kaspera, W.; et al. Triggering anti-GBM immune response with EGFR-mediated photoimmunotherapy. BMC Med. 2022, 20, 16. [Google Scholar] [CrossRef]
- Wu, J.; Jordan, M.; Waxman, D.J. Metronomic cyclophosphamide activation of anti-tumor immunity: Tumor model, mouse host, and drug schedule dependence of gene responses and their upstream regulators. BMC Cancer 2016, 16, 623. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chen, C.; Li, A.; Jing, W.; Sun, P.; Huang, X.; Liu, Y.; Zhang, S.; Du, W.; Zhang, R.; et al. Immunostimulant hydrogel for the inhibition of malignant glioma relapse post-resection. Nat. Nanotechnol. 2021, 16, 538–548. [Google Scholar] [CrossRef]
- Galstyan, A.; Markman, J.L.; Shatalova, E.S.; Chiechi, A.; Korman, A.J.; Patil, R.; Klymyshyn, D.; Tourtellotte, W.G.; Israel, L.L.; Braubach, O.; et al. Blood-brain barrier permeable nano immunoconjugates induce local immune responses for glioma therapy. Nat. Commun. 2019, 10, 3850. [Google Scholar] [CrossRef]
- Wei, J.; Wu, D.; Shao, Y.; Guo, B.; Jiang, J.; Chen, J.; Zhang, J.; Meng, F.; Zhong, Z. ApoE-mediated systemic nanodelivery of granzyme B and CpG for enhanced glioma immunotherapy. J. Control. Release 2022, 347, 68–77. [Google Scholar] [CrossRef]
- Cacciotti, C.; Liu, K.X.; Haas-Kogan, D.A.; Warren, K.E. Reirradiation practices for children with diffuse intrinsic pontine glioma. Neurooncol. Pract. 2021, 8, 68–74. [Google Scholar] [CrossRef]
- Laspidea, V.; Puigdelloses, M.; Labiano, S.; Marrodan, L.; Garcia-Moure, M.; Zalacain, M.; Gonzalez-Huarriz, M.; Martinez-Velez, N.; Ausejo-Mauleon, I.; de la Nava, D.; et al. Exploiting 4-1BB immune checkpoint to enhance the efficacy of oncolytic virotherapy for diffuse intrinsic pontine gliomas. JCI Insight 2022, 7, e154812. [Google Scholar] [CrossRef]
- Gong, Y.; Ye, D.; Chien, C.Y.; Yue, Y.; Chen, H. Comparison of sonication patterns and microbubble administration strategies for focused ultrasound-mediated large-volume drug delivery. IEEE Trans. Biomed. Eng. 2022. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Alteration | Mutated Categories | Prevalence | Description of Mutation | Refs. |
|---|---|---|---|---|
| H3.1K27M | HIST1H3B; Missense mutation | 15% | H3 K27 trimethylation is ablated, generating an inhibition of the polycomb repressive complex 2 target genes, resulting in chromatin disaggregation and cellular aneuploidy. | [38,39,40,41] |
| H3.3K27M | H3F3A; Missense mutation | 70% | ||
| ACVR1 | R206H, R258G, G328E/V/W and G356D; Mutation | 25% | Encoding a serine/threonine kinase (ALK2) receptor with enhanced sensitivity to the ligand activin A, resulting in dysregulation of the BMP/SMAD pathway and increased tumor proliferation | [61,62,63] |
| TP53 | G245S, R175H, R248Q, R248W, R273C, R273H, S241F, V157F; Mutation | 75% | An increased co-occurrence with the H3 K27M mutation, increased DNA and protein instability resulting in decreased apoptosis | [64,65] |
| MYCN | Amplification | 8% | DNA hypermethylation and chromosomal rearrangement leading to aneuploidy | [66,67] |
| ATRX | Depletion | 10% | High co-occurrence with the H3 K27M mutation, causing destabilization of telomeres and altering gene expression in conjunction with the H3 K27M mutation | [45,68,69] |
| Receptor Tyrosine Kinase (RTK) | PDGFR, EGFR, FGFR; Amplification and mutation | 60% | It occurs frequently with the H3 K27M-mutantion | [45,70,71] |
| Cell-cycle regulatory genes controlling RB phosphorylation | CDK4, CDK6, CCND1, CCND2, CCND3; Focal amplifications | 30% | Inactivation of RB relieves negative regulation of the E2F transcription factor, permitting DNA synthesis and cell proliferation | [47,72] |
| Type of Therapy | Treatment | Delivery Method | Patients | Efficacy | Ref. or NCT Number |
|---|---|---|---|---|---|
| ICIs | Ipilimumab/nivolumab | Convection enhanced delivery | Patients (n = 2) | Patient 1: dead; Patient2: progressive disease | [12] |
| Pembrolizumab | Intravenous injection | Recruiting | Unknown | NCT02359565 | |
| Pembrolizumab | Intravenous injection | Patients (n = 5) | Shorter median PFS than expected | PBTC045 | |
| Pidilizumab | Intravenous injection | Active, not recruiting | Unknown | NCT01952769 | |
| CAR T-cell therapy | B7-H3-specific CAR T-cell locoregional therapy | Catheter into the ventricular system | Recruiting | Unknown | NCT04185038 |
| C7R-GD2 CAR T-cell therapy | Intravenous injection | Recruiting | Unknown | NCT04099797 | |
| GD2 CAR T-cell therapy | Intravenous injection and intraventricular delivery | Patients (n = 3) | Patient 1: OS, 13 months; Patient 2: OS, 26 months Patient 3: OS, 20 months. | NCT04196413 | |
| Oncolytic virus | Oncolytic virus: DNX-2401 | Intratumoral injection | Patients (n = 12) | Median OS: 17.8 months (range, 5.9 to 33.5) | NCT03178032 |
| Oncolytic virus: AloCELYVIR | Intravenous injection | Recruiting | Unknown | NCT04758533 | |
| Oncolytic virus: Wild-type Reovirus + Sargramostim | Intravenous injection | Active, not recruiting | Unknown | NCT02444546 | |
| Vaccines | Peptide vaccine: SurVaxM | Subcutaneous injection | Recruiting | Unknown | NCT04978727 |
| Peptide vaccine: H3K27M peptide vaccine | Subcutaneous injection | Not yet recruiting | Unknown | NCT04808245 | |
| DC vaccine: WT1 mRNA-loaded autologous monocyte-derived DCs | Intradermal vaccination | Recruiting | Unknown | NCT04911621 | |
| DC vaccine: TTRNA-DCs | Intradermal vaccination | Recruiting | Unknown | NCT03396575 |
| Treatment Category | Delivery Method | Model | Efficacy | Ref. |
|---|---|---|---|---|
| ICIs: antibody-PD-L1 | Focused ultrasound combined with microbubble-mediated BBB opening (FUS-BBBO) | Mice/no glioma cells | NA | [242] |
| Early ferroptotic cancer cells | In vivo prophylactic tumor vaccination (Injected subcutaneously) | Mice/murine fibrosarcoma MCA205 or glioma GL261 cells | Attenuated the appearance of tumors at the challenge site | [231] |
| Anti-TIM-3 antibody + stereotactic radiosurgery (SRS) | Injected intraperitoneally /Stereotactic radiation | Mice/murine glioma cell line GL261-luc2 | Long-term survival | [232] |
| Zika virus (ZIKV) | Stereotactic injection | Mice/GL261 or CT2A cells | Long-term survival | [233] |
| An interleukin-7-loaded oncolytic adenovirus (oAD-IL7) and a B7H3-targeted CAR-T | Stereotactic injection | Mice/GBM-Luc cells | Prolonged survival and reduced tumor burden. | [234] |
| EGFR-mediated photoimmunotherapy | Light exposure | Mice/GBM cells | Extensive tumor necrosis | [235] |
| Bortezomib and an oncolytic herpes simplex virus-1 (oHSV) | Intraperitoneally injected/intratumorally administrated | Mice/CAL27 cells | Prolonging survival enhance NK cell immunotherapy | [236] |
| An injectable hydrogel system | Intraperitoneally injected/intratumorally administrated | Mice/GL261 | Suppressed tumor recurrence and prolonged the survival | [237] |
| Nanoscale immunoconjugates covalently attached antibodies to CTLA-4 or PD-1 | Tail vein injection | Mice/GL261 | Longer survival | [238] |
| ApoE peptide-functionalized polymersomes encapsulating granzyme B (ApoE-PS-GrB) | Tail vein injection | Mice/LCPN cells | Delayed tumor progression and prolonged survival time | [239] |
| Oncolytic adenovirus: Delta-24-ACT | Intra-tumoral injection | BALB/c mice/murine NP53 and XFM cell lines | Long-term survivors that developed immunological memory against DIPG | [241] |
| Adenoviruses expressing thymidine kinase (TK) and fms-like tyrosine kinase 3 ligand (Flt3L) | Intra-tumoral injection | Mice/ brainstem glioma harboring mACVR1 | Recruitment of antitumor-specific T cells, and increased median survival | [17] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, G.; Qiu, Y.; Zhang, P.; Chen, Z.; Chen, S.; Huang, W.; Wang, B.; Yu, X.; Guo, D. Immunogenic Cell Death Enhances Immunotherapy of Diffuse Intrinsic Pontine Glioma: From Preclinical to Clinical Studies. Pharmaceutics 2022, 14, 1762. https://doi.org/10.3390/pharmaceutics14091762
Liu G, Qiu Y, Zhang P, Chen Z, Chen S, Huang W, Wang B, Yu X, Guo D. Immunogenic Cell Death Enhances Immunotherapy of Diffuse Intrinsic Pontine Glioma: From Preclinical to Clinical Studies. Pharmaceutics. 2022; 14(9):1762. https://doi.org/10.3390/pharmaceutics14091762
Chicago/Turabian StyleLiu, Guohao, Yanmei Qiu, Po Zhang, Zirong Chen, Sui Chen, Weida Huang, Baofeng Wang, Xingjiang Yu, and Dongsheng Guo. 2022. "Immunogenic Cell Death Enhances Immunotherapy of Diffuse Intrinsic Pontine Glioma: From Preclinical to Clinical Studies" Pharmaceutics 14, no. 9: 1762. https://doi.org/10.3390/pharmaceutics14091762